Back to Journals » The Application of Clinical Genetics » Volume 11
Novel mutation in ABBC9 gene associated with congenital hypertrichosis and acromegaloid facial features, without cardiac or skeletal anomalies: a new phenotype
Authors Pachajoa H ![]() , López-Quintero W
, López-Quintero W ![]() , Vanegas S
, Vanegas S ![]() , Montoya CL
, Montoya CL ![]() , Ramírez-Montaño D
, Ramírez-Montaño D
Received 24 October 2017
Accepted for publication 30 November 2017
Published 23 March 2018 Volume 2018:11 Pages 15—21
DOI https://doi.org/10.2147/TACG.S155022
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Harry Pachajoa,1,2 William López-Quintero,3 Sara Vanegas,1 Claudia L Montoya,3 Diana Ramírez-Montaño1
1Department of Basic Medical Sciences, Center for Research on Congenital Anomalies and Rare Diseases (CIACER), Universidad Icesi, Cali, Valle del Cauca, Colombia; 2Pediatric Medical Genetics, Fundación Valle del Lili, Cali, Valle del Cauca, Colombia; 3Dermatology Department, Fundación Valle del Lili, Cali, Valle del Cauca, Colombia
Introduction: Mutations in ABCC9 are associated with Cantú syndrome (CS), a very rare genetic disorder characterized by congenital hypertrichosis, acromegaloid facial appearance (AFA), cardiomegaly, and skeletal anomalies.
Case report: We report an 8-year-old female patient with congenital generalized hypertrichosis and coarse facial appearance but without cardiovascular or skeletal compromise. Whole exome sequencing revealed a novel de novo heterozygous mutation in ABCC9. In addition, the genotype and phenotype of the patient were compared with those of the patients reported in the literature and with other related conditions that include AFA, hypertrichosis and AFA, and CS.
Conclusion: This is the first report of a South-American patient with mutation in ABCC9. We propose that her phenotype is a part of a spectrum of features associated with congenital hypertrichosis and mutations in ABCC9, which differs from CS and related disorders. Whole exome sequencing enabled the identification of the causality of this disease characterized by high clinical and genetic heterogeneity.
Keywords: hypertrichosis, acromegaloid features, AFA syndrome, Cantú syndrome
Corrigendum has been published
Introduction
Hypertrichosis is defined as increase in body hair (lanugo, vellus hair, or terminal hair) that is abnormal for a patient reference group. Hypertrichosis may be classified as congenital or acquired, with generalized or regional hair growth. Congenital hypertrichosis may be isolated or part of a syndrome that is associated with dysmorphic features or metabolic disorders. The genetic basis of hypertrichosis is not well understood. Non-androgen-related excessive growth of terminal hair is associated with several rare genetic conditions.1 Three entities with clinical features of hypertrichosis and acromegaloid facial features have been published: 1) Acromegaloid facial appearance syndrome (AFA, MIM: 102150), 2) Hypertrichosis with acromegaloid facial appearance (HAFA, MIM: #135400), and 3) Cantú syndrome (CS), also known as hypertrichotic osteochondrodysplasia (MIM #239850). CS is the best known of these three genetic disorders. This condition was first described by Cantú et al,2 in 1982, and is characterized by autosomal dominant inheritance, congenital hypertrichosis, distinctive coarse facial features, skeletal abnormalities, and cardiac defects.2,3
About 35 patients with CS have been reported in literature. CS appears to be prevalent among all populations, affecting males and females equally, and the disease seems to typically originate by de novo mutations.4–7 Reported patients with AFA and HAFA syndrome have been documented occasionally with cardiac involvement, like pericardial effusion8 and skeletal malformation, like scoliosis,9 suggesting that cardiovascular and skeletal anomalies are not exclusive to CS. Clinical features of AFA, HAFA and CS are summarized in Table 1. Clinical overlap between CS and other genetic syndromes with hypertrichosis, including lysosomal storage disease, and poor knowledge about this disease may lead to its underdiagnosis.
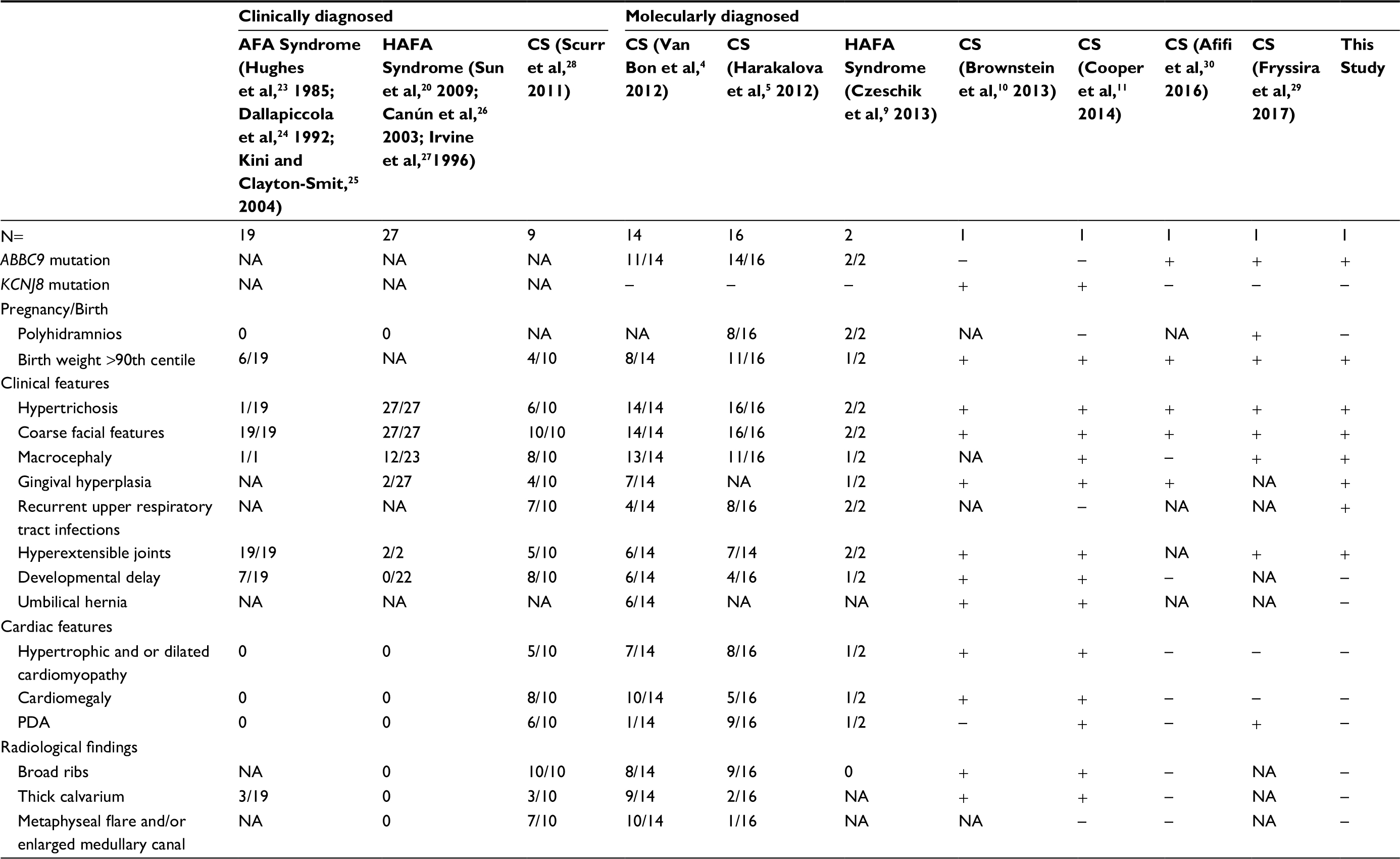 | Table 1 Summary of clinical features of reported patients with AFA, HAFA, and CS Abbreviations: AFA, acromegaloid-like appearance; CS, Cantú syndrome; HAFA, hypertrichosis with acromegaloid facial appearance; NA, not applicable; PDA, patent ductus arteriosus. |
In 2012, Harakalova et al5 performed exome sequencing in 16 individuals with clinical features of CS and described a novel dominant missense mutation in the ABCC9 gene in 14 of them. They concluded that heterozygous missense mutations in ABCC9 cause CS.5 Deletion/duplication type mutations have not yet been reported, and only two cases have been associated with mutations in another gene (KCNJ8).10,11 ABCC9 gene (previously known as SUR2) encodes a transmembrane protein that is part of an ATP-dependent potassium (KATP) channel that couples the metabolic state of a cell with its electrical activity. Two spliced forms with tissue-specific expression have been reported, namely, SUR2A, expressed in cardiac and skeletal muscles, and SUR2B, expressed in vascular smooth muscle and hair follicles. Mutations in ABCC9 gene reduce ATP-mediated K channel inhibition, resulting in dominant channel opening. This effect of this mutation is similar to the side effects produced by treatment with KATP channel agonist (Minoxidil).10 Only two patients with HAFA syndrome have been reported with mutations in ABCC9.9
However, CS is a part of a wide phenotypic spectrum with variable severity including, AFA, HAFA syndromes, and skeletal dysplasia or CS, which is the most severe form. We report a case of an 8-year-old girl from Colombia, with clinical features of congenital HAFA without skeletal abnormalities or cardiac involvement. Whole exome sequencing (WES) was performed in the patient, and a novel heterozygous missense, likely pathogenic variant in ABCC9 was identified.
Case report
We report a case of an 8-year-old girl, from southwest Colombia, who was the first child of a 29-year-old mother and nonconsanguineous 34-year-old father. Pregnancy was uncomplicated, and prenatal ultrasounds were normal, without history of polyhydramnios. Vaginal delivery at the 37th week of gestation was without complications, and the birth weight was 3,650 g (94th centile) and length was 50 cm (83th centile). At birth, the proband presented neonatal respiratory distress, which required monitoring in the neonatal intensive care unit for 8 days. Clinical findings at birth included excessively thick facial hair, mainly in the forehead region, broad nose, wide mouth, full lips, umbilical hernia, and general hypertrichosis moderately distributed on the trunk and limbs. No history of patent ductus arteriosus or another congenital cardiomyopathy was detected.
The patient experienced multiple episodes of respiratory infection during childhood, which improved after turbinoplasty and adenoidectomy at 4 years of age. At 2 years of age, pubic hair appeared, and follow-up was started with a pediatric endocrinologist who ruled out androgenic hormonal disorder (with normal levels of testosterone, α-OH-progesterone, and somatomedin C). Psychomotor development was unaffected. Although she presented language development delay, she exhibited a dysarthria-like speech and mild learning disabilities; however, IQ test results were within the normal range, and she had no motor developmental concerns. Similar clinical pictures were negative in her family history.
At 8 years of age, she was assessed medically. Her weight was 27.6 kg (48th centile) and height was 128 cm (27th centile). Physical examination revealed generalized hypertrichosis mainly on the face, limbs, back region, and genitals (Figure 1). Other findings included low anterior hairline, synophrys, long eyelashes, dolichocephaly, hypoplastic nasal bones, broad nose and lips, dental malocclusion with inferior wide-spaced teeth, bilateral epicanthic folds, and AFA without corneal opacity. Osteomuscular examination revealed right fifth finger clinodactyly, bilateral sandal gap, and dorsal scoliosis.
 | Figure 1 Patient phenotype. Notes: (A) Coarse facial features, AFA, low anterior hairline, synophrys, long eyelashes, epicanthic folds, broad lips, bulbous nose, and broad mouth. Phenotypic similarities with CS. (B) Generalized hypertrichosis predominantly on back and extremities. Abbreviations: AFA, acromegaloid-like appearance; CS, Cantú syndrome. |
At 7 years of age, radiological findings did not reveal any alterations of the extremities, hips, and spine. Radiography of the hand bone age correlated with her biological age. In addition, endocrinological laboratory test results were normal (thyroid-stimulating hormone, free T4, growth hormone, insulin-like growth factor-1, follicle-stimulating hormone, and leutinizing hormone). Echocardiography showed normal biventricular function, normal left ventricular size, no pericardial effusion, no pulmonary hypertension or signs of cardiomyopathy, and electrocardiogram (ECG) was within normal range. Audiometry reported mild hearing loss in the left ear. Metabolic impairment screening studies for mucopolysaccharides (mucopolysaccharide electrophoresis, enzyme activity of α-L-iduronidase, and arylsulfatase B) and quantitative chromatography of amino acids using plasma and urine revealed normal results.
Further investigation was performed using WES by trio approach with massive sequencing platform with Ion Proton™ technology. The library preparation was designed with Ion AmpliSeq Exome technology (Life Technologies, Carlsbad, CA, USA) which captures >97% of Consensus coding DNA sequencing (CCDS) (>19,000 genes and >198,000 exons) and flanking intronic regions (±5 bp). Only variants in the coding region and flanking intronic regions with a minor allele frequency <1.5% were evaluated. Minor allele frequencies were based on the following databases: 1,000 Genomes, dbSNP, Exome Variant Server (ESVor in-house), and Exome Aggregation Consortium (ExAc). A novel missense, likely pathogenic variant in heterozygous state in ABCC9 (NM_005691.3) was identified: c.3625T>C (p.Tyr1209Hys) (Figure 2). This variant has not been previously reported in the literature, and the finding was confirmed by Sanger sequencing (Figure 2) and was compatible with the diagnosis of CS. Variant functional prediction software tools Mutation taster, Condel, SIFT, and FATHMM classified it as a Damaging variant (disease causing) (Table 2). Protein localization of the change is in the transmembrane domain 2 of ABCC9 (Figure 2). No additional variants were identified, including in genes associated with lysosomal storage disease, like mucopolysaccharidoses.
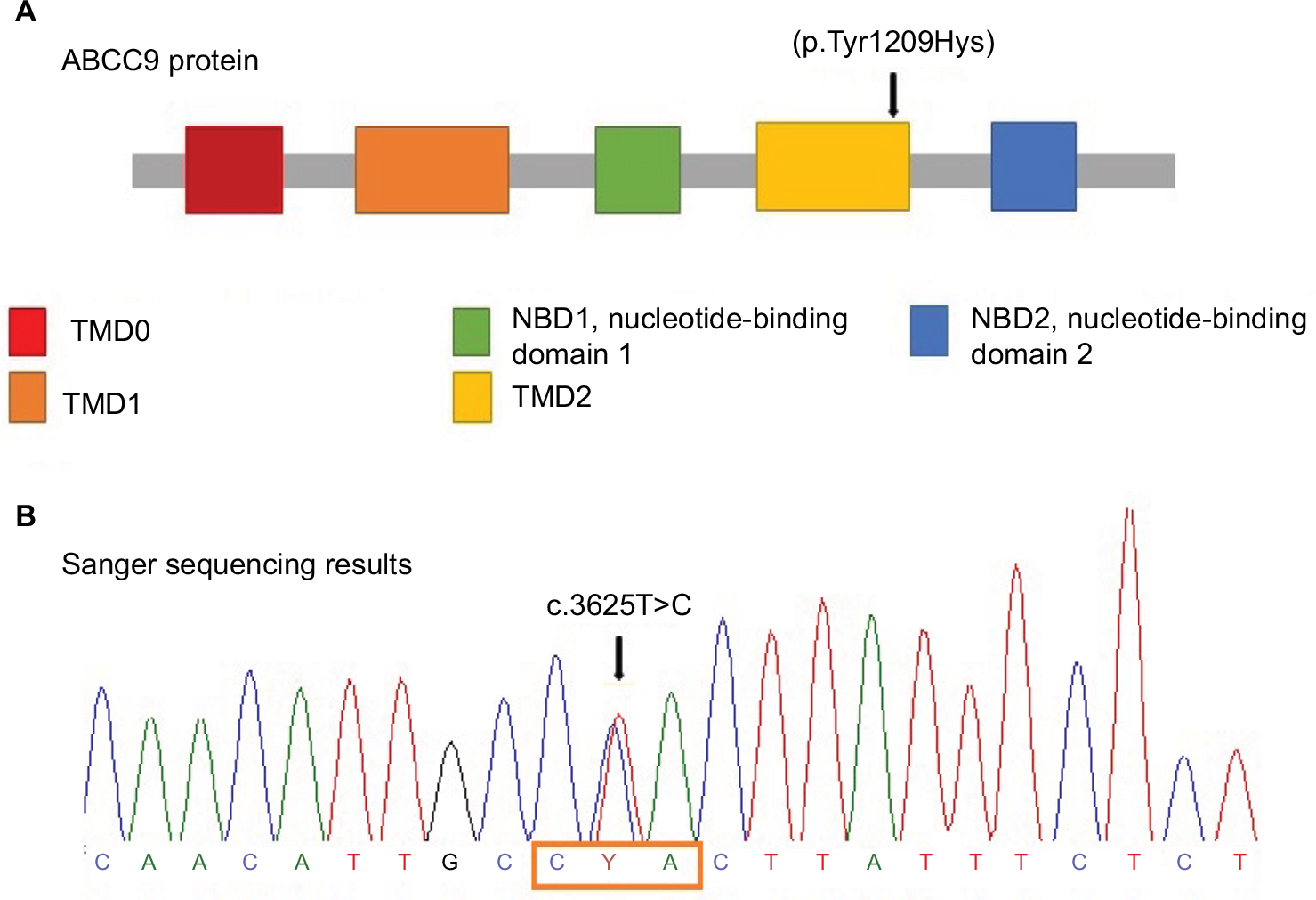 | Figure 2 ABCC9 protein structure and Sanger sequencing results. Notes: (A) ABCC9 protein structure: mutation is located on TMD2 domain. Mutations at this point result in gain of function in the KATP channel resulting in the opening of the potassium channel. The arrow indicates the novel variant that was present in the patient. (B) Sanger sequencing results: The box frames the affected codon and the arrow indicates the heterozygous state of the identified variant. Abbreviation: TMD, transmembrane domain. |
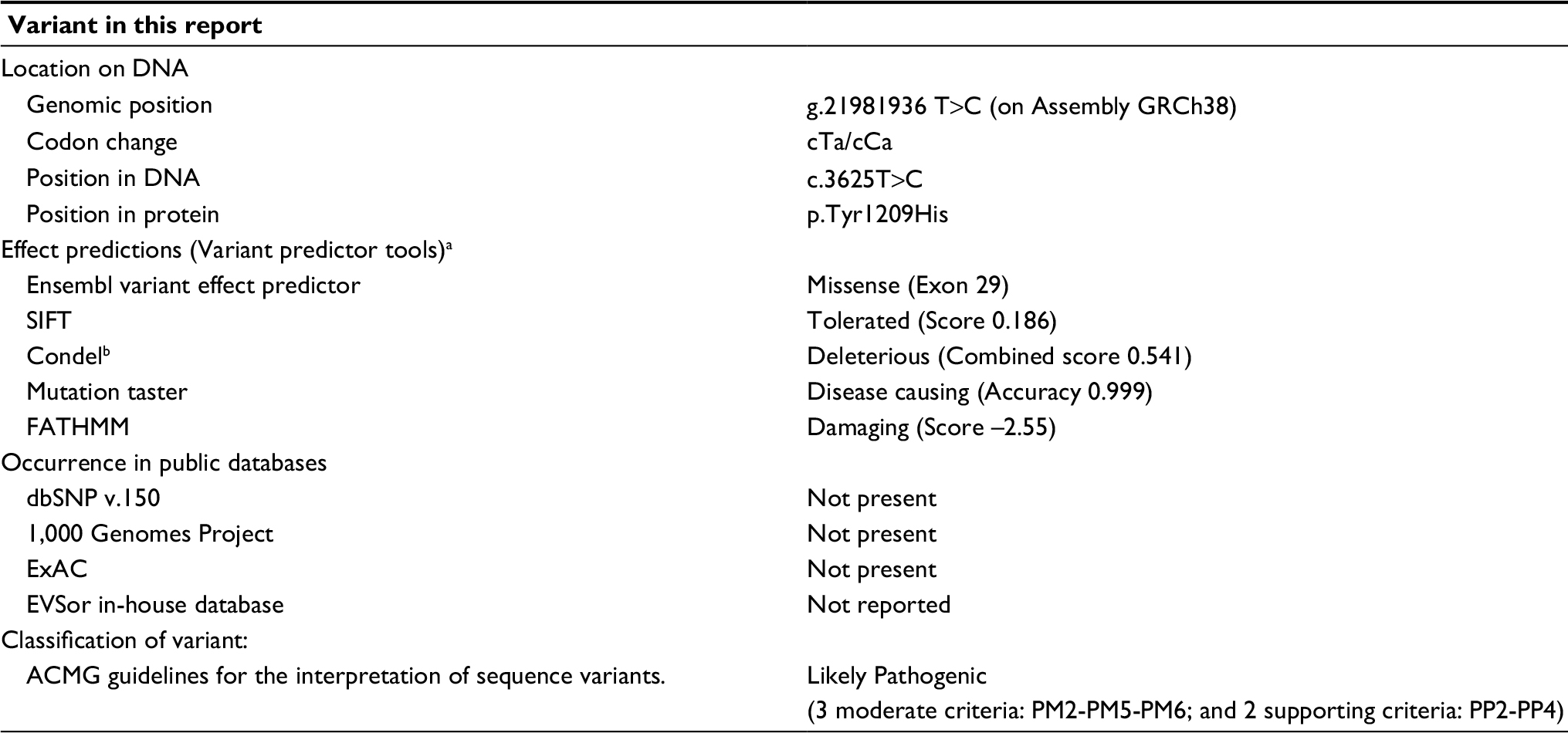 | Table 2 Prediction of novel variant in ABCC9 Notes: aAvailable from: https://varsome.com/variant/hg19/ENST00000261200.4:3Ac.3625T>C. bAvailable from: http://bbglab.irbbarcelona.org/fannsdb/query/condel. Abbreviations: ACMG, American College of Medical Genetics and Genomics; dbSNP, Single-nucleotide polymorphism database (NCBI); EVS, Exome variant service; ExAC, Exome aggregation consortium; PM, pathogenic moderate criteria; PP, pathogenic supporting criteria. |
Ethics and consent to participate
Written informed consent was obtained from patient’s parents for publication of her images and clinical data for scientific purposes. Data were collected in the context of studies performed in accordance with the Declaration of Helsinki Good Clinical Guidelines and protocol #509 “registry of surveillance and survival of congenital defects of the Colombian South-West” approved by the Ethics Committee of Universidad Icesi (Act 192/2011).
Discussion
An important feature of CS is high phenotypic variability. Neonatal characteristics including macrosomia, observed in our patient, soft tissue swelling, and edema have been reported in approximately 50% of the affected individuals.2,3,13,14 During childhood, patients usually have low subcutaneous fat and muscular appearance,4 but our patient did not manifest these features. The phenotypic characteristics in our proband, including coarse facial features, low nasal bridge, epicanthic folds, synophris, and broad mouth and lips, are similar to those described in previously reported cases. In addition, coarse facial features and hypertrichosis are also associated with certain lysosomal storage diseases, such as Hurler and Hunter syndromes. Therefore, a clinical diagnostic workup to rule out these pathologies is necessary.
Cardiac manifestations, including patent ductus arteriosus, ventricular hypertrophy, pulmonary hypertension, pericardial effusion, and increased vascular tortuosity, have been reported in 80% of the patients.4,12–14 Transthoracic echocardiography and electrocardiography results were within the normal range. These findings may suggest HAFA (MIM #135400) as a diagnosis. However, only one individual with HAFA has been identified with a mutation in ABCC9.9 Mutations in this gene are associated with isolated atrial fibrillation, dilated cardiomyopathy,15 myocardial infarction,16 and repolarization syndrome.17 This indicates that ABCC9 plays a key role in KATP channels, which function primarily in the heart and smooth muscles. This phenomenon may explain that mutations could greatly affect the cardiovascular system. Previous reports have described that cardiac manifestations appear early in life, including congenital presentation of hypertrophic and/or dilated cardiomiopathy.4,5 The absence of this feature in our proband was definitive; however, clinical follow-up is necessary regarding possible progressive behavior not previously reported.
Affected individuals with CS have skeletal abnormalities and craniofacial features including thickening of cranial vault, narrow thorax, broad ribs, long bones with metaphyseal widening of long bones, flat-ovoid vertebral bodies, enlarged medullary canals, and coxa valga.4,13,14 Skeletal abnormalities were not found by X-ray imaging. However, clinically, the patient exhibited scoliosis. Similar to cardiac manifestations, absence of skeletal abnormalities in X-ray studies suggest that they are unlikely to appear later in life.4,5 Other neurologic manifestations may include hypotonia, language and motor delays, and light to severe intellectual impairment.4 In our proband, we observed mild dysarthria, without other neurological compromise. Recurrent infections in lower and upper respiratory tract appear in reported patients and in our patient.
ABCC9 gene (also known as SUR2) is located on locus 12p12.1 of human chromosomes and encodes a transmembrane protein of 1,549 amino acids. SUR2 forms the regulatory part of an ATP-sensitive potassium complex that consists of four subunits of a transmembrane pore (KCNJ8). The protein has two splice variants SUR2A (expressed in cardiac and skeletal muscle) and SUR2B (expressed in smooth muscle), which contains three transmembrane cytoplasmic domains (TMD0, TMD1, TMD2), and two nucleotide binding folds (NFB1 and NFB2) (Figure 2). Mutations in CS located in ABCC9 transmembrane domain result from a gain of function in the KATP channel regulated by these subunits.4–6,9 Therefore, missense mutations are believed to cause the opening of the potassium channel, causing metabolic and electric disequilibrium essentially in cardiomyocytes.5,18 We identified a de novo pathogenic missense mutation c.3625T>C (p.Tyr1209Hys), which was not found in the 1,000 Genomes Project, ExAC, EVSor in-house database, and so it was considered as previously not reported. Based on the American College of Medical Genetics and Genomics (AMCG) variant interpretation guideline,19 this variant is classified as likely pathogenic (Table 2), and was validated by Mutation taster, Condel, SIFT, and FATHMM; it is predicted to have deleterious effect. This change occurs in a genomic position highly evolutionarily conserved in vertebrates.
Two similar mutations have been reported in the TMD2 domain, which were associated with AFA and HAFA syndrome. The phenotype of these patients included AFA, gingival hyperplasia, enlarged hands and foot, and arched eyebrows, in combination with congenital severe generalized hypertrichosis.9 In addition, the HAFA phenotype is described as having AFA, congenital hypertrichosis terminalis, and gingival hyperplasia; although the gene is unknown, it has been demonstrated that this condition is a contiguous gene syndrome, comprised on chromosomic region 17q (q24.2-q24.3).20 The data suggest that HAFA might be related with less severe manifestations of CS, with a different genetic cause than ABCC9 and KCNJ8.20 Despite some authors distinguishing AFA, HAFA, and CS, we suggest they should be considered as the same disorder with a variable severity in phenotype rather than as separate conditions.
Furthermore, it is important to mention that WES is an effective diagnostic tool that is useful for a geneticist to identify monogenic conditions. Exome sequencing has greatly influenced the timeframe within which new disease genes are identified. In the last decade, exome sequencing has improved diagnostic performance by more than 25%, leading to identification of disease-causing genes in cases where the diagnosis was previously unknown, and allows a complete characterization of genetic variations.21 Confirmatory identification of the underlying genetic cause of CS mutations in ABCC9 by WES technology has been done in about 28 of 35 patients. This technology helps to clarify the spectrum of this condition and its overlap with other similar syndromes mentioned previously.22
Conclusion
In summary, we report on a Colombian patient with congenital hypertrichosis, acromegaloid facial features, and no skeletal abnormalities or cardiac manifestations, with a novel missense mutation in ABCC9 gene, which may suggest a wide spectrum of phenotypes associated with mutations in this gene. We propose that her mild phenotype could be similar to the patient reported by Czeschik et al9 in 2013, and hence is suggestive of a new genetic condition. However, additional reports are needed to confirm this observation and experimental studies are necessary to demonstrate those findings.
Acknowledgment
We would like to thank the family of the patient for their participation in this study.
Author contributions
All authors contributed toward data analysis, drafting and critically revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Pavone P, Praticò A, Falsaperla R, et al. Congenital generalized hypertrichosis: the skin as a clue to complex malformation syndromes. Ital J Pediatr. 2015;41(1):55. | ||
Cantú JM, Garcia-Cruz D, Sanchez-Corona J, Hernandez A, Nazara Z. A Distinct osteochondrodysplasia with hypertrichosis – individualization of a probable autosomal recessive entity. Hum Genet. 1982;60:36–41. | ||
Garcia-Cruz D, Sánchez-Corona J, Nazará Z, et al. Congenital hypertrichosis, osteochondrodysplasia, and cardiomegaly: further delineation of a new genetic syndrome. Am J Med Genet. 1997;69:138–151. | ||
Van Bon BW, Gilissen C, Grange DK, et al. Cantú syndrome is caused by mutations in ABCC9. Am J Hum Genet. 2012;90:1094–1101. | ||
Harakalova M, van Harssel JJ, Terhal PA, et al. Dominant missense mutations in ABCC9 cause Cantú syndrome. Nature Genet. 2012;44:793–796. | ||
Hiraki Y, Miyatake S, Hayashidani M, et al. Aortic aneurysm and craniosynostosis in a family with Cantú syndrome. Am J Med Genet A. 2014;164A:231–236. | ||
Park JY, Koo SH, Jung YJ, Lim YJ, Chung ML. A patient with Cantú syndrome associated with fatal bronchopulmonary dysplasia and pulmonary hypertension. Am J Med Genet. 2014;164A:2118–2120. | ||
Zalante L, Gasparini P, Savoia A, Lomuto M, Pellicano R. A new case of acromegaloid facial appearance (AFA) síndrome with an expanden phenotype. Clin Dysmorphol. 2000;9:221–222. | ||
Czeschik JC, Voigt C, Goecke TO, et al. Wide clinical variability in conditions with coarse facial features and hypertrichosis caused by mutations in ABCC9. Am J Med Genet A. 2013;161A:295–300. | ||
Brownstein CA, Towne MC, Luquette LJ, et al. Mutation of KCNJ8 in a patient with Cantú syndrome with unique vascular abnormalities - support for the role of K(ATP) channels in this condition. Eur J Med Genet. 2013;56:678–682. | ||
Cooper PE, Reutter H, Woelfle J, et al. Cantú syndrome resulting from activating mutation in the KCNJ8 gene. Hum Mutat. 2014;35:809–813. | ||
Grange DK, Lorch SM, Cole PL, Singh GK. Cantú syndrome in a woman and her two daughters: further confirmation of autosomal dominant inheritance and review of the cardiac manifestations. Am J Med Genet A. 2006;140:1673–1680. | ||
Nevin NC, Mulholland HC, Thomas PS. Congenital hypertrichosis, cardiomegaly and mild osteochondrodysplasia. Am J Med Genet. 1996;66:33–38. | ||
Rosser EM, Kaariainen H, Hurst JA, et al. Three patients with the osteochondrodysplasia and hypertrichosis syndrome – Cantú syndrome. Clin Dysmorphol. 1998;7:79–85. | ||
Bienengraeber M, Olson TM, Selivanov VA, et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic KATP channel gating. Nat Genet. 2004;36:382–387. | ||
Minoretti P, Falcone C, Aldeghi A, et al. A novel Val734Ile variant in the ABCC9 gene associated with myocardial infarction. Clin Chim Acta. 2006;370(1–2):124–128. | ||
Hu D, Barajas-Martínez H, Terzic A, et al. ABCC9 is a novel Brugada and early repolarization syndrome susceptibility gene. Int J Cardiol. 2014;171(3):431–442. | ||
Bryan J, Muñoz A, Zhang X, et al. ABCC8 and ABCC9: ABC transporters that regulate K+ channels. Pflugers Arch. 2007;453(5):703–718. | ||
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. | ||
Sun M, Li N, Dong W, et al. Copy-number mutations on chromosome 17q24.2-q24.3 in congenital generalized hypertrichosis terminalis with or without gingival hyperplasia. Am J Hum Genet. 2009;84:807–813. | ||
Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med. 2014;370:2418–2425. | ||
Bramswig NC, Lüdecke HJ, Alanay Y, et al. Exome sequencing unravels unexpected differential diagnoses in individuals with the tentative diagnosis of Coffin-Siris and Nicolaides-Baraitser syndromes. Hum Genet. 2015;134(6):553–568. | ||
Hughes HE, McAlpine PJ, Cox DW, Philipps S. An autosomal dominant syndrome with ‘acromegaloid’ features and thickened oral mucosa. J Med Genet. 1985;22(2):119–125. | ||
Dallapiccola B, Zelante L, Accadia L, Mingarelli R. Acromegaloid facial appearance (AFA) syndrome: report of a second family. J Med Genet. 1992;29(6):419–422. | ||
Kini U, Clayton-Smith J. Acromegaloid facial appearance syndrome: a further case report. Clin Dysmorphol. 2004;13:251–253. | ||
Canún S, Guevara-Sanginés EG, Elvira-Morales A, Sierra-Romero Mdel C, Rodríguez-Asbun H. Hypertrichosis terminalis, gingival hyperplasia, and a characteristic face: a new distinct entity. Am J Med Genet. 2003;116A(3):278–283. | ||
Irvine AD, Dolan OM, Hadden DR, Stewart FJ, Bingham EA, Nevin NC. An autosomal dominant syndrome of acromegaloid facial appearance and generalised hypertrichosis terminalis. J Med Genet. 1996;33:972–974. | ||
Scurr I, Wilson L, Lees M, et al. Cantu syndrome: report of nine new cases and expansion of the clinical phenotype. Am J Med Genet. 2011;155A:508–518. | ||
Fryssira H, Psoni S, Amenta S, et al. Cantú Syndrome Associated with Ovarian Agenesis. Mol Syndromol. 2017;8(4):206–210. | ||
Afifi HH, Abdel-Hamid MS, Eid MM, Mostafa IS, Abdel-Salam GM. De Novo Mutation in ABCC9 Causes Hypertrichosis Acromegaloid Facial Features Disorder. Pediatr Dermatol. 2016;33(2):e109–e113. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.