Back to Archived Journals » Oncolytic Virotherapy » Volume 4
Novel therapeutic strategies in human malignancy: combining immunotherapy and oncolytic virotherapy
Received 16 March 2015
Accepted for publication 16 April 2015
Published 18 June 2015 Volume 2015:4 Pages 75—82
DOI https://doi.org/10.2147/OV.S54738
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Padma Sampath, Steve H Thorne
Department of Surgery, University of Pittsburgh Cancer Institute, University of Pittsburgh, Pittsburgh, PA, USA
Abstract: Results from randomized clinical trials over the last several years have finally begun to demonstrate the potential of oncolytic viral therapies to treat a variety of cancers. One reason for these successes has been the realization that this platform is most effective when considered primarily as an immunotherapy. Cancer immunotherapy has also made dramatic strides recently with antibodies capable of blocking immune checkpoint inhibitors and adoptive T-cell therapies, notably CAR T-cells, leading a panel of novel and highly clinically effective therapies. It is clear therefore that an understanding of how and when these complementary approaches can most effectively be combined offers the real hope of moving beyond simply treating the disease and toward starting to talk about curative therapies. In this review we discuss approaches to combining these therapeutic platforms, both through engineering the viral vectors to more beneficially interact with the host immune response during therapy, as well as through the direct combinations of different therapeutics. This primarily, but not exclusively focuses on strains of oncolytic vaccinia virus. Some of the results reported to date, primarily in pre-clinical models but also in early clinical trials, are dramatic and hold great promise for the future development of similar therapies and their translation into cancer therapies.
Keywords: oncolytic virus, CAR T-cell, adoptive cell therapy, immune checkpoint inhibitor
Introduction to oncolytic viral therapy
Oncolytic viruses selectively infect and/or lyse cancer cells without causing significant harm to normal tissue. The anti-tumor effects of viruses were first reported over a 100 years ago, with reports of cancer regressions coincident with viral infection or vaccination with live viral vaccines.1 Oncolytic viruses include in vitro passaged wild type viruses (first generation), viruses engineered to selectively replicate in cancer cells (second generation), and may additionally express transgenes to provide additional therapeutic activity (third generation). Early clinical trials used bodily fluids that contained human or animal viruses to treat patients with cancer. Although there was regression, in immunocompromised patients there was morbidity due to viral infection rather than due to the disease itself.2 However with the advent of molecular biology it has become easy to propagate and engineer some viruses and coupled to a better understanding of cancer biology it is possible to logically design tumor-selective viruses.3 Different viruses have different cell specificity and this diversity is being utilized to treat multiple kinds of cancers. Hence vectors based on Adenoviridae, Herpesviridae, Paramyxoviridae, Parvoviridae, Reoviridae, Poxviridae, Retroviridae, and Rhabdoviridae families are all being used in different clinical trials.
Introduction to immunotherapy
Paul Ehrlich suggested in 1909 that there would be more cases of cancer but for the immune system, which is able to eliminate many tumor cells. But it took almost half a century for the concept of immunosurveillance to be recognized and another half a century to prove the concepts of immunosurveillance and immunoediting.4,5 But Coley had already treated patients with mixed cultures of bacteria, thereby prodding the immune system to be activated to eliminate tumors, which heralded the concept of cancer immunotherapy in the 1800s. However cancer immunotherapy, using a person’s immune system to fight cancer, was overshadowed for many years as the immune response was not well enough understood. The administration of IL-2 as an immunotherapy was approved in 1985 by the US Food and Drug Administration (FDA) and has heralded the use of multiple different immunotherapies for the treatment of cancer.6 Recombinant cytokines (including granulocyte macrophage colony-stimulating factor [GM-CSF] and IFN) remained the mainstay of cancer immunotherapy for many years, but recent progress has led to a resurgence in the field. In particular, the development of blockade of checkpoint inhibitors, notably with antibodies against CTLA-47 or PD-1/PD-L18 and the use of TCR engineered or CAR T-cells9,10 and other adoptive cell therapy approaches have demonstrated the potential of this field.
Oncolytic viruses as immunotherapies
The first viral therapies logically designed and engineered to selectively replicate in malignant cells were pioneered in the 1990s with the clinical testing of adenovirus strains, such as ONYX-015.11–13 The basic concept assumed that selective viral infection of cancer cells or replication within these cells would result in amplification of the therapy within the target tissue and that the principal mode of tumor cell killing would be through direct viral-mediated lysis of the infected tumor cells (“onco-lysis”). The immune response raised by the viral infection was commonly considered to be necessary to limit uncontrolled viral infection but also a limitation to effective therapy. This was supported by data in immunodeficient mice, where oncolytic therapies were often more effective.14 As such, a variety of approaches were developed to combine oncolytic viral therapy with partial or transient immunosuppression in order to enhance the viral therapy’s capacity to replicate in the tumor prior to immune-mediated clearance.15–17 This included combining viral therapy with cyclophosphamide or rapamycin, or with natural killer (NK) cell depletion.
Although these pioneering clinical trials with ONXY-015 demonstrated the safety of the platform and the possibility for clinical responses, these responses were only clearly demonstrated in combination with chemotherapy indicating that additional therapeutic activity was needed. The tumor-selective viral replication and gene expression demonstrated with these therapies means they are ideal cancer gene therapy delivery vehicles,18 and so transgene expression represents a key approach to add additional mechanisms of tumor cell killing into the therapies.19 However, the fact that any infected cell would be destroyed as a result of viral replication meant that effective therapeutic transgenes would likely encode secreted proteins or peptides capable of producing a bystander killing effect.20 Also, the transient nature of oncolytic viral transgene expression further restricts the choice of effective therapeutic transgenes. Despite these limitations an extensive panel of transgenes has demonstrated the capacity to enhance oncolytic viral therapeutic effects, at least in pre-clinical models. These have included pro-drug converting enzymes,21,22 anti-angiogenic23–25 and anti-vascular proteins, monoclonal antibodies,26 apoptosis induction,27 and extracellular matrix degrading enzymes.28
However it has become evident that in addition to removing the virus directly, the immune response is also capable of killing infected tumor cells and so may enhance the overall therapeutic activity. Furthermore, when an oncolytic viral therapy produces a complete response in an immunocompetent mouse model, the animals frequently demonstrate the capacity to reject a re-challenge with the same tumor cell line.29 This indicates that an adaptive immune response can be raised by the viral infection within the tumor that is able to cross-protect against tumor associated antigens.
Because the main focus of immunotherapy at the time that oncolytic viral therapies were first described involved systemic delivery of recombinant cytokines, expressing these directly from oncolytic viral therapies was a logical step to take. Tumor-selective production of these cytokines when expressed from oncolytic viruses has the potential to enhance the targeting of the immune response against the tumor and at the same time reduce systemic toxicity relative to use of recombinant cytokines. The simple expression of single cytokines from oncolytic viral therapies therefore represents the first attempted combination of oncolytic virus and immunotherapy.
Single cytokine expression
The expression of single cytokines from oncolytic viral backbones has proven to be a potent approach to enhancing their immunotherapeutic effects.30 An extensive panel of cytokines has been tested, but the most commonly tested have been those that have proven most effective when used as recombinant proteins, notably type I IFN31–33 and GM-CSF.34–39 Although both cytokines have demonstrated the capacity to significantly enhance the therapeutic effects of different oncolytic vectors, the greatest clinical success has been with GM-CSF.34,35,37 Two separate oncolytic viral therapies have demonstrated success in randomized clinical testing, talimogene laherparepvec (T-Vec; Amgen Inc., Thousand Oaks, CA, USA) and pexastimogene devacirepvec (Pexa-Vec; Jennerex, now owned by Sillajen, San Francisco, CA, USA), these represent an oncolytic herpes simplex virus (HSV) strain expressing GM-CSF and an oncolytic vaccinia strain expressing GM-CSF. Randomized Phase III and Phase IIb studies with these viruses respectively have clearly demonstrated the potential of oncolytic viral therapy, and have provided strong support for the argument that enhancing the immunostimulatory effects of these vectors through combination with immunotherapies will be a potent approach to treating cancer.
Therapeutic effects with IFN-expressing vectors have been primarily limited to pre-clinical studies, although a Phase I study with a vesicular stomatitis virus (VSV) expressing IFN has been recently initiated.40 One possibility is that the potent anti-viral effects of IFN detrimentally affect the capacity for oncolytic viral therapies expressing this cytokine to initially infect and replicate within the tumor. Clinical data with vectors expressing IFN will look to answer this question.
The loss of oncolytic activity resulting from the enhanced immune activation seen with cytokine expression can be mitigated in several ways. One approach is to choose a cytokine whose effect is not directly anti-viral. GM-CSF does not directly block viral replication, instead having a more general effect on proliferation of hematopoietic cell lineages.41 Although some concerns have been raised that this might include cell types that have a known tumor-promoting effect, notably monocyte derived suppressor cells,42 the clinical data would indicate that the pro-inflammatory effects of viral infection within the tumor counter any such immunosuppressive effect of GM-CSF expression.
In a similar fashion to GM-CSF, chemokines can influence the overall type and level of immune response without having directly anti-viral effects. As a result several chemokines have been expressed from different oncolytic vectors without appearing to detrimentally affect viral oncolytic activity.43,44 The capacity for chemokine expression to influence the trafficking of particular immune cell subsets into the tumor still results in enhanced overall therapeutic benefit.
An alternative strategy would be to regulate the level or timing of transgene expression. Although this can be partly achieved through careful choice of promoter (such as use of a weaker late promoter that is not expressed until after viral replication has begun), perhaps the most powerful approach is to incorporate exogenous regulation of transgene activity. A variety of approaches is available that can be utilized to control activity of a gene product,45 either through regulation at the gene expression level, mRNA stability or protein stability, with addition of a specific small molecule often used to exogenously control the temporal kinetics of protein activity. In this way a cytokine that when expressed constitutively results in limited initial viral infection of the tumor or premature clearance and loss of oncolytic activity, can have its function suppressed for a controlled period of time after initial delivery.
For example, it was found that several cytokines (including IL-2 or TNF α), when constitutively expressed from an oncolytic vaccinia resulted in significant reductions in the level of viral gene expression from the tumor, despite the fact that overall therapeutic activity was increased.17,46 The immune enhancing effects of constitutive cytokine expression were still able to enhance the overall activity of the virus, despite reducing its oncolytic effects. However, if the level of cytokine activity was exogenously regulated, an initial period of unhindered oncolytic and replicative activity could be allowed to proceed. Once an immune response was naturally initiated and was beginning to limit viral oncolytic activity, a second immunotherapeutic phase of viral activity could be enhanced through exogenously activating cytokine function. This was found to result in significantly enhanced overall therapeutic benefit. Although the additional complexity involved may hinder translation of these approaches, recent descriptions of regulated systems that incorporate common antibiotics as the small molecule component would simplify use in the clinic.47
Other approaches to increasing oncolytic viral immune stimulation
The immune response is a multi-step process, with different cytokines playing key roles at different steps. The effects of any one cytokine may be very different at different steps of the immune response, and may also be concentration-dependent. As a result, there are likely to be limits to the benefits that can be achieved through expression of a single cytokine. Although expression of multiple cytokines, or cytokines and chemokines has been attempted,30 it is likely that the additional immunostimulation may result in limited further increases in therapeutic effects as a result of decreased oncolytic activity.
Several alternative approaches to engineer oncolytic viruses to increase their stimulation of all or specific components of the immune response have been reported (summarized in Table 1).
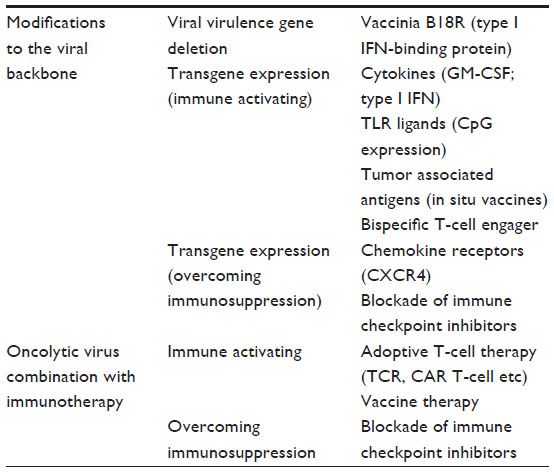 | Table 1 Approaches to enhancing the immunotherapeutic potential of oncolytic virotherapies, through either viral engineering or combination therapies |
Deletion of viral virulence genes
Because many larger DNA viruses contain multiple virulence genes whose products directly inhibit specific steps in the immune response the opportunity exists to delete or mutate one or more of these genes such that the virus loses its ability to control specific immune response steps. For example, vaccinia virus and other poxviruses have evolved distinct strategies to evade either T-cell recognition or humoral responses.48,49 As such strains engineered to lose their capacity to suppress one of these arms of the immune response may demonstrate a shift in the Th1 versus Th2 balance of the immune response raised. However, the majority of the virulence genes tend to interfere with signaling pathways or cytokines involved with activating the innate immune response, notably the IFN pathway. As such, it was demonstrated that deletion of genes that disrupt the IFN response pathway could actually result in greater therapeutic effects than simple expression of IFN from the same viral backbone.50 Consideration of the virus itself is therefore a more subtle but potentially more powerful approach to manipulating the immune response than cytokine expression.
Expression of immune stimulating molecules
Recent advances in the field of tumor vaccines have highlighted the importance of addition of adjuvant. Because oncolytic viruses naturally produce adjuvant when replication in the tumor is achieved, they can selectively induce an inflammatory response in the tumor. However, specific adjuvants capable of binding selected TLRs are becoming more commonly used to enhance cancer vaccines.51 One strategy would therefore be to modify or engineer the oncolytic vectors to further bind specific TLRs. In particular TLR9 activation is often associated with a favorable immune response and so viruses modified to produce greater amounts of CpG motifs in their DNA have been constructed and tested and have displayed enhanced therapeutic effects in pre-clinical models.52
Alternative approaches to enhance the anti-tumor T-cell response have also been tested. In particular, expression of tumor associated antigens directly from the oncolytic virus may result in enhanced in situ vaccination effects and cross-protection of the adaptive immune response against the tumor.53 These “oncolytic vaccines” may allow the raising of an adaptive immune response against additional antigens that would not normally be induced by the virus alone, such as those associated with relapse.54 However, as with all vaccine approaches, it is necessary to know that the antigen being targeted is both present and essential in order for the approach to be successful.
A related approach involves the expression of bispecific T-cell engagers from the virus. These bispecific antibodies bind T-cells and tumor antigens, so locking the T-cells in close proximity to the cancer cell targets.55
Expression of global immune regulators
Many immune responses are mediated through activation of cell signaling pathways. An alternative approach to production of factors that bind at the cell surface to activate these pathways would be to express gene products that are capable of activating steps on these pathways directly. This approach has the potential advantages that the presence of specific receptors is not necessary and the result is often more global than expression of a single cytokine. However over-activation of the immune response may be an issue.
Targeting of immunosuppression within the tumor
Antibodies that prevent activation of immune checkpoint inhibitors, including anti-PD1, anti-PDL1, and anti-CTLA4 represent some of the most promising new cancer therapies of the last 20 years.8,56 This demonstration that blocking of immune checkpoint inhibitors is a critical step in producing and maintaining a robust anti-tumor immune response has provided evidence for the need to both activate the immune response and to prevent premature immune shut-off. Viral infection within the tumor is immune activating. The fact that uncontrolled viral replication has not been reported after extensive testing of many oncolytic viral therapies in the clinic indicates that these vectors are removed by the host immune response. As such they must be capable of transiently and at least partially overcoming the local immunosuppressive environment within the tumor.
The majority of the approaches described to engineer the vectors to boost their immunotherapeutic effects has focused on boosting the immune activating component, however more recently focus has also shifted to enhancing and maintaining the vectors’ capability to overcome the localized immunosuppressive environment. One such approach has been to express a solubilized binding domain from the chemokine receptor CXCR4.57,58 CXCR4 binds the chemokine CXCL12 (SDF-1) and so this acts as a decoy receptor, sequestering the chemokine locally within the tumor microenvironment. CXCL12 is often expressed in the tumor and has been associated with many steps in tumor progression, including induction of metastasis, maintenance of cancer stem cells, and attraction of monocyte derived suppressor cells into the tumor.
Alternatively viral vectors expressing antibodies capable of blocking immune checkpoint inhibitors have also been reported.59 In a similar way to the locally enhanced effects seen after cytokine expression from oncolytic vectors, the advantage of expressing these antibodies from the virus directly is that the virus is able to produce high concentrations of the therapeutic antibody locally within the tumor microenvironment.
Combining oncolytic viruses with other immunotherapies
Amongst cancer therapies immunotherapy perhaps uniquely holds the possibility of producing curative responses. However it is evident that any one immunotherapy is unlikely to be sufficient, and that combinations will be required to optimize their effects.
One of the strengths of oncolytic viral therapies is that they hold the potential to selectively and effectively alter the tumor microenvironment in order to raise an inflammatory response, induce an in situ vaccination, and to overcome local immunosuppressive conditions. However, perhaps the greatest limitation of the approach is that these effects are transient, and repeated systemic delivery with the same viral therapy is likely to be challenging once an anti-viral immune response has been raised. Therefore, combining the capability of an oncolytic virus to transiently modify the tumor microenvironment with a systemic immunotherapy that is capable of producing a long-lasting anti-tumor immune response is an attractive proposition.
Adoptive cell transfer and vaccine therapies
The use of adoptive T-cell transfer and CAR T-cell therapies represents one of the key drivers behind the resurgence of immunotherapy as a viable cancer treatment option.60–62 However these treatments have to date been most effective against leukemia, while solid tumors remain largely resistant to their effects.
Similarly, many therapeutic cancer vaccines have demonstrated the capability of raising a robust systemic adaptive immune response targeting the tumor antigen of interest yet remain limited in their capability to produce effective responses in a clinical setting. One possible reason for this is that the cytotoxic T lymphocyte (CTLs) produced are either unable to efficiently traffic to and extravasate into the tumor, or the cells are capable of entering the tumor, but the suppressive local environment mediates their rapid subversion and they either lose their lytic potential or convert into regulatory T-cells.
The potential for combining these approaches is evident, and has been demonstrated in several pre-clinical models, including the combination of dendritic cell (DC) vaccination with oncolytic viruses expressing chemokines known to attract the T-cells produced into the tumor,43 or the combination of CAR T-cells with oncolytic virus strains expressing both chemokines and cytokines to both attract these cells into the tumor and subsequently maintain their phenotype.63 In another example the capability of an oncolytic virus to raise an in situ vaccination effect can be multiplied through a prime-boost approach with sequential application of serologically distinct oncolytic viral therapies.64
While these data demonstrate the proof-of-principal for combining the approaches, it is likely that considerable further improvement can be made through logical engineering of the viral vectors and/or the T-cell therapy to most beneficially synergize with each other.
As an alternative to using the virus to help direct the T-cells into the tumor it has also been demonstrated that some cells, including immune cells can be infected with oncolytic virus ex vivo and then be used as carrier vehicles to deliver the virus into the tumor.65,66 Because the viral particle is essentially inert outside of an infected cell, it is not capable of directed trafficking to the tumor, meaning only a very small percentage of any systemically delivered inoculum will infect the tumor. This effect is amplified in previously immunized individuals. Some cells, including some therapeutic immune cell types, have been shown to be effective at trafficking to the tumor after systemic delivery. Therefore, the use of pre-infected cells as delivery vehicles is one approach to achieving increased delivery of the virus to the tumor. This approach can even lead to evasion of immune recognition (in a “Trojan horse” mechanism). However the benefits of combining a carrier cell and an oncolytic viral payload are amplified when immune cells are used as the carrier. Uninfected immune cells that are themselves therapeutic and delivered together with the pre-infected immune cells can more efficiently target the tumor in the context of an ongoing viral infection, meaning their effects are amplified and their ability to attract additional immune cells to the tumor are increased.
Blockade of immune checkpoint inhibitors
Because the effects of an ongoing oncolytic viral infection within the tumor appear to be primarily pro-inflammatory, it is logical that they should be combined with blockade of immune checkpoint inhibitors in a synergistic fashion. This has been demonstrated in several ways; as mentioned previously the virus itself can be used to express the antibodies (such as anti-PD1, anti-PDL1 or anti-CTLA4), in order to increase their concentration locally within the tumor. This can help overcome systemic toxicities (as seen with anti-CTLA4) or to increase the concentration locally where it will be most needed (as with anti-PD1/PDL1).
Alternatively, it has been shown that combination therapies with Newcastle disease virus (NDV)67 or vaccinia68 and anti-CTLA4 can effectively treat several cancer types. However, it was observed that the benefits produced during combination of anti-CTLA4 with oncolytic vaccinia virus were dependent on the viral strains and the regimen used. Combinations beginning on the same day resulted in loss of therapeutic benefit. This was attributed to an early induction of anti-viral immunity reducing the effectiveness of the viral therapy. Synergy was instead seen when the antibody treatment was begun around 3 days after initial viral delivery. This recapitulates some of the work seen with exogenous regulation of cytokine transgenes expressed form oncolytic viral therapies.46 In both cases allowing an initial period of unhindered viral replication in the tumor before addition of the immunotherapy resulted in the greatest therapeutic benefit. Considerations of timing of oncolytic virus-immunotherapy combinations may therefore be crucial as these approaches are moved into a clinical setting.
Initial clinical testing of the oncolytic HSV strain expressing GM-CSF, T-VEC with anti-CTLA4 antibody (ipilimumab) has begun and early clinical results presented at the 2014 ASCO (American Society of Clinical Oncology) conference were impressive. Full clinical results are eagerly anticipated.
Future directions
Combining different immunotherapies with oncolytic viruses is an attractive proposition and initial studies have borne this out with reports of robust responses in pre-clinical models. To date the only clinical data reported has been preliminary results of the combination of T-VEC and ipilimumab, but again the data so far are impressive. Obviously it will be critical to see if these data are borne out in larger scale clinical testing.
However, much of this data is primarily proof-of principal in nature and so the scope for further improvement is large. It appears that a focused and logically designed approach to optimizing the design and engineering of both the viral and immunotherapeutic component (especially when CAR T-cells are incorporated) would lead to further dramatic improvements in therapeutic effects. Because of the genetic basis of both of these approaches the scope for iterative cycles of enhanced therapeutic effect is essentially unlimited.
However, perhaps the critical bottleneck in the application of these approaches will be the clinical translation. Out of this new and exciting generation of immunotherapies several antibodies to block immune checkpoint inhibitors (anti-CTLA-4 and PD1 inhibitors) are currently all that have been approved, while no oncolytic virus is currently approved in the US or Europe. Therefore designing trials combining these approaches may be difficult. It is hoped that the FDA will provide assistance in bringing these promising approaches more rapidly to the treatment of patients.
Conclusion
Cancer immunotherapy and oncolytic viral therapies represent two of the most promising new platforms for the treatment of cancer. Furthermore their mechanisms of action mean that combining these approaches would be likely to provide significant additional therapeutic benefit. This has to date been explored primarily in pre-clinical testing. It appears that there is significant scope for further engineering of the oncolytic viral components specifically to enhance these interactions. This will likely lead to new and further improved therapeutic approaches.
Disclosure
SHT has a financial interest in Western Oncolytics. The authors have no other conflicts of interest to disclose.
References
Dock G. Rabies virus vaccination in a patient with cervical carcinoma. American Journal of Medical Science. 1904;127:563. | |
Kelly E, Russell SJ. History of oncolytic viruses: genesis to genetic engineering. Mol Ther. 2007;15(4):651–659. | |
Cattaneo R, Miest T, Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6(7):529–540. | |
Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21(2):137–148. | |
Vesely MD, Schreiber RD. Cancer immunoediting: antigens, mechanisms, and implications to cancer immunotherapy. Ann N Y Acad Sci. 2013;1284:1–5. | |
Rosenberg SA. Decade in review-cancer immunotherapy: entering the mainstream of cancer treatment. Nat Rev Clin Oncol. 2014;11(11):630–632. | |
Hodi FS, O’Day SJ, McDermott DF, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–723. | |
Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–2454. | |
Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314(5796):126–129. | |
Gill S, June CH. Going viral: chimeric antigen receptor T-cell therapy for hematological malignancies. Immunol Rev. 2015;263(1):68–89. | |
Heise C, Sampson-Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med. 1997;3(6):639–645. | |
Khuri F, Nemunaitis J, Ganly I, et al. A controlled trial of Onyx-015, an E1B gene-deleted adenovirus, in combination with chemotherapy in patients with recurrent head and neck cancer. Nat Med. 2000;6(8):879–885. | |
Nemunaitis J, Khuri F, Ganly I, et al. Phase II trial of intratumoral administration of ONYX-015, a replication-selective adenovirus, in patients with refractory head and neck cancer. J Clin Oncol. 2001;19(2):289–298. | |
Gnant MF, Puhlmann M, Alexander HR Jr, Bartlett DL. Systemic administration of a recombinant vaccinia virus expressing the cytosine deaminase gene and subsequent treatment with 5-fluorocytosine leads to tumor-specific gene expression and prolongation of survival in mice. Cancer Res. 1999;59(14):3396–3403. | |
Lun XQ, Jang JH, Tang N, et al. Efficacy of systemically administered oncolytic vaccinia virotherapy for malignant gliomas is enhanced by combination therapy with rapamycin or cyclophosphamide. Clin Cancer Res. 2009;15(8):2777–2788. | |
Alvarez-Breckenridge CA, Yu J, Price R, et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat Med. 2012;18(12):1827–1834. | |
Chen H, Sampath P, Hou W, Thorne SH. Regulating cytokine function enhances safety and activity of genetic cancer therapies. Mol Ther. 2013;21(1):167–174. | |
Thorne SH, Hwang TH, O’Gorman WE, et al. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J Clin Invest. 2007;117(11):3350–3358. | |
Kirn DH, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9(1):64–71. | |
Hermiston T. Gene delivery from replication-selective viruses: arming guided missiles in the war against cancer. J Clin Invest. 2000;105(9):1169–1172. | |
Niculescu-Duvaz I, Springer CJ. Introduction to the background, principles, and state of the art in suicide gene therapy. Mol Biotechnol. 2005;30(1):71–88. | |
Hedley D, Ogilvie L, Springer C. Carboxypeptidase-G2-based gene-directed enzyme-prodrug therapy: a new weapon in the GDEPT armoury. Nat Rev Cancer. 2007;7(11):870–879. | |
Kuo CJ, Farnebo F, Yu EY, et al. Comparative evaluation of the antitumor activity of antiangiogenic proteins delivered by gene transfer. Proc Natl Acad Sci U S A. 2001;98(8):4605–4610. | |
Thorne SH, Tam BY, Kirn DH, Contag CH, Kuo CJ. Selective intratumoral amplification of an antiangiogenic vector by an oncolytic virus produces enhanced antivascular and anti-tumor efficacy. Mol Ther. 2006;13(5):938–946. | |
Tysome JR, Wang P, Alusi G, et al. Lister vaccine strain of vaccinia virus armed with the endostatin-angiostatin fusion gene: an oncolytic virus superior to dl1520 (ONYX-015) for human head and neck cancer. Hum Gene Ther. 2011;22(9):1101–1108. | |
Buckel L, Advani SJ, Frentzen A, et al. Combination of fractionated irradiation with anti-VEGF expressing vaccinia virus therapy enhances tumor control by simultaneous radiosensitization of tumor associated endothelium. Int J Cancer. 2013;133(12):2989–2999. | |
Fernandes MS, Gomes EM, Butcher LD, et al. Growth inhibition of human multiple myeloma cells by an oncolytic adenovirus carrying the CD40 ligand transgene. Clin Cancer Res. 2009;15(15):4847–4856. | |
Kim JH, Lee YS, Kim H, Huang JH, Yoon AR, Yun CO. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. J Natl Cancer Inst. 2006;98(20):1482–1493. | |
Contag CH, Sikorski R, Negrin RS, et al. Definition of an enhanced immune cell therapy in mice that can target stem-like lymphoma cells. Cancer Res. 2010;70(23):9837–9845. | |
Lichty BD, Breitbach CJ, Stojdl DF, Bell JC. Going viral with cancer immunotherapy. Nat Rev Cancer. 2014;14(8):559–567. | |
Shashkova EV, Spencer JF, Wold WS, Doronin K. Targeting Interferon-alpha Increases Antitumor Efficacy and Reduces Hepatotoxicity of E1A-mutated Spread-enhanced Oncolytic Adenovirus. Mol Ther. 2007;15(3):598–607. | |
Li H, Peng KW, Dingli D, Kratzke RA, Russell SJ. Oncolytic measles viruses encoding interferon beta and the thyroidal sodium iodide symporter gene for mesothelioma virotherapy. Cancer Gene Ther. 2010;17(8):550–558. | |
Kirn DH, Wang Y, Le Boeuf F, Bell J, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4(12):e353. | |
Park BH, Hwang T, Liu TC, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9(6):533–542. | |
Heo J, Reid T, Ruo L, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19(3):329–336. | |
Liu BL, Robinson M, Han ZQ, et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003;10(4):292–303. | |
Andtbacka RH, Collichio FA, Amatruda T, Senzer N, Chesney J, Delman KA, et al. OPTiM: A randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected stage IIIB/C and IV melanoma. J Clin Oncol. 2013;31(Suppl):LBA9008. | |
Chang J, Zhao X, Wu X, et al. A Phase I study of KH901, a conditionally replicating granulocyte-macrophage colony-stimulating factor: armed oncolytic adenovirus for the treatment of head and neck cancers. Cancer Biol Ther. 2009;8(8):676–682. | |
Breitbach C, Bell JC, Hwang T-H, Kirn DH, Burke J. The emerging therapeutic potential of the oncolytic immunotherapeutic Pexa-Vec (JX-594). Oncolytic Virother. 2015;4:25–31. | |
Willmon CL, Saloura V, Fridlender ZG, et al. Expression of IFN-beta enhances both efficacy and safety of oncolytic vesicular stomatitis virus for therapy of mesothelioma. Cancer Res. 2009;69(19):7713–7720. | |
Thorne SH. The role of GM-CSF in enhancing immunotherapy of cancer. Immunotherapy. 2013;5(8):817–819. | |
Kohanbash G, McKaveney K, Sakaki M, et al. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-alpha. Cancer Res. 2013;73(21):6413–6423. | |
Li J, O’Malley M, Urban J, et al. Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer. Mol Ther. 2011;19(4):650–657. | |
Li J, O’Malley M, Sampath P, Kalinski P, Bartlett DL, Thorne SH. Expression of CCL19 from oncolytic vaccinia enhances immunotherapeutic potential while maintaining oncolytic activity. Neoplasia. 2012;14(12):1115–1121. | |
Thorne SH. Enhancing biological therapy through conditional regulation of protein stability. Expert Rev Mol Med. 2010;12:e2. | |
Banaszynski LA, Sellmyer MA, Contag CH, Wandless TJ, Thorne SH. Chemical control of protein stability and function in living mice. Nat Med. 2008;14(10):1123–1127. | |
Sando R 3rd, Baumgaertel K, Pieraut S, et al. Inducible control of gene expression with destabilized Cre. Nat Methods. 2013;10(11):1085–1088. | |
Buller RM, Palumbo GJ. Poxvirus pathogenesis. Microbiol Rev. 1991; 55(1):80–122. | |
Smith GL, Benfield CT, Maluquer de Motes C, et al. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol. 2013;94(Pt 11):2367–2392. | |
Wang LC, Lynn RC, Cheng G, et al. Treating tumors with a vaccinia virus expressing IFNbeta illustrates the complex relationships between oncolytic ability and immunogenicity. Mol Ther. 2012;20(4):736–748. | |
Zhu X, Fallert-Junecko BA, Fujita M, et al. Poly-ICLC promotes the infiltration of effector T cells into intracranial gliomas via induction of CXCL10 in IFN-alpha and IFN-gamma dependent manners. Cancer Immunol Immunother. 2010;59(9):1401–1409. | |
Cerullo V, Diaconu I, Romano V, et al. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol Ther. 2012;20(11):2076–2086. | |
Zhang J, Tai LH, Ilkow CS, et al. Maraba MG1 virus enhances natural killer cell function via conventional dendritic cells to reduce postoperative metastatic disease. Mol Ther. 2014;22(7):1320–1332. | |
Kottke T, Boisgerault N, Diaz RM, et al. Detecting and targeting tumor relapse by its resistance to innate effectors at early recurrence. Nat Med. 2013;19(12):1625–1631. | |
Albelda SM, Thorne SH. Giving oncolytic vaccinia virus more BiTE. Mol Ther. 2014;22(1):6–8. | |
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271(5256):1734–1736. | |
Gil M, Seshadri M, Komorowski MP, Abrams SI, Kozbor D. Targeting CXCL12/CXCR4 signaling with oncolytic virotherapy disrupts tumor vasculature and inhibits breast cancer metastases. Proc Natl Acad Sci U S A. 2013;110(14):E1291–E1300. | |
Gil M, Komorowski MP, Seshadri M, et al. CXCL12/CXCR4 blockade by oncolytic virotherapy inhibits ovarian cancer growth by decreasing immunosuppression and targeting cancer-initiating cells. J Immunol. 2014;193(10):5327–5337. | |
Dias JD, Hemminki O, Diaconu I, et al. Targeted cancer immunotherapy with oncolytic adenovirus coding for a fully human monoclonal antibody specific for CTLA-4. Gene Ther. 2012;19(10):988–998. | |
June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 2007;117(6):1466–1476. | |
Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3(95):95ra73. | |
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric Antigen Receptor-Modified T Cells in Chronic Lymphoid Leukemia. N Engl J Med. 2011;365(8):725–733. | |
Nishio N, Diaconu I, Liu H, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 2014;74(18):5195–5205. | |
Tysome JR, Li X, Wang S, et al. A novel therapeutic regimen to eradicate established solid tumors with an effective induction of tumor-specific immunity. Clin Cancer Res. 2012;18(24):6679–6689. | |
Thorne SH, Negrin RS, Contag CH. Synergistic antitumor effects of immune cell-viral biotherapy. Science. 2006;311(5768):1780–1784. | |
Cole C, Qiao J, Kottke T, et al. Tumor-targeted, systemic delivery of therapeutic viral vectors using hitchhiking on antigen-specific T cells. Nat Med. 2005;11(10):1073–1081. | |
Zamarin D, Holmgaard RB, Subudhi SK, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci Transl Med. 2014;6(226):226ra32. | |
Rojas JJ, Sampath P, Hou W, Thorne SH. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin Cancer Res. In press 2015. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.