Back to Journals » International Journal of Nanomedicine » Volume 13
Novel nanocrystal-based solid dispersion with high drug loading, enhanced dissolution, and bioavailability of andrographolide
Authors Ma Y, Yang Y, Xie J, Xu J, Yue P, Yang M
Received 31 January 2018
Accepted for publication 9 April 2018
Published 28 June 2018 Volume 2018:13 Pages 3763—3779
DOI https://doi.org/10.2147/IJN.S164228
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Linlin Sun
Yueqin Ma,1,2,* Yang Yang,2,* Jin Xie,1 Junnan Xu,1 Pengfei Yue,1 Ming Yang1
1Key Laboratory of Modern Preparation of TCM, Ministry of Education, Jiangxi University of Traditional Chinese Medicine, Nanchang, Jiangxi, 330004, 2Department of Medicine and Pharmacy, 94th Hospital of People’s Liberation Army, Nanchang, Jiangxi, 330002, China
*These authors contributed equally to this work
Objective: The current study sought to design a quickly dissolving, high drug loading nanocrystal-based solid dispersion (NC-SD) in order to improve the dissolution of poorly soluble drugs.
Methods: The NC-SD was prepared by means of combination of homogenization and spray-drying. Polymer hydroxypropylmethylcellulose (HPMC) was used as baseline dispersant for NC-SD of the model drug – andrographolide (AG). Three superdisintegrants cohomogenized with HPMC were used as codispersant for AG-NC-SD and compared to common water-soluble dispersants – mannitol and lactose. The dissolution characterization and oral bioavailability of AG-NC-SD were evaluated.
Results: The AG-NC-SD with the higher concentration of HPMC exhibited fast dissolution due to the enhanced wettability of HPMC. The water-soluble codispersants (mannitol and lactose) did not completely prevent AG-NC from aggregation during spray-drying. To achieve much faster AG release, cohomogenized superdisintegrants at a level of 20% must be used along with 25% HPMC. Compared with water-soluble dispersants like mannitol and lactose, superdisintegrants with high swelling capacity were much more effective dispersants for enhancing fast redispersion/dissolution of AG-NC-SD via a swelling-triggered erosion/disintegration mechanism. Surfactant-free AG-NC-SD with 15% cohomogenized sodium carboxymethyl starch combined with 15% HPMC and 10% lactose enhanced the dissolution further, without comprising drug loading, exhibited a barely compromised dissolution rate compared to precursor NC suspensions (f2>50), and possessed drug loading up to 67.83%±1.26%. The pharmacokinetics results also demonstrated that the AG-NC-SD significantly improved the bioavailability in vivo of AG (P<0.05), compared with to the coarse AG.
Conclusion: This study illustrates that a quickly dissolving, high drug load, surfactant-free NC-SD can be prepared by using a superdisintegrant as codispersant, and provides a feasible strategy to improve the oral bioavailability of poorly soluble drugs.
Keywords: nanocrystal-based solid dispersion, nanocrystals, andrographolide, superdisintegrant, dissolution, bioavailability
Introduction
Nanocrystals (NCs) in the form of aqueous nanosuspensions are considered one of the most valuable approaches to formulation of poorly soluble drugs (Biopharmaceutical Classification System class II drugs), which has numerous advantages in increasing the solubility and dissolution velocity of poorly soluble drugs attributed to their small particles <1 μm in diameter and large specific surfaces.1–3 Due to physical stability issues associated with drug-NC suspensions, such as particle-size growth and aggregation upon milling and prolonged storage,4,5 and in view of patient compliance/preference, NC suspensions have to be converted into dried NC-aggregated particles by means of specialized techniques – spray-drying or freeze-drying – which has been increasing in attractiveness.6,7 However, the main challenge is that dried NC-aggregated particles must have the ability to recover their original nanosuspensions and improve the dissolution of poorly soluble drugs.8–10
Combinations of soluble polymers, sugars, and surfactants have been shown to be effective in stabilizing drug nanosuspensions.11 This combination allowed for fast recovery of drug NCs from NC-aggregated particles.12,13 However, despite their effectiveness as dispersants, surfactants could pose several challenges, such as aggregation of drug nanoparticles in suspensions during milling/storage and low drug loading capacity (most <20%, seldom >50%).14,15 For example, van Eerdenbrugh et al combined microcrystalline cellulose and D-α-tocopheryl polyethylene glycol 1000 succinate to use as dispersants for redispersible itraconazole NCs. D-α-Tocopheryl polyethylene glycol 1000 succinate and microcrystalline cellulose had to be used at ≥50% w:w (50% drug loading) and 200% (31.3% drug loading) with respect to the drug to achieve immediate itraconazole release.16 Azad et al17 used a hydroxypropylmethylcellulose (HPMC)/polyvinylpyrrolidone (PVP)–sodium dodecyl sulfate combination to achieve fast dissolution from itraconazole nanocomposites, with drug loading of 14.9%. Fu et al prepared spray-dried allisartan isoproxil NCs that had relative high drug loading of 61.7% and excellent redispersibility, but it was mainly attributed to the addition of surfactant sodium dodecyl sulfate of 6.17%.18 Additional challenges associated with the use of anionic surfactants include incompatibilities with other ionic molecules, sensitivity to pH, salt, or temperature changes, gastrointestinal tract irritation, and even toxicity when used in excess.19–21 In view of the aforementioned issues, surfactant usage should be minimized to mitigate all potential negative impact. Sugars (eg, sucrose, lactose), sugar alcohols (eg, mannitol, sorbitol), and water-soluble polymers (eg, hydroxypropylcellulose, HPMC, PVP, and polyethylene glycol) have been used widely in NC-aggregated particle formulations as dispersants.22,23 However, in the absence of surfactants, sugars and sugar alcohols may have to be added in high concentrations up to 1,000% w:w with respect to drug NCs to ensure fast release from NC-composite particles.24–26 Such a high concentration of dispersants could minimize the loading capacity of drug-NC particles. Combination of excellent dispersants used as the matrix of NCs is very crucial to reduce irreversible drug-NC aggregation during solidification and enhance drug loading.27
Solid dispersion (SD) is an effective dispersion technique to improve the dissolution rate of poorly soluble drugs, which are usually dispersed in the SD matrix in the molecule or amorphous state.28 Unfortunately, the poor physical stability and low drug loading (<20%) of SD have severely limited the widespread application of this technology.29 Combinations of NCs and SD technology may be a very interesting strategy for solidification of NC suspensions. In this work, to enhance drug loading and redispersibility of NCs, an NC-based SD (NC-SD) was developed by dispersing NCs into special matrix formers, which was expected to have dual advantages of NCs and SD. As shown in Figure 1, superdisintegrants, ie, crospovidone (CPD), croscarmellose sodium (CMS), and sodium carboxymethyl starch (SCS), are cross-linked semisynthetic or synthetic polymers with high swelling capacities, which are used widely for fast disintegration of pharmaceutical tablets.30,31 Bhakay et al32 coated wet comilled griseofulvin–CMS suspensions on Pharmatose via a fluidized bed processor, and yielded griseofulvin nanocomposites with relatively low drug loading (<13% w:w) for enhancing drug-nanoparticle release/dissolution. Chaulang et al used SCS as a matrix for furosemide SD at a ratio of 1:1, and the SD prepared significantly improved dissolution (5.8-fold) with relatively high drug loading (33.3%).33 Fujii et al used CPD as a carrier to prepare indomethacin SD with <40% loading, and the amorphous indomethacin SD prepared improved dissolution significantly.34 Therefore, superdisintegrants might be good alternative dispersants for production of NC-SD with excellent dissolution and high drug loading of poorly soluble drugs.
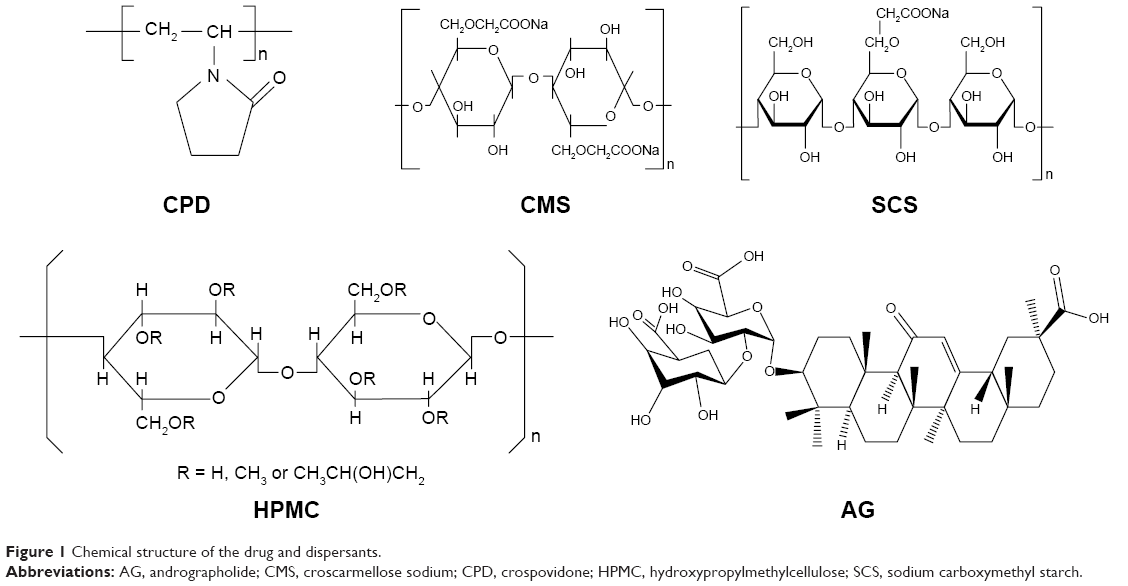 | Figure 1 Chemical structure of the drug and dispersants. |
As the model drug in this work, andrographolide (AG; Figure 1) has been reported to possess pharmacological activities, including antibacterial, anti-inflammatory, and particularly anti-respiratory infection and anti-dysentery activity.35,36 Unfortunately, AG exhibits poor solubility in water and relatively high permeability, the poor dissolution most responsible for its poor bioavailability.37,38 To overcome these issues, several formulation approaches have been developed, such as SD, nanocomposite particles, and dried nanosuspensions. Although the bioavailability of AG has been improved to some extent, its drug loading is not very high, ie, 20% for AG-PVP SD,39 no more than 50% for nanocomposite particles,40,41 and 23% for dried nanosuspensions.42 Our current study firstly elucidated an attempt to design an NC-SD with a combination of polymers stabilizer and superdisintegrants as matrix dispersant, in order to enhance the drug loading, redispersibility, and bioavailability of drug NCs. The main purposes of this study were to prepare an AG-NC suspension (AG-NS), and convert it into an AG-NC-SD by spray-drying. The polymer HPMC, alone and in combination with superdisintegrants (CPD, CMS, and SCS; Figure 1) or polyols (mannitol and lactose), was used as codispersant, and performance compared for preparation of an AG-NC-SD. We also aimed to evaluate the particle size and morphology of the AG-NC-SD by laser diffractometry and scanning electron microscopy, investigate the solid state of bulk AG and the AG-NC-SD by means of differential scanning calorimetry (DSC) and X-ray diffraction analyses, and evaluate the dissolution characterization and oral bioavailability of the AG-NC-SD.
Methods
SCS (DST type A) was purchased from Chineway Pharmaceutical (Shanghai, China). CMS (Kiccolate), and CPD (Kollidon CL-F) were donated by Fengli Jingqiu Pharmaceutical (Beijing, China). HPMC was donated by Anhui Sunhere Pharmaceutical Excipients (Huainan, China). AG (purity >95%) were purchased from Zelang Medical Technology (Nanjing, China). Lactose and mannitol were obtained from Damao Chemical (Tianjin, China).
Preparation of AG NC-SD
Formulations of the AG-NS are listed in Table 1. Selection of homogenization conditions and formulations was guided by our prior work on homogenization.40,41 According to formulations as listed in Table 1, AG 10% (w:v), and HPMC% (w:w, relative weight to drug) were first dispersed in 100 mL water as dispersion medium. The suspensions obtained were further disintegrated by a high-shear homogenizer (FA25; Fluko, Shanghai, China) at 19,000 rpm for 5 minutes. Then, the prepared coarse AG suspension was homogenized at 1,000 bar for 30 cycles with a piston-gap high-pressure homogenizer (AH-1000D; ATS Engineering, Brampton, ON, Canada). Then, AG-NSs were obtained. In addition, two conventional soluble dispersants (mannitol and lactose) and three superdisintegrants (CPD, CMS, and SCS) was used as codispersants. They were dispersed in AG-NS and further homogenized at 600 bar for 30 cycles. After that, the treated AG-NSs were spray-dried with a mini-spray-dryer (B290; Büchi Labortechnik AG, Flawil, Switzerland). Process parameters were inlet temperature 120°C, outlet temperature 57°C–83°C, aspiration rate 55%, pump speed 10%, and atomizing airflow 50 mmHg. The AG-NC-SD was deposited at the bottom of the collector. Then, the AG-NC-SD collected was further evaluated.
 | Table 1 Formulations and drug loading of the NC-SD |
Particle size of AG-NS
Diameters of freshly redispersed AG-NC-SD were measured with a Mastersizer Microplus (Malvern Instruments, Malvern, UK) with a working range of 0.05–550 mm. Analysis of diffraction patterns was done according to the Mie model (particle refractive index 1.67). Average 10%, 50%, and 90% volume percentiles (D10, D50, and D90) were determined to assess variation in particle sizes. All measurements were performed in triplicate.
Viscosity of AG-NS
The viscosity of the selected dispersion solutions was determined at 30°C by an LVDV-II + Pro viscometer (Brookfield Engineering Laboratories, Middleboro, MA, USA). Measurements were performed in triplicate, and the average was calculated.
Determination of drug wettability
The wettability of different dispersants solutions on AG was evaluated by the contact angle of the drug:43
|
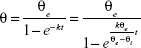
where θ is the contact angle, θe the quasiequilibrium contact angle, θi the initial contact angle, t the measuring time, and k the wetting index, which is used to evaluate wetting ability. Contact angle was characterized by the sessile-drop technique with a goniometer (OCA20; DataPhysics, Filderstadt, Germany). Compressed disks of the AG powders were made at 30 MPa compression force by means of a laboratory powder press (769YP-15A; Keqi High and New Technology, Tianjin, China). The tangent of the droplet on the disk surface was measured to determine the contact angle.
Redispersibility of AG-NC-SD
The redispersibility index (RDI) of AG-NC-SD was calculated:43
|
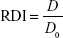
where D0 is the volume-weighted (D50) mean particle size of the precursor AG-NS directly prior to spray-drying and D the corresponding value of redispersed NCs from spray-dried AG-NC-SD. An RDI of near 1 indicates that AG-NC-NP can recover completely back to the original AG-NS after rehydration.
Scanning electron microscopy
Morphology of the spray-dried AG-NC-SD was evaluated using scanning electron microscopy (X650; Hitachi, Tokyo, Japan). Samples were glued and solidified on metal sample plates by means of double-sided carbon tape. All samples were gold-coated (thickness ~15–20 nm) by means of a sputter coater (Fisons Instruments, Ipswich, UK). Electrical potential was set as 2 kV at 25 mA for 10 minutes.
DSC
A DSC analyzer (Diamond; PerkinElmer, Waltham, MA, USA) was used to determine the peak melting temperature of the coarse AG, dispersants, and representative AG-NC-SD formulations (sample [S]4, S7, S10, S12, and S18). Powder samples of 6–7 mg were weighed before being placed in a sealed perforated aluminum pan and heated from 20°C to 260°C at a rate of 10°C/min. Nitrogen was used as the purge gas and protective gas at flow rates of 50 mL/min and 150 mL/min, respectively.
X-ray powder diffraction (XRPD)
The crystallinity of coarse AG, dispersants, and the representative AG-NC-SD formulations (S4, S7, S10, S12, and S18) was analyzed using XRPD (Panalytical, Almelo, Netherlands). Samples were scanned for 2θ at 5°–90° at a scan rate of 0.2°C/min. Measurements were performed at a voltage of 40 kV and 25 mA.
Drug loading determination and drug release evaluation of AG-NC-SD
The concentration of AG in the sample was assayed by high-performance liquid chromatography (HPLC) according to a reported method.44 Chromatographic separation was achieved on Hypersil ODS2 (25 mm×4 mm, 5 μm). Chromatographic conditions were set as detection wavelength 225 nm, mobile phase methanol–water (60:40 v:v), injection volume 20 μL, flow rate 1 mL/min, and temperature 30°C.
Dissolution experiments on AG-NC-SD were performed using an RC8 dissolution apparatus (Guoming Medicine and Equipment, Tianjin, China) according to the 2015 Chinese pharmacopoeia paddle method and compared with those of AG-NS and coarse AG. A representative AG-NC-SD containing an equivalent AG dose of 20 mg was weighed and added to 900 mL pure water. The dissolution medium was maintained at 37.0°C±0.1°C, and a paddle speed of 100 rpm was used. After addition of the AG-NC-SD, a 2 mL sample was withdrawn and equal blank medium compensated immediately. Withdrawn samples were filtered through a 0.22 μm-filter membrane and determined by HPLC.
Dissolution profiles are expressed as mean ± SD. The similarity factor (f2)45 was used to assess differences in dissolution profiles of AG-NC-SDs (S1–S18):
|
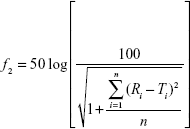
where Ri represents the dissolution concentration of the compared formulation at a predetermined time, Ti the dissolution concentration of the developed formulation at a predetermined time, and n the number of dissolution time points. Generally, if f2 is much more than 50, the dissolution profile of the developed formulation is not significantly different from that of the reference formulation.
In vivo pharmacokinetic study of AG-NC-SD
Pharmacokinetic characterization of the AG-NC-SD (S18) was compared with crude AG and the precursor AG-NS (S1). This study protocol was approved by the institutional ethics committee of Jiangxi University of Traditional Chinese Medicine. Animal welfare and experimental procedures were performed strictly in accordance with the Guide for the Care and Use of Laboratory Animals and the ethics regulations of the university.
The rats were housed at 25°C±2°C with 50%±10% relative humidity and a 12-hour light–dark cycle. They were permitted free access to food and water until 12 hours prior to experiments. Male Wistar rats with an average weight of 200±20 g were fasted for 12 hours before administration. Eighteen rats were randomly divided into three groups of six animals each. AG-NC-SD, AG-NS, and crude AG dispersed in water were administered orally at a dose of 20 mg/kg. At predetermined intervals of 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 9, 12, 15, and 24 hours, 0.25 mL blood samples were collected by retro orbital puncture. Plasma was obtained from whole blood in heparinized tubes via centrifugation at 4,000 rpm for 15 minutes and frozen at −20°C until analysis. AG plasma concentrations were determined by a validated HPLC–tandem mass-spectrometry method.42 Glipizide was used as the internal standard (IS). Full-scan spectra showed parent ions at m/z 395.10 for AG and m/z 444.1 for IS. An elution system of acetonitrile:0.1% formic acid solution (15:85 v:v) was used at a flow rate of 0.2 mL/min. Retention times for AG and IS were 2.8 and 3.2 minutes, respectively. The main pharmacokinetic parameters were calculated by DAS 2.0 software (Mathematical Pharmacology Professional Committee of China). Analysis of variance was carried out using SPSS Statistics 21 software. Differences were considered statistically significant at P<0.05.
Results and discussion
Influence of dispersants on preparation of AG-NC-SD
The freshly prepared AG NSs were spray-dried into the NC-SD. Theoretical and actual drug contents of the AG-NC-SD are presented in Table 1. The results showed that AG-NZ-SDs with high drug loading (50%–85%) were prepared by spray-drying, and most of the AG-NC-SDs had higher drug loading than those reported in the literature. Particle sizes (D10, D50, and D90) of the AG-NS after homogenization and redispersed AG-NS from spray-dried AG-NC-SD are shown in Figure 2A and B, respectively. Particle sizes of the AG-NS (S1–S18) after homogenization were 0.112–0.141 μm (D10), 0.428–0.846 μm (D50), and 0.786–25.23 μm (D90). Particle sizes of the redispersed AG-NS after spray-drying were 0.127–0.536 μm (D10), 0.452–2.821 μm (D50), and 0.881–40.78 μm (D90). These results indicated that the different dispersants significantly influenced the particle size of AG-NS after homogenization and spray-drying.
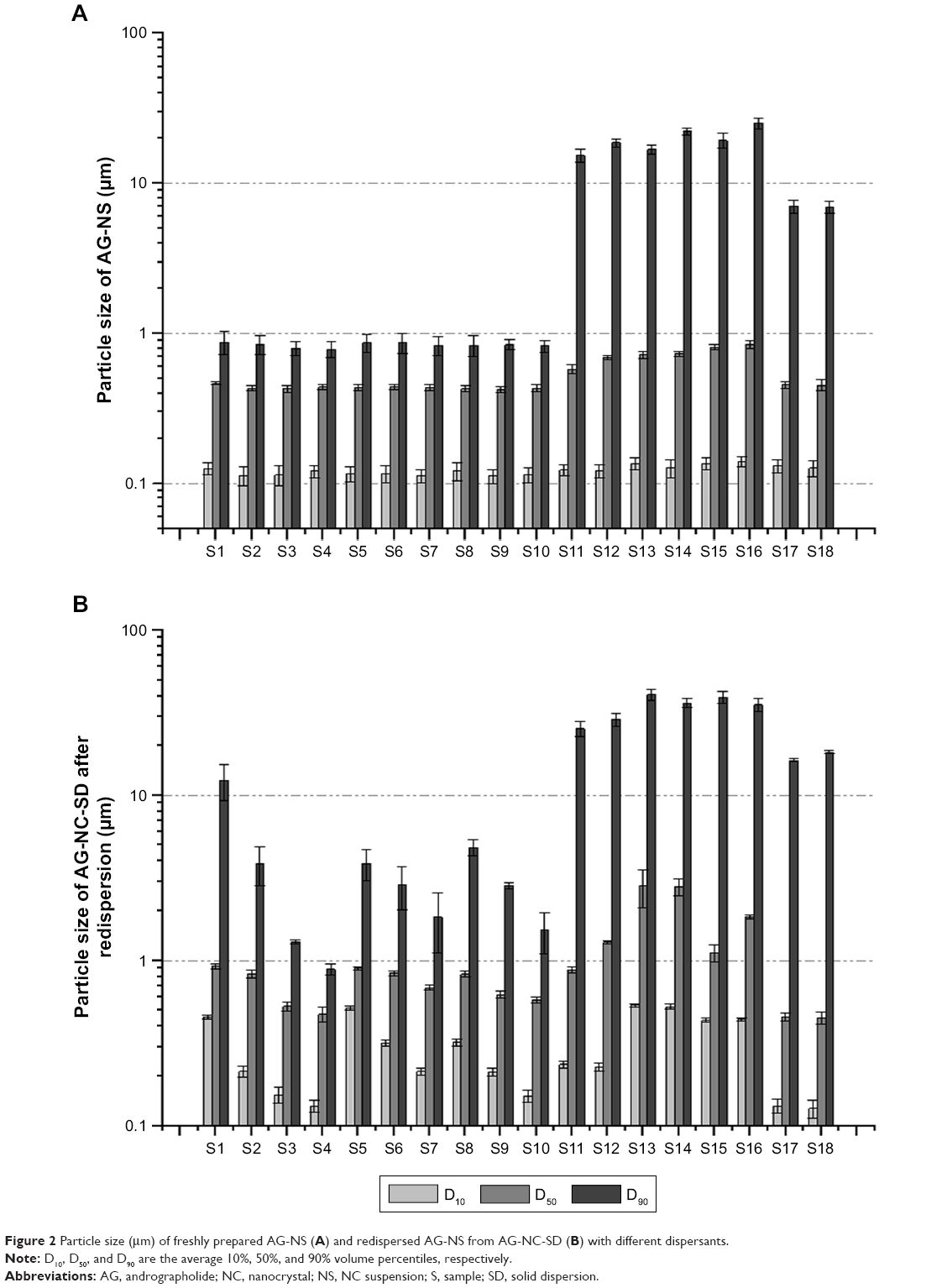 | Figure 2 Particle size (μm) of freshly prepared AG-NS (A) and redispersed AG-NS from AG-NC-SD (B) with different dispersants. |
Roles of HPMC
Figure 2A shows that AG-NS (S1) stabilized by only 15% HPMC (w:w relative to drug) was successfully prepared by homogenization, which resulted in the formation of NCs with D50 particle size of 467±16 nm, and D90 size of 876±11 nm (<1 μm). With increased HPMC concentration from 15% to 85% (S2–S4), AG-NS particle size decreased (S2 432 nm, S3 428 nm, S4 426 nm), which indicated that 15% concentration of HPMC as a steric stabilizer was sufficient to prevent aggregation of AG-NC during homogenization. This was related to the wettability and viscosity of HPMC. Table 2 shows viscosity and wettability properties of various dispersant solutions. The results demonstrated that 15% HPMC solution exhibited a higher wettability-index value (k=0.212) compared to pure water (0.059), which likely adsorbed on the surface of the AG-NC, and imparted some steric stability and enhanced wettability. However, with increased HPMC concentrations in solution, the viscosity of dispersion solutions increased substantially, which could have driven a decrease of particle-size-reduction efficiency of AG-NC during homogenization, and hence AG-NC particle size with 85% HPMC was not remarkably different than that with 25% and 50% HPMC.
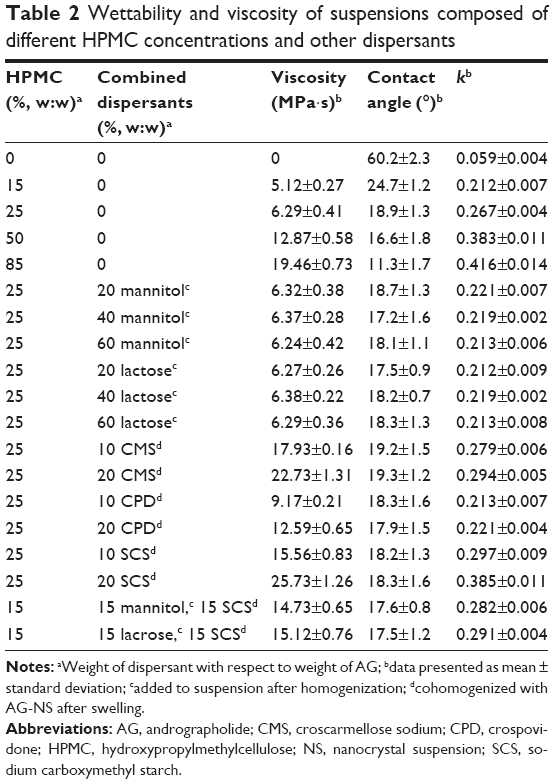 | Table 2 Wettability and viscosity of suspensions composed of different HPMC concentrations and other dispersants |
Figure 2B shows that the particle size of redispersed AG-NS from AG-NS-SD (S1, S2, S3, and S4) after rehydration was obviously increased, and RDI values of AG-NS-SD (S1–S4) after rehydration were 1.98±0.08, 1.87±0.06, 1.23±0.05, and 1.21±0.02, respectively. This meant that NCs in AG-NS-SD formed large aggregated particles after spray-drying, particularly at 15% HPMC used as dispersant (S1). Smaller drug aggregates were formed at the higher HPMC concentrations of 25% (S2) and 50% (S3) because of enhanced wettability (higher k-values, Table 2), but 85% HPMC concentration completely prevented AG-NC particle aggregation. This might have been related to enhanced viscosity of suspensions and increase HPMC concentrations. Table 2 shows that HPMC solution viscosity increased from 5.12±0.27 mPa·s to 19.46±0.73 mPa·s and concentration from 15% to 85%. At relatively higher concentrations (S3 and S4), a thicker layer of the adsorbed polymer and stronger adsorption could have mitigated NC aggregation.46,47 Therefore, 25% HPMC concentration was used as a baseline dispersant and combined with other codispersants.
Roles of mannitol and lactose combined with HPMC
It can be seen from Figure 2A that AG-NS particle sizes (S5–S10) stabilized by 25% HPMC (w:w relative to drug) and combined with either sucrose or lactose were 467±16 nm (D50), and 876±11 nm (D90), which were not different than AG-NS stabilized by only 25% HPMC. With increased mannitol or lactose concentration of 20%–60%, AG-NS particle size did not seem to be different. This indicated that mannitol and lactose had no effect on nanosizing of AG-NS during homogenization compared to HPMC alone as dispersant (S2). The addition of either mannitol or lactose did not increase the viscosity or wettability of the suspensions (as listed in Table 2), but the particle size of the redispersed suspensions from AG-NS-SD (S5–S10) after rehydration increased remarkably (Figure 2B), even at 60% mannitol or lactose concentration, and RDI values of AG-NS-SD (S7 and S10) were 1.578±0.06 and 1.348±0.05, respectively. These meant that to ensure acceptable drug loading, a combination of HPMC and mannitol or lactose at the concentrations applied could not completely prevent AG-NC particles from aggregation during spray-drying.
Roles of superdisintegrants combined with HPMC
To improve the redispersibility and dissolution of AG from AG-NC-SD, roles of the superdisintegrants (SCS, CPD, and CMS) combined with HPMC were investigated. The physical properties and morphology of these superdisintegrants48–50 are provided in Table 3. As per Figure 2A, AG-NS (S11–S16) particle size stabilized by 25% HPMC (w:w relative to drug) and combined with superdisintegrants as codispersants was significantly larger (D50>800 nm, D90>10 μm) AG-NS (S1–S10) particles stabilized by 25% HPMC. AG-NS particles stabilized by 15% HPMC and 10% SCS and combined with 10% mannitol (S17) and lactose (S18) were also much larger than those of S2, particularly for D90. It was thought that the cohomogenized suspensions of AG and superdisintegrants seemed to be a multimodal mixture of large swollen superdisintegrant particles and AG-NC, as evidenced by the large D90 values corresponding to AG-NS (S12–S18). Note that superdisintegrants (CMS, CPD and SCS) were not insoluble polymers in water, D50 values of the as-received SCS, CPD, and CMS particles were about 57±4, 36±1, and 53±2 μm, but D50 and D90 values of the SCS, CPD, and CMS particles swollen in 25% HPMC solution (before cohomogenization), were 102.6±0.3 and 265.5±0.2, 50.1±0.1 and 135.8±0.1, and 76.6±0.2 and 148.8±0.3 μm, respectively. As such, the large swollen particles of the superdisintegrants were able to be pulverized into small particles during the cohomogenization of AG–superdisintegrant.
 | Table 3 Physical properties of the superdisintegrants |
Particles of AG-NS stabilized by 25% HPMC and combined with superdisintegrants (S11–S16) were relatively large, but this did not mean that AG-NS formed any large aggregations. Due to the presence of cohomogenized superdisintegrant swollen particles, AG-NC particle size in the suspensions could not be accurately determined via laser diffraction, but this was able to be demonstrated by means of in vitro dissolution if AG-NC did not form large aggregated particles. The dissolution profiles of the freshly prepared AG-NS (S1, S2, S11–S18) stabilized by HPMC alone and combined with 25% HPMC are illustrated in Figure 3. The results demonstrated that the dissolution curve of AG-NS stabilized by 25% HPMC combined with CPD (S13 and S14) seemed to be different from those of S1 and S2 at the initial stage of dissolution, but dissolution profiles of other formulations (S11, S12, S15–S18) were not different from those of S1 or S2 (f2>50). This indicated that cohomogenization of superdisintegrants and AG-NS did not result in the aggregation of AG-NC in suspensions. For AG-NS stabilized by CPD, the relatively slow dissolution at the initial stage might have been related to the adsorption property of CPD.51
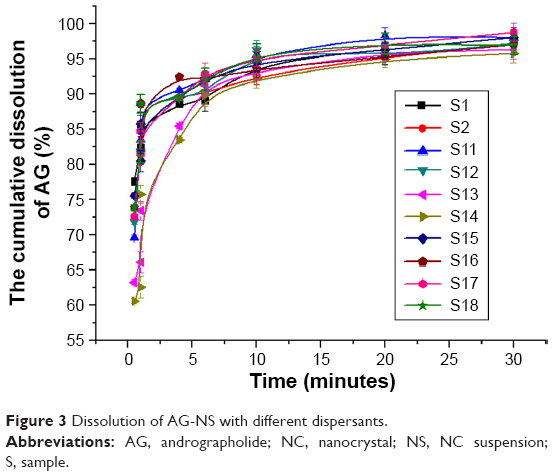 | Figure 3 Dissolution of AG-NS with different dispersants. |
It is also worth mentioning that addition of superdisintegrants enhanced the stability of AG-NS (data not shown). Unlike the addition of mannitol or lactose, cohomogenized 10% SCS or 10% CMS with 25% HPMC (Table 2) significantly raised the viscosity of the AG-NS because of the removal of free water in the suspension via absorption by highly swellable superdisintegrants. On the other hand, cohomogenized 10% CPD did not seem to raise the viscosity of suspensions (Table 1), which was related with the low swelling capacity of CPD (5.4 cm3/g compared to 23.6 cm3/g for SCS and 13.5 cm3/g for CMS), as shown in Table 3. The increase in superdisintegrant concentration led to high suspension viscosity. When extra 85% HPMC was used as the dispersant, the suspension also possessed relatively high viscosity, and the wettability of SCS and CMS was better than that of CPD at applied concentrations (Table 2). As such, the viscosity and wettability of polymers also might have been responsible for the enhanced dissolution of AG-NS.
Roles of dispersants in dissolution of AG-NC-SD
Drug-dissolution profiles from coarse AG and spray-dried AG-NC-SD with various dispersants were measured. As discussed in previous sections, the AG-NSs (S1–S10) stabilized with 25% HPMC alone and with additional mannitol/lactose exhibited similar particle size to drug NCs during homogenization. AG-NSs stabilized with 25% HPMC and additional cohomogenized superdisintegrants (S11–S18) exhibited similar dissolution behavior to those of AG-NSs stabilized with 25% HPMC (f2>50), even though the presence of large particles was determined by laser diffraction. Therefore, the major impact of various dispersants on drug release in the process of dissolution is expected to be related to their capacity to release drug NCs from the AG-NC-SD after spray-drying.
Differences in dissolution profiles from the AG-NC-SD containing 25% HPMC with either of these additional dispersants alone (mannitol, lactose, and cohomogenized superdisintegrants) compared to those from coarse AG for comparison are illustrated in Figure 4. Coarse AG particles with D50 of 15.5 μm had extremely poor dissolution, with only 30.1% of AG dissolved after 30 minutes. The AG-NC-SD led to an enhanced drug-dissolution rate (Figure 4A), regardless of the dispersants used. For AG-NC-SD with either 15% HPMC (S1) or 25% HPMC (S2), the release of AG was significantly improved compared to coarse AG (f2<50), ie, 43.5% for S1 and 56.7% for S2 at 30 minutes. However, drug release from S1 and S2 was still unsatisfactory compared to that of AG-NS (f2<50). Enhanced-speed release was achieved when 50% (S3) and 85% HPMC (S4) were used as dispersants, ie, 83.8% for S3 and 86.6% for S4 within 5 minutes. All these results could be explained by the larger surface area of AG-NC compared with coarse AG and enhanced wettability of AG owing to the presence of dispersants (85% HPMC > 50% HPMC >25% HPMC > 15% HPMC, seen in Table 2). Despite the enhanced wetting efficiency with higher HPMC concentrations (Table 2) and smaller AG-NC aggregates in the spray-drying, 85% HPMC exhibited only slight improvement in AG release compared with 50% HMPC.
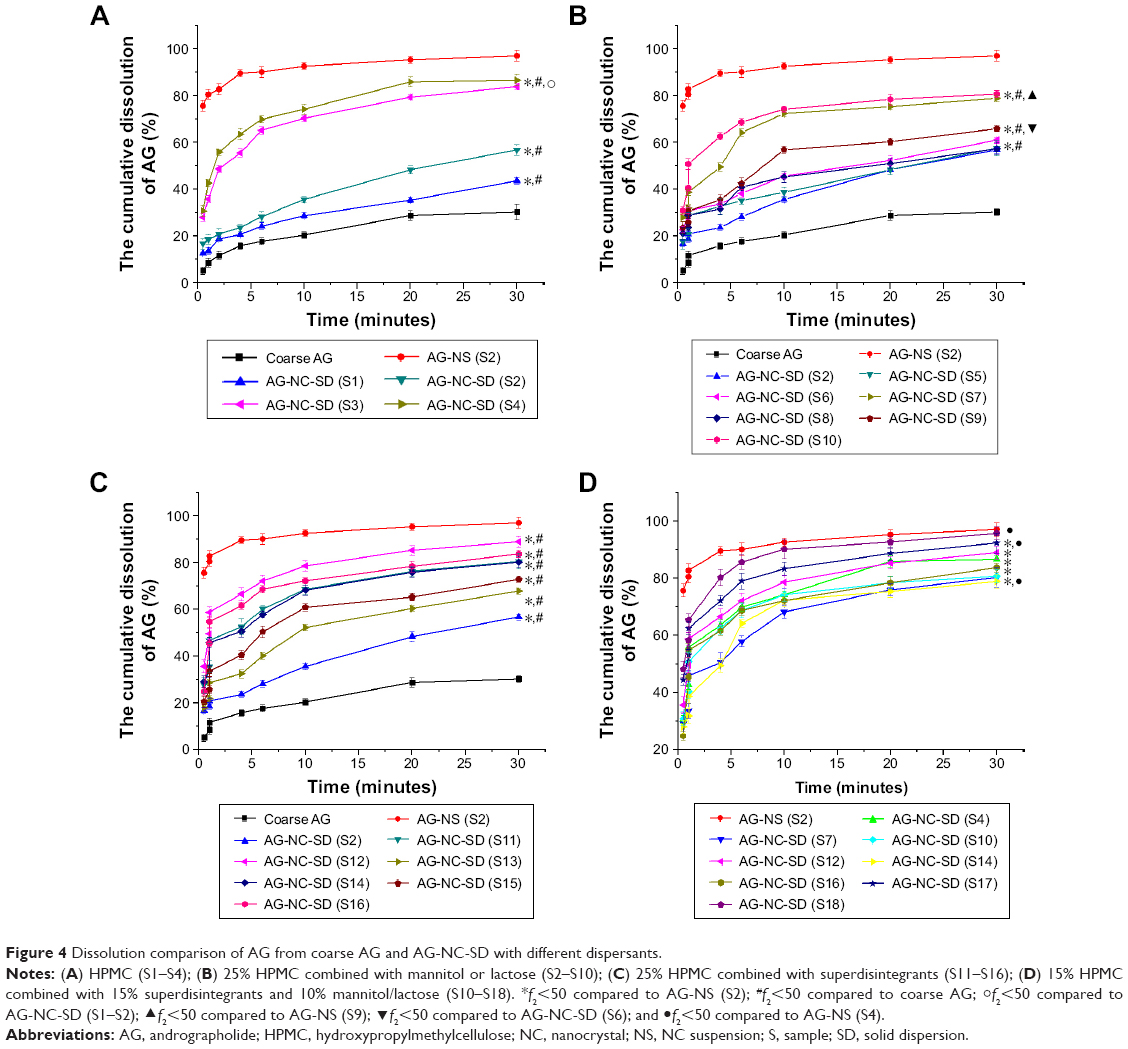 | Figure 4 Dissolution comparison of AG from coarse AG and AG-NC-SD with different dispersants. |
The impact of the polyol dispersants (mannitol/lactose) at 20%–60% level, along with the baseline 25% HPMC, is shown in Figure 4B. Upon addition of 20% and 40% concentrations of mannitol (S5 and S6) or lactose (S8 and S9), a slight improvement (f2<50) in drug release was observed (Figure 4B) compared to HPMC alone (S2), ie, 58.9% for S6 and 60.8% for S8 at 60 minutes. When 60% mannitol (S7, drug loading 78.95%) and lactose (S10, drug loading 80.67%) were used, the dissolution performance of AG-NC-SD improved significantly (f2<50) compared with those of AG-NC-SD (S2). However, the dissolution profiles of S7 and S10 were significantly different (f2<50) from that of the precursor AG-NS (S2) before spray-drying and also much slower (f2<50) compared to that of AG-NC-SD (S4) at equal concentrations of dispersant. Therefore, mannitol or lactose combined with HPMC 60% still did not completely prevent AG-NC from aggregation during spray-drying, which was consistent with the results shown in Figure 2B.
The impact of HPMC combined with swellable cohomogenized dispersants (SCS/CPD/CMS) at 10% with baseline 25% HPMC is shown in Figure 4C. Dissolution profiles were obviously improved compared to AG-NC-SD with 25% HPMC (S2, f2<50). With 20% superdisintegrant used as codispersants, SCS (S12), CPD (S14), and CMS (S16) were briefly cohomogenized with AG-NS and spray-dried into the SD, and the dissolution performance of AG-NS was significantly improved (f2<50). This indicated that superdisintegrants can lead to faster AG release and thus be better dispersants than mannitol or lactose at the concentrations applied. More interestingly, their desirable impact positively correlated with their swelling capacity (SCS > CMS > CPD). Finally, it was noted that the dissolution performance of AG-NC-SD with 20% SCS was best of all the formulations.
In general, the codispersants besides the baseline 25% HPMC led to improvement in dissolution of AG-NC. Conventional soluble dispersants (mannitol and lactose) combined with HPMC allowed for drug release from the AG-NC-SD via erosion/dissolution, which was initiated by dissolution of the dispersant. On the other hand, the combination of briefly cohomogenized superdisintegrants with soluble HPMC facilitated erosion/disintegration of the AG-NC-SD matrix via both HPMC erosion/dissolution and swelling-induced degradation.32 The swellable superdisintegrant particles embedded in NC-SD facilitated the matrix erosion/disintegration. Apparently, simultaneous action of both mechanisms was additive and beneficial.
Optimum dispersants of AG-NC-SD with high drug loading and excellent dissolution
The performances of cohomogenized superdisintegrants for quickly dissolving AG-NC-SD are illustrated in Figure 4C. Their superior performance over conventional codispersants like mannitol and lactose was clear. At high dispersant concentration, AG-NC-SD (S12 and S16) exhibited much faster release than S7 and S10 (f2<50). However, dissolution of AG-NC-SD (S14) was not satisfactory, which might have been related to the adsorption properties of CPD.48 Additionally, the AG-NC-SD stabilized by 85% HPMC (S4) also possessed enhanced drug dissolution (f2<50). Even so, the dissolution rate of the AG-NC-SD (S4, S10, S12, and S16) seemed to be slower than AG-NS (S2) (f2<50). This seemingly anomalous behavior could be due to gelling-induced slow erosion/dissolution of the polymeric matrix at this high polymer concentration, which has been reported elsewhere.52 Furthermore, drug loading of AG-NZ-SD (S4, S10) was unacceptable, ie, 50.38% for S4 and 52.87% for S10. To achieve maximum-speed dissolution and drug loading of AG-NC-SD at lower concentrations, 15% HPMC and 15% SCS combined with 10% sucrose or lactose were used as synergistic dispersants for AG-NZ-SD, which led to the fast dissolution of AG. Enhanced release was achieved (f2<50), ie, 92.31% for S17 and 96.76% for S18 within 30 minutes (Figure 4D). Also, the dissolution of AG-NC-SD (S18) was not significantly different from S2 (f2>50). It was concluded that 15% HPMC combined with 10% lactose and 15% SCS as dispersants effectively improved the dissolution performance of AG-NC-SD, which could be ascribed to smaller AG aggregates after spray-drying (Figure 2) and increased wettability and appropriate viscosity (Table 2), which ensured fast water penetration and excellent swelling ability of superdisintegrants and facilitated the fast disintegration and diffusion of AG-NC-SD. More interestingly, S17 and S18 drug loading was up to 67.83%, which far exceeded the drug loading of conventional formulations.39–42
Morphology of AG-NC-SD
The morphology images of AG and representative AG-NC-SD are shown in Figure 5. The coarse AG was plate-shaped AG particles with particle size of 10–60 μm (Figure 5A). The spray-dried AG-NC-SDs with 25% HPMC (S1) seemed to be NC-aggregated particles (Figure 5B). This further demonstrated that 25% HPMC did not prevent AG-NC from aggregation during spray-drying. Spray-dried AG-NC-SDs with 50% and 85% HPMC (S3 and S4) exhibited a wrinkled-sphere shape (Figure 5C and D). The surface wrinkles might be attributed to the collapsed HPMC matrix, which was initially inflated due to internal vapor formation during spray-drying. Polymers have been reported to be film-forming agents that can enrich surfaces and form spray-dried particles with wrinkled morphology.53
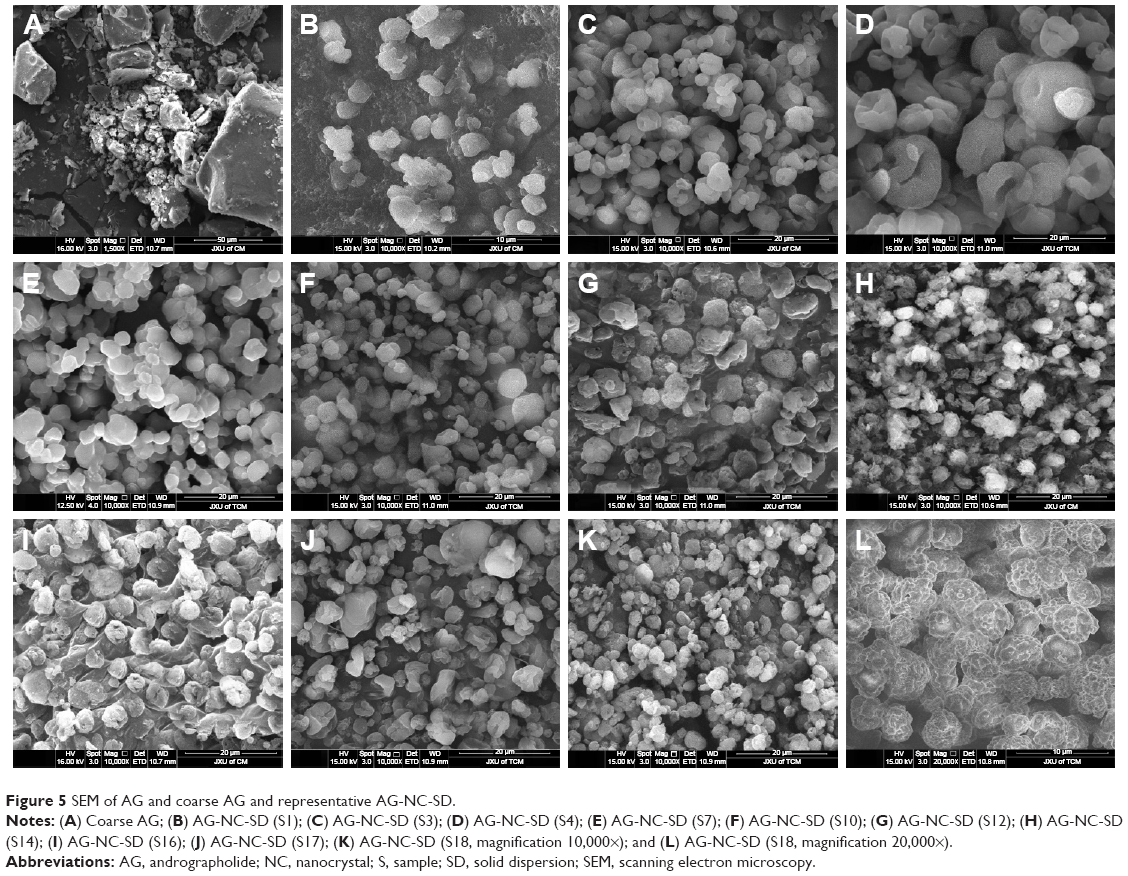 | Figure 5 SEM of AG and coarse AG and representative AG-NC-SD. |
Spray-dried AG-NC-SDs with 60% mannitol (S7) appeared to be have adhesive particles (Figure 5E), which can be related to the fact that mannitol has a low glass-transition temperature of 11°C and tends easily toward glass transition shortly after spray-drying.54 Table 3 shows that CMS particles were fibrous, CPD particles spongy, and SCS particles round, but the spray-dried AG-NC-SDs containing lactose and superdisintegrants as codispersants (S10, S12, S14, and S16) were near-spherical (Figure 5F–I) and the circularization of AG-NC-SDs with SCS combined with either mannitol (S17) or lactose (S18) was excellent (Figure 5J–K). Therefore, 15% HPMC combined with 10% lactose and 15% SCS could be beneficial dispersants for AG-NC-SD during spray-drying.
Crystalline state of AG-NC-SD
The crystalline state of coarse AG, dispersants, and AG-NC-SD was examined by means of DSC and XRPD. The melting temperature of coarse AG was found to be 230°C (Figure 6A). Wide plane peaks of for HPMC and SCS were observed (Figure 6B and E). The endothermic melting peak of mannitol was 165°C (Figure 6C) and the melting peak of lactose was 150°C, with the decomposition peak at 220°C (Figure 6D). DSC thermograms of the representative spray-dried AG-NZ-SD exhibited a notable endothermic event associated with the melting of crystalline AG (Figure 6F–J). The peak melting temperature was lowered in the spray-dried AG-NC-SDs. Also there was a slight reduction in fusion enthalpy for spray-dried AG-NC-SDs compared to coarse AG. The reduction observed in melting points and enthalpy might have been due to particle-size reduction and/or defect crystal formation of the drug during homogenization.18 According to the Gibbs–Thomson equation,55 the melting temperature of a material is proportional to its cohesive energy, which could indicate that NCs with reduced cohesive energy require less energy for melting, thus resulting in a reduction in melting temperature and enthalpy compared to the coarse AG crystals.
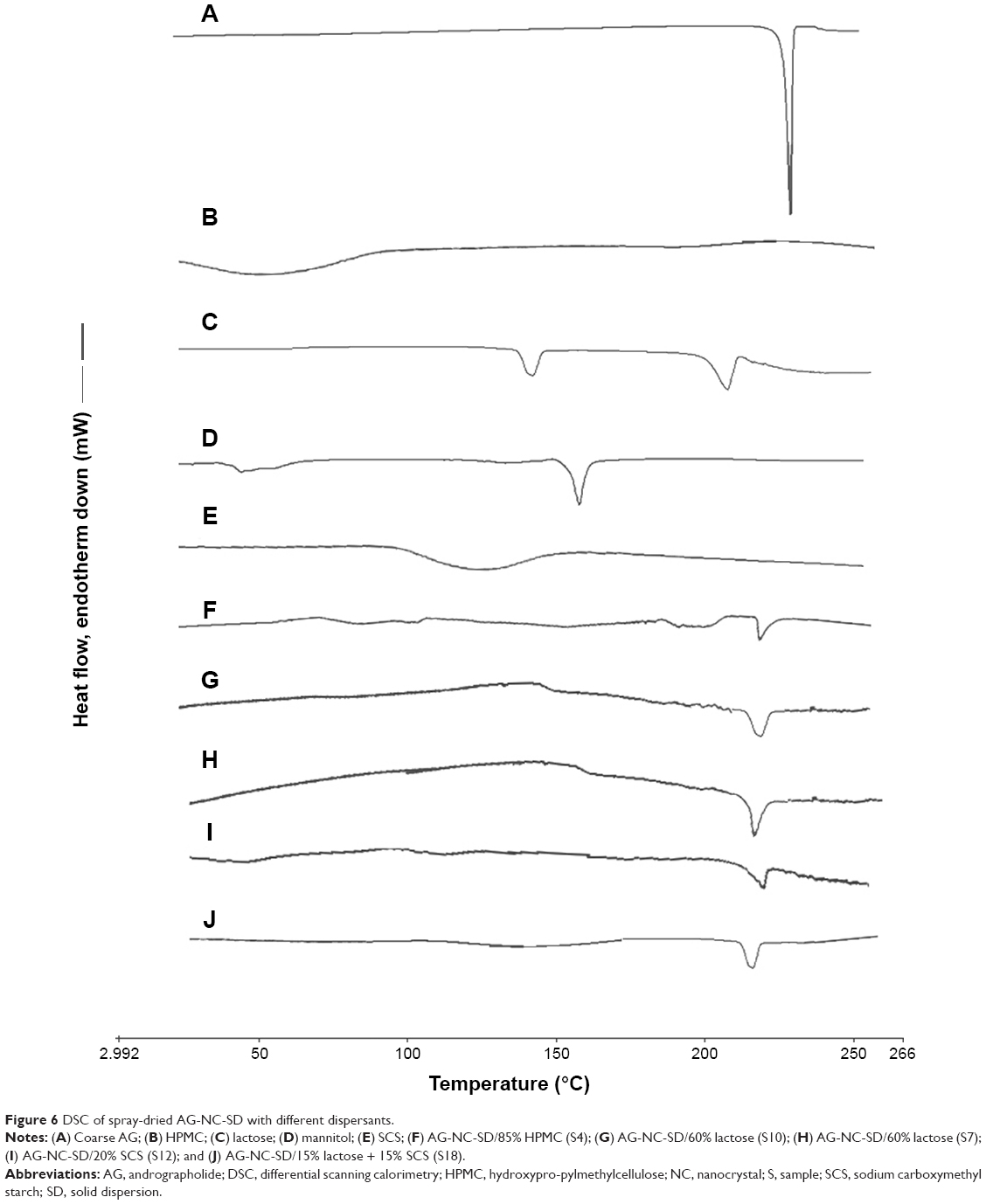 | Figure 6 DSC of spray-dried AG-NC-SD with different dispersants. |
The effect of homogenization on AG crystallinity was also investigated via X-ray diffraction (Figure 7). Coarse AG exhibited crystalline peaks at 2θ of 12.2°, 13.3°, 15.4°, and 25.9° (Figure 7A). Lactose exhibited a crystalline peak at 2θ of 19.3° (Figure 7D). Mannitol had a crystalline peak at 24.9° (Figure 7E). AG in spray-dried AG-NC-SDs (Figure 7G–J) showed distinct crystalline peaks similar to coarse AG, as could be seen from XRPD. AG peak positions remained unchanged compared to coarse AG, but with lower peak intensity, which could be attributed to the aforementioned crystal-defect formation and/or particle-size reduction during homogenization–drying. The XRPD and DSC results overall suggested that the crystalline nature of AG in AG-NC-SD was largely preserved after homogenization, despite the formation of defects, differently from the conventional AG-SD with amorphous or molecule drug.39,42
 | Figure 7 XRD of spray-dried AG-NC-SD with different dispersants. |
As illustrated in Figure 8, in three forms of AG-NC-SD, AG in NC state dispersed homogeneously in the amorphous matrix formers of different dispersants. The polymers HPMC, sugar/sugar alcohol, and cohomogenized superdisintegrants played an important role in the formation and dissolution of NC-SD. On one hand, the dispersants prevented drug NCs from aggregation during homogenization and spray-drying via a steric-barrier effect; on the other hand, they enhanced fast redispersion and dissolution of NC-SD triggered by penetration/erosion/swelling after hydration. Owing to their swelling capacity, wet cohomogenized superdisintegrants were much more effective dispersants than conventional, water-soluble dispersants like mannitol and lactose. HPMC 15% combined with 10% lactose and 15% SCS (S18) could be effective dispersants for AG-NC-SD. To demonstrate the performance in vivo of this formulation strategy, the pharmacokinetics of AG-NC-SD (S18) were further evaluated.
 | Figure 8 Matrix structure and dissolution of nanocrystal-based solid dispersion. |
In vivo pharmacokinetic evaluation of AG-NC-SD
Pharmacokinetic studies of AG-NC-SD were undertaken in rats to investigate whether the AG-NC-SD (S18) was able to improve oral bioavailability of AG in vivo compared with precursor AG-NS and crude AG. The plasma-concentration versus time curves of the AG-NC-SD, AG-NS, and crude AG are shown in Figure 9. It could be seen that plasma concentrations of AG-NC-SD were significantly higher within 5 hours after administration compared to crude AG (P<0.05), but not significantly different from AG-NS (P>0.05). The main pharmacokinetic parameters of AG-NC-SD, AG-NS, and crude AG are listed in Table 4. Compared with the time to maximum concentration of crude AG (1.11 hours), AG-NC-SD (0.44 hours), and AG-NS (0.36 hours) were significantly decreased (P<0.05). Mean peak concentrations of the AG-NC-SD (323.423±43.527 μg/L) and AG-NS (346.741±38.163 μg/L) were significantly increased (P<0.05) compared with crude AG (52.506±10.652 μg/L). As listed in Table 4, the AUC0–8 of the AG-NC-SD (1,794.738±311.213 μg/L·hour) was 4.72 times (P<0.05) as high as that of crude AG (379.521±124.233 μg/L·hour). However, the main pharmacokinetic parameters (maximum concentration and AUC0–8) of AG-NC-SD were not significantly different from AG-NS (P>0.05), which was also consistent with the dissolution studies (Figure 4). As illustrated in Figure 10, AG-NC-SD recovered easily back to the AG-NS in the gastrointestinal tract after oral administration, dependent on the synergetic effect of penetration/swelling/erosion from lactose and SCS. It could be speculated that the reduced particle size of NCs with an increase in surface area significantly increased the dissolution rate of AG and increased the adhesiveness of gut lumen, which could have resulted in a high concentration gradient and rapid absorption of the AG molecules across the gastrointestinal membrane (Figure 10). Reduced particle size plays an important role in dissolution and absorption from gastrointestinal tissue.56,57
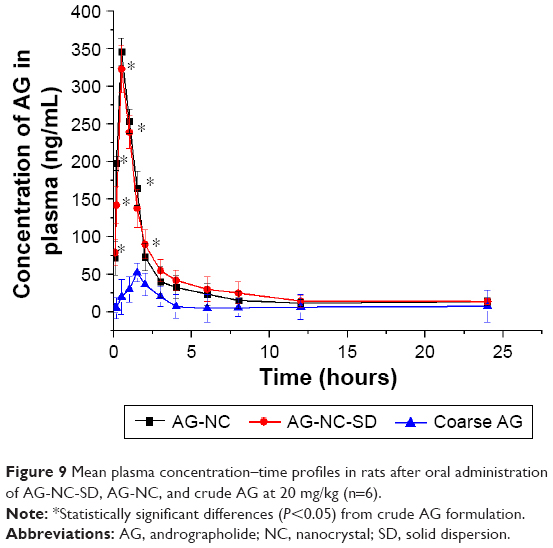 | Figure 9 Mean plasma concentration–time profiles in rats after oral administration of AG-NC-SD, AG-NC, and crude AG at 20 mg/kg (n=6). |
 | Table 4 Pharmacokinetic parameters of AG in rats after oral administration of AG-NC-SD, AG-NS, and crude AG at 20 mg/kg (n=6) |
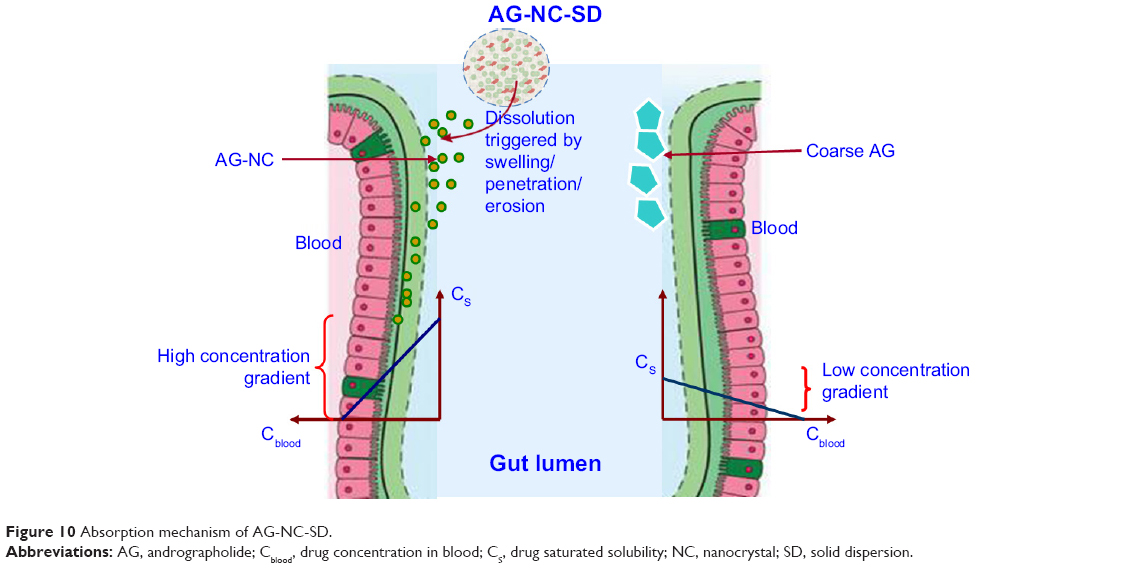 | Figure 10 Absorption mechanism of AG-NC-SD. |
Conclusion
A novel NC-SD using surfactant-free dispersants as a matrix was successfully prepared by combining homogenization and spray-drying technology, with the goal of enhancing the drug loading and improving the speed of drug dissolution. Owing to their swelling capacity, the superdisintegrants were much more effective dispersants than conventional, water-soluble dispersants like mannitol and lactose. To achieve much faster AG release without surfactants, cohomogenized superdisintegrants (SCS) at 15% must be used along with 15% HPMC and 10% lactose. Erosion/disintegration of the AG-NC-SD was triggered by the dissolution of soluble polymer and sugar/sugar alcohol, as well as the swelling-induced disintegration provided by the superdisintegrants. The crystal state of AG presented in AG-NC-SD remained unchanged during the preparation. The AG-NC-SD exhibited a fast dissolution rate and significantly improved the bioavailability of AG. This study has demonstrated that quickly dissolving, high drug loading, surfactant-free NC-SDs could be prepared by combined superdisintegrants as codispersants, and also provide a feasible strategy to improve the oral bioavailability of poorly soluble drug.
Acknowledgments
The authors would like to acknowledge the financial support from the Scientific Research Foundation for the National Natural Science Foundation of China (81560656 and 81760715), the Fund of Distinguished Young Scientists of Jiangxi Province (20162BCB23033), and the Natural Science Fund of Jiangxi Province (20161BAB205221).
Disclosure
The authors report no conflicts of interest in this work.
References
Merisko-Liversidge E, Liversidge GG, Cooper ER. Nanosizing: a formulation approach for poorly-water-soluble compounds. Eur J Pharm Sci. 2003;18(2):113–120. | ||
Keck CM, Müller RH. Drug nanocrystals of poorly soluble drugs produced by high pressure homogenisation. Eur J Pharm Biopharm. 2006;62(1):3–16. | ||
Yue PF, Li Y, Wan J, et al. Process optimization and evaluation of novel baicalin solid nanocrystals. Int J Nanomedicine. 2013;8:2961–2973. | ||
Bilgili E, Li M, Afolabi A. Is the combination of cellulosic polymers and anionic surfactants a good strategy for ensuring physical stability of BCS class II drug nanosuspensions? Pharm Dev Technol. 2016;21(4):499–510. | ||
Knieke C, Rawtani A, Davé RN. Concentrated fenofibrate nanoparticle suspensions from melt emulsification for enhanced drug dissolution. Chem Eng Technol. 2014;37(1):157–167. | ||
Toziopoulou F, Malamatari M, Nikolakakis I, Kachrimanis K. Production of aprepitant nanocrystals by wet media milling and subsequent solidification. Int J Pharm. 2017;533(2):324–334. | ||
Malamatari M, Somavarapu S, Taylor KM, Buckton G. Solidification of nanosuspensions for the production of solid oral dosage forms and inhalable dry powders. Expert Opin Drug Deliv. 2016;13(3):435–450. | ||
Zhang Y, Feng J, McManus SA, et al. Design and solidification of fast-releasing clofazimine nanoparticles for treatment of cryptosporidiosis. Mol Pharm. 2017;14(10):3480–3488. | ||
Ma YQ, Zhang ZZ, Li G, et al. Solidification drug nanosuspensions into nanocrystals by freeze-drying: a case study with ursodeoxycholic acid. Pharm Dev Technol. 2016;21(2):180–188. | ||
Alaei S, Ghasemian E, Vatanara A. Spray drying of cefixime nanosuspension to form stabilized and fast dissolving powder. Powder Technol. 2016;288:241–248. | ||
Wang Y, Zheng Y, Zhang L, et al. Stability of nanosuspensions in drug delivery. J Control Release. 2013;172(3):1126–1141. | ||
Nekkanti V, Pillai R, Venkateshwarlu V, Harisudhan T. Development and characterization of solid oral dosage form incorporating candesartan nanoparticles. Pharm Dev Technol. 2009;14(3):290–298. | ||
Niwa T, Miura S, Danjo K. Design of dry nanosuspension with highly spontaneous dispersible characteristics to develop solubilized formulation for poorly water-soluble drugs. Pharm Res. 2011;28(9):2339–2349. | ||
Sarnes A, Kovalainen M, Häkkinen MR, et al. Nanocrystal-based per-oral itraconazole delivery: superior in vitro dissolution enhancement versus Sporanox is not realized in in vivo drug absorption. J Control Release. 2014;180:109–116. | ||
de Smet L, Saerens L, de Beer T, et al. Formulation of itraconazole nanococrystals and evaluation of their bioavailability in dogs. Eur J Pharm Biopharm. 2014;87(1):107–113. | ||
van Eerdenbrugh B, Vercruysse S, Martens JA, et al. Microcrystalline cellulose, a useful alternative for sucrose as a matrix former during freeze-drying of drug nanosuspensions: a case study with itraconazole. Eur J Pharm Biopharm. 2008;70(2):590–596. | ||
Azad M, Moreno J, Bilgili E, Davé R. Fast dissolution of poorly water soluble drugs from fluidized bed coated nanocomposites: impact of carrier size. Int J Pharm. 2016;513(1–2):319–331. | ||
Hou Y, Shao J, Fu Q, Li J, Sun J, He Z. Spray-dried nanocrystals for a highly hydrophobic drug: increased drug loading, enhanced redispersity, and improved oral bioavailability. Int J Pharm. 2017;516(1–2):372–379. | ||
Liversidge GG, Cundy KC. Particle size reduction for improvement of oral bioavailability of hydrophobic drugs – I: absolute oral bioavailability of nanocrystalline danazol in beagle dogs. Int J Pharm. 1995;125(1):91–97. | ||
Suzuki M, Machida M, Adachi K, et al. Histopathological study of the effects of a single intratracheal instillation of surface active agents on lung in rats. J Toxicol Sci. 2000;25(1):49–55. | ||
Lebhardt T, Roesler S, Uusitalo HP, Kissel T. Surfactant-free redispersible nanoparticles in fast-dissolving composite microcarriers for dry-powder inhalation. Eur J Pharm Biopharm. 2011;78(1):90–96. | ||
Chin WW, Parmentier J, Widzinski M, Tan EH, Gokhale R. A brief literature and patent review of nanosuspensions to a final drug product. J Pharm Sci. 2014;103(10):2980–2999. | ||
Kesisoglou F, Panmai S, Wu Y. Nanosizing: oral formulation development and biopharmaceutical evaluation. Adv Drug Deliver Rev. 2007;59(7):631–644. | ||
Kim S, Lee J. Effective polymeric dispersants for vacuum, convection and freeze drying of drug nanosuspensions. Int J Pharm. 2010;397(1–2):218–224. | ||
Kho K, Hadinoto K. Effects of excipient formulation on the morphology and aqueous re-dispersibility of dry-powder silica nano-aggregates. Colloids Surf A Physicochem Eng Aspects. 2010;359(1–3):71–81. | ||
Hu J, Ng WK, Dong Y, Shen S, Tan RB. Continuous and scalable process for water-redispersible nanoformulation of poorly aqueous soluble APIs by antisolvent precipitation and spray-drying. Int J Pharm. 2011;404(1–2):198–204. | ||
Hong J, Liu Y, Xiao Y, et al. High drug payload curcumin nanosuspensions stabilized by mPEG-DSPE and SPC: in vitro and in vivo evaluation. Drug Deliv. 2017;24(1):109–120. | ||
Lee TW, Boersen NA, Hui HW, Chow SF, Wan KY, Chow AH. Delivery of poorly soluble compounds by amorphous solid dispersions. Curr Pharm Des. 2014;20(3):303–324. | ||
Tian B, Zhang L, Pan Z, Gou J, Zhang Y, Tang X. A comparison of the effect of temperature and moisture on the solid dispersions: aging and crystallization. Int J Pharm. 2014;475(1–2):385–392. | ||
Omidian H, Park K. Swelling agents and devices in oral drug delivery. J Drug Deliv Sci Technol. 2008;18(2):83–93. | ||
Agrawal A, Dudhedia M, Deng W, et al. Development of tablet formulation of amorphous solid dispersions prepared by hot melt extrusion using quality by design approach. AAPS PharmSciTech. 2016;17(1):214–232. | ||
Bhakay A, Azad M, Vizzotti E, Davé RN, Bilgili E. Enhanced recovery and dissolution of griseofulvin nanoparticles from surfactant-free nanocomposite microparticles incorporating wet-milled swellable dispersants. Drug Dev Ind Pharm. 2014;40(11):1509–1522. | ||
Chaulang G, Patel P, Hardikar S, Kelkar M, Bhosale A, Bhise S. Formulation and evaluation of solid dispersions of furosemide in sodium starch glycolate. Trop J Pharm Res. 2009;8(1):43–51. | ||
Fujii M, Okada H, Shibata Y, Teramachi H, Kondoh M, Watanabe Y. Preparation, characterization, and tableting of a solid dispersion of indomethacin with crospovidone. Int J Pharm. 2005;293(1–2):145–153. | ||
Wang T, Liu B, Zhang W, Wilson B, Hong JS. Andrographolide reduces inflammation-mediated dopaminergic neurodegeneration in mesencephalic neuron-glia cultures by inhibiting microglial activation. J Pharmacol Exp Ther. 2004;308(3):975–983. | ||
Shen Y, Chen C, Chiou W. Andrographolide prevents oxygen radical production by human neutrophils: possible mechanism(s) involved in its anti-inflammatory effect. Br J Pharmacol. 2002;135(2):399–406. | ||
Panossian A, Hovhannisyan A, Mamikonyan G, et al. Pharmacokinetic and oral bioavailability of andrographolide from Andrographis paniculata fixed combination kan jang in rats and human. Phytomedicine. 2007;7(5):351–364. | ||
Ye L, Wang T, Tang L, et al. Poor oral bioavailability of a promising anticancer agent andrographolide is due to extensive metabolism and efflux by P-glycoprotein. J Pharm Sci. 2011;100(11):5007–5017. | ||
Bothiraja C, Shinde MB, Rajalakshmi S, Pawar AP. Evaluation of molecular pharmaceutical and in vivo properties of spray-dried isolated andrographolide-PVP. J Pharm Pharmacol. 2009;61(11):1465–1472. | ||
Xu J, Ma Y, Xie Y, et al. Design and evaluation of novel solid self-nanodispersion delivery system for andrographolide. AAPS PharmSciTech. 2017;18(5):1572–1584. | ||
Chen Y, Liu Y, Xu J, et al. Design and evaluation of nanocomposite microparticles to enhance dissolution and oral bioavailability of andrographolide. Powder Technol. 2018;323:219–229. | ||
Qiao H, Chen L, Rui T, et al. Fabrication and in vitro/in vivo evaluation of amorphous andrographolide nanosuspensions stabilized by d-α-tocopheryl polyethylene glycol 1000 succinate/sodium lauryl sulfate. Int J Nanomedicine. 2017;12:1033–1046. | ||
Yue PF, Li G, Dan JX, et al. Study on formability of solid nanosuspensions during solidification – II: novel roles of freezing stress and cryoprotectant property. Int J Pharm. 2014;475(1–2):35–48. | ||
Gu Y, Ma J, Liu Y, Chen B, Yao S. Determination of andrographolide in human plasma by high-performance liquid chromatography/mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;854(1–2):328–331. | ||
Stevens RE, Gray V, Dorantes A, Gold L, Pham L. Scientific and regulatory standards for assessing product performance using the similarity factor, f2. AAPS J. 2015;17(2):301–306. | ||
Lee J. Drug nano- and microparticles processed into solid dosage forms: physical properties. J Pharm Sci. 2003;92(10):2057–2068. | ||
Bilgili E, Afolabi A. A combined microhydrodynamics–polymer adsorption analysis for elucidation of the roles of stabilizers in wet stirred media milling. Int J Pharm. 2012;439(1–2):193–206. | ||
Shah U, Augsburger L. Evaluation of the functional equivalence of crospovidone NF from different sources – I: physical characterization. Pharm Dev Technol. 2001;6(1):39–51. | ||
Azad M, Afolabi A, Patel N, Davé R, Bilgili E. Production of colloidal suspensions of superdisintegrants via wet stirred media milling. Particuology. 2014;14(3):76–82. | ||
Azad M, Afolabi A, Bhakay A, Leonardi J, Davé R, Bilgili E. Enhanced physical stabilization of fenofibrate nanosuspensions via wet co-milling with a superdisintegrant and an adsorbing polymer. Eur J Pharm Biopharm. 2015;94:372–385. | ||
Schulz M, Fussnegger B, Bodmeier R. Adsorption of carbamazepine onto crospovidone to prevent drug recrystallization. Int J Pharm. 2010;391(1–2):169–176. | ||
Knieke C, Azad MA, To D, Bilgili E, Davé RN. Sub-100 micron fast dissolving nanocomposite drug powders. Powder Technol. 2015;271:49–60. | ||
Kim HJ, Chen XD, Pearce D. Surface characterization of four industrial spray dried dairy powders in relation to chemical composition, structure and wetting property. Colloids Surf B Biointerfaces. 2002;26(3):197–212. | ||
Sou T, Orlando L, McIntosh MP, Kaminskas LM, Morton DA. Investigating the interactions of amino acid components on a mannitol-based spray-dried powder formulation for pulmonary delivery: a design of experiment approach. Int J Pharm. 2011;421(2):220–229. | ||
Wu W, Nancollas GH. A new understanding of the relationship between solubility and particle size. J Solution Chem. 1998;27(6):521–531. | ||
Lu Y, Qi J, Dong X, Zhao W, Wu W. The in vivo fate of nanocrystals. Drug Discov Today. 2017;22(4):744–750. | ||
Chen C, Wang L, Cao F, et al. Formulation of 20(S)-protopanaxadiol nanocrystals to improve oral bioavailability and brain delivery. Int J Pharm. 2016;497(1–2):239–247. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.