Back to Journals » Neuropsychiatric Disease and Treatment » Volume 16
NR4A2 Exacerbates Cerebral Ischemic Brain Injury via Modulating microRNA-652/Mul1 Pathway
Received 1 June 2020
Accepted for publication 27 August 2020
Published 6 October 2020 Volume 2020:16 Pages 2285—2296
DOI https://doi.org/10.2147/NDT.S265601
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Jun Chen
Qiandong Liu, Qinglin Dong
Department of Emergency, People’s Hospital of Rizhao, Rizhao 276800, Shandong, People’s Republic of China
Correspondence: Qinglin Dong
Department of Emergency, People’s Hospital of Rizhao, No. 126, Tai’an Road, Donggang District, Rizhao 276800, Shandong, People’s Republic of China
Tel/Fax +86-0633-3365983
Email [email protected]
Background: Nuclear receptor subfamily group A member 2 (NR4A2), a transcription factor, was suggested to be involved in the pathogenesis of ischemic stroke. Nevertheless, the specific role of NR4A2 in ischemic brain injury has yet to be elucidated. Our aim was to probe the mechanisms behind the repression of microRNA (miRNA) expression resulting from NR4A2 regulation in ischemic brain injury.
Methods: A rat model with transient global cerebral ischemia (tGCI) was established, followed by HE staining and immunohistochemistry for verification. Subsequently, NR4A2 expression in rat brain tissues was detected by RT-qPCR, Western blot and immunohistochemistry. Then, PC12 cells were treated with NR4A2 alteration and subjected to oxygen-glucose deprivation (OGD) for cerebral ischemia simulation. Cell viability, apoptosis and cycle distribution were detected by CCK-8 and flow cytometry, respectively. miR-652 expression in rat brain tissues and cells was then detected by RT-qPCR, and then the targeting mRNAs of miR-652 were predicted through bioinformatic websites. Finally, the effect of miR-652 and mitochondrial E3 ubiquitin ligase 1 (Mul1) on the PC12 cell activity after OGD treatment was verified by rescue experiments.
Results: NR4A2 and Mul1 were expressed highly in brain tissues of rats with tGCI, while miR-652 was expressed poorly. NR4A2 inhibited the expression of miR-652 by transcription, thus blocking the inhibition of miR-652 on Mul1 to repress PC12 cell activity and promote apoptosis and G0/G1 cell cycle arrest.
Conclusion: The transcription factor NR4A2 mediates the expression of Mul1 through transcriptional repression of miR-652, thus promoting ischemic brain injury.
Keywords: ischemic brain injury, NR4A2, microRNA-652, Mul1, transient global cerebral ischemia
Introduction
Stroke, the second major cause of mortality and the leading cause of disability among adults worldwide, occurs as a consequence of disruption in the blood supply to the brain, culminating in death or everlasting neurological deficits.1 Most patients who went through stroke do not benefit from the current therapies including thrombolysis and thrombectomy due to limited therapeutic time window and secondary brain injury.2 Application of exogenous antioxidants has been demonstrated as beneficial against brain ischemic injury, but its efficacy remains unstable. Moreover, the detrimental connections between immune cells, glial cells, and matrix components during the pathology of stroke lead to inflammation that develops to fibrosis.3–5 Consequently, identifying novel targets and appropriate therapeutic windows are necessary to overcome ischemic brain injury.6
The nuclear receptor subfamily 4 group A member 2 (NR4A2), also termed as nuclear receptor-related 1, belongs to the family of orphan nuclear receptors with no recognized ligand and is acknowledged as a transcription factor with a distinctive physiological role.7 Notably, NR4A2 was found to be a player in recombinant tissue-type plasminogen activator treatment after ischemic stroke.8 More specifically, the deletion of NR4A2 sustained mitochondrial homeostasis and prolonged neuronal survival, thus alleviating cerebral ischemia-reperfusion injury.9 Interestingly, NR4A2 was validated to be a target for multiple microRNAs (miRNAs), such as miR-18310 and miR-381-3p.11 Nevertheless, no studies have examined the role of NR4A2, as a transcription factor in regulating the expression of miRNAs. miRNAs, single-stranded non-coding RNA with 20–24 nucleotides, have been widely reported to modulate various molecular processes, involving edema formation, apoptosis as well as inflammation after stroke.12,13 Moreover, miR-652 was observed to protect rats against injuries caused by cerebral ischemia/reperfusion through directly targeting NOX2.14 Our prediction using the bioinformatic tool JASPAR revealed that there is a binding relationship between miR-652 and NR4A2. Therefore, we postulated that NR4A2 may regulate the expression of miR-652 expression by functioning as a transcription factor. Furthermore, other three online websites displayed that mitochondrial E3 ubiquitin ligase 1 (Mul1) is a target of miR-652. A recent study reported that Mul1 enhanced brain injury by impairing mitochondrial dynamics in rats with ischemic stroke.15 In light of these findings, we hypothesized that NR4A2 knockdown has the potency to reduce ischemic brain injury by restoring miR-652 expression and then decreasing Mul1 expression. To validate this proposition, we investigated the roles of NR4A2 knockdown on the expression patterns of miR-652 and Mul1 and on proliferation and apoptosis of oxygen-glucose deprivation (OGD)-treated PC12 cells.
Materials and Methods
Animal Experiments
Twelve adult male Sprague-Dawley rats (weight 250–300 g and age 13–15 weeks) were from Dossy Biological Technology Co., Ltd. (Chengdu, Sichuan, China). All animals were maintained in conditions with a 12 h light/12 h dark cycle, 25 ± 2°C room temperature and 50-65% humidity. The study was implemented following the Guide for the Care and Use of Laboratory Animals proposed by the National Institutes of Health and approved by the Animal Care and Use Committee of People’s Hospital of Rizhao. A transient global cerebral ischemia (tGCI) model was developed in rats using bilateral common carotid artery occlusion in combination with arterial hypotension. Rats were anesthetized by an intraperitoneal injection of pentobarbital sodium at 50 mg/kg prior to the surgery. Then, the animals were fixed in the supine position, and the hair was removed from the chin and the neck. The right femoral artery and right jugular vein were intubated for physiological monitoring and blood extraction. An incision about 3 cm long was created in the middle of the neck, and the bilateral common carotid artery was separated and placed on a 4/0 nylon suture below each carotid artery. The blood was extracted through the right jugular vein (2–2.5 mL/100g) using a heparinized syringe to maintain the mean arterial pressure at 35–45 mmHg. Subsequently, bilateral common carotid artery occlusion was performed for 20 min using a microartery clamp. The clamp was then removed, and the blood collected was re-injected into the right jugular vein at a rate of 1.5 mL/min. Rats in the sham group underwent the same procedure except for bilateral common carotid artery occlusion and arterial hypotension. During the operation and 1.5 h after the operation, the rectal temperature was measured and maintained at about 37°C with an electric hot plate.
Neurologic Examinations
Neural dysfunction was evaluated after tGCI surgery according to the defect grading system introduced by Longa et al.16 A scale of 0 to 4 was used to assess tGCI behavioral and motor changes in rats after surgery. When the tail was suspended, the rats extended the two forelimbs to the floor, which indicated normal behavior (0). When the contralateral forelimb was on the lateral side, the rats scored 1 and without no other abnormalities were observed. The rats were placed on the ground to move freely and their behaviors were observed. Those spontaneously moving in all directions but circling to the left were scored 2, those falling to the left were scored 3, while those who were very weak and had a depressed level of consciousness were scored 4.
2,3,5-Triphenyltetrazolium Chloride (TTC) Staining
Rats were euthanized by an intraperitoneal injection of pentobarbital sodium at 120 mg/kg, and then the brain tissues were cut into coronal sections. The brain sections were stained in 2% TTC at 37°C for a period of 30 min and fixed with 4% paraformaldehyde for 24 h. The images were captured under an optical microscope (Olympus Optical Co., Ltd., Tokyo, Japan). The injured tissues were not stained, and the red area was normal tissues. The proportion (percentage) of the infarct area = the infarct area (light color area)/the total area of the transverse section × 100%.
Hematoxylin-Eosin (HE) Staining
Rats (n = 6) were euthanized at 72 h after tGCI, and the brain was immediately removed and fixed with 4% paraformaldehyde for 48 h. After fixation, brain samples were embedded in paraffin and cut along the coronary plane at 4 mm posterior to the coronary artery, and continuously cut into coronary sections (5 μm thickness) at the dorsal hippocampus for HE staining.
Immunohistochemical Staining
The brain sections were soaked in 0.24% H2O2 and treated with tris-buffered saline containing 3% normal goat serum and 0.2% Triton X-100. Subsequently, the sections were probed overnight with the primary antibodies against cytochrome C (Cyto C, #05-497, Sigma-Aldrich Chemical Company, St Louis, MO, USA) and NR4A2 (#N6431, 1:1000, Sigma-Aldrich) at 4°C and with biotinized secondary antibody. Finally, immunoreactivity was developed in diaminobenzidine. Section analysis was conducted by a confocal microscope.
Nissl Staining
Paraffinized brain samples were treated with Nissl staining solution (Sangon Biotech, Shanghai, China). After staining, purple staining was observed, which showed the basic neural structure of the brain. A large number of Nissl bodies were observed, indicating that neural cells have high protein synthesis ability. However, when the neural cells were damaged, the number of Nissl bodies decreased significantly. The number of stained cells from randomly selected regions was analyzed with Image-Pro Plus 6.0.
Reverse Transcription Quantitative Polymerase Chain Reaction (RT-qPCR)
Total RNA was isolated from rat brain tissues or OGD-treated cells using a Total RNA Kit (Takara, Shiga, Japan), and complementary DNA (cDNA) was synthesized using 5x Primescript reverse transcription reagents (Takara, Shiga, Japan) as described by the manufacturer. Real-time (RT)-PCR was carried out using SYBR Premix ExTaq™ (Tli RNaseH Plus, Takara, Japan) for quantitative analysis on a 7500 real-time PCR system (Applied Biosystems). The primers were: miR-652 forward: 5ʹ-GGCGCCACTAGGGTTGT-3ʹ; reverse: 5ʹ-GAACATGTCTGCGTATCTC-3ʹ; NR4A2 forward: 5ʹ-CCGCCGAAATCGTTGTCAGTAC-3ʹ; reverse: 5ʹ-TTCGGCTTCGAGGGTAAACGAC-3ʹ; Mul1 forward: 5ʹ-GTGTGTGCCTTATGCTGTCATCG-3ʹ, reverse: 5ʹ-GGTAGTTCGGTTCCACACCATC-3ʹ; U6 forward: 5ʹ-CTCGCTTCGGCAGCACAT-3ʹ, reverse: 5ʹ-TTTGCGTGTCATCCTTGCG-3ʹ; GAPDH forward: 5ʹ-GTCTCCTCTGACTTCAACAGCG-3ʹ, reverse: 5ʹ-ACCACCCTGTTGCTGTAGCCAA-3ʹ.
Western Blot Analysis
Total protein in brain tissues or cells was extracted using radioimmunoprecipitation assay lysis buffer (Beyotime Biotechnology Co., Ltd., Shanghai, China) supplemented with protease inhibitor cocktail (Beyotime) and phosphatase inhibitor cocktail (Beyotime). The protein concentration in the supernatant was quantified by a bicinchoninic acid assay protein assay kit (Sigma) after a 10-min centrifugation at 12,000 ×g. The proteins were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transblotted to polyvinylidene fluoride membranes (Roche Diagnostics, Indianapolis, IN, USA). The membrane was then incubated overnight with antibodies against Cyto C (1:1000, #05-497, Sigma), Bcl2 (ab196495, 1:2000, Abcam), and β-actin (1:10,000, Cell Signaling Technologies, Beverly, MA, USA) at 4°C overnight after 1 h of blocking in 5% skim milk or bovine serum albumin. Afterwards, the membrane was probed with a horseradish peroxidase-labeled secondary antibody (ab205718, 1:20,000, Abcam) at ambient temperature for 1 h. The intensity of the band signals was measured using enhanced chemiluminescence (Amersham Pharmacia, Piscataway, NJ, USA). Image J was used to perform densitometric analysis.
Cell Culture
PC12 cells (ATCC, Manassas, VA, USA) were grown in DMEM (Gibco, Carlsbad, CA, USA) containing 10% fetal bovine serum (Gibco), 100 kU/L penicillin and 100 mg/L streptomycin (Sigma). The cells were incubated in a humidified incubator with 95% air and 5% CO2 at 37°C. The culture medium was refreshed 3 times a week, and cells were passaged at 1:3.
As previously described,17 PC12 cells were exposed to OGD to mimic ischemic injury in vitro. The cells were washed three times with phosphate-buffered saline before OGD. Then, the NR4A2 or Mul1 overexpression vector, short hairpin RNAs (shRNAs) targeting NR4A2 or Mul1, or miR-652 mimic/inhibitor were delivered into cells for 24 h, whose successful was verified by RT-qPCR. Subsequently, the cells were cultured in a humidified modular hypoxia incubation chamber (Billups-Rothenberg, Del Mar, CA, USA) containing 5% CO2 and 95% N2 for a period of 6 h. The chamber was maintained in an incubator for at 37°C. Control cultures were kept in an incubator under atmospheric oxygen levels.
Cell Counting Kit-8 (CCK-8)
CCK-8 (Dojindo Molecular Technologies, Kumamoto, Japan) assay was applied according to the manufacturer’s instructions. PC12 were harvested and seeded in 96-well plates at 1 × 105 cells/well. After treatment, CCK-8 reagents were added, and the optical density (OD) value at 450 nm wavelength was evaluated using a microplate reader (Tecan, Mannedorf, Switzerland).
Flow Cytometry
Annexin V/fluorescein isothiocyanate (FITC) kits (BD Biosciences) were utilized as per the manufacturer’s protocols. Following OGD treatment and transfection, PC12 cells were cultured in a 6-well plate. The cells were incubated with 100 μL binding buffer and 5 μL FITC-labeled Annexin V (20 μg/mL) in darkness for 15 min at ambient temperature. Then, 5 μL propidium iodide (PI; 50 μg/mL) was added for a 5-min incubation in the dark. Thereafter, a 400-μL binding buffer was added and immediately loaded onto a FACScan for flow cytometric quantitative detection (within 1 h).
RNA Pull-Down
The 3ʹ-biotinylated miR-652 (TGCCTACTGAGCTGATATCAGT) or miR-652-mut (TGCCTACTCAGCTGATATCAGT) (20 nmol/L) were transfected into 293 cells. The pull-down assay was performed in biotin-coupled RNA complexes after incubation with streptavidin-coated magnetic beads (Life Technology, Carlsbad, CA, USA) for 24 h. Finally, the enrichment of NR4A2 was calculated based on RT-qPCR results.
Chromatin Immunoprecipitation (ChIP)
In short, cross-linked cell lysates were sonicated and immunoprecipitated. Purification of immunoprecipitation DNA was carried out with a universal DNA purification kit (TIANGEN Biotech Co., Ltd., Beijing, China). Then, 1% of each sample was separated as “input”. NR4A2 antibody (1:400) was applied as the primary antibody. Purified DNA was subjected to qPCR analysis.
Dual-Luciferase Reporter Assay
The targeting mRNAs of miR-652 were predicted through bioinformatic websites including TargetScan (http://www.targetscan.org/vert_72/), miRDB (http://mirdb.org/index.html) and RNA22 (https://cm.jefferson.edu/rna22/). The luciferase activity was detected using a Dual-Luciferase® Reporter Assay System (E1910, Promega Corporation, Madison, WI, USA).
Statistical Analysis
All data were processed by a SPSS 22.0 (IBM, Chicago, IL, USA) statistical software. The measurement data are displayed as a form of mean ± standard deviation (SD). Data were compared using unpaired t-test for comparison between two groups, and one-way or two-way analysis of variance (ANOVA) along with Tukey’s post hoc test for three or more groups. Differences were considered significant when p < 0.05.
Results
High Expression of NR4A2 is Identified in Brain Tissues of Rats with Ischemic Brain Injury
We first developed a cerebral ischemic rat model by tGCI surgery. It was observed that the neurological deficit was significantly increased (Figure 1A) in rats after tGCI surgery by neurological deficit score, and the cerebral infarct size in the rats after surgery became significantly larger (Figure 1B) by TTC staining. Subsequently, we used HE staining to detect the level of pathological injury in brain tissues and found that brain tissues from rats after tGCI showed cell morphological changes with blurred cell boundaries and increased pathological levels (Figure 1C). Moreover, immunohistochemical staining of Cyto C in brain tissues found that after tGCI treatment, the expression of Cyto C was increased significantly (Figure 1D), along with a significant decline in the number of Nissl bodies in the hippocampus of rat brain tissues after tGCI (Figure 1E). Then, we detected the mRNA expression of NR4A2 by RT-qPCR, which revealed that NR4A2 mRNA expression in rat brain tissues was significantly enhanced after tGCI treatment (Figure 1F). Consistently, the Western blot and immunohistochemical staining displayed a same trend (Figure 1GH).
 |
Figure 1 NR4A2 is upregulated by ischemic brain injury. (A) Rat neurological scores in tGCI and sham-operated rats; (B) detection of infarct area in rat brain tissues by TTC staining; (C) pathological structure of rat hippocampus by HE staining; (D) immunohistochemical staining of Cyto C in brain tissues of rats; (E) Nissl staining positive cell ratio in hippocampus of rats; (F) the mRNA expression of NR4A2 in rat brain tissues by RT-qPCR; (G) the protein expression of NR4A2 in rat brain tissues by Western blot; (H) immunohistochemical staining of NR4A2 in rat brain tissues. All values were expressed as means ± SD (n = 6). Comparison was performed using unpaired t-test. #p < 0.05, ##p < 0.01 vs sham-operated rats. |
Silencing of NR4A2 Increases PC12 Cell Activity After OGD Treatment
Thus, PC12 cells were exposed under OGD to simulate ischemic brain injury in vitro. We first applied RT-qPCR and Western blot to detect the expression of NR4A2 in PC12 cells after OGD treatment, and we noted that the expression of MR4A2 was significantly enhanced after OGD treatment (Figure 2A and B). We then overexpressed or knocked down NR4A2 expression in PC12 cells. The success transfection was confirmed by RT-qPCR and Western blot (Figure 2C and D). The cell viability was examined by CCK-8 assays. After overexpression of NR4A2, cell activity was significantly inhibited, while reducing NR4A2 expression in PC12 cells further promoted cell activity (Figure 2E). We used flow cytometry to detect apoptosis levels and monitored that NR4A2 significantly promoted PC12 apoptosis as well as G0/G1 cell cycle arrest (Figure 2F and G).
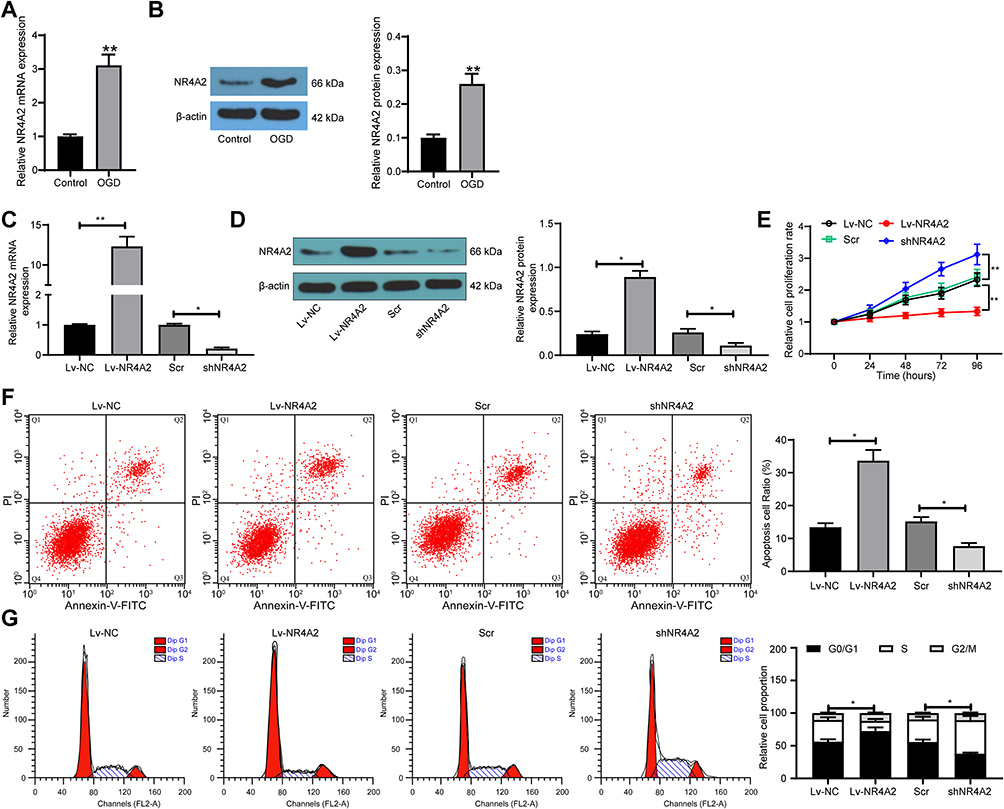 |
Figure 2 Silencing of NR4A2 increases PC12 cell viability after OGD treatment. (A) NR4A2 in PC12 cells exposed to OGD by RT-qPCR; (B) the protein expression of NR4A2 in PC12 cells exposed to OGD by Western blot. NR4A2 was overexpressed or silenced in PC12 cells; (C) the mRNA expression of NR4A2 in PC12 cells after transfection by RT-qPCR; (D) the protein expression of NR4A2 in PC12 cells after transfection by Western blot; (E) PC12 cell proliferation determined by CCK-8 assays; (F) apoptosis level of PC12 cells by flow cytometry; (G) cell cycle distribution of PC12 cells detected by flow cytometry. All values were expressed as means ± SD from three independent experiments. Unpaired t-test (panel (A and B), one-way (panel (C, D and F)) or two-way ANOVA (panel (E and G) followed by Tukey’s post hoc test was utilized for comparison. *p < 0.05, **p < 0.01 vs PC12 cells transfected with Lv-NC or Scr. |
NR4A2 Transcriptionally Represses miR-652 Expression
Subsequently, RT-qPCR assay indicated that miR-652 was significantly lower in rat brain tissues after tGCI surgery relative to sham-operated rat brain tissues. In PC12 cells, overexpression of NR4A2 significantly inhibited miR-652 expression, whereas inhibition of NR4A2 expression promoted miR-652 expression (Figure 3A). We then predicted the binding relationship between NR4A2 and miR-652 promoters through the JASPAR website (Figure 3B and C). Afterwards, ChIP-qPCR validated that after overexpression of NR4A2, the enriched miR-652 fragments were significantly increased, while after NR4A2 knockdown, the miR-652 fragments enriched by NR4A2 antibodies were significantly reduced (Figure 3D).
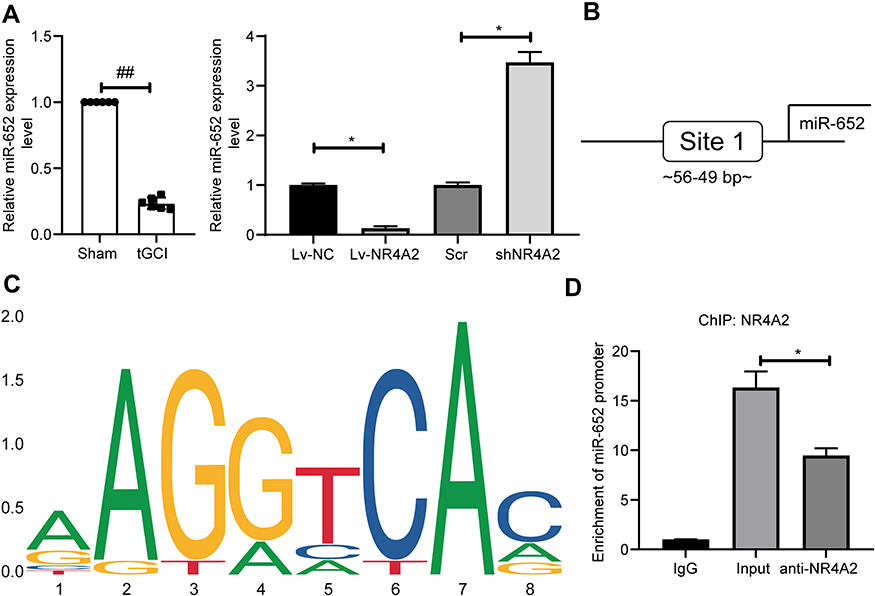 |
Figure 3 NR4A2 regulates miR-652 expression through transcriptional inhibition. (A) miR-652 expression in rat brain tissues and PC12 cells measured by RT-qPCR; (B) the binding sites between NR4A2 and miR-652 promoter predicted by the JASPAR website; (C) conserved binding sequences between NR4A2 and miR-652; (D) ChIP-qPCR detection of NR4A2 binding to miR-652 promoters. All values were expressed as means ± SD from three independent experiments. Unpaired t-test (panel (A) left) or one-way (panel (A) right and (D) followed by Tukey’s post hoc test was utilized for comparison. ##p < 0.01 vs sham-operated rats; *p < 0.05 vs PC12 cells transfected with Lv-NC or Scr or the IgG group. |
miR-652 Inhibitor Blocks the Protective Effect of NR4A2 Silencing on PC12 Cells After OGD Treatment
We subsequently co-transfected PC12 cells with NR4A2 + miR-652 mimic or shNR4A2 + miR-652 inhibitor. The cells were successfully established according to the RT-qPCR detection (Figure 4A). CCK-8 experimental results demonstrated that further overexpression of miR-652 in cells overexpressing NR4A2 significantly promoted cell activity, while further knockdown of miR-652 expression in cells with low expression of NR4A2 inhibited cell activity (Figure 4B). Moreover, we detected cell cycle and apoptosis by flow cytometric analysis and observed that overexpression of miR-652 significantly alleviated G0/G1 cell cycle arrest caused by NR4A2 overexpression and inhibited cell apoptosis (Figure 4C and D). We further used Western blot to determine the expression of Cyto C and Bcl-2 in PC12 cells, and found that overexpression of miR-652 suppressed the expression of Cyto C in cells and promoted the Bcl-2 expression, but miR-652 knockdown resulted in the opposite effects (Figure 4E).
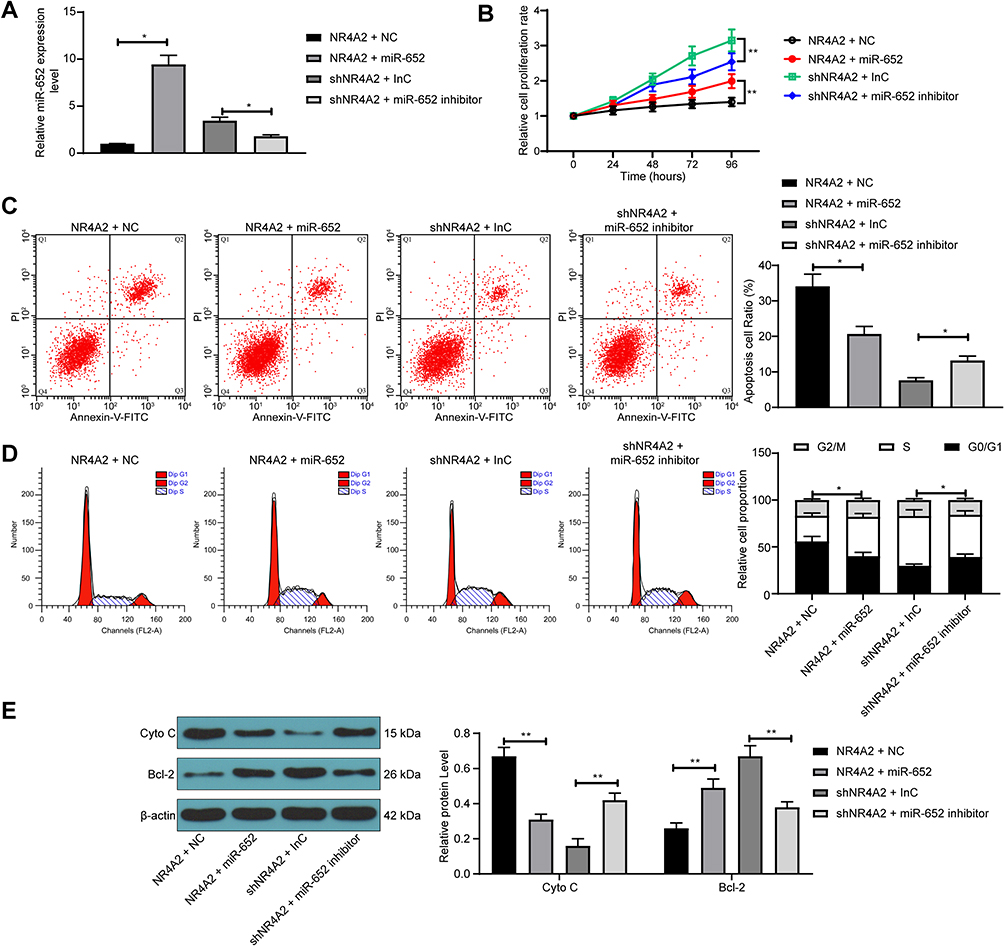 |
Figure 4 miR-652 downregulation reverses the protective effect of NR4A2 shRNA on PC12 cells after OGD treatment. PC12 cells were co-transfected with NR4A2 + miR-652 or shNR4A2 + miR-652 inhibitor. (A) miR-652 expression in PC12 cells after co-transfection measured by RT-qPCR; (B) PC12 cell proliferation determined by CCK-8 assays; (C) apoptosis level of PC12 cells by flow cytometry; (D) cell cycle distribution of PC12 cells detected by flow cytometry; (E) the protein expression of Cyto C and Bcl-2 in PC12 cells evaluated by Western blot. All values were expressed as means ± SD from three independent experiments. One-way (panel (A and C)) or two-way ANOVA (panel (B, D and E)) followed by Tukey’s post hoc test was applied for comparison. *p < 0.05, **p < 0.01 vs PC12 cells transfected with NR4A2 + NC or shNR4A2 + InC. |
Mul1 is a Target Gene of miR-652
To further validate the role of miR-652 in ischemic brain injury, we predicted the targeting mRNAs of miR-652 by bioinformatic websites TargetScan (http://www.targetscan.org/vert_72/), miRDB (http://mirdb.org/index.html), and RNA22 (https://cm.jefferson.edu/rna22/). The Venn map plotted indicated that only Mul1 was in the intersection (Figure 5A). Thus, we examined the binding relationship between miR-652 and Mul1 by dual-luciferase assays. The luciferase activity in 293T cells transfected with Mul1-MT was significantly inhibited by miR-652 mimic (Figure 5B). Moreover, RNA pull-down results illustrated that biotin-labeled miR-652 enriched significantly more Mul1 fragments (Figure 5C), which indicated the binding relationship between miR-652 and Mul1. Subsequently, we found that after tGCI treatment, the expression of Mul1 in rat brain tissues was significantly increased (Figure 5D). In addition, in PC12 cells, overexpression of NR4A2 notably increased Mul1 expression, while inhibition of NR4A2 remarkably repressed its expression. Yet further overexpression or inhibition of miR-652 in PC12 cells significantly reversed the role of Lv-NR4A2 or sh-NR4A2 (Figure 5E and F).
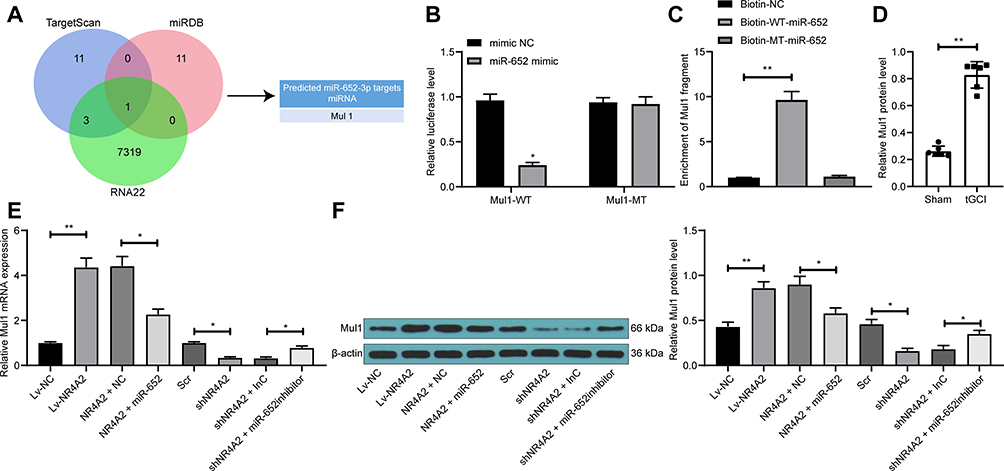 |
Figure 5 miR-652 directly targets Mul1. (A) Targeting mRNAs of miR-652 predicted through bioinformatic websites TargetScan (http://www.targetscan.org/vert_72/), miRDB (http://mirdb.org/index.html) and RNA22 (https://cm.jefferson.edu/rna22/); (B) relative luciferase activity of cells co-transfected with Mul1-MT or Mul1-WT with miR-652 mimic or mimic NC; (C) biotin-labeled miR-652 enriched Mul1 fragment detected by RNA pull-down; (D) Mul1 protein expression in rat brain tissues tested by Western blot; (E) Mul1 mRNA expression in PC12 cells after transfection evaluated by RT-qPCR; (F) Mul1 protein expression in PC12 cells after transfection tested by Western blot. All values were expressed as means ± SD from three independent experiments. One-way (panel (C, D and E)) or two-way ANOVA (panel (B) followed by Tukey’s post hoc test was applied for comparison. *p < 0.05, **p < 0.01 vs cells transfected with mimic NC, Biotin-NC, Lv-NC, NR4A2 + NC, Scr, shNR4A2 + InC or sham-operated rats. |
Overexpression of Mul1 Promotes Apoptosis of PC12 Cells After OGD Treatment
To determine the role of Mul1 in ischemic brain injury, we knocked down Mul1 expression in cells that overexpressing NR4A2, and overexpressed Mul1 expression in cells that under-expressing NR4A2. The Western blot results indicated the successful delivery (Figure 6A). As shown in Figure 6B-E, overexpression of Mul1 in the presence of shNR4A2 suppressed the cell proliferation of PC12 cells after OGD treatment, promoted apoptosis and cell cycle arrest in the G0/G1 phase, and significantly promoted Cyto C expression and inhibited Bcl-2 expression, whereas the opposite experimental results were presented due to knockdown of Mul1 and overexpression of NR4A2.
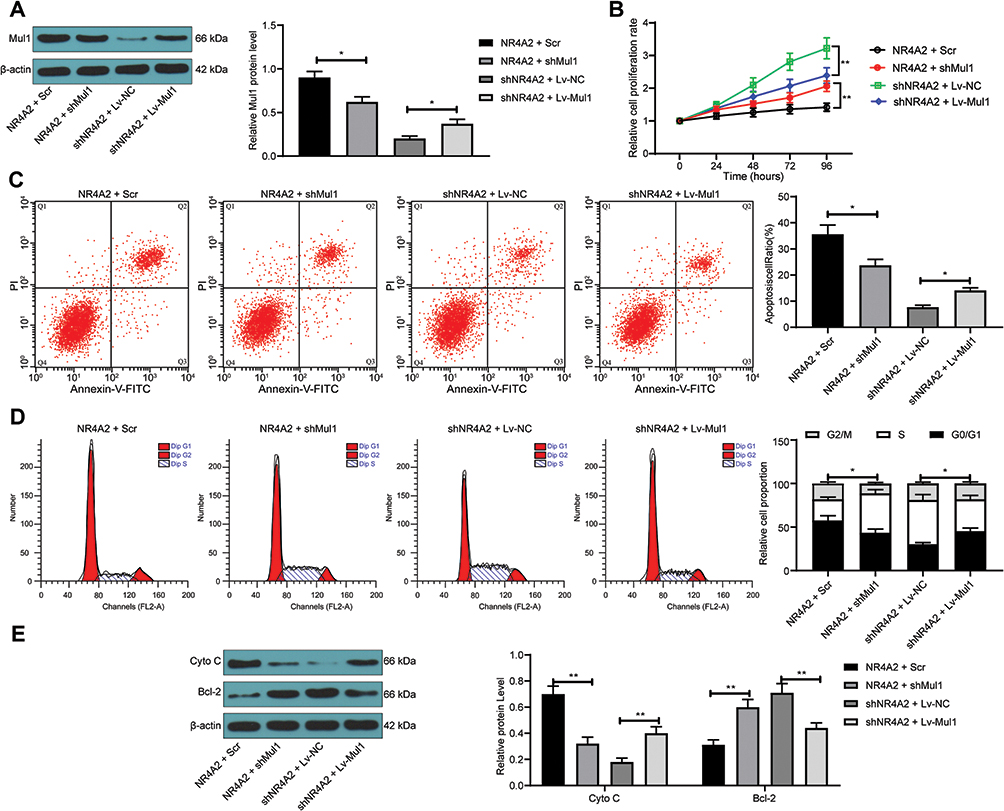 |
Figure 6 Overexpression of Mul1 promotes apoptosis of PC12 cells after OGD treatment. PC12 cells were co-transfected with NR4A2 + shMul1 or shNR4A2 + Lv-Mul1. (A) Mul1 protein expression in PC12 cells after transfection tested by Western blot; (B) PC12 cell proliferation determined by CCK-8 assays; (C) apoptosis level of PC12 cells by flow cytometry; (D) cell cycle distribution of PC12 cells detected by flow cytometry; (E) the protein expression of Cyto C and Bcl-2 in PC12 cells evaluated by Western blot. All values were expressed as means ± SD from three independent experiments. One-way (panel (A and C) or two-way ANOVA (panel (B, D and E)) followed by Tukey’s post hoc test was applied for comparison. *p < 0.05, **p < 0.01 vs PC12 cells transfected with NR4A2 + Scr or shNR4A2 + Lv-NC. |
Discussion
Stroke represents one of the leading causes of fatalities and physical disability in a world range, and non-coding RNAs are endogenous molecules that function importantly in the pathophysiology and retrieval events following ischemic stroke.18 Recently, a transcription factor, translationally controlled tumor protein was found to attenuate neurobehavior and oxidative stress injury in rats with cerebral palsy by elevating miR-200a expression to restrain the expression of myelin transcription factor 1-like.19 Accordingly, we postulated that NR4A2, another transcription factor modulated the expression of miR-652 to mediate the expression of Mul1, a putative target of miR-652 indirectly. In this article, we conclude that NR4A2 is a transcription factor-mediated OGD-induced injury in vitro, which was highly expressed in rats with ischemic brain injury, and knockdown of NR4A2 enhanced the PC12 cell viability. Then, we identified that miR-652 directly interacted with and was negatively regulated by NR4A2, and miR-652 was poorly expressed in rats with ischemic brain injury. Further data indicated Mul1 as a target of miR-652.
Initially, our present study observed that miR-652 expression was decreased, and mRNA and protein expression of NR4A2 was increased in the brain tissues of rats with tGCI. Moreover, NR4A2 modulated miR-652 expression through transcriptional inhibition, with the prediction of bioinformatic website JASPAR and verification of ChIP-qPCR assays. Silencing of NR4A2 promoted proliferation and inhibited apoptosis of PC12 cells, which were reversed by miR-652 inhibitor. Previously, Beard et al reported that NR4A2 was upregulated in cancers and facilitated cell proliferation, migration as well as chemoresistance.20 Additionally, silencing of NR4A2 contributed to an enhancement in hepatic stellate cell proliferation and a decline in cell percentage in S phase.21,22 As for its association with miRNAs, NR4A2 was downregulated by miR-204, miR-93 and miR-302d in cultured dopaminergic neurons.23 Besides, NR4A2 was found to be conversely regulated by miR-137 to expedite endothelial progenitor cell proliferation in mice with cerebral ischemic stroke.24 After identifying the interacting relationship between NR4A2 and miR-652, we carried out functional rescue experiments and found that upregulated miR-652 in PC12 cells overexpressing NR4A2 resulted in lower Cyto C and higher Bcl-2 expression than cells overexpressing NR4A2 and NC. The mitochondrial pathway is driven by the secretion of pro-apoptotic intramitochondrial factors, including Cyto C, while Bcl-2 is the major protein that represses cell apoptosis.25 Similarly, miR-652-3p was reduced in the cardiac tissues of mice with I/R injury, and miR-652-3p served as a negative modulator and suppressed mitochondrial fission and apoptosis in cardiomyocytes.26
In addition, we found that Mul1, overexpressed in the brain tissues of rats with tGCI, was a possible target of miR-652. Besides, Mul1 was positively regulated by NR4A2, while negatively modulated by miR-652 in PC12 cells stimulated with OGD. Transient mitochondrial hyperfusion is notable in cells under exposure to starvation, hypoxia and toxic insults, while the Mul1 may function in the maintenance and improvement of neuronal mitochondrial integrity under these conditions.27 Also, Mul1 mutation causes prolonged rhythm of locomotor activity, which may elucidate the mechanisms of action underlying some neurological and behavioral symptoms in Parkinson’s disease.28 Moreover, Mul1 upregulation in heart was observed to contribute to ischemia/reperfusion injury in rats.29 Since Mul1 is able to connect to numerous mitochondrial proteins and apoptosis-associated molecules, it is easy to realize the tight linking between Mul1 and apoptosis.30,31 While we established that Mul1 repressed cell viability, while enhanced apoptosis in PC12 cells with NR4A2 knockdown, as evidenced by promoted Cyto C expression and reduced Bcl-2. In line with our findings, hypoxia-reoxygenation treatment caused cellular injury in cultured HT22 cells, which occurred concomitant with the upregulation of Mul1, whereas knockdown of Mul1 alleviated injury induced by hypoxia-reoxygenation through reducing apoptosis.32
Conclusion
In conclusion, we prove a putative role of NR4A2 as a negative molecular target in regulating PC12 cell viability through transcriptional downregulation of miR-652 and upregulation of Mul1. Hereby, it is proposed that silencing of NR4A2 may contribute to the improved PC12 cell viability in ischemic brain injury. Interestingly, silencing of NR4A2 expression with siRNA downregulated IL-17 expression and upregulated the expression of IL-10, an anti-inflammatory factor.33 Similarly, NR4A2 protected dopaminergic neurons against neuroinflammation insults by constricting the release of neurotoxic mediators by microglia,34 while its role in regulating inflammation in ischemic stroke remains largely unknown. Therefore, our future attention may place it. Still, our findings here are limited to OGD cells in vitro, the effect of NR4A2/miR-652/Mul1 axis on ischemic brain injury in vivo needs further study.
Disclosure
The authors declare no potential conflicts of interest for this work.
References
1. Khoshnam SE, Winlow W, Farzaneh M, Farbood Y, Moghaddam HF. Pathogenic mechanisms following ischemic stroke. Neurol Sci. 2017;38(7):1167–1186.
2. Zou X, Yang XJ, Gan YM, et al. Neuroprotective effect of phthalide derivative CD21 against ischemic brain injury: involvement of MSR1 mediated DAMP peroxiredoxin1 clearance and TLR4 signaling inhibition. J Neuroimmune Pharmacol. 2020. doi:10.1007/s11481-020-09911-0
3. Amruta N, Rahman AA, Pinteaux E, Bix G. Neuroinflammation and fibrosis in stroke: the good, the bad and the ugly. J Neuroimmunol. 2020;346:577318. doi:10.1016/j.jneuroim.2020.577318
4. Tuttolomondo A, Di Raimondo D, Pecoraro R, et al. HLA and killer cell immunoglobulin-like receptor (KIRs) genotyping in patients with acute ischemic stroke. J Neuroinflammation. 2019;16(1):88. doi:10.1186/s12974-019-1469-5
5. Tuttolomondo A, Pecoraro R, Casuccio A, et al. Peripheral frequency of CD4+ CD28- cells in acute ischemic stroke: relationship with stroke subtype and severity markers. Medicine. 2015;94(20):e813. doi:10.1097/MD.0000000000000813
6. Rodriguez C, Agulla J, Delgado-Esteban M. Refocusing the brain: new approaches in neuroprotection against ischemic injury. Neurochem Res. 2020. doi:10.1007/s11064-020-03016-z
7. Jakaria M, Haque ME, Cho DY, Azam S, Kim IS, Choi DK. Molecular Insights into NR4A2(Nurr1): an emerging target for neuroprotective therapy against neuroinflammation and neuronal cell death. Mol Neurobiol. 2019;56(8):5799–5814. doi:10.1007/s12035-019-1487-4
8. Merino-Zamorano C, Hernandez-Guillamon M, Jullienne A, et al. NURR1 involvement in recombinant tissue-type plasminogen activator treatment complications after ischemic stroke. Stroke. 2015;46(2):477–484. doi:10.1161/STROKEAHA.114.006826
9. Zhang Z, Yu J. Nurr1 exacerbates cerebral ischemia-reperfusion injury via modulating YAP-INF2-mitochondrial fission pathways. Int J Biochem Cell Biol. 2018;104:149–160. doi:10.1016/j.biocel.2018.09.014
10. Shao CZ, Xia KP. Sevoflurane anesthesia represses neurogenesis of hippocampus neural stem cells via regulating microRNA-183-mediated NR4A2 in newborn rats. J Cell Physiol. 2019;234(4):3864–3873. doi:10.1002/jcp.27158
11. Liu L, Yao J, Li Z, et al. miR-381-3p knockdown improves intestinal epithelial proliferation and barrier function after intestinal ischemia/reperfusion injury by targeting nurr1. Cell Death Dis. 2018;9(3):411. doi:10.1038/s41419-018-0450-z
12. Tan JR, Koo YX, Kaur P, et al. microRNAs in stroke pathogenesis. Curr Mol Med. 2011;11(2):76–92. doi:10.2174/156652411794859232
13. Xu W, Gao L, Zheng J, et al. The roles of microRNAs in stroke: possible therapeutic targets. Cell Transplant. 2018;27(12):1778–1788. doi:10.1177/0963689718773361
14. Zuo ML, Wang AP, Song GL, Yang ZB. miR-652 protects rats from cerebral ischemia/reperfusion oxidative stress injury by directly targeting NOX2. Biomed Pharmacother. 2020;124:109860. doi:10.1016/j.biopha.2020.109860
15. Ren KD, Liu WN, Tian J, et al. Mitochondrial E3 ubiquitin ligase 1 promotes brain injury by disturbing mitochondrial dynamics in a rat model of ischemic stroke. Eur J Pharmacol. 2019;861:172617. doi:10.1016/j.ejphar.2019.172617
16. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi:10.1161/01.STR.20.1.84
17. Zhu Q, Zhang Y, Liu Y, et al. MLIF alleviates SH-SY5Y neuroblastoma injury induced by oxygen-glucose deprivation by targeting eukaryotic translation elongation factor 1A2. PLoS One. 2016;11(2):e0149965. doi:10.1371/journal.pone.0149965
18. Heydari E, Alishahi M, Ghaedrahmati F, Winlow W, Khoshnam SE, Anbiyaiee A. The role of non-coding RNAs in neuroprotection and angiogenesis following ischemic stroke. Metab Brain Dis. 2020;35(1):31–43. doi:10.1007/s11011-019-00485-2
19. He X, Liu Z, Pang Y, Xu W, Zhao L, Li H. Downregulation of transcription factor TCTP elevates microRNA-200a expression to restrain Myt1L expression, thereby improving neurobehavior and oxidative stress injury in cerebral palsy rats. Cell Cycle. 2020;19(8):855–869. doi:10.1080/15384101.2020.1717044
20. Beard JA, Tenga A, Hills J, et al. The orphan nuclear receptor NR4A2 is part of a p53-microRNA-34 network. Sci Rep. 2016;6:25108. doi:10.1038/srep25108
21. Chen P, Li J, Huo Y, et al. Orphan nuclear receptor NR4A2 inhibits hepatic stellate cell proliferation through MAPK pathway in liver fibrosis. PeerJ. 2015;3:e1518. doi:10.7717/peerj.1518
22. Chen P, Li J, Huo Y, et al. Adenovirus-mediated expression of orphan nuclear receptor NR4A2 targeting hepatic stellate cell attenuates liver fibrosis in rats. Sci Rep. 2016;6:33593. doi:10.1038/srep33593
23. Pereira LA, Munita R, Gonzalez MP, Andres ME. Long 3ʹUTR of Nurr1 mRNAs is targeted by miRNAs in mesencephalic dopamine neurons. PLoS One. 2017;12(11):e0188177. doi:10.1371/journal.pone.0188177
24. Liu XL, Wang G, Song W, Yang WX, Hua J, Lyu L. microRNA-137 promotes endothelial progenitor cell proliferation and angiogenesis in cerebral ischemic stroke mice by targeting NR4A2 through the Notch pathway. J Cell Physiol. 2018;233(7):5255–5266. doi:10.1002/jcp.26312
25. Yin F, Zhou H, Fang Y, et al. Astragaloside IV alleviates ischemia reperfusion-induced apoptosis by inhibiting the activation of key factors in death receptor pathway and mitochondrial pathway. J Ethnopharmacol. 2020;248:112319. doi:10.1016/j.jep.2019.112319
26. Wang K, Gan TY, Li N, et al. Circular RNA mediates cardiomyocyte death via miRNA-dependent upregulation of MTP18 expression. Cell Death Differ. 2017;24(6):1111–1120. doi:10.1038/cdd.2017.61
27. Puri R, Cheng XT, Lin MY, Huang N, Sheng ZH. Mul1 restrains Parkin-mediated mitophagy in mature neurons by maintaining ER-mitochondrial contacts. Nat Commun. 2019;10(1):3645. doi:10.1038/s41467-019-11636-5
28. Doktor B, Damulewicz M, Pyza E. Effects of MUL1 and PARKIN on the circadian clock, brain and behaviour in Drosophila Parkinson’s disease models. BMC Neurosci. 2019;20(1):24. doi:10.1186/s12868-019-0506-8
29. Wang SJ, Chen H, Tang LJ, et al. Upregulation of mitochondrial E3 ubiquitin ligase 1 in rat heart contributes to ischemia/reperfusion injury. Can J Physiol Pharmacol. 2020;98(5):259–266. doi:10.1139/cjpp-2019-0285
30. Cilenti L, Di Gregorio J, Ambivero CT, Andl T, Liao R, Zervos AS. Mitochondrial MUL1 E3 ubiquitin ligase regulates Hypoxia Inducible Factor (HIF-1alpha) and metabolic reprogramming by modulating the UBXN7 cofactor protein. Sci Rep. 2020;10(1):1609. doi:10.1038/s41598-020-58484-8
31. Peng J, Ren KD, Yang J, Luo XJ. Mitochondrial E3 ubiquitin ligase 1: A key enzyme in regulation of mitochondrial dynamics and functions. Mitochondrion. 2016;28:49–53. doi:10.1016/j.mito.2016.03.007
32. Guo S, Zhang YY, Peng JJ, et al. Natural compound methyl protodioscin protects rat brain from ischemia/reperfusion injury through regulation of Mul1/SOD2 pathway. Eur J Pharmacol. 2019;849:50–58. doi:10.1016/j.ejphar.2019.01.057
33. Trudler D, Levy-Barazany H, Nash Y, Samuel L, Sharon R, Frenkel D. Alpha synuclein deficiency increases CD4(+) T-cells pro-inflammatory profile in a Nurr1-dependent manner. J Neurochem. 2020;152(1):61–71. doi:10.1111/jnc.14871
34. Chen XX, Qian Y, Wang XP, et al. Nurr1 promotes neurogenesis of dopaminergic neuron and represses inflammatory factors in the transwell coculture system of neural stem cells and microglia. CNS Neurosci Ther. 2018;24(9):790–800. doi:10.1111/cns.12825
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.