Back to Journals » Degenerative Neurological and Neuromuscular Disease » Volume 6
Novel targets for Huntington’s disease: future prospects
Received 24 December 2015
Accepted for publication 9 February 2016
Published 4 May 2016 Volume 2016:6 Pages 25—36
DOI https://doi.org/10.2147/DNND.S83808
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Müller
Sarah L Mason,1 Roger A Barker1,2
1John van Geest Centre for Brain Repair, 2Department of Clinical Neuroscience, University of Cambridge, Cambridge, UK
Abstract: Huntington’s disease (HD) is an incurable, inherited, progressive, neurodegenerative disorder that is characterized by a triad of motor, cognitive, and psychiatric problems. Despite the noticeable increase in therapeutic trials in HD in the last 20 years, there have, to date, been very few significant advances. The main hope for new and emerging therapeutics for HD is to develop a neuroprotective compound capable of slowing down or even stopping the progression of the disease and ultimately prevent the subtle early signs from developing into manifest disease. Recently, there has been a noticeable shift away from symptomatic therapies in favor of more mechanistic-based interventions, a change driven by a better understanding of the pathogenesis of this disorder. In this review, we discuss the status of, and supporting evidence for, potential novel treatments of HD that are currently under development or have reached the level of early Phase I/II clinical trials.
Keywords: disease modification, transcript dysregulation, glial modulation, cell death
Introduction
Huntington’s disease (HD) is an incurable, inherited, progressive, neurodegenerative disorder that is characterized by a triad of motor, cognitive, and psychiatric problems,1 although it is now widely recognized that there are clinical features that extend beyond these domains, such as abnormalities in sleep2,3 and metabolism.4
The genetic basis of HD is an unstable CAG expansion within exon 1 of the huntingtin gene, located on the short arm of chromosome 4,5 which leads to the production of abnormal mutant huntingtin (mHtt) that is ubiquitously expressed throughout the human body. As such, the pathology of HD is not exclusively restricted to the brain,6 but is nevertheless most clearly seen at this site, with many structural scanning studies in premanifest gene carriers revealing significant striatal atrophy prior to the onset of clinical features. In some cases, these changes are found a decade or more in advance of patients’ estimated time for disease onset,7–16 and the extent of striatal (especially caudate) atrophy increases as patients approach clinical onset and start to develop motor symptoms and signs.10,15 Of course, changes in a large number of extrastriatal sites have also been reported, in some cases in premanifest patients, including atrophy of the globus pallidus,12,14 thalamus,12,16 amygdala,17 and insula,14 but none shows the same level of correlation with disease onset as those associated with striatal volume loss.11
HD is a relatively rare condition with considerably lower prevalence than many other neurodegenerative diseases, such as Alzheimer’s or Parkinson’s disease. Approximately 2.71 people per 100,000 worldwide18 are affected by the disease, with approximately 30,000 people in the USA and 38,000 people in Europe currently living with HD, and a further 250,000 people worldwide who are at risk of having inherited the mutated gene.19 Numbers appear to be increasing, as revealed in a recent study which estimated that more than 5,700 people in the UK over the age of 21 years had clinical features of HD in 2010, almost twice as many as that reported in 1990.20 The reason for this increase is unclear, although it is likely to reflect advances in the diagnostic process and the greater availability and improved management of patients in specialist clinics with subsequently increased life expectancies. The exact prevalence of the disease is hard to determine because despite genetic testing being available in most countries, only between 4% and 24% of at-risk individuals choose to be tested for the expansion.21 Social stigma and the potential for discrimination by both employers and insurance companies probably contribute to the reasons underlying the poor uptake of predictive testing in the UK, along with the current lack of disease-modifying therapies and the inadequacy of information provided by general practitioners.22 However, regardless of the reasons, the increased prevalence of HD results in an increase in the economic burden of the disease, which is already substantial. It has been estimated that the direct medical costs of a patient with late-stage HD in the USA is between $22,582 and $37,495 per year, much of which can be attributed to nursing home care costs.23 Given this, new therapeutic options are greatly needed, of both the symptomatic and disease-modifying type.
Clinical features of HD
The dominant motor feature of HD is the presence of distinctive choreic movements that are relentless during waking hours, cannot be voluntarily suppressed, and worsen in stressful situations.24 These are accompanied by problems of gait, speech, and swallowing. In the later stages of the disease, these choreic features tend to give way to bradykinesia and rigidity, resulting in increased dependence on others, and in the final stages of the disease immobility. In juvenile cases, more often, the chorea is not seen, and the patients develop a progressive parkinsonian state with profound bradykinesia, tremor, and myoclonus.
Although HD has traditionally been thought of as a movement disorder, cognitive and psychiatric disturbances are also seen in nearly all cases of HD, begin early in the course of the disease, and in many cases appear ahead of any overt motor features.25,26 The cognitive impairment in HD is classified as a subcortical dementia, typically with cognitive deficits that are consistent with disruption to the functional integrity of the frontostriatal circuitry.27 In early disease, the cognitive difficulties experienced by patients tend to be highly specific with a characteristic pattern of impairment, although the severity of this does vary between individuals or within individual families. Cognitive decline is slowly progressive and tends to correlate with the extent of atrophy in cortical28,29 and subcortical structures,28 eventually leading to global cognitive impairment in most, but not all, cases.30
HD is also associated with a wide range of psychiatric symptoms, including affective disorders, irritability, apathy, and psychosis.31,32 The exact underlying pathology responsible for these aspects of the disease is currently unknown. As psychiatric symptoms tend to cluster in certain HD families, even in non-gene-carrying relatives,33,34 it has been suggested that affective changes may be a response to the emotional stress of living in an HD family. However, while such a relationship may play a part, it is unlikely to be the whole explanation.
Recent studies have shown that it is the cognitive and psychiatric difficulties experienced by patients, and not the movement disorder, that put the largest burden on HD families, and which are most predictive of the need for nursing home care.35–37 Therefore, treating these aspects of the disease, rather than the motor features, may have the greatest impact on quality of life for both the patient and their carer/family.
Existing treatments
The course of HD is relentless: following disease onset, the clinical features will progress over a course of 20 years and will ultimately result in death.38 Despite the noticeable increase in HD therapeutic trials in the last 20 years, there have been very few significant breakthroughs.39 Currently, few high-quality clinical trials have been conducted, although this is changing with the establishment of such networks as the Huntington’s Study Group and the European Huntington’s Disease Network, who liaise directly with industry partners to ensure the quality and relevance to HD of all new trial protocols. The majority of drugs presently used in clinical practice are chosen not based on reproducible verifiable evidence, but more on anecdotal clinical experience, with the only empirical evidence to support their use coming from case reports or small open-label studies. This is especially the case for both the cognitive and psychiatric aspects of the disease. In a recent study of European prescription practices, 84% of the registered 2,128 patients with HD were prescribed symptomatic treatments, of which 50% were for depression compared to 28% that received antichoreic medication. Patients were also commonly treated for irritability and aggression (13%), sleep disturbances (9%), and psychosis (7%), indicating that nonmotor aspects of the disease represent the major burden of the condition.40
To date, tetrabenazine is still the only drug licensed for use in this disease, although antipsychotic medications, such as olanzapine or sulpiride, are commonly prescribed off label to help control aberrant movements, with the added benefit that they help to stabilize mood, improve sleep, and increase weight.39 However, the ultimate hope for new and emerging HD therapeutics is to develop a neuroprotective compound capable of slowing down or even stopping the progression of the disease and ultimately preventing the subtle early signs from developing into manifest disease. Until recently, few compounds that had demonstrated promise in preclinical studies went on to be the subject of major clinical trials and those that have, have been unsuccessful. However, this may change as research moves more toward drug screening with regard to specific mechanistic-based interventions.41
This change has been driven in part by a better understanding of the underlying pathogenic pathways in HD. These pathogenic processes include transcriptional dysregulation; excitotoxic death of neurons mediated by glutamate and N-methyl-d-aspartic acid (NMDA) receptors, possibly through a primary glial pathology; neuronal dysfunction and death due to perturbations in mitochondrial function; loss of trophic support to specific populations of cells; inappropriate activation of apoptotic pathways; and/or dysfunction of intracellular protein-clearance mechanisms, such as autophagy and the ubiquitin–proteasome system.
Currently, disease-modifying therapies can be thought of as taking one of two broad approaches: 1) targeting the processes by which mHtt is believed to cause cell death or 2) lowering the level of mHtt in the brain. In this review, we focus our discussion on novel drug targets for HD that have sufficient preclinical data to be developed to the point of early clinical trials but have not been developed beyond a Phase II study. Our discussion is limited to pharmacological interventions; for further information on cell-based therapies, the reader is referred to recent reviews.42,43
Search methods
To identify the ongoing and recently completed trials in HD, a PubMed search was undertaken with the following search terms: “Huntington’s disease” and either “clinical trial” or “randomised clinical trial” and the generic name for the drug of interest. Only full-text, English-language articles on drugs at Phase II of development or earlier were selected. In addition, the US National Institutes of Health trials database (clinicaltrials.gov) and EU Clinical Trials Register (clinicaltrialsregister.eu) websites were searched to identify any ongoing or recently completed Phase I/II trials. Finally, the reference list of each publication was searched in an attempt to get a comprehensive summary of the literature available. Table 1 details the trials listed as ongoing in these databases, while Table 2 details those trials classified as completed.
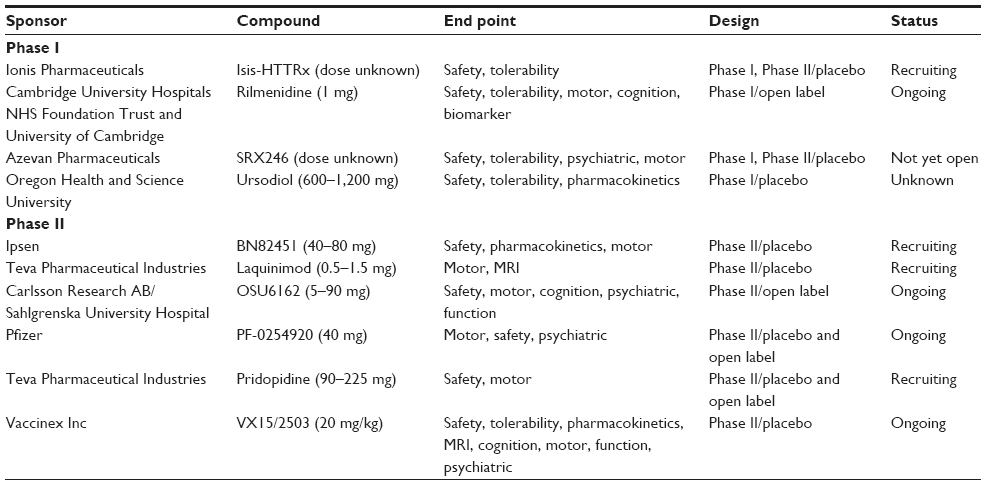 | Table 1 Ongoing trials |
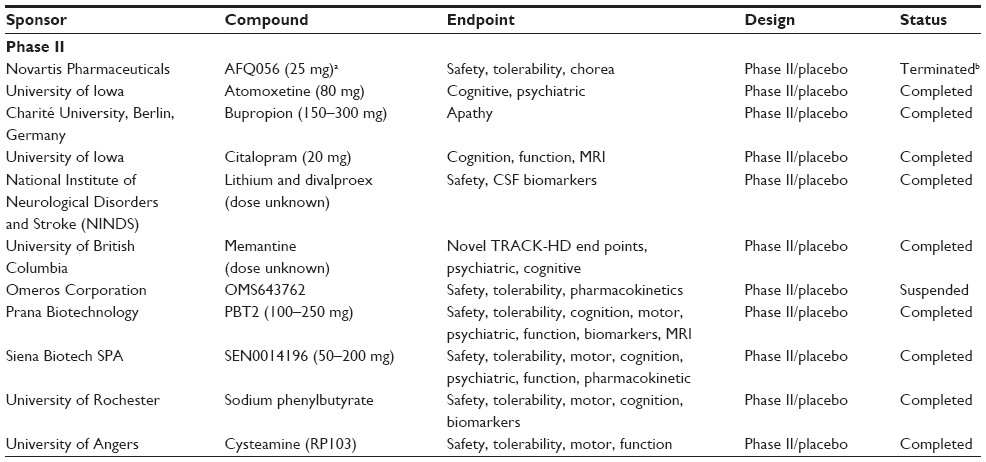 | Table 2 Completed trials |
Symptomatic treatments
It has been speculated that a major underlying cause of the involuntary movements in HD results from a relative overactivity of the dopaminergic system in the striatum. Both OSU6162 and pridopidine are drugs with a similar chemical and pharmacological profile that both act on the monoaminergic receptors in the brain. They have been shown to work on the dopaminergic and serotonergic receptor subpopulations and facilitate behavior by either stimulating or inhibiting the receptor depending upon whether the baseline levels of dopamine are too low or too high, respectively.44–46 Pridopidine has already been subject to both Phase II and Phase III studies, and while there was evidence of a statistically significant, dose-dependent improvement on the Unified Huntington’s Disease Rating Scale after 26 weeks of treatment, the primary outcome measure for the study, a modified motor score, was not met.47,48 Consequently, the drug has now been bought by Teva Pharmaceuticals, who have taken it back to a Phase II study looking at the safety and efficacy of higher doses of pridopidine on motor, cognitive, and psychiatric function.
The related agent, OSU6162, was well tolerated in a smaller clinical trial, with improvement on several nonmotor variables, including depression, although it did not improve motor performance in this short (8-week) double-blind crossover study in 18 patients with HD.49 OSU6162 is now the subject of a larger and longer open-label safety study across multiple diseases, of which HD is one.
One attraction of these drugs relates to their theoretical potential to provide beneficial symptomatic relief for patients across the full disease spectrum, as the changes in dopamine neurotransmission are biphasic in HD, with a relative increase in dopamine levels in early disease that transitions to a state of relative dopamine depletion later on in the condition.50 Given this, compounds that can act across these changes in dopamine transmission have the potential to provide unique benefits that are simply not possible with the drugs that are currently used in the clinic. However, to date clinical studies have focused on patients with mid-to late-stage disease, so it is still unknown whether either pridopidine or OSU6162 can truly provide symptomatic benefit across all stages of HD.
An alternative target to the dopaminergic system is the glutamatergic receptor, which is also expressed in disease-relevant areas of the central nervous system (CNS). Early evidence from rodents that received excitotoxic amino acid lesions of the striatum indicated that NMDA-receptor antagonists may be of benefit in HD.51,52 For this reason, NMDA-receptor antagonists, such as amantadine, have been used to manage some of the features of HD,39 with only limited success.
A novel drug that targets another aspect of this glutamatergic system is AFQ056, an mGluR5 antagonist. It is believed that the aberrant movements in HD may be partially caused by disinhibition of the glutamatergic inputs/neurons in the striatum/ subthalamic nucleus.53 Therefore, it has been postulated that mGluR5 antagonists may be helpful at reducing chorea in HD. While this hypothesis was not supported by the results from a recent randomized placebo-controlled trial, there was some evidence of improved motor function on a novel speeded tapping task, although the clinical relevance of this to everyday motor performance is still not fully understood.54 However, this was a very short study (32 days) with a small number of patients (n=42 randomized to either drug or placebo), and patients were only dosed at the maximum dose (150 mg) for 7 days. Therefore, the study was underpowered to demonstrate efficacy, and the results should be interpreted with caution. Furthermore, patients were permitted to be on existing antipsychotic, antidepressant, and benzodiazepine treatments throughout the trial, making it difficult to draw any firm conclusions from this study.
The development of symptomatic motor treatments for HD has now extended to include functional neurosurgery. Deep brain stimulation (DBS) has been identified as a potential treatment in some patients with refractory drug-resistant chorea. The identification of the target of DBS in HD is often complicated by atrophy of, and structural changes within, the basal ganglia that occur early in the disease course and evolve over time. The internal globus pallidus is the most commonly selected target, and there is accumulating evidence to suggest that DBS of this region may improve chorea,55–61 although this has to be measured against the fact that it can worsen other motoric aspects of this disease, such as dysarthria and bradykinesia.56 Stimulation of the internal pallidus has been shown to be both safe and to have a positive effect on quality of life.61 However, these data should be interpreted with caution, as all studies are open label and conducted in groups of no more than seven patients. Furthermore, the patients selected for such interventions are obviously not representative of the HD population as a whole. To truly assess the efficacy of this treatment a much larger, well-controlled trial needs to be conducted.
Aside from the motoric aspects of HD, it is now widely acknowledged that the cognitive and psychiatric aspects of HD are profoundly disabling and strongly influence the extent and duration of time that patients can continue to function independently. As such, work has been invested in developing novel drugs to help manage these aspects of the disease.
SRX246 is a highly selective V1A receptor antagonist that has been trialed as a way to treat the neuropsychiatric symptoms of HD, in particular, irritability and aggression. This is based on the premise that social and emotional behavior in humans is mediated by the pituitary hormone arginine vasopressin (AVP),62 and that AVP is reduced from the very early stages of HD in both humans63 and rodent models.64 However, the relationship between vasopressin and the psychiatric aspects of HD has not been unequivocally established as yet, and thus needs to be investigated further. The dominant AVP-receptor subtype in the CNS is the V1A receptor, which can be found throughout the limbic system and in several cortical regions, all of which support its role in emotion regulation. As such, this receptor represents a potential therapeutic target for treating stress-related neuropsychiatric symptoms, such as aggression and depression.65 SRX246 has been shown to be capable of penetrating the blood–brain barrier in preclinical models66 and has also been shown to block the effect of intranasal vasopressin on the neural response to angry faces in healthy participants67 in brain regions relevant to HD, such as the precuneus, anterior cingulate, and putamen. A Phase I safety, tolerability, and efficacy study of SRX246, sponsored by Azevan Pharmaceuticals, is due to start shortly in the hope that this may prove to be a useful drug for controlling the behavioral problems seen in HD. Furthermore, SRX246 may also have the potential to improve patients’ abilities to process their own emotions and those of others, an abnormality that is apparent even in premanifest disease68 and causes profound problems with social interactions.
Other attempts to ameliorate the psychiatric features of HD have so far proven unsuccessful. Bupropion is a dopamine–norepinephrine-reuptake inhibitor that has been shown to rescue depressive-like features in the R6/1 mouse model of HD at the premanifest stage.69,70 A recent Phase II placebo controlled crossover trial looked at the efficacy of bupropion in treating apathy in HD. The trial finished in late 2014, but the results are yet to be published. Similarly, atomoxetine has been trialed in HD, as it is a nonstimulant norepinephrine-reuptake inhibitor that is commonly used to treat adults and children with attention deficit/hyperactivity disorder. Given the similarity between the cognitive deficits seen in HD and these developmental conditions, a small randomized, placebo-controlled, double-blind, crossover study to evaluate the safety and efficacy of atomoxetine was conducted in 20 adults with early HD over a 10-week period. Unfortunately, there was no benefit of the drug on attention or executive function, although it was well tolerated. However, considering the small number of participants in this trial, it is possible that the study was not sufficiently powered to see an effect, and thus no real conclusions can be drawn on the ultimate utility of this agent.
In light of the disappointing results presented herein and the fact that there are many CNS sites of interaction between the glutamatergic and dopaminergic systems in the brain, it may be that targeting both dopamine- and glutamate-receptor dysfunction in HD provides the best strategy for symptom relief. This approach is yet to be tested in any HD clinical trial.
Disease-modifying therapies
In the absence of a cure, the ultimate aim in HD research is to develop a disease-modifying compound capable of slowing the progression of this disease. Well-timed administration of such a drug would delay disease onset and potentially prevent its manifestation entirely. For the first time, several potential disease-modifying drugs are currently in the early stages of clinical development targeting a range of possible neuropathological mechanisms, but specifically transcriptional dysregulation, the immune system, apoptotic and excitotoxic cell-death pathways, and the actual production of mHtt.
Modifying transcriptional dysregulation
Transcriptional regulation is abnormal in HD and believed to be a critical aspect driving the pathology of the disease. As such, modulating transcription through histone acetylation has been proposed as a potential therapeutic target in HD. These agents work by inhibiting the enzyme histone deacetyltransferase, which prevents the removal of acetyl groups from the DNA-transcriptional process. Sodium phenylbutyrate is a histone deacetyltransferase inhibitor that has been shown to be well tolerated in HD at a maximum dose of 15 g/day,71 but the study was not powered to demonstrate efficacy. Currently, no further clinical development of this agent has been pursued.
Similarly, selisistat (SEN0014196), a selective inhibitor of SirT1, works by modulating mHtt gene expression through histone hypoacetylation and decreased acetyltransferase activity. Inhibition of SirT1 increases acetylation of specific lysine residues and thereby increases the rate of mHtt-specific autophagocytic clearance.72 At a preclinical level, selisistat has been shown to be cytoprotective and neuroprotective in the Drosophila and R6/2 mouse model of HD, with evidence to show it increases life span and delays psychomotor abnormalities and inclusion formation.73 This drug has now been demonstrated to be safe and well tolerated in healthy adults74 and early stage patients with HD as a result of a European Commission-funded FP7 grant: the Paddington study (http://cordis.europa.eu/result/rcn/156494_en.html). Thus, Sienna Biotech SPA have pursued the development of this drug with a further Phase II study, which has recently been completed, although the results are yet to be published.
At this stage, it is not possible to draw any conclusions on the validity of this approach, given that there is no signal of efficacy with either drug. However, both drugs are well tolerated and warrant further investigation, given the preclinical data.
Immune/glial modulation
One pathogenic pathway that has been implicated in HD is activation of the innate immune system as a result of the cell-autonomous effects of mHtt in monocytes and microglia. Therapies are being tested that target this process in HD, including the immunomodulator laquinimod and the SemA4D inhibitor VX15/2503. Although the exact mechanisms of action for laquinimod are not known, it can reduce nuclear factor kappaB (NFκB) activity in astrocytes and by so doing may restore brain-derived neurotrophic factor (BDNF) levels within the brain; it may also act on the MAPK-signaling pathway, leading to a reduction in the phosphorylation of p38 and JNK.75 A Phase II, double-blind, placebo-controlled trial assessing the safety and efficacy of three doses of laquinimod is currently recruiting participants and will conclude in 2017. Targeting the same system but with a different mechanism of action, the VX15/2503 antibody is designed to block the functional activity of the protein SemA4D, an axon-guidance molecule, and subsequently reduce microglia and astrocyte activity in the HD brain as a way of slowing the disease process. A new Phase II trial adopting this strategy, sponsored by Vaccinex, is under way in HD and will use imaging markers in combination with the standard HD-related end points to assess the safety, tolerability, and efficacy of this vaccine, with results also anticipated in 2017.
Inhibiting mediators of cell death
Memantine is an NMDA-receptor antagonist that improves cognition in other neurodegenerative diseases, such as Parkinson’s disease.76 The loss of cells, especially in the striatum in HD, has been linked to NMDA-mediated cytotoxicity, and evidence from primate models suggests that memantine is capable of reducing this cytotoxicity,77 as is also seen in transgenic mouse models of HD.78 Open-label clinical trials have been undertaken and suggest that the use of memantine may slow the progression of the disease79 and provide symptomatic benefit motorically;80 however, both these studies were open label and involved only a small number of patients.
Since the degeneration of striatal medium spiny neurons is pivotal in HD pathology, strategies for reversing this cell loss include cell replacement, while an alternative involves using agents to enhance endogenous neurogenesis and regenerate the damaged tissue. However, while this will increase the neuronal population in the striatum, it does nothing to change the mechanisms underlying the initial cause of the degeneration, leaving the new neurons vulnerable to the same pathological processes. To combat this, it has been suggested that combined treatments may be better, and one possibility would involve using memantine and cell-replacement therapies to deal with the striatal pathology of HD.81 To our knowledge, this approach has not been tested experimentally.
The enzyme PDE10 is responsible for regulating intracellular signaling by hydrolyzing cyclic nucleotides and is specifically and highly expressed in striatal medium spiny neurons.82 Inhibition of PDE10A leads to an increased accumulation of cyclic adenosine monophosphate and cyclic guanosine monophosphate in the striatum83 and increased phosphorylation of a number of signaling-related proteins, such as Glu1 subunits84 and extracellular signal-regulated kinases.85 Furthermore, given that the mHtt protein interacts with the CBP/CREB-binding protein,86 it has been postulated that increasing cyclic adenosine monophosphate could lead at least partially to a restoration of this pathway. In preclinical studies, acute administration of a PDE10A antagonist reduced striatal excitotoxicity in a model of HD induced by quinolinic acid injection,87 while chronic administration improved both striatal and cortical morphological changes and motor and cognitive function,88 including spatial and recognition memory.89 Consequently, these drugs have been taken to the clinic with two independent trials of chronic administration of PDE10. One involves a randomized, double-blind, placebo-controlled trial of PF-0254920, which was started in early 2015 in a group of 260, and a second study involves a randomized, double-blind, placebo-controlled trial of OMS643762 in 120 patients with HD. Both studies are looking at the relative efficacy of different doses of the drug on motor, cognitive, and psychiatric performance, with additional imaging end points in the PF-0254920 study. The results of neither study are yet available.
BN82451 is a novel compound from a family of small molecules designed to engage with multiple target pathways,90 of which the three major ones are, neuronal death, namely, excitotoxicity, oxidative stress, and inflammation, although it is also believed to have mitochondria-protective properties. All of these pathways have been implicated in the neuropathology of HD. Following preclinical testing that demonstrated a significant amelioration of motor deficits and extended survival in R6/2 mice,91 a Phase II safety and tolerability study was begun in a small population of patients with manifest HD, which was due to finish in late 2015. The study aimed to collect additional data on the pharmacokinetics of this drug, as well as look for any sign of efficacy on the motor profile of HD. However, participants were being dosed for only 4 weeks, and it is questionable whether it is possible to truly evaluate all of this over such a short period in such small number of patients.
Other ongoing trials that are yet to report their results include a study of rilmenidine, an antihypertensive agent that was previously approved for use by the US Food and Drug Administration for this purpose. It is known to cross the blood–brain barrier and is believed to induce autophagy through an mTOR-independent pathway.92 Evidence from rodent models of HD suggests that this agent is capable of reducing soluble mHtt but not aggregates in the brain.93 A small investigator-led open-label feasibility study looking at the effect of rilmenidine in early HD is ongoing in Cambridge, UK, with the results anticipated in 2016. Another study is looking at ursodiol, which is an exogenous form of ursodeoxycholic acid, the precursor to the bile acid taursodeoxycholic acid, which is synthesized in the liver. Ursodiol is believed to function as an antiapoptotic agent, and as such has been looked at in patients with HD. A Phase I safety, tolerability, and pharmacokinetic study of the drug was started in 2007, and was due to finish in 2009; to our knowledge, the results have never been published.
Cysteamine has also been trialed in HD. Cysteamine is the reduced form of cystamine, which is a transglutaminase inhibitor that has been shown to be neuroprotective in the R6/2 and other transgenic mouse models of HD. In these studies, when administered prior to the onset of clinical features, the agent offered striatal neuroprotection with reduced Htt-inclusion formation and reduced transglutaminase activity.94,95 In the more slowly progressing R6/1 mouse model of HD, cysteamine was also shown to be neuroprotective, but the authors postulated that in this case it also worked by increasing BDNF.96 This drug has previously been shown to be well tolerated in patients with HD,97,98 and recently a new 36-month, delayed-onset trial of cysteamine reported its preliminary findings via a press release. This reported that cysteamine slowed the rate of motor deterioration with a 25% reduction on the Unified Huntington’s Disease Rating Scale compared to placebo (http://www.raptorpharma.com/pipeline/rp103-dr-cysteamine-for-huntingtons-disease). Given these positive results, it is hoped that researchers continue to explore this therapeutic avenue in a larger, more comprehensive study.
“Huntingtin-lowering” approaches
Regardless of the exact cause, the neuropathological abnormalities in HD are directly related to the accumulation of mHtt into intracellular aggregates (inclusions) that can be found in both the cortical and striatal neurons.99 While it is widely accepted that the aggregation process is central to HD pathogenesis, it is yet to be established whether the aggregates have a causal role in neurotoxicity (for a comprehensive review of the subject, see Rubinsztein and Carmichael100 or Arrasate and Finkbeiner).101 Extensive work is under way to better understand the changes that occur in HD at a molecular level, including where within the cell the aggregates form and how their presence affects the behavior and survival of the neuron. Better understanding of these processes will allow researchers to identify new therapeutic approaches for reducing the buildup of aggregates or toxic species of mHtt. To date, no such approach has made the transition from preclinical to clinical studies, although an extensive amount of animal work is ongoing in this area.102
Nevertheless, any therapy that directly inhibits the expression of Htt would theoretically attack the primary disease mechanism and thus modify the pathological course of HD. Isis 443139 is an antisense oligonucleotide that has been designed to do exactly that, by targeting Htt messenger RNA to reduce its synthesis, and by so doing lead to a reversal of the clinical signs and symptoms of HD. Until recently, there were problems with this whole approach, including issues of delivery across the blood–brain barrier and their capacity to distribute the therapeutic agent across the whole CNS and “treat” all affected cells.103 However, Ionis (formerly Isis) Pharmaceuticals has developed a strategy to overcome some of the problems by delivering specific antisense oligonucleotides that target Htt messenger RNA via an intrathecal route with a system that allows good brain penetration of their therapeutic agent. This approach has previously been employed without significant side effects in patients with amyotrophic lateral sclerosis caused by SOD1 gene mutations.104 Isis 443139 is currently the subject of an international, Phase I, first-in-human, safety trial, and the first patient was dosed with this agent in October 2015. The results of this study are anticipated in late 2017. Alternative approaches to lowering mHtt expression include the use of zinc-finger protein repressors and RNA interference, but development of these approaches is still at the preclinical stage.
It is, however, worth acknowledging that as yet, the safety of all these approaches is unestablished, especially with respect to the extent to which the therapeutic agent targets only mHtt and not the normal wild-type protein.105 Indeed, the long-term consequence of lowering wild-type Htt is unknown. While the preclinical data have indicated that HTT knockdown is well tolerated,106–109 the true safety profile of this whole approach will only be established through human trials. Furthermore, these types of experimental therapeutic strategies have the potential to produce sustained side effects without the option to withdraw the therapy, making it even more important that a considered and cautionary approach is taken with any trials.
Apart from these mHtt-specific approaches, alternative ways of treating the protein aggregation in HD have been entertained. For example, in both rodent models of HD and patient studies, it has been shown that aggregation of mHtt is promoted by increased concentrations of redox-active metals, such as iron and copper.110,111 However, in R6/2 mice exposed to PBT2, a moderate-affinity 8-hydroxyquinoline transition metal ligand that acts as a chaperone for copper zinc and iron, led to improvements in motor performance, with increased body and brain weight and a prolonged life span.112 A recent Phase II, randomized, double-blind, placebo-controlled trial into the safety of PBT2 concluded that the drug was well tolerated. However, there was no evidence of an improvement on cognition, which was the secondary end point for the study, although there was a modest improvement in the Trail Making Test Part B in the highest-dose group (250 mg).113
Cell transplantation and regeneration
Cell-replacement therapies aim to counteract the disease process in HD by replacing cells lost to the disease process, such as the striatal projection neurons. An alternative but related approach seeks to protect vulnerable cells (typically those targeted for cell replacement) through the cellular or viral delivery of neuroprotective neurotrophic factors (for an in-depth review, see Clelland et al).42 Data from excitotoxically lesioned animals have demonstrated the potential value of these approaches,114 and data from early open-label clinical trials of fetal striatal cell transplantation in mild-to-moderate HD have demonstrated the safety of the technique115,116 and also gave some signal of efficacy by virtue of a reduction in the rate of decline in some patients,117–119 with improvement of symptoms120 and reduction in disease pathology.121 However, these studies were all small, unblinded, and lacked adequate controls and are thus susceptible to placebo effects and biases. There have also been data showing that the grafted cells are subject to the same disease process of the host brain, which ultimately will compromise their capacity to repair the HD brain.122
In one of the very early studies in this area, five patients with HD who had undergone bilateral fetal striatal cell transplants116 demonstrated increases in fluorodeoxyglucose positron-emission tomography activity at the site of graft implantation postsurgery, although the exact origin of this signal was not clear (grafted tissue versus host response). During a 6-year follow-up period, activation increased at the site of the grafting in contrast to controls, which saw a 7% reduction in local metabolic activity over the same time period. This was accompanied by an improvement across motor, neuropsychological, and functional domains.123 Chorea and performance on untimed cognitive tasks remained stable at the 6-year time point, but an increase in dystonia and a decline in cognitive performance for timed tasks were noted.124 In contrast, other studies have shown no benefit with fetal striatal cell transplantation on patients over a 3- to 10-year postoperative period using a large battery of motor, cognitive, and functional assessments.125
While it has been shown that using fetal cell grafts in HD is safe, the method’s true efficacy cannot be assessed without a large systematic clinical trial, although a much larger trial using this approach is now in the final stages of being prepared for reporting (http://www.repair-hd.eu). Regardless of the results of this trial, the problems with using human fetal cells as donor tissue have led a number of groups to work toward making a stem cell-based striatal medium spiny neuron for grafting in HD.126
Conclusion
There has been a clear shift away from symptomatic therapies toward more mechanistic, disease-modifying treatments in HD, looking at a number of possible approaches. This has led to many small trials exploring the safety and efficacy of these different therapeutic strategies. This is both encouraging and exciting, and there is no doubt that the number and quality of clinical trials in HD have increased over the last 5 years in this area. For the first time, there are a number of large, well-designed, properly controlled, industry-sponsored trials ongoing looking at several different treatment targets. Nevertheless, HD remains a rare disease with a limited number of patients, which has an impact on trial design, and while the quality of trial protocols has improved, the burden that these place on individual patients has increased typically by virtue of the length of the study and the intensity of the study visits. This has meant that more advanced patients are finding it harder to complete such studies and are more likely to drop out before completing the trial. Therefore, while research into treatments to help patients in the premanifest or early stages of the disease is promising, there is still a distinct lack of research going into providing more effective treatments to help patients with disease in the more advanced stages.
Acknowledgments
RB was partially funded by the NIHR Cambridge Biomedical Research Centre and the Cambridge University NHS Foundation, and SM was funded by Cambridge and Peterborough Mental Health Partnership NHS Trust.
Disclosure
The authors report no conflicts of interest in this work.
References
Bates G, Harper P, Jones L. Huntington’s Disease. 3rd ed. Oxford: Oxford University Press; 2002. | |
Goodman AO, Rogers L, Pilsworth S, et al. Asymptomatic sleep abnormalities are a common early feature in patients with Huntington’s disease. Curr Neurol Neurosci Rep. 2011;11(2):211–217. | |
Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES. Disintegration of the sleep-wake cycle and circadian timing in Huntington’s disease. J Neurosci. 2005;25(1):157–163. | |
Goodman AO, Murgatroyd PR, Medina-Gomez G, et al. The metabolic profile of early Huntington’s disease – a combined human and transgenic mouse study. Exp Neurol. 2008;210(2):691–698. | |
[No authors listed]. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993;72(6):971–983. | |
Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57(5):369–384. | |
Aylward EH, Brandt J, Codori AM, Mangus RS, Barta PE, Harris GJ. Reduced basal ganglia volume associated with the gene for Huntington’s disease in asymptomatic at-risk persons. Neurology. 1994;44(5):823–828. | |
Aylward EH, Codori AM, Barta PE, Pearlson GD, Harris GJ, Brandt J. Basal ganglia volume and proximity to onset in presymptomatic Huntington disease. Arch Neurol. 1996;53(12):1293–1296. | |
Aylward EH, Codori AM, Rosenblatt A, et al. Rate of caudate atrophy in presymptomatic and symptomatic stages of Huntington’s disease. Mov Disord. 2000;15(3):552–560. | |
Aylward EH, Sparks BF, Field KM, et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology. 2004;63(1):66–72. | |
Campodonico JR, Aylward E, Codori AM, et al. When does Huntington’s disease begin? J Int Neuropsychol Soc. 1998;4(5):467–473. | |
Harris GJ, Codori AM, Lewis RF, Schmidt E, Bedi A, Brandt J. Reduced basal ganglia blood flow and volume in pre-symptomatic, gene-tested persons at-risk for Huntington’s disease. Brain. 1999; 122(Pt 9):1667–1678. | |
Kipps CM, Duggins AJ, Mahant N, Gomes L, Ashburner J, McCusker EA. Progression of structural neuropathology in preclinical Huntington’s disease: a tensor based morphometry study. J Neurol Neurosurg Psychiatry. 2005;76(5):650–655. | |
Thieben MJ, Duggins AJ, Good CD, et al. The distribution of structural neuropathology in pre-clinical Huntington’s disease. Brain. 2002; 125(Pt 8):1815–1828. | |
Paulsen JS, Hayden M, Stout JC, et al. Preparing for preventive clinical trials: the Predict-HD study. Arch Neurol. 2006;63(6):883–890. | |
Paulsen JS, Magnotta VA, Mikos AE, et al. Brain structure in preclinical Huntington’s disease. Biol Psychiatry. 2006;59(1):57–63. | |
Dogan I, Eickhoff SB, Schulz JB, et al. Consistent neurodegeneration and its association with clinical progression in Huntington’s disease: a coordinate-based meta-analysis. Neurodegener Dis. 2013;12(1):23–35. | |
Pringsheim T, Wiltshire K, Day L, Dykeman J, Steeves T, Jette N. The incidence and prevalence of Huntington’s disease: a systematic review and meta-analysis. Mov Disord. 2012;27(9):1083–1091. | |
Leegwater-Kim J, Cha JH. The paradigm of Huntington’s disease: therapeutic opportunities in neurodegeneration. NeuroRx. 2004; 1(1):128–138. | |
Evans SJ, Douglas I, Rawlins MD, Wexler NS, Tabrizi SJ, Smeeth L. Prevalence of adult Huntington’s disease in the UK based on diagnoses recorded in general practice records. J Neurol Neurosurg Psychiatry. 2013;84(10):1156–1160. | |
Tibben A. Predictive testing for Huntington’s disease. Brain Res Bull. 2007;72(2–3):165–171. | |
Morrison PJ. Accurate prevalence and uptake of testing for Huntington’s disease. Lancet Neurol. 2010;9(12):1147. | |
Divino V, Dekoven M, Warner JH, et al. The direct medical costs of Huntington’s disease by stage: a retrospective commercial and Medicaid claims data analysis. J Med Econ. 2013;16(8):1043–1050. | |
Kremer B. Clinical neurology of Huntington’s disease In: Bates GP, Harper P, Jones L, editors. Huntington’s Disease. 3rd ed. Oxford: Oxford University Press; 2002:267–277. | |
Stout JC, Paulsen JS, Queller S, et al. Neurocognitive signs in prodromal Huntington disease. Neuropsychology. 2011;25(1):1–14. | |
Tabrizi SJ, Scahill RI, Owen G, et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington’s disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013;12(7):637–649. | |
Ho AK, Sahakian BJ, Brown RG, et al. Profile of cognitive progression in early Huntington’s disease. Neurology. 2003;61(12):1702–1706. | |
Starkstein SE, Brandt J, Bylsma F, Peyser C, Folstein M, Folstein SE. Neuropsychological correlates of brain atrophy in Huntington’s disease: a magnetic resonance imaging study. Neuroradiology. 1992;34(6):487–489. | |
Backman L, Robins-Wahlin TB, Lundin A, Ginovart N, Farde L. Cognitive deficits in Huntington’s disease are predicted by dopaminergic PET markers and brain volumes. Brain. 1997;120(Pt 12):2207–2217. | |
Begeti F, Tan AY, Cummins GA, et al. The Addenbrooke’s Cognitive Examination – Revised accurately detects cognitive decline in Huntington’s disease. J Neurol. 2013;260(11):2777–2785. | |
Caine ED, Shoulson I. Psychiatric syndromes in Huntington’s disease. Am J Psychiatry. 1983;140(6):728–733. | |
Craufurd D, Thompson JC, Snowden JS. Behavioral changes in Huntington disease. Neuropsychiatry Neuropsychol Behav Neurol. 2001;14(4):219–226. | |
Folstein SE, Franz ML, Jensen BA, Chase GA, Folstein MF. Conduct disorder and affective disorder among the offspring of patients with Huntington’s disease. Psychol Med. 1983;13(1):45–52. | |
Baxter LR Jr, Mazziotta JC, Pahl JJ, et al. Psychiatric, genetic, and positron emission tomographic evaluation of persons at risk for Huntington’s disease. Arch Gen Psychiatry. 1992;49(2):148–154. | |
Hamilton JM, Salmon DP, Corey-Bloom J, et al. Behavioural abnormalities contribute to functional decline in Huntington’s disease. J Neurol Neurosurg Psychiatry. 2003;74(1):120–122. | |
Nehl C, Paulsen JS. Cognitive and psychiatric aspects of Huntington disease contribute to functional capacity. J Nerv Ment Dis. 2004; 192(1):72–74. | |
Williams JK, Barnette JJ, Reed D, et al. Development of the Huntington Disease Family Concerns and Strategies Survey from focus group data. J Nurs Meas. 2010;18(2):83–99. | |
Foroud T, Gray J, Ivashina J, Conneally PM. Differences in duration of Huntington’s disease based on age at onset. J Neurol Neurosurg Psychiatry. 1999;66(1):52–56. | |
Mason SL, Barker RA. Advancing pharmacotherapy for treating Huntington’s disease: a review of the existing literature. Expert Opin Pharmacother. 2016;17(1):41–52. | |
Priller J, Ecker D, Landwehrmeyer B, Craufurd D. A Europe-wide assessment of current medication choices in Huntington’s disease. Mov Disord. 2008;23(12):1788. | |
Ross CA, Aylward EH, Wild EJ, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10(4):204–216. | |
Clelland CD, Barker RA, Watts C. Cell therapy in Huntington disease. Neurosurg Focus. 2008;24(3–4):E9. | |
Wright BL, Barker RA. Established and emerging therapies for Huntington’s disease. Curr Mol Med. 2007;7(6):579–587. | |
Rung JP, Rung E, Helgeson L, et al. Effects of (-)-OSU6162 and ACR16 on motor activity in rats, indicating a unique mechanism of dopaminergic stabilization. J Neural Transm (Vienna). 2008;115(6):899–908. | |
Burstein ES, Carlsson ML, Owens M, et al. II. In vitro evidence that (-)-OSU6162 and (+)-OSU6162 produce their behavioral effects through 5-HT2A serotonin and D2 dopamine receptors. J Neural Transm (Vienna). 2011;118(11):1523–1533. | |
Carlsson ML, Burstein ES, Kloberg A, et al. I. In vivo evidence for partial agonist effects of (-)-OSU6162 and (+)-OSU6162 on 5-HT2A serotonin receptors. J Neural Transm (Vienna). 2011;118(11):1511–1522. | |
de Yebenes JG, Landwehrmeyer B, Squitieri F, et al. Pridopidine for the treatment of motor function in patients with Huntington’s disease (MermaiHD): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol 2011;10(12):1049–1057. | |
Huntington Study Group HART Investigators. A randomized, double-blind, placebo-controlled trial of pridopidine in Huntington’s disease. Mov Disord. 2013;28(10):1407–1415. | |
Kloberg A, Constantinescu R, Nilsson MK, et al. Tolerability and efficacy of the monoaminergic stabilizer (-)-OSU6162 (PNU-96391A) in Huntington’s disease: a double-blind cross-over study. Acta Neuropsychiatr. 2014;26(5):298–306. | |
Schwab LC, Garas SN, Drouin-Ouellet J, Mason SL, Stott SR, Barker RA. Dopamine and Huntington’s disease. Expert Rev Neurother. 2015; 15(4):445–458. | |
Levine MS, Klapstein GJ, Koppel A, et al. Enhanced sensitivity to N-methyl-D-aspartate receptor activation in transgenic and knockin mouse models of Huntington’s disease. J Neurosci Res. 1999;58(4):515–532. | |
Cepeda C, Hurst RS, Altemus KL, et al. Facilitated glutamatergic transmission in the striatum of D2 dopamine receptor-deficient mice. J Neurophysiol. 2001;85(2):659–670. | |
André VM, Cepeda C, Levine MS. Dopamine and glutamate in Huntington’s disease: a balancing act. CNS Neurosci Ther. 2010; 16(3):163–178. | |
Reilmann R, Rouzade-Dominguez ML, Saft C, et al. A randomized, placebo-controlled trial of AFQ056 for the treatment of chorea in Huntington’s disease. Mov Disord. 2015;30(3):427–431. | |
Biolsi B, Cif L, Fertit HE, Robles SG, Coubes P. Long-term follow-up of Huntington disease treated by bilateral deep brain stimulation of the internal globus pallidus. J Neurosurg. 2008;109(1):130–132. | |
Delorme C, Rogers A, Lau B, et al. Deep brain stimulation of the internal pallidum in Huntington’s disease patients: clinical outcome and neuronal firing patterns. J Neurol. 2016;263(2):290–298. | |
Gonzalez V, Cif L, Biolsi B, et al. Deep brain stimulation for Huntington’s disease: long-term results of a prospective open-label study. J Neurosurg. 2014;121(1):114–122. | |
Hebb MO, Garcia R, Gaudet P, Mendez IM. Bilateral stimulation of the globus pallidus internus to treat choreathetosis in Huntington’s disease: technical case report. Neurosurgery. 2006;58(2):E383. | |
Moro E, Lang AE, Strafella AP, et al. Bilateral globus pallidus stimulation for Huntington’s disease. Ann Neurol. 2004;56(2):290–294. | |
Velez-Lago FM, Thompson A, Oyama G, et al. Differential and better response to deep brain stimulation of chorea compared to dystonia in Huntington’s disease. Stereotact Funct Neurosurg. 2013;91(2):129–133. | |
Wojtecki L, Groiss SJ, Ferrea S, et al. A prospective pilot trial for pallidal deep brain stimulation in Huntington’s disease. Front Neurol. 2015;6:177. | |
Garrison JL, Macosko EZ, Bernstein S, Pokala N, Albrecht DR, Bargmann CI. Oxytocin/vasopressin-related peptides have an ancient role in reproductive behavior. Science. 2012;338(6106):540–543. | |
Gabery S, Halliday G, Kirik D, Englund E, Petersen A. Selective loss of oxytocin and vasopressin in the hypothalamus in early Huntington disease: a case study. Neuropathol Appl Neurobiol. 2015;41(6):843–848. | |
Wood NI, Goodman AO, van der Burg JM, et al. Increased thirst and drinking in Huntington’s disease and the R6/2 mouse. Brain Res Bull. 2008;76(1–2):70–79. | |
Meyer-Lindenberg A, Tost H. Neural mechanisms of social risk for psychiatric disorders. Nat Neurosci. 2012;15(5):663–668. | |
Fabio KM, Guillon CD, Lu SF, et al. Pharmacokinetics and metabolism of SRX246: a potent and selective vasopressin 1a antagonist. J Pharm Sci. 2013;102(6):2033–2043. | |
Lee RJ, Coccaro EF, Cremers H, et al. A novel V1a receptor antagonist blocks vasopressin-induced changes in the CNS response to emotional stimuli: an fMRI study. Front Syst Neurosci. 2013;7:100. | |
Mason SL, Zhang J, Begeti F, et al. The role of the amygdala during emotional processing in Huntington’s disease: from pre-manifest to late stage disease. Neuropsychologia. 2015;70:80–89. | |
Renoir T, Argyropoulos A, Hannan AJ. Antidepressant-like effect of the norepinephrine-dopamine reuptake inhibitor bupropion in a mouse model of Huntington’s disease with dopaminergic dysfunction. J Huntingtons Dis. 2012;1(2):261–266. | |
Renoir T, Argyropoulos A, Chevarin C, Lanfumey L, Hannan AJ. Sexually dimorphic dopaminergic dysfunction in a transgenic mouse model of Huntington’s disease. Pharmacol Biochem Behav. 2014;127:15–20. | |
Hogarth P, Lovrecic L, Krainc D. Sodium phenylbutyrate in Huntington’s disease: a dose-finding study. Mov Disord. 2007;22(13):1962–1964. | |
Jeong H, Then F, Melia TJ Jr, et al. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009;137(1):60–72. | |
Smith MR, Syed A, Lukacsovich T, et al. A potent and selective sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington’s disease. Hum Mol Genet. 2014;23(11):2995–3007. | |
Westerberg G, Chiesa JA, Andersen CA, et al. Safety, pharmacokinetics, pharmacogenomics and QT concentration-effect modelling of the SirT1 inhibitor selisistat in healthy volunteers. Br J Clin Pharmacol. 2015;79(3):477–491. | |
Mrzljak L. Development of kynurenine monooxygenase (KMO) inhibitor CHDI-340246 for the treatment of Huntington’s disease: a progress update. Poster presented at: CHDI Foundation 8th Annual HD Therapeutics Conference; April 8–11, 2013; Venice, Italy. | |
Wang HF, Yu JT, Tang SW, et al. Efficacy and safety of cholinesterase inhibitors and memantine in cognitive impairment in Parkinson’s disease, Parkinson’s disease dementia, and dementia with Lewy bodies: systematic review with meta-analysis and trial sequential analysis. J Neurol Neurosurg Psychiatry. 2015;86(2):135–143. | |
Carter RL, Chen Y, Kunkanjanawan T, et al. Reversal of cellular phenotypes in neural cells derived from Huntington’s disease monkey-induced pluripotent stem cells. Stem Cell Rep. 2014;3(4):585–593. | |
Wu J, Tang T, Bezprozvanny I. Evaluation of clinically relevant glutamate pathway inhibitors in in vitro model of Huntington’s disease. Neurosci Lett. 2006;407(3):219–223. | |
Beister A, Kraus P, Kuhn W, Dose M, Weindl A, Gerlach M. The N-methyl-D-aspartate antagonist memantine retards progression of Huntington’s disease. J Neural Transm Suppl. 2004;(68):117–122. | |
Ondo WG, Mejia NI, Hunter CB. A pilot study of the clinical efficacy and safety of memantine for Huntington’s disease. Parkinsonism Relat Disord. 2007;13(7):453–454. | |
Anitha M, Nandhu MS, Anju TR, Jes P, Paulose CS. Targeting glutamate mediated excitotoxicity in Huntington’s disease: neural progenitors and partial glutamate antagonist – memantine. Med Hypotheses. 2011;76(1):138–140. | |
Coskran TM, Morton D, Menniti FS, et al. Immunohistochemical localization of phosphodiesterase 10A in multiple mammalian species. J Histochem Cytochem. 2006;54(11):1205–1213. | |
Schmidt CJ, Chapin DS, Cianfrogna J, et al. Preclinical characterization of selective phosphodiesterase 10A inhibitors: a new therapeutic approach to the treatment of schizophrenia. J Pharmacol Exp Ther. 2008;325(2):681–690. | |
Grauer SM, Pulito VL, Navarra RL, et al. Phosphodiesterase 10A inhibitor activity in preclinical models of the positive, cognitive, and negative symptoms of schizophrenia. J Pharmacol Exp Ther. 2009;331(2):574–590. | |
Siuciak JA, Chapin DS, Harms JF, et al. Inhibition of the striatum-enriched phosphodiesterase PDE10A: a novel approach to the treatment of psychosis. Neuropharmacology. 2006;51(2):386–396. | |
Steffan JS, Kazantsev A, Spasic-Boskovic O, et al. The Huntington’s disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci U S A. 2000;97(12):6763–6768. | |
Giampa C, Patassini S, Borreca A, et al. Phosphodiesterase 10 inhibition reduces striatal excitotoxicity in the quinolinic acid model of Huntington’s disease. Neurobiol Dis. 2009;34(3):450–456. | |
Giampa C, Laurenti D, Anzilotti S, Bernardi G, Menniti FS, Fusco FR. Inhibition of the striatal specific phosphodiesterase PDE10A ameliorates striatal and cortical pathology in R6/2 mouse model of Huntington’s disease. PloS One. 2010;5(10):e13417. | |
Giralt A, Saavedra A, Carreton O, et al. PDE10 inhibition increases GluA1 and CREB phosphorylation and improves spatial and recognition memories in a Huntington’s disease mouse model. Hippocampus. 2013;23(8):684–695. | |
Chabrier PE, Auguet M. Pharmacological properties of BN82451: a novel multitargeting neuroprotective agent. CNS Drug Rev. 2007; 13(3):317–332. | |
Klivenyi P, Ferrante RJ, Gardian G, Browne S, Chabrier PE, Beal MF. Increased survival and neuroprotective effects of BN82451 in a transgenic mouse model of Huntington’s disease. J Neurochem. 2003;86(1):267–272. | |
Aad G, Abbott B, Abdallah J, et al. Search for diphoton events with large missing transverse energy in 7 TeV proton-proton collisions with the ATLAS detector. Phys Rev Lett. 2011;106(12):121803. | |
Rose C, Menzies FM, Renna M, et al. Rilmenidine attenuates toxicity of polyglutamine expansions in a mouse model of Huntington’s disease. Hum Mol Genet. 2010;19(11):2144–2153. | |
Dedeoglu A, Kubilus JK, Jeitner TM, et al. Therapeutic effects of cystamine in a murine model of Huntington’s disease. J Neurosci. 2002;22(20):8942–8950. | |
Fox JH, Barber DS, Singh B, et al. Cystamine increases L-cysteine levels in Huntington’s disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. J Neurochem. 2004;91(2):413–422. | |
Borrell-Pagès M, Canals JM, Cordelières FP, et al. Cystamine and cysteamine increase brain levels of BDNF in Huntington disease via HSJ1b and transglutaminase. J Clin Invest. 2006;116(5):1410–1424. | |
Dubinsky R, Gray C. CYTE-I-HD: phase I dose finding and tolerability study of cysteamine (Cystagon) in Huntington’s disease. Mov Disord. 2006;21(4):530–533. | |
Prundean A, Youssov K, Humbert S, Bonneau D, Verny C. A phase II, open-label evaluation of cysteamine tolerability in patients with Huntington’s disease. Mov Disord. 2015;30(2):288–289. | |
DiFiglia M, Sapp E, Chase KO, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277(5334):1990–1993. | |
Rubinsztein DC, Carmichael J. Huntington’s disease: molecular basis of neurodegeneration. Expert Rev Mol Med. 2003;5(20):1–21. | |
Arrasate M, Finkbeiner S. Protein aggregates in Huntington’s disease. Exp Neurol. 2012;238(1):1–11. | |
Kim S, Kim KT. Therapeutic approaches for inhibition of protein aggregation in Huntington’s disease. Exp Neurobiol. 2014;23(1):36–44. | |
Aronin N, DiFiglia M. Huntingtin-lowering strategies in Huntington’s disease: antisense oligonucleotides, small RNAs, and gene editing. Mov Disord. 2014;29(11):1455–1461. | |
Miller TM, Pestronk A, David W, et al. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. Lancet Neurol. 2013;12(5):435–442. | |
Ismailoglu I, Chen Q, Popowski M, Yang L, Gross SS, Brivanlou AH. Huntingtin protein is essential for mitochondrial metabolism, bioenergetics and structure in murine embryonic stem cells. Dev Biol. 2014;391(2):230–240. | |
Kordasiewicz HB, Stanek LM, Wancewicz EV, et al. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron. 2012;74(6):1031–1044. | |
McBride JL, Pitzer MR, Boudreau RL, et al. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol Ther. 2011;19(12):2152–2162. | |
Grondin R, Kaytor MD, Ai Y, et al. Six-month partial suppression of huntingtin is well tolerated in the adult rhesus striatum. Brain. 2012;135(Pt 4):1197–1209. | |
Stiles DK, Zhang Z, Ge P, et al. Widespread suppression of huntingtin with convection-enhanced delivery of siRNA. Exp Neurol. 2012;233(1):463–471. | |
Fox MD, Snyder AZ, Vincent JL, Raichle ME. Intrinsic fluctuations within cortical systems account for intertrial variability in human behavior. Neuron. 2007;56(1):171–184. | |
Xiao G, Fan Q, Wang X, Zhou B. Huntington disease arises from a combinatory toxicity of polyglutamine and copper binding. Proc Natl Acad Sci U S A. 2013;110(37):14995–15000. | |
Cherny RA, Ayton S, Finkelstein DI, Bush AI, McColl G, Massa SM. PBT2 reduces toxicity in a C. elegans model of polyQ aggregation and extends lifespan, reduces striatal atrophy and improves motor performance in the R6/2 mouse model of Huntington’s disease. J Huntingtons Dis. 2012;1(2):211–219. | |
Huntington Study Group Reach2HD Investigators. Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: a phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015;14(1):39–47. | |
Dunnett SB, Rosser AE. Stem cell transplantation for Huntington’s disease. Exp Neurol. 2007;203(2):279–292. | |
Rosser AE, Barker RA, Harrower T, et al. Unilateral transplantation of human primary fetal tissue in four patients with Huntington’s disease: NEST-UK safety report ISRCTN no 36485475. J Neurol Neurosurg Psychiatry. 2002;73(6):678–685. | |
Bachoud-Lévi A, Bourdet C, Brugières P, et al. Safety and tolerability assessment of intrastriatal neural allografts in five patients with Huntington’s disease. Exp Neurol. 2000;161(1):194–202. | |
Madrazo I, Franco-Bourland RE, Castrejon H, Cuevas C, Ostrosky-Solis F. Fetal striatal homotransplantation for Huntington’s disease: first two case reports. Neurol Res. 1995;17(4):312–315. | |
Philpott LM, Kopyov OV, Lee AJ, et al. Neuropsychological functioning following fetal striatal transplantation in Huntington’s chorea: three case presentations. Cell Transplant. 1997;6(3):203–212. | |
Kopyov OV, Jacques S, Lieberman A, Duma CM, Eagle KS. Safety of intrastriatal neurotransplantation for Huntington’s disease patients. Exp Neurol. 1998;149(1):97–108. | |
Hauser RA, Furtado S, Cimino CR, et al. Bilateral human fetal striatal transplantation in Huntington’s disease. Neurology. 2002; 58(5):687–695. | |
Freeman TB, Cicchetti F, Hauser RA, et al. Transplanted fetal striatum in Huntington’s disease: phenotypic development and lack of pathology. Proc Natl Acad Sci U S A. 2000;97(25):13877–13882. | |
Cisbani G, Cicchetti F. The fate of cell grafts for the treatment of Huntington’s disease: the post-mortem evidence. Neuropathol Appl Neurobiol. 2014;40(1):71–90. | |
Bachoud-Levi AC, Remy P, Nguyen JP, et al. Motor and cognitive improvements in patients with Huntington’s disease after neural transplantation. Lancet. 2000;356(9246):1975–1979. | |
Bachoud-Lévi AC, Gaura V, Brugières P, et al. Effect of fetal neural transplants in patients with Huntington’s disease 6 years after surgery: a long-term follow-up study. Lancet Neurol. 2006;5(4):303–309. | |
Barker RA, Mason SL, Harrower TP, et al. The long-term safety and efficacy of bilateral transplantation of human fetal striatal tissue in patients with mild to moderate Huntington’s disease. J Neurol Neurosurg Psychiatry. 2013;84(6):657–665. | |
Kelly CM, Dunnett SB, Rosser AE. Medium spiny neurons for transplantation in Huntington’s disease. Biochem Soc Trans. 2009; 37(Pt 1):323–328. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.