Back to Journals » The Application of Clinical Genetics » Volume 9
Novel treatment options for lysosomal acid lipase deficiency: critical appraisal of sebelipase alfa
Authors Su K, Donaldson E, Sharma R
Received 9 January 2016
Accepted for publication 10 May 2016
Published 17 October 2016 Volume 2016:9 Pages 157—167
DOI https://doi.org/10.2147/TACG.S86760
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Martin Maurer
Kim Su,1 Emma Donaldson,1 Reena Sharma2
1Division of Gastroenterology/Hepatology, 2The Mark Holland Metabolic Unit, Salford Royal Hospital NHS Foundation Trust, Salford, UK
Abstract: Lysosomal acid lipase deficiency (LAL-D) is a rare disorder of cholesterol metabolism with an autosomal recessive mode of inheritance. The absence or deficiency of the LAL enzyme gives rise to pathological accumulation of cholesterol esters in various tissues. A severe LAL-D phenotype manifesting in infancy is associated with adrenal calcification and liver and gastrointestinal involvement with characteristic early mortality. LAL-D presenting in childhood and adulthood is associated with hepatomegaly, liver fibrosis, cirrhosis, and premature atherosclerosis. There are currently no curative pharmacological treatments for this life-threatening condition. Supportive management with lipid-modifying agents does not ameliorate disease progression. Hematopoietic stem cell transplantation as a curative measure in infantile disease has mixed success and is associated with inherent risks and complications. Sebelipase alfa (Kanuma) is a recombinant human LAL protein and the first enzyme replacement therapy for the treatment of LAL-D. Clinical trials have been undertaken in infants with rapidly progressive LAL-D and in children and adults with later-onset LAL-D. Initial data have shown significant survival benefits in the infant group and improvements in biochemical parameters in the latter. Sebelipase alfa has received marketing authorization in the United States and Europe as long-term therapy for all affected individuals. The availability of enzyme replacement therapy for this rare and progressive disorder warrants greater recognition and awareness by physicians.
Keywords: sebelipase alfa, LAL deficiency, Wolman disease
Introduction
Lysosomal acid lipase deficiency (LAL-D) is an ultra-rare autosomal recessive lysosomal storage disorder. It is progressive in nature and characterized by the absence or deficiency of LAL. This enzyme is responsible for the metabolism of cholesterol esters and triglycerides derived from low-density lipoproteins (LDLs). Impaired activity of LAL results in pathological accumulation of cholesterol in various tissues.1,2 The severe infantile form of the disease presents as gross intestinal malabsorption, adrenal calcification, hepatomegaly, and early mortality.1–4 The late-onset presentation in children and adults includes dyslipidemia and liver fibrosis with associated consequences of premature atherosclerosis and liver cirrhosis.4,5 Traditionally, there have been no curative therapies for LAL-D, and treatment strategies are supportive in nature, centering upon optimizing nutritional status, the use of lipid-modifying agents to minimize atherosclerosis, and managing the consequences of disease progression (eg, liver transplantation for end-organ failure).
The development of enzyme replacement therapies (ERTs) for lysosomal storage disorders was first exemplified in the development of treatment for type 1 Gaucher disease.6,7 ERTs are currently available for other lysosomal storage disorders, but until recently, there has been no ERT equivalent for LAL-D.
Sebelipase alfa (Kanuma; Alexion Pharmaceuticals, Cheshire, CT, USA) is a recombinant human LAL that has gained marketing authorization for the treatment of LAL-D in Europe and the United States. The availability of enzyme replacement for this condition will fundamentally change management strategies for physicians treating patients with LAL-D. Although this is a rare condition and likely underrecognized in the health care setting, the existence of replacement therapy necessitates increased awareness of the clinical manifestations of LAL-D from the general and specialist physician. Here, we review the pathophysiology, clinical features, and treatment of LAL-D, as well as provide an overview of sebelipase alfa and future directions in LAL-D management.
Pathophysiology of LAL-D
LAL is a glycoprotein located in the lysosomal compartment of mammalian cells, most prominent in macrophages and hepatocytes. LAL catalyzes the hydrolysis of cholesterol esters and triglycerides, providing cells with a source of free cholesterol. The underlying pathophysiology of LAL-D relates to disturbed lysosomal cholesterol metabolism. The resultant accumulation of cholesterol in affected tissues gives rise to accompanying clinical phenotypes, such as hepatomegaly and liver fibrosis from hepatic infiltration, or malabsorption secondary to gastrointestinal involvement.1–5
Cholesterol is essential in many physiological processes. These include its role as a precursor molecule in steroid hormone synthesis, bile acid synthesis, and as a component of cellular membranes. The LAL enzyme is able to generate free cholesterol for cellular use through metabolizing exogenously derived cholesterol. Exogenous cholesterol is obtained from LDL, which enters hepatocytes via receptor-mediated endocytosis. Upon reaching the lysosomal compartment, LAL catalyzes the hydrolysis of cholesterol esters and triglyceride components into free cholesterol and free fatty acids, respectively.8 The generation of free cholesterol inhibits de novo cholesterol synthesis through feedback inhibition of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the key limiting factor in cholesterol synthesis. Free cholesterol end products also inhibit the activation of sterol regulatory element–binding proteins, transcription factors that activate genes responsible for cholesterol synthesis.9
In the absence or reduction of LAL activity, higher levels of cholesterol esters and triglycerides accumulate in the lysosome, with resultant reduction in intracellular free cholesterol. This impairs the negative feedback mechanism acting upon HMG-CoA reductase and sterol regulatory element–binding protein, with resultant synthesis of cholesterol and accumulation in organs giving rise to pathological effects that manifest clinically as either infantile LAL-D or cholesterol ester storage disease (CESD).10
Genetics of LAL-D
LAL-D is an autosomal recessive condition. The LIPA gene, consisting of a 2.5 kb cDNA sequence spanning ten exons and nine introns, encodes this enzyme. The LIPA gene localizes to the long arm of chromosome 10 at position 10q23.31.11–13 LAL-D may manifest in homozygotes or compound heterozygotes harboring mutations in the LIPA gene. Currently over 40 mutations have been described in LAL-D, with differing functional defects including instability at the messenger RNA (mRNA) level, altered protein/catalytic site structure, and reduced or complete absence of enzyme activity.14
Phenotype severity in LAL-D has been linked with the activity of LIPA gene product. Early-onset LAL-D in infancy, also known as Wolman disease, is more severe and associated with rapid progression and early mortality. LIPA mutations seen in infantile LAL-D are associated with complete absence or severely diminished LAL activity.10,13,14 There is a larger repertoire of mutations in infantile LAL-D, for example, complete inactivation of catalytic function due to truncation mutations or the absence of correctly spliced mRNA. This contrasts with LAL-D of later onset, historically known as CESD, which is associated with residual LAL enzyme activity and may present with a more variable phenotype. In contrast to infantile LAL-D, over 50% of described LIPA mutations in CESD are associated with an exon 8 splice junction mutation (E8SJM). This mutation encodes a defective LAL with no enzymatic activity, but a small fraction of normally spliced mRNA is present, giving rise to residual LAL activity.5
Epidemiology of LAL-D
Epidemiological data are limited owing to the rarity of diagnoses and few reported cases in the literature. Studies into the prevalence of LAL-D reveal disparity between the frequency of LIPA mutations reported in the literature and the number of reported cases. Underdiagnosis is likely.15
LIPA mutations in infantile LAL-D are heterogeneous. Unlike the E8SJM in CESD, there is no dominant LIPA mutation in this phenotype. Increased frequency of Wolman disease in the Los Angeles Iranian Jewish community has been reported based on the screening of an exon 4 mutation.16 The carrier frequency was estimated to be 3%, with an estimated 1 in 4,200 live births predicted to be homozygous of the mutation. In contrast to this, an Australian study estimated the prevalence of Wolman disease to be 1.9 per million.17 These two populations are ethnically distinct from each other, and it is possible that high prevalence in certain groups is due to founder effect. Further studies will be highly relevant in establishing the prevalence of LAL-D in different populations and across ethnicities.
In the CESD phenotype, prevalence and carrier frequency of the most common E8SJM LIPA mutation has been estimated for different populations (Table 1), with highest estimates in Caucasian populations. Although the authors found no African-American individuals with this mutation, there are case reports of LIPA mutations associated with the infantile phenotype in African individuals.18 Again, further epidemiological data will be significant for verification of the existing data.
 | Table 1 Estimated E8SJM allele frequency and CESD prevalence among different populations Abbreviations: CESD, cholesterol ester storage disease; E8SJM, exon 8 splice junction mutation. |
Clinical presentation of LAL-D
The differences in LAL activity associated with infantile LAL-D and late-onset CESD give rise to a spectrum of clinical phenotypes. The rarity of cases means that our knowledge is predominantly based on published case reports, and long-term survival data are lacking. Manifestation in infancy is severe and associated with absent or severely reduced LAL activity with early mortality.6,11,20–23 LIPA gene mutations that retain greater residual activity give rise to milder phenotypes (CESD) presenting later in childhood or adulthood. LAL-D can be overlooked or misdiagnosed when presenting features are indistinguishable from nonalcoholic fatty liver disease (NAFLD) or dyslipidemia.24–26
Clinical presentation in infancy
Characterization of early-onset LAL-D presenting in infancy was first described by Dr Wolman in the 20th century.1 Clinical features are severe and manifest within the first few weeks of life. It is associated with rapid clinical deterioration and early mortality, usually by 1 year of age. Symptoms in infantile LAL-D may manifest as malabsorption, growth failure, vomiting, diarrhea/steatorrhea, adrenal failure, and abdominal distention. Examination and biochemical findings include hepatosplenomegaly, adrenal calcification, dyslipidemia, abnormal liver function tests (LFTs), and anemia. Histological findings in affected patients reveal pathological accumulation of cholesterol and triglycerides in multiple organs.1–3
Survival beyond 1 year of age is rare in infantile disease, although in certain cases survival has been prolonged in those who have undergone liver transplantation and/or hematopoietic stem cell transplantation (HSCT). A recent study reported median survival of 3.7 months in patients with LAL-D diagnosed before the age of 2. Those who underwent treatment (HSCT and/or liver transplant) reported median survival of 8.6 months, but in the untreated group this was only 3 months. Of those who survived beyond 12 months, the majority had received HSCT or liver transplantation and HSCT.3
Other rare lysosomal and nonlysosomal disorders such as Niemann–Pick disease A, Gaucher’s disease, galactosemia, hemophagocytic lymphohistiocytosis, and disorders of amino acid metabolism are to be considered in differential diagnoses while considering LAL-D presenting in infancy.3
Clinical presentation in childhood and adulthood
Longitudinal case reviews of LAL-D diagnosed beyond infancy reveal that LAL-D is predominantly a pediatric condition, with paucity of diagnoses in those over the age of 40.4,5 Presentation of LAL-D in childhood and adulthood is most variable, in contrast with the infantile variant.
Given the spectrum of phenotypes, CESD may present to endocrinologists/cardiologists with dyslipidemia, or to gastroenterologists with deranged LFTs or frank liver cirrhosis and associated complications.24–27 Physicians in metabolic, cardiovascular, and gastrointestinal disciplines (as well as the wider pool of physicians) are encouraged to be aware of this condition, especially pertinent now that enzyme replacement is available in order to reduce delay or misdiagnosis of what may now be a treatable condition.
LAL-D should be considered in differential diagnosis when the detection of abnormal liver function may be noted on chance routine testing in asymptomatic individuals or for viral hepatitis screening.28 Hepatomegaly is commonly present, and patients may possess features (including histological) that are reminiscent of NAFLD/nonalcoholic steatohepatitis (NASH), or cryptogenic cirrhosis with associated complications.24,29
LAL-D has been described in the setting of patients presenting with hyperlipidemia or type IIb hyperlipoproteinemia and may be misdiagnosed as familial hyperlipidemic disorders.25,26,30
Histological abnormalities
Widespread histological abnormalities are seen in the tissues of patients with LAL-D. The earliest case reports documented autopsy findings of cholesterol and triglyceride deposits in various organs, with changes most marked in the liver, spleen, gastrointestinal tract, adrenals, lymph nodes, and bone marrow.1,2,31
The cholesterol content of the liver and spleen is increased. Liver cholesterol content can be markedly elevated, and histology may range from the presence of cholesterol ester crystals within hepatocytes and reticuloendothelial cells (Kupffer cells, macrophages) to microvesicular steatosis, fibrotic scarring, and frank cirrhosis (Figures 1–3). The use of specific lysosomal markers cathepsin D, lysosomal associate membrane protein 1 and 2 in liver immunohistochemistry may be of diagnostic benefit in differentiating LAL-D from other forms of liver disease given that microvesicular steatosis is not a reliable marker for CESD.32
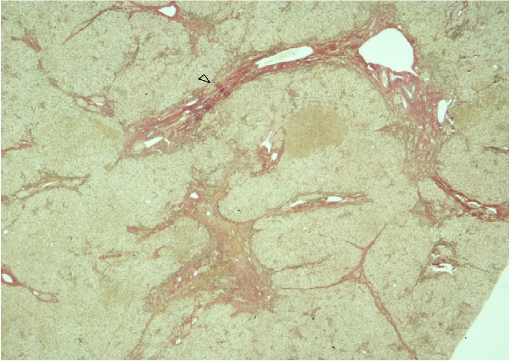 | Figure 1 Bridging fibrosis on Von Giesson (elastic) stain. Notes: Liver biopsy from a patient diagnosed with lysosomal acid lipase deficiency at the age of 40 years. The arrowhead denotes bridging fibrosis. |
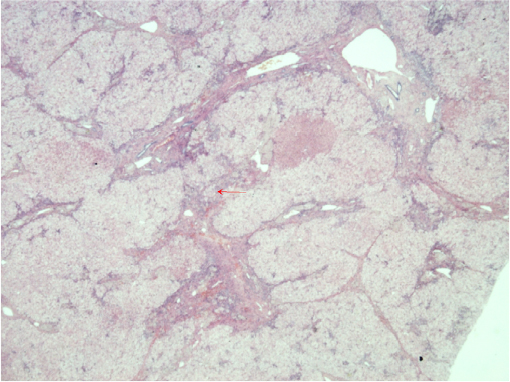 | Figure 2 Bridging fibrosis H and E stain. Notes: Liver biopsy from a patient diagnosed with lysosomal acid lipase deficiency at the age of 40 years. The red arrow denotes bridging fibrosis. Abbreviation: H and E, hemotoxylin and eosin. |
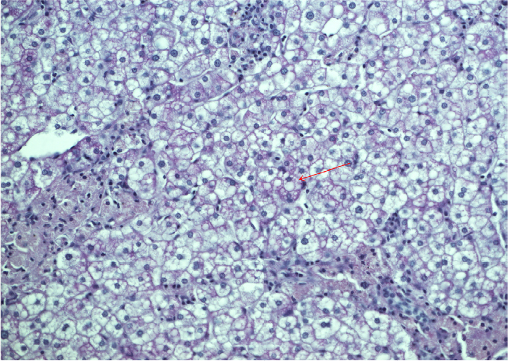 | Figure 3 Microvesicular steatohepatosis on PAS stain. Notes: Liver biopsy from a patient diagnosed with lysosomal acid lipase deficiency at the age of 40 years. The red arrow denotes microvesicular steatosis. Abbreviation: PAS, periodic acid–Schiff. |
In the spleen, reticuloendothelial cells become transformed into lipid-laden foam cells, manifesting as splenomegaly.
Changes in the gastrointestinal tract are seen in the mucosa and, to lesser extent, the submucosa of the small intestines. Prominence of foam-laden vacuolated macrophages infiltrating the lamina propria occurs, with distortion of the architecture and dysfunction in absorptive and enzymatic activity of the small intestinal enterocytes.
Adrenal gland enlargement is seen in infantile disease, with lipid accumulation, necrosis, and calcification of zona reticularis.
Biochemical derangements
Commonly noted biochemical derangements in LAL-D include deranged LFTs with elevated alanine transaminase (ALT) and/or aspartate aminotransferase (AST), which can deteriorate as disease progresses. Hyperbilirubinemia and elevated gamma glutamyl transferase may also be seen. Dyslipidemia is a common feature, with elevated levels of triglycerides, total cholesterol, and low high-density lipoprotein. High-density lipoprotein levels may be reduced.3
Radiological abnormalities
The earliest case studies of infantile disease reported adrenal gland calcification on X-ray imaging. Computed tomography (CT) and magnetic resonance imaging scan can identify adrenal enlargement necrosis and calcification. The radiological imaging can demonstrate hepatomegaly (low attenuation on CT indicates fat infiltration, but this is not diagnostic as it is also seen in NAFLD and NASH), splenomegaly, and lymphadenopathy (Figure 4).33 Ultrasound scan in pediatric case reports have demonstrated bowel wall thickening in the small intestines.34
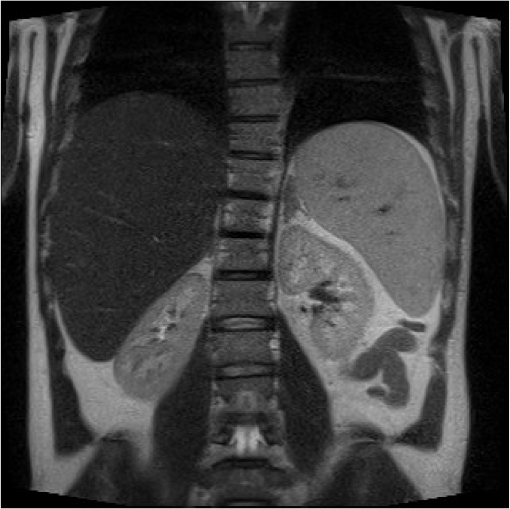 | Figure 4 Hepatosplenomegaly and liver steatosis on MRI scan of an adult patient diagnosed with cholesterol ester storage disease. Abbreviation: MRI, magnetic resonance imaging. |
Survival and morbidity
One study has reported mean time from clinical/biochemical abnormalities to diagnosis was 9 years (median: 5.8 years). In contrast to the NAFLD/NASH population, this cohort of patients has accelerated progression to fibrosis: of 31 patients with liver biopsies, 85% had features of fibrosis and/or cirrhosis, and all were under 18 years of age.5 The finding of significant liver fibrosis contrasts with the adult NAFLD population: an estimated 3%–15% of NALFD will progress to NASH.35 The high proportion of LAL-D patients who may have significant liver disease is also reflected in the acid lipase replacement investigating safety and efficacy (ARISE) trial:36 25 out of 32 (78%) liver biopsies revealed bridging fibrosis and/or cirrhosis (Ishak score ≥3). Significantly eight of the ten cirrhotic subjects did not have a diagnosis of cirrhosis, showing that advanced liver disease can remain asymptomatic.36 There is no longitudinal survival data for LAL-D, although there are reports of individuals diagnosed in old age (80 years), presumably with milder phenotypical variants of LAL-D.30
Dyslipidemia is a common biochemical abnormality in CESD, but definitive studies of increased cardiovascular mortality in LAL-D are not available. There are documented cases of premature atherosclerosis and cardiovascular disease in patients with CESD, but these are observed associations.24,29,37
Clinical diagnosis of LAL-D
In individuals where LAL-D is a differential, diagnosis can be made on a blood spot38 or peripheral leukocyte LAL activity. Liver biopsy is not the first-line diagnostic investigation for LAL-D. However, biopsies are undertaken if there is clinically suspected liver disease or to exclude other etiologies of chronic liver disease. Liver biopsies are often obtained in confirmed LAL-D prior to treatment, and may be repeated if clinical deterioration is noted. Genetic analysis of LIPA mutations is not a compulsory part of the diagnostic workup. There is no genotype–phenotype correlation in CESD: the LIPA mutation does not predict the clinical course/outcome.
Clinical management of LAL-D
Current management of LAL-D is largely supportive. Nutritional supplementation has been used in infantile disease, including fat-free enteral and parenteral nutrition, but with no morbidity or mortality benefits. Low cholesterol/reduced saturated fat diets have been used in pediatric and adult CESD, although this has also demonstrated low efficacy.39,40
Lipid-modifying agents
HMG-CoA reductase inhibitors (statins) have been administered to reduce endogenous cholesterol synthesis in pediatric and adult cases of CESD. These have been used alone or in conjunction with other lipid-modifying agents (ezetimibe, cholestyramine), but results have been variable.41
Some case reports have noted improved lipid profile with statin therapy (predominantly lovastatin although simvastatin has also been administered). Evaluation of lipid parameters on statin therapy has varied from 6 months to 9 years postcommencement. The addition of ezetimibe to lovastatin has demonstrated additional improvements to total cholesterol and LDL levels in certain cases.40,42,43
However, despite their apparent lipid-modifying properties, it remains unclear whether statins ameliorate premature atherosclerosis or reduces cardiovascular disease in this group of patients. Importantly, statins have shown no benefit in modifying the progression of liver disease associated with CESD.4 Although some reports have noted improved liver histology such as reduced hepatocyte vacuolation, no comment was made on the progression or otherwise of liver fibrosis.24 Others have reported reduced fat infiltration on radiological imaging, but without coexisting liver histology.42 Clinical improvement of hepatosplenomegaly has been reported, but methods of assessment vary between reports.39 Furthermore, others have shown no CT-based improvement in liver volumes with statin therapy.24 Repeated liver histology during statin treatment has been undertaken in several cases, but all revealed progression of underlying disease.4
Hematopoietic stem cell therapy
Stem cell/bone marrow transplantation for LAL-D is potentially curative, as normal LAL activity will be derived from donor cells. Stem cell/bone marrow transplantation for LAL-D is potentially curative as normal LAL activity will be derived from donor cells, but outcomes of HSCT have been mixed.44 Successful cases of bone marrow and umbilical cord blood transplantation have been documented up to 4 and 11 years posttransplantation, with normalization of LAL, improved lipid parameters, resolution of symptoms, regression of hepatosplenomegaly, and histological improvement in the liver.45,46
Deaths have also been reported after transplantation: two of four patients in one case series died due to sinusoidal obstruction syndrome.46 Another revealed normalization of peripheral LAL levels, but death occurred secondary to progression of underlying LAL-D, with subsequent histology revealing failure of engraftment in the liver.47
Although HSCT has proved successful in select cases, this is not a ubiquitous treatment strategy. Failure of engraftment, risks associated with the procedure, and emergence of post-transplant complications means that alternatives are needed.
Liver transplantation
Similar to HSCT, liver transplantation is also associated with risks and complications. This procedure has been undertaken in LAL-D patients (in both infants and adults). Liver transplantation for an infant with infantile LAL-D was undertaken prior to bone marrow transplantation, but recurrence of cirrhosis in the graft and delayed bone marrow transplantation were felt to have contributed to death.48
Liver transplantation undertaken for later-onset LAL-D (CESD) has been undertaken in cases complicated by end-stage cirrhosis.49–51 Follow-up of affected children and adolescents who had undergone this procedure has been documented, varying from 10 months to 4 years postprocedure. These cases demonstrated good clinical progress, normalization of LFTs, and improved lipid parameters. One case of transplantation in an adult female with LAL-D reported satisfactory graft function 2 years postprocedure and improved lipid profile, although peripheral LAL levels remained low. Evidence for long-term mortality and morbidity post-transplantation are currently limited.52
Current strategies remain unsatisfactory in halting the progress of LAL-D. Finite resources, specific criteria for transplantation, and the risks posed by these procedures preclude their universal availability to affected individuals.
There is also concern that transplanted grafts can potentially develop liver disease, as the total amount of peripheral LAL activity continues to be low in patients who have received liver transplantation.37
Enzyme replacement strategies and preclinical data
The development of an LAL enzyme replacement enables correction of metabolic defects that characterize LAL-D and reduce pathological accumulation of cholesterol in affected tissues. Preclinical studies have shown that this is feasible. In vivo models of LAL deficiency have demonstrated that LAL replacement targets the enzyme to the destined compartment and ameliorates the pathological effects of LAL deficiency. In the mouse model of LAL-D, infusion of recombinant human LAL revealed histological improvement in several organs (liver, spleen, and intestines), with histological improvement noted in Kupffer cells.53
Other methods of targeting recombinant human LAL in in vivo systems have been published. These include the production of human LAL in Chinese hamster ovary cells and adenovirus-mediated transfer of the LAL cDNA into hepatocytes.54,55 These systems have not progressed beyond preclinical stages.
Sebelipase alfa
Sebelipase alfa (Kanuma) is the first enzyme replacement developed by Synageva Biopharma Ltd (now Alexion Pharmaceuticals) for LAL-D. Clinical trials have been conducted in infants displaying the rapidly progressive form of LAL-D and in children and adults with later-onset phenotype. Data show significantly enhanced survival in affected infants and correction of biochemical abnormalities in older subjects.
Sebelipase alfa is a recombinant human LAL; hence, the amino acid sequence is identical to that of the human protein. It is a glycoprotein with a molecular weight of 55 kDa. Carbohydrate-containing moieties of the drug include N-acetylglucosamine, mannose-6-phosphate (M6P), and other mannose-terminated N-linked glycan structures. The presence of glycan moieties confers specificity, allowing targeted delivery of LAL to the lysosomal compartment by means of the macrophage mannose receptor on reticuloendothelial cells and the M6P receptor. The recombinant protein is synthesized in genetically modified hens (Gallus gallus) with secretion of the protein into the egg white.56,57
Sebelipase alfa licensing and indications
Currently, sebelipase alfa has received marketing authorization in Europe, the United States, and Japan.58 It is classified as an orphan medicine owing to the rarity of LAL-D. Treatment can be initiated upon the diagnosis of LAL-D in affected individuals of all ages. It is administered as an intravenous infusion, and the recommended dose for infants under 6 months is 1 mg/kg every week, while in adults and older children with later-onset LAL-D (CESD), this is 1 mg/kg every 2 weeks.
Pharmacodynamics
Sebelipase alfa localizes to the lysosomal compartment via receptor-mediated uptake by reticuloendothelial cells and other cells expressing the M6P receptor. The therapeutic effect of the drug is evidenced by the effect on liver transaminases and lipid profile in trial subjects. LAL-CL01, the first-in-human sebelipase alfa study, revealed a significant reduction in ALT and AST levels from baseline in 100% and 67% of subjects, respectively, at day 28.57
Preclinical data
Data derived from preclinical models have demonstrated the efficacy and acceptable safety profile in mammals.
In the rat model of LAL deficiency, sebelipase alfa-treated animals showed improved parameters compared to placebo. Sebelipase alfa (5 mg/kg) administration weekly for 4 weeks resulted in reduced lipid content in Kupffer cells and hepatocytes, improved transaminases, and reduced liver weight in the treatment group.59
Toxicology studies reveal no significant findings after short-term (4 weeks) administration or long-term (6 months) repeated dosing. Short-term dosing at up to 50 mg/kg per week has been used in murine and primate models (human equivalent doses of ~8 and 16 mg/kg respectively), while 30 mg/kg per week long-term dosing in primates (human equivalent dose of ~10 mg/kg) did not reveal concerning findings based on clinical observations.
Initial clinical data
The first-in-human Phase I/II study of sebelipase alfa (LAL-CL01) was conducted in nine adult subjects with clinical manifestations of LAL-D (deranged ALT/AST, lipid profile, and/or hepatomegaly).57 Three dosing regimens (0.35, 1, and 3 mg/kg) infused intravenously on a weekly basis were investigated in sequential cohorts. Results were encouraging and revealed good safety profile and significant reduction in ALT and AST levels from baseline in 100% and 67% of subjects, respectively, at day 28.
Efficacy
In infant-onset LAL-D
Sebelipase alfa has shown significant survival benefit in infants with LAL-D. LAL-CL03 is an open-label, single-arm, Phase II/III trial conducted in nine infants under 6 months of age, with the primary outcome measure of survival at 12 months.60 Six subjects (67%) receiving sebelipase alfa achieved the primary end point; this was compared with a historical cohort of patients with similar characteristics of which 0% survived to 12 months. Clinical features in these nine infants included deranged ALT and AST (median 145 and 125 IU/L, respectively), hepatomegaly (nine subjects), splenomegaly (eight subjects), and adrenal calcification (six subjects) at baseline. Resolution of symptoms, reduced organomegaly, and improved biochemical parameters were all noted in the treatment group. Deaths occurred in four patients, but none were attributed to the drug.
Children and adults
LAL-CL04 was a continuation of LAL-CL01 over a period of 52 weeks, with a total of eight subjects (one subject was no longer eligible for the study due to development of decompensated liver disease).61 This study revealed that the initial improvement in transaminases was sustained over the longer dosing period (Table 2). Sebelipase alfa was infused weekly in three dosage groups (0.35 mg/kg, 1 mg/kg or 3 mg/kg). Thereafter two dosing groups (1 mg/kg or 3 mg/kg) were administered on a fortnightly basis for up to 52 weeks. At 52 weeks, ALT and AST in all subjects had normalized (mean ALT 34 IU/L, mean AST 32 IU/L), and improvements in lipid profiles were seen (after an initial rise at the end of LAL-CL01 period). No differences between 1 and 3 mg/kg doses could be distinguished.
 | Table 2 LAL-CL04 biochemical parameters at 52 weeks Note: aOne subject received sebelipase for 38 weeks. Abbreviations: ALT, alanine transaminase; AST, aspartate aminotransferase; HDL, high-density lipoprotein; LDL, low-density lipoprotein; SD, standard deviation. |
The Phase III of ARISE/LAL-CL02 trial was a double-blind, placebo-controlled trial conducted with 66 subjects (children and adults) comparing sebelipase alfa (dosage 1 mg/kg every 2 weeks) to placebo.36 At the end of the 20-week closed-label period, all subjects received sebelipase alfa in the open-label extension. The primary end point of ALT normalization at the end of the 20-week closed-label period was reached in 33% of the treatment group, compared to 7% in the placebo group. Other secondary outcomes were also significantly improved in the treatment group (Table 3). During the open-label period, the former placebo group experienced statistically significant falls in ALT and LDL cholesterol and elevation in high-density lipoprotein levels.
 | Table 3 Primary and secondary end points in ARISE study Notes: *Total number of patients =57 (sebelipase alfa =32; placebo =25). From N Engl J Med, Burton BK, Balwani M, Feillet F, et al. A Phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency, 2015;373(11):1010–1020. Copyright© 2015 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society.36 Abbreviations: ALT, alanine transaminase; ARISE, acid lipase replacement investigating safety and efficacy; AST, aspartate aminotransferase; HDL, high-density lipoprotein; LDL, low-density lipoprotein; MRI, magnetic resonance imaging; SD, standard deviation. |
Safety profile of sebelipase alfa
Current information derived from clinical trials of sebelipase alfa has revealed it is relatively well tolerated. Additional trials in infants and adults remain ongoing and will supplement existing data (Table 4).
 | Table 4 Known adverse drug reactions for sebelipase alfa Note: Data from a previous study.62 |
LAL-CL01, the first-in-human Phase I/II study of sebelipase alfa, was conducted in nine subjects with three dosing regimens (0.35, 1, and 3 mg/kg) infused intravenously on a weekly basis. Each subject received four doses of the drug, and monitoring revealed it to be well tolerated with no reported serious adverse events. Adverse events in two subjects were felt to be treatment-related.57
The LAL-CL04 extension to this trial reported one subject having experienced an infusion-related reaction, although subsequent reintroduction of sebelipase did not have any further adverse effects.60 Additional trials also support the acceptable safety profile of sebelipase alfa. ARISE/LAL-CL02 reported three serious adverse reactions, including one infusion-associated event. Most hypersensitivity reactions were recorded in infant subjects.36
Immunogenicity
Experience with other ERTs in other lysosomal diseases suggest that ERTs are immunogenic, and the development of antidrug antibodies can affect drug efficacy.62–64 In the case of sebelipase alfa, this seems more common in infants. Four infants in LAL-CL03 developed antidrug antibodies, mainly within the first 2 months of exposure. Antibodies in two of these subjects showed in vitro inhibition of enzyme activity and inhibition of cellular uptake of LAL. In three of the subjects, antidrug titers decreased to undetectable levels over the study period. Adult subjects participating in LAL-CL04 trial did not demonstrate antidrug antibodies over the study period of 52 weeks.60
Five subjects in ARISE had detectable antidrug antibodies, mostly within 3 months of exposure, but antibody titers were low and did not affect efficacy or impact safety.36
Discussion
The availability of ERT for LAL-D is promising. Initial studies have shown survival improvement in infantile disease and improvement in biochemical parameters for late-onset disease in children and adults. However, several aspects relating to long-term outcome are unknown. We do not know the effect of ERT on the progression of liver fibrosis. The primary end point of the ARISE trial was normalization of serum ALT, but LFTs are not reliable correlates of fibrosis or cirrhosis, and transaminases in advanced liver disease may be within the normal range. Cirrhosis may take years to develop, and longitudinal data will be valuable in this assessment. Regression of fibrosis has been shown in other liver diseases with treatment of the underlying etiology.65 It will be interesting to know if this is the case with sebelipase alfa.
LAL-D subjects with decompensated cirrhosis and severe liver disease (Child-Pugh Score C) were not included in trials. The burden of end-stage liver disease in this group is significant, with limited treatment options.66,67 In addition, subjects with liver comorbidities were not part of the inclusion criteria for these trials. It would be of interest to know the additive effect of LAL-D upon liver disease progression given that steatosis is known to act synergistically with other factors in fibrosis progression.68
Available sebelipase alfa trials have demonstrated significant improvements in the lipid profiles of participating subjects. Whether this will ameliorate premature atherosclerosis and significantly affect mortality and/or morbidity in terms of cardiovascular disease remains to be seen. No studies have addressed the effects of lipid-lowering agents on LAL-D progression/mortality.
There are no recommendations for sebelipase alfa dosing in pregnancy, and the effects of this drug in human fetal development are unknown. However, data derived from animal studies have shown no detrimental effects on fetal development.62
Not surprisingly, some patients especially in infantile LAL-D group have shown antibody response. Like other ERTs, sebelipase alfa being a foreign protein is potentially immunogenic, and this aspect of treatment should always be considered by treating clinicians.
Currently ongoing clinical trials are seeking to address some of the issues raised. An open-label, single-arm, Phase II study in infants under 8 months with rapidly progressive LAL-D (NCT02193867, sponsored by Alexion) seeks to assess long-term parameters (up to 3 years). Primary outcome measures include safety profile, changes in biochemical and physical parameters, and the use of concurrent medications. Secondary outcomes include survival at 12 months and beyond, growth status, and pharmacokinetic characterization.
Another Phase II, open-label trial (NCT02112994, sponsored by Alexion) seeks to assess the safety and efficacy of sebelipase alfa in a broader population of infants over 8 months, children, and adults. Secondary outcome measures include assessment of liver severity (Child-Pugh and United Kingdom End Stage Liver Disease [UKELD] score as well as histopathology) up to 96 weeks, immunogenicity, and effect of concurrent medications.
Conclusion
Sebelipase alfa is the first of its kind for ERT in LAL-D. Current data demonstrate significant survival impact in infants with rapidly progressive disease and marked improvements in biochemical parameters in the older LAL-D group. Longitudinal data will be relevant in assessing long-term impact on disease progression and mortality, as well as in characterization of adverse events and immunogenicity. Although LAL-D is an uncommon condition and rarely described in the literature, genetic analyses estimate a higher prevalence than case reports would suggest. The rarity and clinical spectrum of the LAL-D phenotype makes it likely that it is underdiagnosed. Enhancing awareness of LAL-D in physicians is essential given the availability of ERT for a life-threatening disorder and may considerably impact detection rates and supplement existing knowledge of the natural history, progression, and epidemiology of this uncommon condition.
Disclosure
Dr Reena Sharma is principle investigator for sebelipase alfa CL04 trial and has received travel grants and bursaries from Synageva and Alexion. The other authors report no conflicts of interest in this work.
References
Abramov A, Schorr S, Wolman M. Generalized xanthomatosis with calcified adrenals. AMA J Dis Child. 1956;91(3):282–286. | ||
Wolman M, Sterk VV, Gatt S, Frenkel M. Primary familial xanthomatosis with involvement and calcification of the adrenals. Report of two more cases in siblings of a previously described infant. Pediatrics. 1961;28:742–757. | ||
Jones SA, Vannayannopoulos V, Schneider E, et al. Rapid progression and mortality of lysosomal acid lipase deficiency presenting in infants. Genet Med. 2016;18(5):452–458. | ||
Bernstein DL, Hulkova H, Bialer MG, Desnick RJ. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an under diagnosed disease. J Hepatol. 2013;58(6):1230–1243. | ||
Burton BK, Deegan PB, Enns GM, et al. Clinical features of lysosomal acid lipase deficiency. J Pediatr Gastroenterol Nutr. 2015;61(6):619–625. | ||
Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency – macrophage-targeted glucocerebrosidase for Gaucher’s disease. N Engl J Med. 1991;324(21):1464–1470. | ||
Desnick RJ, Schuchman EH. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu Rev Genomics Hum Genet. 2012;13:307–335. | ||
Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. Role of lysosomal acid lipase in the metabolism of plasma low-density lipoprotein. Observations in cultured fibroblasts from a patient with cholesteryl ester storage disease. J Biol Chem. 1975;250(21):8487–8495. | ||
Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. 2002;109(9):1125–1131. | ||
Hoeg JM, Demosky SJ, Pescovits OH, Brewer HB. Cholesteryl ester storage disease and Wolman disease: phenotypic variants of lysosomal acid cholesteryl ester hydrolase deficiency. Am J Hum Genet. 1984;36(6):1190–1203. | ||
Anderson RA, Sando GN. Cloning and expression of cDNA encoding human lysosomal acid lipase/cholesteryl ester hydrolase: similarities to gastric and lingual lipases. J Biol Chem. 1991;266(33):22479–22484. | ||
Anderson RA, Rao N, Byrum RS, et al. In situ localization of the genetic locus encoding the lysosomal acid lipase/cholesteryl esterase (LIPA) deficient in Wolman disease to chromosome 10q23.2–q23.3. Genomics. 1993;15(1):245–247. | ||
Aslanidis C, Ries S, Fehringer P, Buchler C, Klima H, Schmitz G. Genetic and biochemical evidence that CESD and Wolman disease are distinguished by residual lysosomal acid lipase activity. Genomics. 1996;33(1):85–93. | ||
Anderson RA, Byrum RS, Coates PM, Sando GN. Mutations at the lysosomal acid cholesteryl ester hydrolase gene locus in Wolman disease. Proc Natl Acad Sci U S A. 1994;91(7):2718–2722. | ||
Muntoni S, Wiebusch H, Jansen-Rust M, et al. Prevalence of cholesteryl ester storage disease. Arterioscler Thromb Vasc Biol. 2007;27(8):1866–1868. | ||
Valles-Ayoub Y, Esfandiarifard S, No D, et al. Wolman disease (LIPA p.G87V) genotype frequency in people of Iranian-Jewish ancestry. Genet Test Mol Biomarkers. 2011;15(6):395–398. | ||
Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281(3):249–254. | ||
Ries S, Aslanidis C, Fehringer P, Carel JC, Gendrel D, Schmitz G. A new mutation in the gene for lysosomal acid lipase leads to Wolman disease in an African kindred. J Lipid Res. 1996;37(8):1761–1765. | ||
Scott SA, Liu B, Nazarenko I, et al. Frequency of the cholesteryl ester storage disease common LIPA E8SJM mutation (c.894G>A) in various racial and ethnic groups. Hepatology. 2013;58(3):958–965. | ||
Patrick AD, Lake BD. Deficiency of an acid lipase in Wolman’s disease. Nature. 1969;222(5198):1067–1068. | ||
Burke JA, Schubert WK. Deficient activity of acid lipase in cholesterol ester storage disease. J Lab Clin Med. 1971;78(6):988–989. | ||
Sloan HR, Fredrickson DS. Enzyme deficiency in cholesteryl ester storage disease. J Clin Invest. 1972;51(7):1923–1926. | ||
Anderson RA, Bryson GM, Parks JS. Lysosomal acid lipase mutations that determine phenotype in Wolman and cholesterol ester storage disease. Mol Genet Metab. 1999;68(3):333–345. | ||
Gasche C, Aslanidis C, Kain R, et al. A novel variant of lysosomal acid lipase in cholesteryl ester storage disease associated with mild phenotype and improvement on lovastatin. J Hepatol. 1997;27(4):744–750. | ||
Decarlis S, Agostoni C, Ferrantw F, Scarlino S, Riva E, Giovannini M. Combined hyperlipidaemia as a presenting sign of cholesteryl ester storage disease. J Inherit Metab Dis. 2009;32(Suppl 1):S11–S13. | ||
Stitziel NO, Fouchier SW, Siouke B, et al; National Heart, Lung and Blood Institute GO Exome Sequencing Project. Exome sequencing and directed clinical phenotyping diagnose cholesterol ester storage disease presenting as autosomal recessive hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2013;33(12):2909–2914. | ||
Elleder M, Ledvinova J, Cieslar P, Kuhn R. Subclinical course of cholesterol ester storage disease (CESD) diagnosed in adulthood. Report on two cases with remarks on the nature of the liver storage process. Virchows Arch A Pathol Anat Histopathol. 1990;416(4):357–365. | ||
Chatrath H, Keilin S, Attar BM. Cholesterol ester storage disease (CESD) diagnosed in an asymptomatic adult. Dig Dis Sci. 2009;54(1):168–173. | ||
Elleder M, Chlumská A, Hyánek J, et al. Subclinical course of cholesteryl ester storage disease in an adult with hypercholesterolemia, accelerated atherosclerosis, and liver cancer. J Hepatol. 2000;32(3):528–534. | ||
Pisciotta L, Fresa R, Bellocchio A, et al. Cholesteryl ester storage disease (CESD) due to novel mutations in the LIPA gene. Mol Genet Metab. 2009;97(2):143–148. | ||
Crocker AC, Vawter GF, Neuhauser EB, Rosowsky A. Wolman’s disease: three new patients with a recently described lipidosis. Pediatrics. 1965;35(4):627–640. | ||
Hulkova H, Elleder M. Distinctive histopathological features that support a diagnosis of cholesterol ester storage disease in liver biopsy specimens. Histopathology. 2012;60(7):1107–1113. | ||
Fulcher AS, Narla LD, Hingsbergen EA. Pediatric case of the day: Wolman disease (primary familial xanthomatosis with involvement and calcification of the adrenal glands). Radiographics. 1998;18(2):533–535. | ||
Nchimi A, Rausin L, Khamis J. Ultrasound appearance of bowel wall in Wolman’s disease. Pediatr Radiol. 2003;33(4):284–285. | ||
Anstee QM, Targher G, Day CP. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat Rev Gastroenterol Hepatol. 2013;10(6):330–344. | ||
Burton BK, Balwani M, Feillet F, et al. A Phase 3 trial of sebelipase alfa in lysosomal acid lipase deficiency. N Engl J Med. 2015;373(11):1010–1020. | ||
Ambler GK, Hoare M, Brais R, et al. Orthotopic liver transplantation in an adult with cholesterol ester storage disease. JIMD Rep. 2013;8:41–46. | ||
Hamilton J, Jones I, Srivastava R, Galloway P. A new method for the measurement of lysosomal acid lipase in dried blood spots using the inhibitor Lalistat 2. Clin Chim Acta. 2012;413(15–16):1207–1210. | ||
Glueck CJ, Lichtenstein P, Tracy T, Speirs J. Safety and efficacy of treatment of pediatric cholesteryl ester storage disease with lovastatin. Pediatr Res. 1992;32(5):559–565. | ||
Tadiboyina VT, Liu DM, Miskie BA, Wang J, Hegel RA. Treatment of dyslipidemia with lovastatin and ezetimibe in an adolescent with cholesterol ester storage disease. Lipids Health Dis. 2005;28(4):26. | ||
Leone L, Ippoliti PF, Antonicelli R. Use of simvastatin plus cholestyramine in the treatment of lysosomal acid lipase deficiency. J Pediatr. 1991;119(6):1008–1009. | ||
Tarantino MD, McNamara DJ, Granstrom P, Ellefson RD, Unger EC, Udall JN. Lovastatin therapy for cholesterol storage disease in two sisters. J Pediatr. 1991;118(1):131–135. | ||
Rassoul F, Richter V, Lohse P, Naumann A, Purschwitz K, Keller E. Long-term administration of the HMG-CoA reductase inhibitor lovastatin in two patients with cholesteryl ester storage disease. Int J Clin Pharmacol Ther. 2001;39(5):199–204. | ||
Jyonouchi H, Cyong JC, Shen FW, Fernandes G, Day NK, Good RA. Alteration of murine serum lipase activity after allogeneic bone marrow transplantation. Clin Immunol Immunopathol. 1982;22(1):94–104. | ||
Stein J, Garty BZ, Dror Y, Fenig E, Zeigler M, Yaniv I. Successful treatment of Wolman disease by unrelated umbilical cord blood transplantation. Eur J Pediatr. 2007;166(7):663–666. | ||
Tolar J, Petryk A, Khan K, et al. Long-term metabolic, endocrine, and neuropsychological outcome of hematopoietic cell transplantation for Wolman disease. Bone Marrow Transplant. 2009;43(1):21–27. | ||
Gramatges MM, Dvorak CC, Regula DP, Enns GM, Weinberg K, Agarwal R. Pathological evidence of Wolman’s disease following hematopoietic stem cell transplantation despite correction of lysosomal acid lipase activity. Bone Marrow Transplant. 2009;44(7):449–450. | ||
Krivit W, Peters C, Dusenbery K, et al. Wolman disease successfully treated by bone marrow transplantation. Bone Marrow Transplant. 2000;26(5):567–570. | ||
Ferry GD, Whisennand HH, Finegold MJ, Alpert E, Glombicki A. Liver transplantation for cholesteryl ester storage disease. J Pediatr Gastroenterol Nutr. 1991;12(3):376–378. | ||
Arterburn JN, Lee WM, Wood RP, Shaw BW, Markin RS. Orthotopic liver transplantation for cholesterol ester storage disease. J Clin Gastroenterol. 1991;13(4):482–485. | ||
Leone L, Ippoliti PF, Antonicelli R, Balli F, Gridelli B. Treatment and liver transplantation for cholesterol ester storage disease. J Pediatr. 1995;127(3):509–510. | ||
Hansen K, Horslen S. Metabolic liver diseases in children. Liver Transpl. 2008;14(5):713–733. | ||
Du H, Schiave S, Levine M, Mishra J, Heur M, Grabowski G. Enzyme therapy for lysosomal acid lipase deficiency in the mouse. Hum Mol Genet. 2001;10(16):1639–1648. | ||
Du H, Levine M, Ganesa C, Witte DP, Cole ES, Grabowski G. The role of mannosylated enzyme and the mannose receptor in enzyme replacement therapy. Am J Hum Genet. 2005;77(6):1061–1074. | ||
Du H, Heur M, Witte DP, Ameis D, Grabowski GA. Lysosomal acid lipase deficiency: correction of lipid storage by adenovirus-mediated gene transfer in mice. Hum Gene Ther. 2002;13(11):1361–1372. | ||
Stahl PD, Rodman JS, Miller MJ, Schlesinger PH. Evidence of receptor-mediated binding of glycoproteins, glycoconjugates and lysosomal glycosidases by alveolar macrophages. Proc Natl Acad Sci U S A. 1978;75(3):1399–1403. | ||
Balwani M, Breen C, Enns GM, et al. Clinical effect and safety profile of recombinant human lysosomal acid lipase in patients with cholesteryl ester storage disease. Hepatology. 2013;58(3):950–957. | ||
Shirley M. Sebelipase alfa. First global approval. Drugs. 2015;75(16):1935–1940. | ||
Leavitt M, Burt AD, Hu D, et al. Recombinant lysosomal acid lipase normalizes liver weight, transaminases and histopathological abnormalities in an in vivo model of cholesteryl ester storage disease. J Hepatol. 2011;54(Suppl 1):S358. | ||
Jones S, Plantaz D, Vara R, et al. Impact of sebelipase alfa on survival and liver function in infants with rapidly progressive lysosomal acid lipase deficiency. J Hepatol. 2015;62(Suppl 2):S811. | ||
Valayannopoulos V, Malinova V, Honzík T, et al. Sebelipase alfa over 52 weeks reduces serum transaminases, liver volume and improves serum lipids in patients with lysosomal acid lipase deficiency. J Hepatol. 2014;61(5):1135–1142. | ||
European Medicines Agency. Kanuma product information. London, UK: European Medicines Agency. Available from: http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/004004/WC500192715.pdf. Accessed December 19, 2015. | ||
Harmatz P. Enzyme replacement therapies and immunogenicity in lysosomal storage diseases: is there a pattern? Clin Ther. 2015;37(9):2130–2134. | ||
Jameson E, Jones S, Wraith JE. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst Rev. 2013;11:CD009354. | ||
Baruteau J, Broomfield A, Crook V, et al. Successful desensitisation in a patient with CRIM-positive infantile-onset Pompe disease. JIMD Rep. 2014;12:99–102. | ||
Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115(2):209–218. | ||
D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44(1):217–231. | ||
Powell EE, Jonsson JR, Clouston AD. Steatosis: co-factor in other liver diseases. Hepatology. 2005;42(1):5–13. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.