Back to Journals » OncoTargets and Therapy » Volume 12
NRAS Contributes to Retinoblastoma Progression Through SNHG16/miR-183-5p/NRAS Regulatory Network
Authors Sun G, Su G ![]() , Liu F, Han W
, Liu F, Han W
Received 25 September 2019
Accepted for publication 21 November 2019
Published 6 December 2019 Volume 2019:12 Pages 10703—10715
DOI https://doi.org/10.2147/OTT.S232470
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Geoffrey Pietersz
Guangli Sun,1 Gang Su,2 Fang Liu,1 Wenjie Han1
1Department of Ophthalmology, The First Affiliated Hospital of Zhengzhou University, Henan, People’s Republic of China; 2Department of Cardiovascular Surgery, The First Affiliated Hospital of Zhengzhou University, Henan, People’s Republic of China
Correspondence: Gang Su
Department of Cardiovascular Surgery, The First Affiliated Hospital of Zhengzhou University, No. 1, East Jianshe Road, Erqi District, Zhengzhou, Henan 450000, People’s Republic of China
Tel/Fax +86-371-66913114
Email [email protected]
Purpose: The oncogene of wild type neuroblastoma RAS viral oncogene homolog (NRAS) has been found to involve in the tumorigenesis of cancers. However, the role of NRAS in retinoblastoma (RB) progression remains largely unknown.
Methods: The expression levels of NRAS, miR-183-5p and small nucleolar RNA host gene 16 (SNHG16) were measured using quantitative real-time polymerase chain reaction assay or Western blot assay, respectively. Cell proliferation and apoptosis were analyzed with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay or flow cytometry, respectively. Transwell assay was used to determine cell migration and invasion abilities. The interaction between miR-183-5p and NRAS or SNHG16 was analyzed using bioinformatics analysis and dual-luciferase reporter assay.
Results: NRAS was elevated in RB tissues and cell lines, knockdown of NRAS could inhibit proliferation, migration and invasion but induced apoptosis in vitro and suppressed tumor growth in vivo. NRAS was confirmed to be a target of miR-183-5p and was negatively regulated by miR-183-5p in RB cells. Moreover, overexpressed NRAS reversed miR-183-5p mediated inhibition on RB cell progression. Besides that, SNHG16 directly interacted with miR-183-5p and reduced miR-183-5p expression in RB cells. The suppression of RB cell progression induced by SNHG16 silencing could be partially attenuated by the inhibition of miR-183-5p. Besides that, SNHG16 could regulate NRAS expression through competitively binding to miR-183-5p in RB cells.
Conclusion: NRAS functioned as an oncogene to contribute to RB progression by SNHG16/miR-183-5p/NRAS regulatory network, indicating a novel and promising therapeutic target for RB.
Keywords: SNHG16, miR-183-5p, NRAS, retinoblastoma, progression
Introduction
Retinoblastoma (RB) is the most common intraocular malignancy in infants and young children approximately representing 2.5% to 4% of all pediatric malignancies, and has a high risk of death.1 Due to the overwhelming majority of RB occurs in a very early stage of childhood,2 therefore, curing the disease as well as preserving vision are needed to be considered during the therapy of RB. Recent evidence exhibits that innovations in molecular genetics and targeted treatment have significantly improved the overall survival in children with RB.3
The neuroblastoma RAS viral oncogene homolog (NRAS) gene is an oncogene, which has the potential to result in normal cells to become cancerous cells when mutated.4 NRAS gene encodes a protein called NRAS protein, a member of RAS sarcoma (Ras) small GTPases, which is involved in the activation of both MAPK and PI3K/AKT/MYC signaling pathway5 and participates in the transmitting signals within cells by acting as GDP/GTP-regulated switches, thereby instructing cell growth, division, maturation, and taking on specialized functions.6,7 Mutational NRAS has been reported to play a critical role in the development of several types of cancer. These mutations cause constitutive activation of NRAS proteins, which directly induce uncontrollable growth and division of cells to affect malignant differentiation and proliferation.8 Recently, wild type RAS, including NRAS, Kirsten rat sarcoma viral oncogene homolog (KRAS), Harvey rat sarcoma viral oncogene homolog (HRAS), was also found to play important roles in both RAS mutant and RAS wild type cancers.9 For example, Peter Dietrich et al revealed that wild-type RAS isoform KRAS contributed to acquire resistance to RAF inhibitors in melanoma and hepatocellular carcinoma (HCC) and might be a promising therapeutic target.10,11 Besides that, they also indicated NRAS was a novel prognostic marker, affected proliferation and clonogenicity as well as enhanced sorafenib resistance in HCC.12 The retinoblastoma susceptibility gene Rb gene is a well-recognized tumor suppressor gene, which biallelic mutation is classically thought to be the initiate of RB.13 Previous evidence has indicated the genetic interaction between the Rb gene and NRAS in the development of tumor tumorigenesis.14,15 Additionally, Yang and Mei discovered that NRAS existed in RB samples, and might involve in the RB progression.16 However, the role of wild-type NRAS in RB progression remains largely unknown.
Up to date, thousands of non-coding RNAs (ncRNAs) have been identified within cells. ncRNAs are a class of RNAs lacking protein-coding function and have been observed to involve in diverse cellular processes, such as chromatin remodeling, transcription, post-transcriptional modifications and signal transduction, and play key roles in the development of many diseases, including cancers.17,18 Long non-coding RNAs (lncRNAs) and microRNAs (miRNAs) are two major types of ncRNAs. In RB, several lncRNAs and miRNAs have been investigated to function as oncogenic drivers or tumor suppressors to implicate in the development of RB.19–21
In this study, we focused the attention on the expression pattern of wild type NRAS in RB, explored the precise function as well as the regulatory network of wild type NRAS in the RB progression.
Materials and Methods
Tissue Samples
30 human RB tissues specimens and 30 normal retina tissues were obtained from The First Affiliated Hospital of Zhengzhou University. All clinical specimens did not receive any antitumor therapy before surgical resection and were immediately preserved in liquid nitrogen. This study was conducted based on the Declaration of Helsinki and was permitted by the research ethics committees of The First Affiliated Hospital of Zhengzhou University. All patients had provided the written informed consents.
Cell Culture and Transfection
Human retinal epithelial cell ARPE-19 and human RB cell lines Y-79, WERI-Rb-1 were purchased from American Type Culture Collection (APTT, Rockville, MD, USA). Human RB cell lines 67BR and SO-Rb50 were obtained from BioVector NTCC Inc. (Beijing, China). Four RB cell lines were maintained with RPMI 1640 medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, Gibco). ARPE-19 was cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) containing with 10% FBS, and 1% penicillin/streptomycin. All cells were incubated at 37°C with 5% CO2.
Small interfering RNA (siRNA) targeting NRAS (si-NRAS), siRNA targeting SNHG16 (si-SNHG16), siRNA negative control (si-NC), the short hairpin RNA (shRNA) targeting NRAS (sh-NRAS), shRNA scramble control (sh-NC), pcDNA (NC), pcDNA-NRAS overexpression vector (NRAS), pcDNA-SNHG16 overexpression vector (SNHG16), the miR-183-5p mimic, miR-183-5p inhibitor (miR-183-5p or anti-miR-183-5p) and the corresponding negative control (miR-NC or anti-miR-NC) were synthesized by Genepharma (Shanghai, China). Lipofectamine 3000 transfection reagent (Invitrogen, Carlsbad, CA, USA) was applied to transfect these oligonucleotides or vectors into Y-79 and SO-Rb50 cells.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Trizol reagent (Invitrogen) was utilized to isolate total RNAs from RB tissue and cells according to the standard introductions. For mRNA expression detection, the PrimeScript™ RT Reagent Kit (Takara, Dalian, China) was utilized to synthesize the complementary DNA (cDNA). For miRNA analysis, cDNA was generated using the miScript Reverse Transcription kit (Qiagen, Valencia, CA, USA). Then qPCR was performed with SYBR Premix Ex Taq (Qiagen). The fold change of miRNA or mRNA was calculated using 2−ΔΔCT methods and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or U6. All experiments were performed in triplicate. The special primers for miR-183-5p or U6 were purchased from GeneCopoeia. The primer sequences for SNHG16, NRAS and GAPDH are listed as followed: SNHG16 F, 5’-CAGAATGCCATGGTTTCCCC-3’, and R 5’-TGGCAAGAGACTTCCTGAGG-3’; NRAS F, 5’-ATGAGGACAGGCGAAGGCT-3’, and R, 5’-TGAGTCCCATCATCACTGCTG-3’;GAPDH F, 5’-ATTCCATGGCACCGTCAAGGCTGA-3’, and R, 5’-TTCTCCATGGTGGTGAAGACGCCA-3’.
Western Blot
RIPA lysis buffer (Beyotime, Shanghai, China) was used to isolate proteins from transfection cells, and then the content was quantified by a bicinchoninic acid (BCA) method following the recommendations of manufacturer. An equal amount of the extracts were separated by 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). After blocked with 5% non-fat milk for 1 h, the membrane was interacted with primary antibodies against NRAS and GAPDH, and then incubated with the HRP-conjugated secondary antibody. Finally, immunoreactive signals were visualized using Enhanced Chemiluminescence.
Cell Viability
Transfection cells were seeded into 96-well plates with triplicate repeat for each condition and cultured for overnight at 37°C. Then each well was incubated with 10 μL of MTT solution ((3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Sigma, St. Louis, MO, USA) for another 4 h. After removing the supernatants, 150 μL DMSO (Sigma) was added into each well. The absorbance value of each well was measured at 490 nm.
Cell Apoptosis
Annexin V-FITC/PI apoptosis detection kit (BD Biosciences, San Jose, CA, USA) was used to test cell apoptosis according to the standard protocol. In Brief, resuspended cells were stained with 10 μL Annexin V-FITC and PI in the dark for 15 min. Finally, the apoptotic cells were analyzed by FlowJo software. The experiment was repeated three times.
Transwell Assay
Briefly, transfection cells were seeded into the transwell upper chamber and the lower chamber was filled with 10% FBS medium. After incubation for 24 h, invaded cells were stained and counted with a microscope. For invasion assay, the upper transwell membrane was pre-coated with Matrigel (BD Biosciences). Experiments were performed three times.
Dual-Luciferase Reporter Assay
The WT or MUT SNHG16/NRAS containing miR-183-5p sequences were synthesized and cloned into the pmiR-RB-Report (Promega, Shanghai, China). Then, 10 μg constructed vector was co-transfected with miR-183-5p mimics or miR-NC for 48 h. Finally, a luciferase reporter assay kit (Promega) was applied to analyze the relative luciferase activity.
Xenograft Experiments in vivo
The study was permitted by the Animal Research Committee of The First Affiliated Hospital of Zhengzhou University and undertaken according to the guidelines of the National Animal Care and Ethics Institution. BALB/c nude mice (female, 4–6 weeks) were used to establish the xenograft assays. The nude mice were subcutaneously inoculated with Y-79 cells, which were infected with lentivirus containing sh-NRAS or sh-NC. After 7 days following the inoculation, the tumor size was examined every 5 days and the tumor volume was calculated. After 32 days, all mice were killed and tumor masses were weighted and harvested for further molecular analysis.
Statistical Analysis
All statistical data from triplicate repeat independently experiment was exhibited as a mean ± standard deviation (SD). Student’s t test or one-way analysis of variance (ANOVA) was performed to analyze the differences among two or multiple groups using GraphPad Prism 7 software (GraphPad Inc., San Diego, CA, USA). The correlation analysis was conducted using Pearson’s chi-square test. P < 0.05 indicated a statistical significance.
Results
The Expression of NRAS Is Up-Regulated in RB Tissues and Cell Lines
To investigate the possible role of NRAS in RB, the expression of NRAS was detected and we found that compared with the normal tissues and retinal epithelial cell ARPE-19, the expression of NRAS at mRNA and protein levels was significantly increased in RB tissues and cell lines including Y-79, SO-Rb50, WERI-Rb-1, and 67BR (Figure 1A–D).
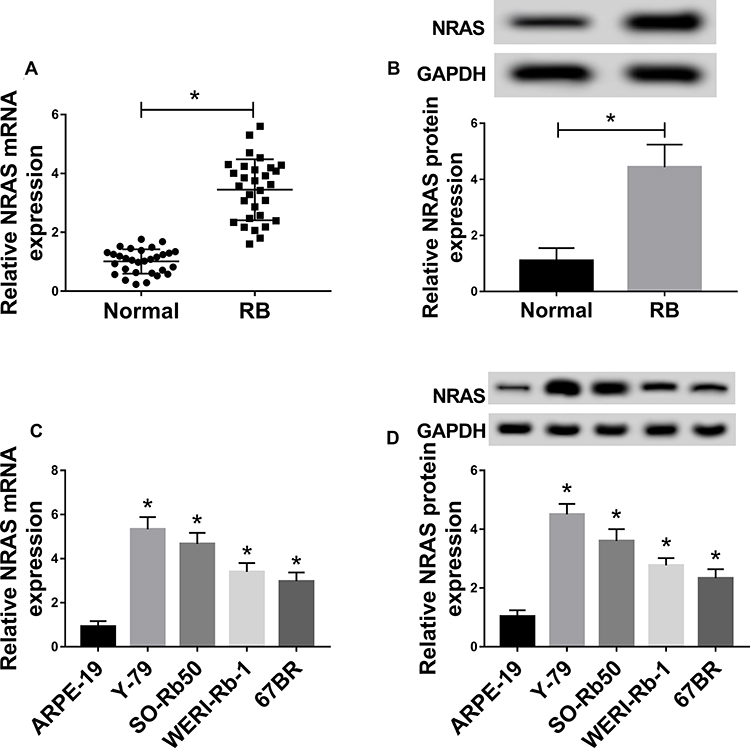 |
Figure 1 The expression of NRAS was up-regulated in RB tissues and cell lines. (A,B) The expression of NRAS in RB tissues and normal tissues was detected by qRT-PCR or Western blot. (C,D) qRT-PCR or Western blot was used to measure the expression of NRAS in retinal epithelial cell ARPE-19 and RB cell lines including Y-79, SO-Rb50, WERI-Rb-1, and 67BR. *P<0.05. |
NRAS Silence Suppresses Cell Progression in RB
To explore the potential biological functions of NRAS in RB cells, Y-79 and SO-Rb50 cells were transfected with si-NC or si-NRAS. After transfection, a significant reduction of NRAS expression was observed in the group transfected with si-NRAS (Figure 2A and B). Subsequently, MTT assay results showed that knockdown of NRAS suppressed the proliferation of Y-79 and SO-Rb50 cells (Figure 2C and D). Immediately, flow cytometry analysis indicated NRAS silence induced apoptosis in RB cells (Figure 2E). After that, transwell assay was performed and we found NRAS deletion obviously reduced the migration and invasion of Y-79 and SO-Rb50 cells (Figure 2F and G). Taken together, NRAS silence could suppress cell progression in RB cells.
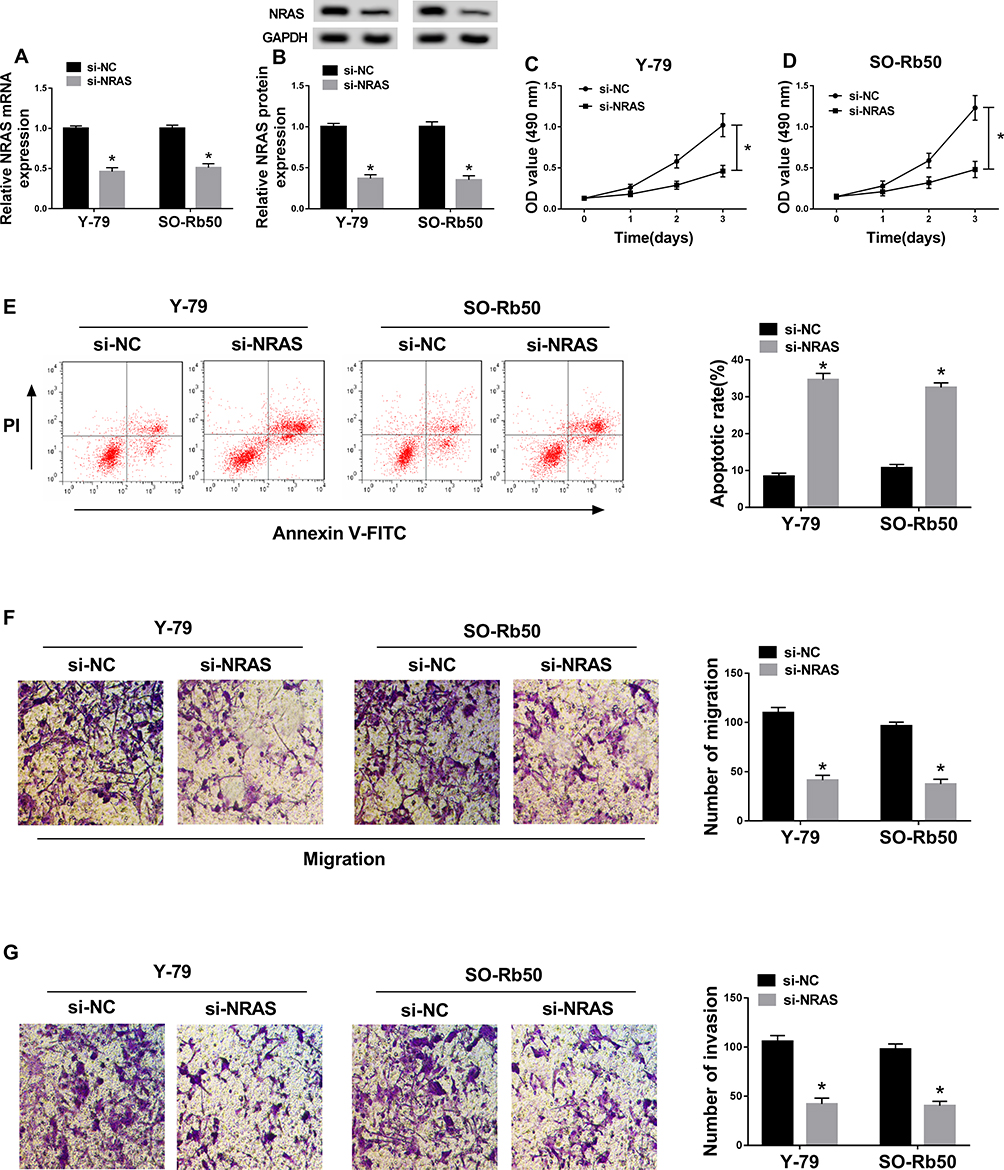 |
Figure 2 NRAS silence suppressed cell progression in RB cells. Y-79 and SO-Rb50 cells were transfected with si-NC or si-NRAS. (A,B) The expression of NEAS was detected in Y-79 and SO-Rb50 cells after transfection using qRT-PCR or Western blot, respectively. (C,D) MTT assay was used to analyze cell proliferation in RB. (E) Cell apoptosis was detected by flow cytometry analysis. (F,G) Transwell assay was applied to determine cell migration and invasion abilities in Y-79 and SO-Rb50 cells. *P<0.05. |
NRAS Is a Target of miR-183-5p in RB Cells
To further elucidate the regulatory network of NRAS in RB progression, the miRNAs target were predicted using TargetScan online database and miR-183-5p was predicted to contain the binding sites of NRAS (Figure 3A). To verify this prediction, a dual-luciferase reporter assay was conducted and results indicated miR-183-5p obviously reduced the luciferase activity of NRAS-WT reporter, while there was no markedly change in NRAS-MUT reporter in Y-79 and SO-Rb50 cells (Figure 3B and C). Besides that, miR-183-5p mimic transfection inhibited NRAS expression, while miR-183-5p inhibitor promoted NRAS expression (Figure 3D–G). In the meanwhile, miR-183-5p was demonstrated to be down-regulated in RB tissues and cell lines (Figure 3H and I) and co-expression analysis exhibited a markedly negative correlation between NRAS and miR-183-5p expression (Figure 3J), further suggesting the direct interaction between NRAS and miR-183-5p. Thus, these results showed that miR-183-5p directly interacted with NRAS and negatively regulated NRAS expression in RB cells.
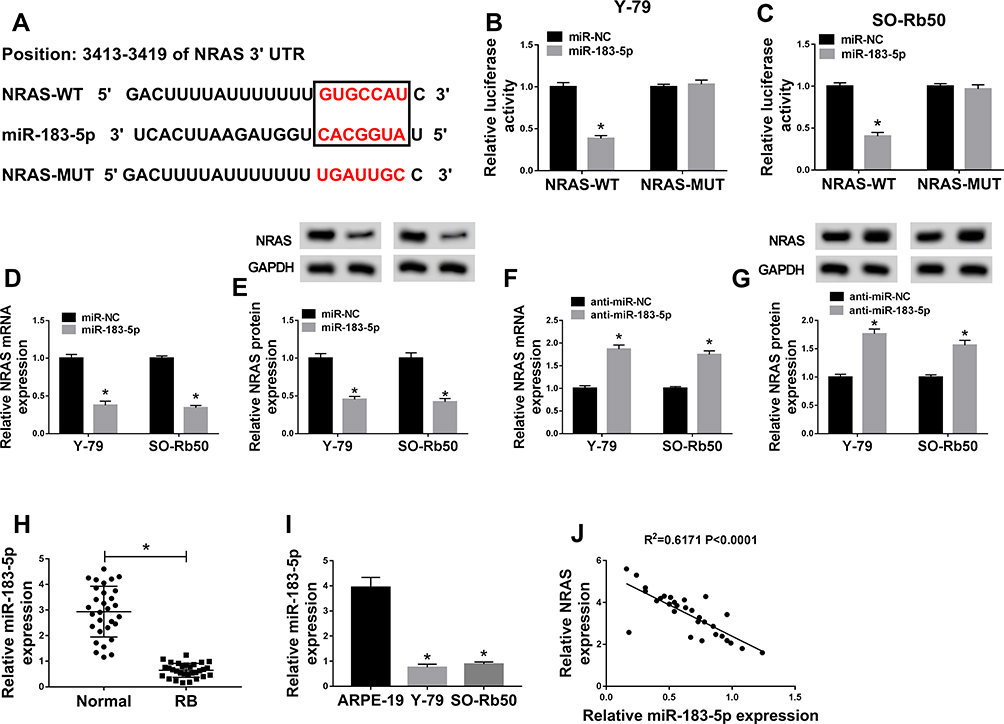 |
Figure 3 NRAS was a target of miR-183-5p in RB cells. (A) The binding sites between NRAS and miR-183-5p were predicted. (B,C) The Luciferase activity was analyzed in Y-79 and SO-Rb50 cells co-transfected with NRAS-WT or NRAS-MUT and miR-NC, miR-183-5p mimic. (D–G) The level of NRAS at mRNA and protein levels was detected in Y-79 and SO-Rb50 cells transfected with miR-183-5p or anti-miR-183-5p or their corresponding control (miR-NC or anti-miR-NC) using qRT-PCR or Western blot, respectively. (H) The expression of miR-183-5p was measured in RB tissues and normal tissues by qRT-PCR. (I) Analysis of miR-183-5p expression in ARPE-19, Y-79 and SO-Rb50 was performed using qRT-PCR. (J) The correlation between miR-183-5p and NRAS was analyzed by Pearson’s chi-square test. *P<0.05. |
Overexpressed NRAS Reverses miR-183-5p Mediated Inhibition on RB Progression
Y-79 and SO-Rb50 cells were transfected with miR-NC, miR-183-5p, miR-183-5p + pcDNA, miR-183-5p + NRAS to explore the regulatory function of miR-183-5p and NRAS in RB progression. Then we found a significant decrease of NRAS mRNA and protein expression in Y-79 and SO-Rb50 cells after miR-183-5p transfection, while these reductions could be restored by NRAS re-expression (Figure 4A–D), indicating the successful transfection. Subsequently, rescue assay was performed and the results showed that overexpressed NRAS could reverse miR-183-5p induced repression on proliferation (Figure 4E and F), migration and invasion (Figure 4I–L), enhancement on apoptosis (Figure 4G and H) in Y-79 and SO-Rb50 cells. In all, we knew that NRAS was involved in miR-183-5p mediated regulation on RB progression.
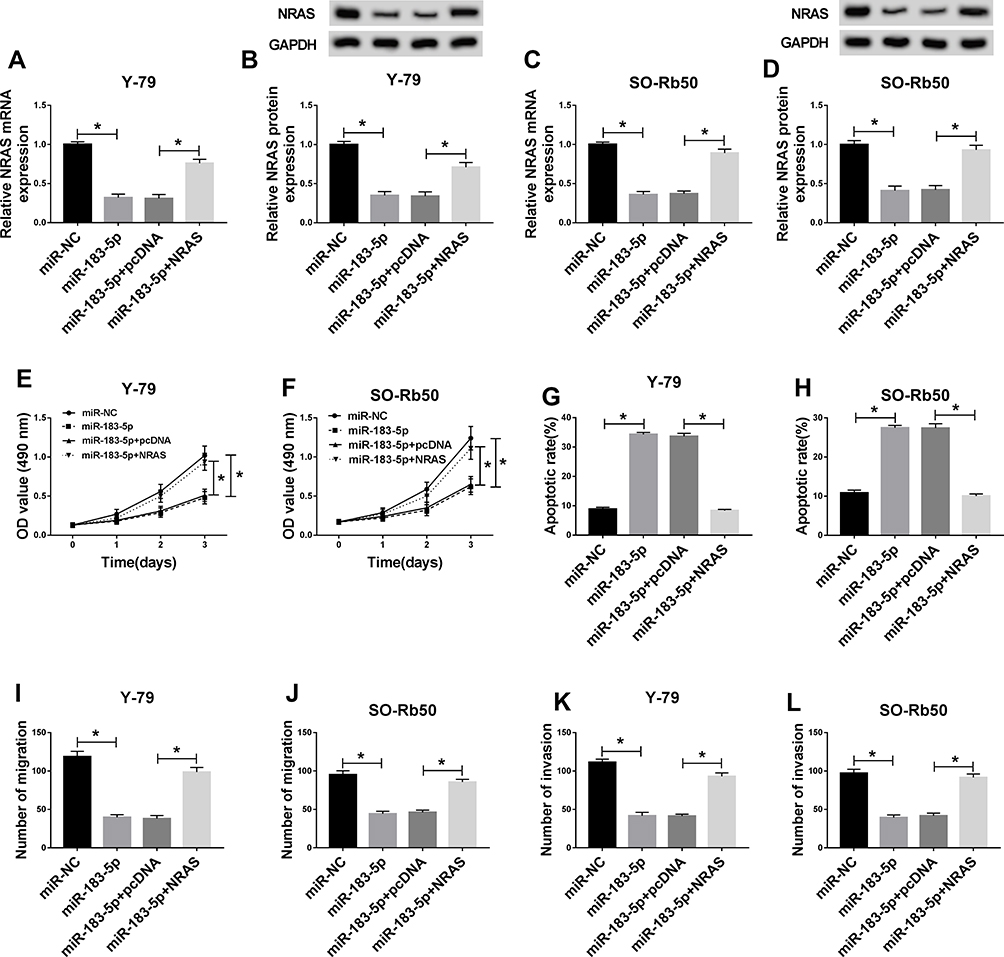 |
Figure 4 Overexpressed NRAS reversed miR-183-5p mediated inhibition on RB progression. Y-79 and SO-Rb50 cells were transfected with miR-NC, miR-183-5p, miR-183-5p + pcDNA, miR-183-5p + NRAS. (A–D) The expression of NRAS was detected in Y-79 and SO-Rb50 cells after transfection using qRT-PCR or Western blot, respectively. (E,F) Cell proliferation was determined by MTT assay. (G,H) Cell apoptosis was measured by flow cytometry. (I–L) The migration and invasion of Y-79 and SO-Rb50 cells were examined by Transwell assay. *P<0.05. |
SNHG16 Is a Sponge of miR-183-5p in RB Cells
Based on the prediction of Starbase2.0 program, SNHG16 was predicted to be a sponge of miR-183-5p with putative binding sites (Figure 5A). After that, a dual-luciferase reporter assay was performed to validate this hypothesis, and the results showed that overexpressed miR-183-5p obviously reduced the luciferase activity of SNHG16-WT reporter but not of SNHG16-MUT reporter in Y-79 and SO-Rb50 cells (Figure 5B and C). Moreover, the expression of miR-183-5p was inhibited by overexpressed SNHG16 but promoted by decreased SNHG16 in Y-79 and SO-Rb50 cells (Figure 5D and E). Afterwards, up-regulated SNHG16 expression was found in RB tissues and cell lines (Figure 5F and G), and a weakly negative correlation between SNHG16 and miR-183-5p was demonstrated in RB samples (Figure 5H). These data indicated that SNHG16 negatively interacted with miR-183-5p in RB cells.
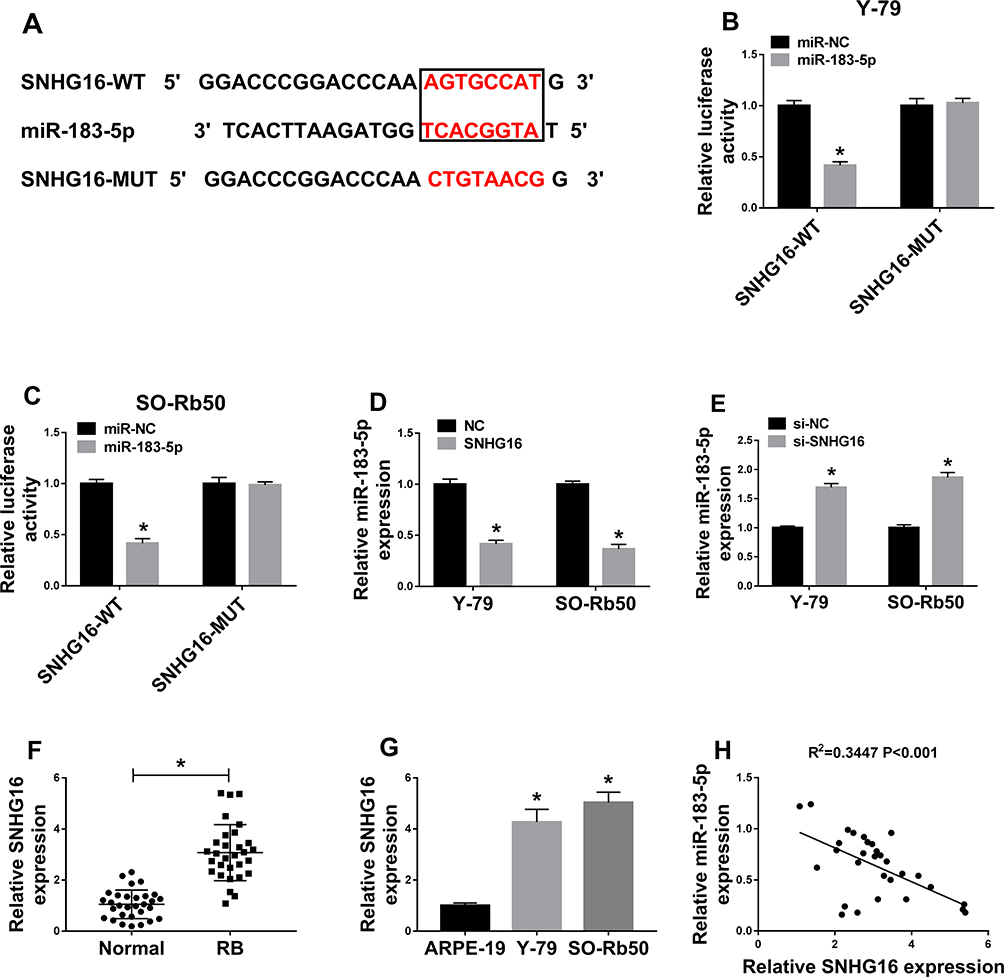 |
Figure 5 SNHG16 was a sponge of miR-183-5p in RB cells. (A) The binding sites between SNHG16 and miR-183-5p were listed. (B,C) The Luciferase activity was analyzed in Y-79 and SO-Rb50 cells co-transfected with SNHG16-WT or SNHG16-MUT and miR-NC, miR-183-5p mimic. (D,E) The level of SNHG16 was measured in Y-79 and SO-Rb50 cells transfected with SNHG16 or si-SNHG16 or their corresponding control (NC or si-NC) using qRT-PCR. (F,G) The expression of SNHG16 was measured in RB tissues and cell lines by qRT-PCR. (H) The correlation between miR-183-5p and SNHG16 was analyzed by Pearson’s chi-square test. *P<0.05. |
SNHG16 Deletion Suppresses Cell Progression by Directly Binding to miR-183-5p in RB Cells
To investigate the role and the underlying mechanism of SNHG16 in RB progression, Y-79 and SO-Rb50 cells were transfected with si-NC, si-SNHG16, si-SNHG16 + anti-miR-NC, si-SNHG16 + anti-miR-183-5p. Then we found that miR-183-5p expression was increased by si-SNHG16 transfection, which was reduced by miR-183-5p inhibitor transfection in Y-79 and SO-Rb50 cells (Figure 6A and B), indicating successful transfection. Afterwards, we demonstrated that SNHG16 silence could inhibit proliferation (Figure 6C and D), migration and invasion (Figure 6G–J), but induce apoptosis (Figure 6E and F) in Y-79 and SO-Rb50 cells, while these effects could be rescued by the inhibition of miR-183-5p. Therefore, we illustrated that SNHG16 deletion could suppress cell progression by directly binding to miR-183-5p in RB cells.
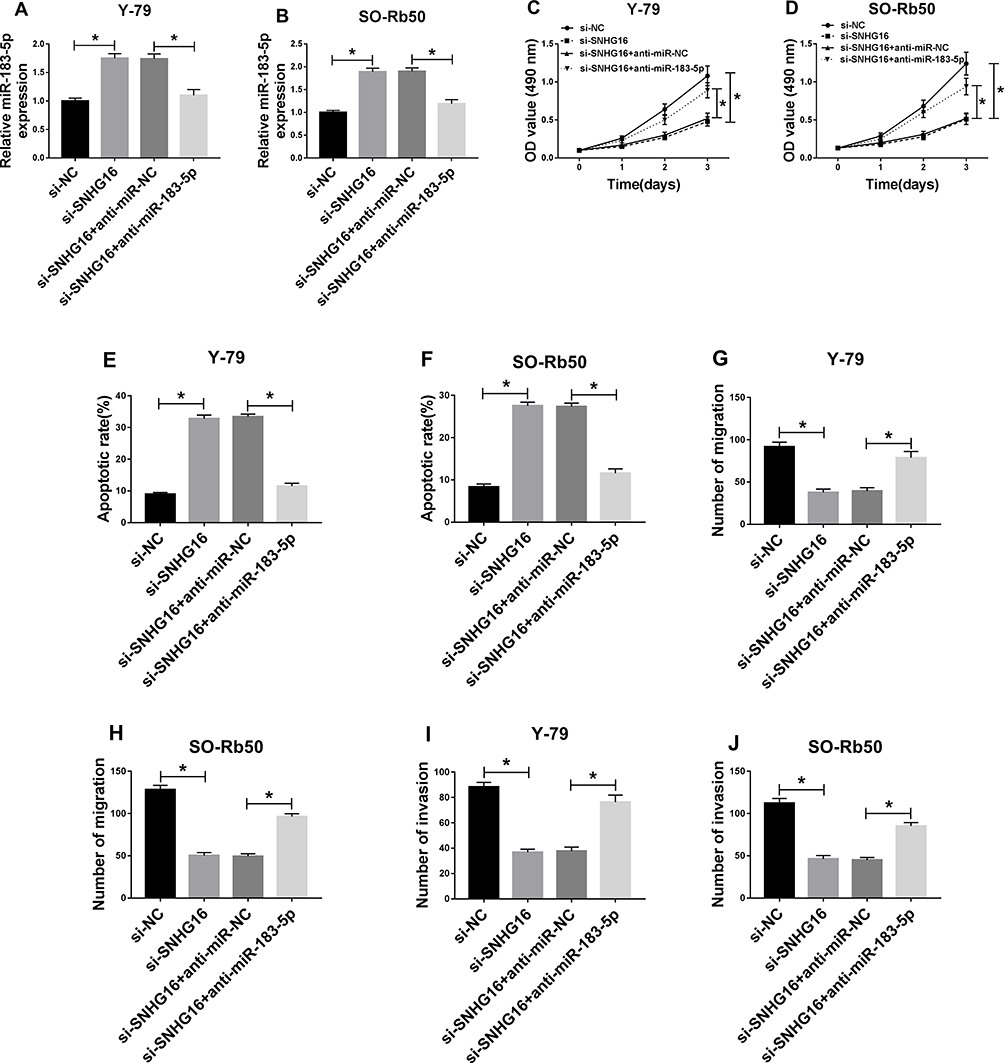 |
Figure 6 SNHG16 deletion suppressed cell progression by directly binding to miR-183-5p in RB cells. Y-79 and SO-Rb50 cells were transfected with si-NC, si-SNHG16, si-SNHG16 + anti-miR-NC, si-SNHG16 + anti-miR-183-5p. (A,B) The level of miR-183-5p was detected using qRT-PCR in Y-79 and SO-Rb50 cells. (C,D) Cell proliferation was analyzed by MTT assay. (E,F) Flow cytometry was used to test cell apoptosis. (G–J) Cell migration and invasion abilities were analyzed by Transwell assay. *P<0.05. |
SNHG16 Regulates NRAS Expression Through Competitively Binding to miR-183-5p in RB Cells
Based on the miR-183-5p/NRAS axis, we further wanted to investigate whether SNHG16 could regulate NRAS expression by binding to miR-183-5p. Then we discovered that the expression levels of NRAS, whether mRNA or protein, were reduced by SNHG16 knockdown, while these reductions could be restored by miR-183-5p inhibition in Y-79 and SO-Rb50 cells (Figure 7A–D). These data indicated that SNHG16 could regulate NRAS expression by serving as a sponge of miR-183-5p in RB cells.
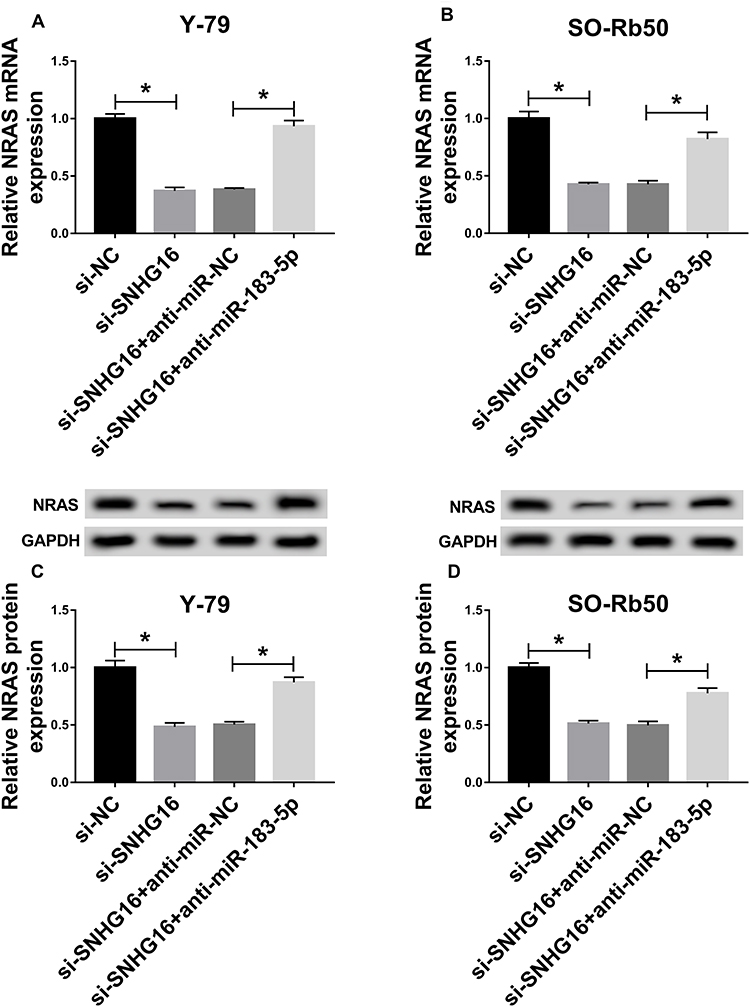 |
Figure 7 SNHG16 regulated NRAS expression through competing for miR-183-5p in RB cells. Y-79 and SO-Rb50 cells were transfected with si-NC, si-SNHG16, si-SNHG16 + anti-miR-NC, si-SNHG16 + anti-miR-183-5p. (A–D) The mRNA and protein expression levels of NRSA were detected in Y-79 and SO-Rb50 cells using qRT-PCR or Western blot, respectively. *P<0.05. |
Knockdown of NRAS Inhibits Tumor Growth in vivo
To further elucidate the carcinogenesis role of NRAS in vivo, xenograft models were established using Y-79 cells stably injected with lentivirus containing sh-NRAS or sh-NC. Subsequently, results showed NRAS deletion inhibited tumor growth in vivo, demonstrated by the suppression of the tumor volume and weight in the sh-NRAS group compared with these in the sh-NC group (Figure 8A and B). Moreover, molecular analysis was performed and results indicated knockdown of NRAS significantly reduced the levels of NRAS and SNHG16 but increased the level of miR-183-5p in the sh-NRAS group compared to sh-NC group (Figure 8C–F). These data suggested that knockdown of NRAS suppressed RB tumor growth in vivo.
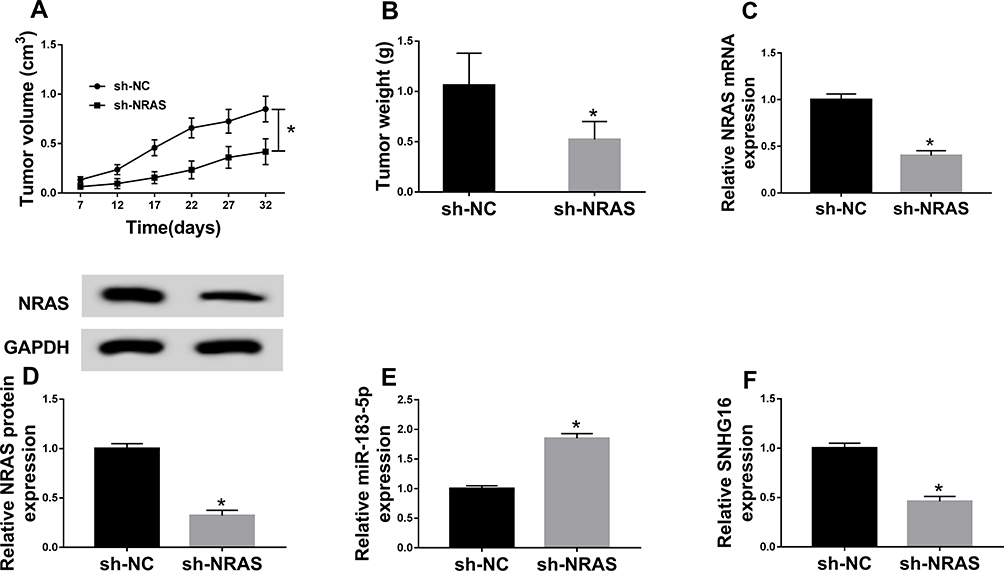 |
Figure 8 Knockdown of NRAS inhibits tumor growth in vivo. Xenograft models were established using Y-79 cells stably infected with lentivirus containing sh-NRAS or sh-NC. (A) Tumor volume was calculated every 5 days. (B) Tumor weight was analyzed in each group. (C–F) The levels of NRAS, miR-183-5p and SNHG16 were measured in two groups by qRT-PCR or Western blot, respectively. *P<0.05. |
Discussion
RAS proto-oncogenes, including NRAS, KRAS, and HRAS, have been identified to act as the most frequently activated oncogenes in diverse human cancers. NRAS proteins are key crossroads of cellular signal transduction that relay cell surface signals from outside of the cell to cell nucleus, thereby leading to the control of various cellular processes.22 Previous studies have indicated the alterations of NRAS in various cancers and the potential roles in the modulation of cell survival, proliferation, metastasis, drug resistance, tumor growth as well as angiogenesis in cancers.23,24 For instance, Wen et al indicated NRAS deletion promoted miR-26a mediated sensitivity of the gastric cancer cell to cisplatin.25 Liu et al showed NRAS acted as a target of miR-29a to involve in overexpressed miR-29a induced chemosensitivity of lung cancer cells to cisplatin.26 Additionally, miR-22 repressed cell proliferation and migration and sensitized the cell to paclitaxel by targeting NRAS in breast cancer.27 All the findings suggest NRAS deletion may be a promising therapeutic strategy in tumors. However, the functions of NRAS in RB cells are still widely unknown.
In this study, we focused attention on NRAS in RB cells, and we found compared with the normal tissues and retinal epithelial cell ARPE-19, NRAS was significantly elevated in RB tissues and cell lines, indicating the potential regulatory roles in RB. Subsequently, functional experiments were performed using small interfering RNA (siRNA) targeting NRAS (si-NRAS) and results showed that NRAS silencing mediated the inhibition of cell proliferation, migration and invasion, and the enhancement of cell apoptosis in RB. Besides that, in vivo experiments also showed knockdown of NRAS suppressed tumor growth. Therefore, we demonstrated that wild type NRAS functioned as an oncogene to promote RB progression.
Accumulating evidence has suggested that abnormal expression of miRNAs is strongly implicated in the progression of cancers by the regulation of target proteins, including RB. For example, miR-613 functioned as a tumor suppressor to repress RB malignancy by directly targeting E2F5.28 MiR-433 negatively modulated cell proliferation, metastasis, and cell cycle by targeting Notch1 and PAX6 inhibition.29 MiRNA-183 inhibited RB cell progression by negatively regulating LRP6 expression.30 In the current study, NRAS was predicted and confirmed to be a target of miR-183-5p and was negatively regulated by miR-183-5p in RB cells. MiR-183-5p was decreased in RB tissues and cell lines, and functional assay indicated that increased the expression of miR-183-5p significantly inhibited RB cell progression, while these effects could be reversed by highly expressed NRAS. Thus, we knew that NRAS was involved in miR-183-5p mediated inhibition on RB progression.
LncRNAs are also important components involved in cancer progression. Up to date, a variety of lncRNAs have been demonstrated to implicate in the regulation of RB. For example, MALAT1 stimulated the autophagy of RB cells via the miR-124/STX17 regulation network to regulate RB progression.31 AFAP1-AS1 served as an oncogene to modulate cell proliferation, metastasis and cell cycle to affect RB development.32 Among these lncRNAs, SNHG16 has also been reported to be increased and exert pro-tumor effects in numerous tumors, such as osteosarcoma, gastric cancer, and non-small lung cancer.33–35 Besides that, Xu et al recently exhibited that SNHG16 deletion inhibited RB cell progression in vitro as well as tumor growth in vivo.36 However, the complex molecular mechanisms underlying SNHG16 mediated regulation in RB remain unclear. In this work, an obvious increase of SNHG16 expression in RB tissues and cells was found, and SNHG16 silence inhibited RB cell proliferation, migration and invasion but induced apoptosis, thereby suppressing RB development, which was consistent with the previous study. After that, the potential targets of SNHG16 were predicted and we found SNHG16 directly bound to miR-183-5p and negatively modulated miR-183-5p level in RB cells. Immediately, rescue experiments were conducted and we demonstrated that SNHG16 deletion suppressed cell progression by directly binding to miR-183-5p in RB. Furthermore, expression analysis showed SNHG16 could regulate NRAS expression by binding to miR-183-5p in RB cells. Thus, SNHG16/miR-183-5p/NRAS regulatory network in RB development was identified.
In conclusion, our results showed that wild type NRAS was up-regulated in RB cells and knockdown of NRAS could suppress RB cell progression. Besides that, molecular mechanism exploration indicated that NRAS might mediate oncogenic effects in RB cells via SNHG16/miR-183-5p/NRAS regulatory network, which provided a novel therapeutic approach for RB.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Mcevoy JD, Dyer MA .Genetic and epigenetic discoveries in human retinoblastoma. Crit Rev Oncog.2015;20:217–225. doi:10.1615/CritRevOncog.v20.i3-4
2. Mendoza PR, Grossniklaus HE. The biology of retinoblastoma. Prog Mol Biol Transl Sci. 2015;134:503–516. doi:10.1016/bs.pmbts.2015.06.012
3. Grossniklaus HE. Retinoblastoma. Fifty years of progress. The LXXI Edward Jackson memorial lecture. Am J Ophthalmol. 2014;158:875–891. doi:10.1016/j.ajo.2014.07.025
4. Munoz-Couselo E, Adelantado EZ, Ortiz C, et al. NRAS-mutant melanoma: current challenges and future prospect. Onco Targets Ther. 2017;10:3941–3947. doi:10.2147/ott.s117121
5. Whitwam T, Vanbrocklin MW, Russo ME, et al. Differential oncogenic potential of activated RAS isoforms in melanocytes. Oncogene. 2007;26:4563–4570. doi:10.1038/sj.onc.1210239
6. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465. doi:10.1038/nrc1097
7. Charbel C, Fontaine RH, Malouf GG, et al. NRAS mutation is the sole recurrent somatic mutation in large congenital melanocytic nevi. J Invest Dermatol. 2014;134:1067–1074. doi:10.1038/jid.2013.429
8. Mandala M, Merelli B, Massi D. Nras in melanoma: targeting the undruggable target. Crit Rev Oncol Hematol. 2014;92:107–122. doi:10.1016/j.critrevonc.2014.05.005
9. Zhou B, Der CJ, Cox AD. The role of wild type RAS isoforms in cancer. Semin Cell Dev Biol. 2016;58:60–69. doi:10.1016/j.semcdb.2016.07.012
10. Dietrich P, Kuphal S, Spruss T, et al. Wild-type KRAS is a novel therapeutic target for melanoma contributing to primary and acquired resistance to BRAF inhibition. Oncogene. 2018;37:897–911. doi:10.1038/onc.2017.391
11. Dietrich P, Koch A, Fritz V, et al. Wild type Kirsten rat sarcoma is a novel microRNA-622-regulated therapeutic target for hepatocellular carcinoma and contributes to sorafenib resistance. Gut. 2018;67:1328–1341. doi:10.1136/gutjnl-2017-315402
12. Dietrich P, Gaza A, Wormser L, et al. Neuroblastoma RAS viral oncogene homolog (NRAS) is a novel prognostic marker and contributes to sorafenib resistance in hepatocellular carcinoma. Neoplasia (New York, NY). 2019;21:257–268. doi:10.1016/j.neo.2018.11.011
13. Soliman SE, Racher H, Zhang C, et al. Genetics and molecular diagnostics in retinoblastoma–an update. Asia Pac J Ophthalmol (Phila). 2017;6:197–207. doi:10.22608/apo.201711
14. Takahashi C, Ewen ME. Genetic interaction between Rb and N-ras: differentiation control and metastasis. Cancer Res. 2006;66:9345–9348. doi:10.1158/0008-5472.can-06-1250
15. Williams JP, Stewart T, Li B, et al. The retinoblastoma protein is required for Ras-induced oncogenic transformation. Mol Cell Biol. 2006;26:1170–1182. doi:10.1128/mcb.26.4.1170-1182.2006
16. Yang Y, Mei Q .miRNA signature identification of retinoblastoma and the correlations between differentially expressed miRNAs during retinoblastoma progression. Mol Vis.2015;21:1307–1317. doi:10.3390/molecules21101307
17. Anastasiadou E, Jacob LS, Slack FJ. Non-coding RNA networks in cancer. Nat Rev Cancer. 2018;18:5–18. doi:10.1038/nrc.2017.99
18. Li PF, Chen SC, Xia T, et al. Non-coding RNAs and gastric cancer. World J Gastroenterol. 2014;20:5411–5419. doi:10.3748/wjg.v20.i18.5411
19. Golabchi K, Soleimani-Jelodar R, Aghadoost N, et al. MicroRNAs in retinoblastoma: potential diagnostic and therapeutic biomarkers. J Cell Physiol. 2018;233:3016–3023. doi:10.1002/jcp.26070
20. Quan LJ, Wang WJ. FEZF1-AS1 functions as an oncogenic lncRNA in retinoblastoma. Biosci Rep. 2019. doi:10.1042/bsr20190754
21. Hu C, Liu S, Han M, et al. Knockdown of lncRNA XIST inhibits retinoblastoma progression by modulating the miR-124/STAT3 axis. Biomed Pharmacother. 2018;107:547–554. doi:10.1016/j.biopha.2018.08.020
22. Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012;120:3397–3406. doi:10.1182/blood-2012-05-378596
23. Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi:10.1126/science.296.5573.1655
24. Shaw LM. Identification of insulin receptor substrate 1 (IRS-1) and IRS-2 as signaling intermediates in the alpha6beta4 integrin-dependent activation of phosphoinositide 3-OH kinase and promotion of invasion. Mol Cell Biol. 2001;21:5082–5093. doi:10.1128/mcb.21.15.5082-5093.2001
25. Wen L, Cheng F, Zhou Y, et al. MiR-26a enhances the sensitivity of gastric cancer cells to cisplatin by targeting NRAS and E2F2. Saudi J Gastroenterol. 2015;21:313–319. doi:10.4103/1319-3767.166206
26. Liu X, Lv X, Yang Q, et al. MicroRNA-29a functions as a tumor suppressor and increases cisplatin sensitivity by targeting NRAS in lung cancer. Technol Cancer Res Treat. 2018;17:1533033818758905. doi:10.1177/1533033818758905
27. Song YK, Wang Y, Wen YY, et al. MicroRNA-22 suppresses breast cancer cell growth and increases paclitaxel sensitivity by targeting NRAS. Technol Cancer Res Treat. 2018;17:1533033818809997. doi:10.1177/1533033818809997
28. Zhang Y, Zhu X, Zhu X, et al. MiR-613 suppresses retinoblastoma cell proliferation, invasion, and tumor formation by targeting E2F5. Tumour Biol. 2017;39:1010428317691674. doi:10.1177/1010428317691674
29. Li X, Yang L, Shuai T, et al. MiR-433 inhibits retinoblastoma malignancy by suppressing Notch1 and PAX6 expression. Biomed Pharmacother. 2016;82:247–255. doi:10.1016/j.biopha.2016.05.003
30. Wang J, Wang X, Li Z, et al. MicroRNA-183 suppresses retinoblastoma cell growth, invasion and migration by targeting LRP6. FEBS J. 2014;281:1355–1365. doi:10.1111/febs.12659
31. Huang J, Yang Y, Fang F, et al. MALAT1 modulates the autophagy of retinoblastoma cell through miR-124-mediated stx17 regulation. J Cell Biochem. 2018;119:3853–3863. doi:10.1002/jcb.26464
32. Hao F, Mou Y, Zhang L, et al. LncRNA AFAP1-AS1 is a prognostic biomarker and serves as oncogenic role in retinoblastoma. Biosci Rep. 2018;38. doi:10.1042/bsr20180384
33. Wang X, Hu K, Chao Y, et al. LncRNA SNHG16 promotes proliferation, migration and invasion of osteosarcoma cells by targeting miR-1301/BCL9 axis. Biomed Pharmacother. 2019;114:108798. doi:10.1016/j.biopha.2019.108798
34. Han W, Du X, Liu M, et al. Increased expression of long non-coding RNA SNHG16 correlates with tumor progression and poor prognosis in non-small cell lung cancer. Int J Biol Macromol. 2019;121:270–278. doi:10.1016/j.ijbiomac.2018.10.004
35. Lian D, Amin B, Du D, et al. Enhanced expression of the long non-coding RNA SNHG16 contributes to gastric cancer progression and metastasis. Cancer Biomark. 2017;21:151–160. doi:10.3233/cbm-170462
36. Xu C, Hu C, Wang Y, et al. Long noncoding RNA SNHG16 promotes human retinoblastoma progression via sponging miR-140-5p. Biomed Pharmacother. 2019;117:109153. doi:10.1016/j.biopha.2019.109153
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.