Back to Journals » Drug Design, Development and Therapy » Volume 14
Novel HDAC/Tubulin Dual Inhibitor: Design, Synthesis and Docking Studies of α-Phthalimido-Chalcone Hybrids as Potential Anticancer Agents with Apoptosis-Inducing Activity
Authors Mourad AAE ![]() , Mourad MAE
, Mourad MAE ![]() , Jones PG
, Jones PG
Received 9 April 2020
Accepted for publication 19 June 2020
Published 3 August 2020 Volume 2020:14 Pages 3111—3130
DOI https://doi.org/10.2147/DDDT.S256756
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Tuo Deng
Ahmed A E Mourad,1 Mai A E Mourad,2 Peter G Jones3
1Pharmacology and Toxicology Department, Faculty of Pharmacy, Port-Said University, Port-Said, Egypt; 2Medicinal Chemistry Department, Faculty of Pharmacy, Port-Said University, Port-Said, Egypt; 3Institute of Inorganic and Analytical Chemistry, Braunschweig, Germany
Correspondence: Ahmed A E Mourad Tel +201069233766
Email [email protected]
Introduction: In order to develop novel anticancer HDAC/tubulin dual inhibitors, a novel series of α-phthalimido-substituted chalcones-based hybrids was synthesized and characterized by IR, 1H NMR, 13C NMR, mass spectroscopy and X-ray analysis.
Methods: All the synthesized compounds were evaluated for their in vitro anticancer activity against MCF-7 and HepG2 human cancer cell lines using MTT assay. To explore the mechanism of action of the synthesized compounds, in vitro β-tubulin polymerization and HDAC 1 and 2 inhibitory activity were measured for the most potent anticancer hybrids. Further, cell cycle analysis was also evaluated.
Results: The trimethoxy derivative 7j showed the most potent anticancer activity, possessed the most potent β-tubulin polymerase and HDAC 1 and 2 inhibitory activity and efficiently induced cell cycle arrest at both G2/M and preG1phases in the MCF-7 cell line.
Keywords: chalcones, α-phthalimido, anticancer, cell cycle, histone deacetylase inhibitor, tubulin polymerase inhibitor
Summary
- Novel series of α-phthalimido-chalcone hybrids were synthesized and identified by different spectroscopic techniques.
- The newly synthesized compounds experienced pfotent anticancer activity with IC50 at micromolar concentrations against MCF-7 and Hep G2 cell lines, particularly, 7j hybrid exhibited 2.57 and 4.51 fold superior anticancer activity than CA-4 against the tested cell lines, respectively.
- The most potent anticancer compounds were investigated as HDAC/tubulin dual inhibitors.
- Compound 7j inhibit in vitro β-tubulin polymerization and HDAC 1 and2 activity at IC50 lower than that of CA-4 and entinostat.
- Cell cycle analysis was done for the most active compounds and it was clear that the tested compounds successfully arrested cell cycle at G2/M phase and induced preG1 apoptosis compared to the reference compounds.
- Molecular docking studies were carried out to explore the binding pattern of the most active synthesized compounds toward colchicine binding site in tubulin protein and HDAC2 active site.
Introduction
Cancer is basically a group of fatal diseases recognized by uncontrolled cell growth.1 While chemotherapy remains the main treatment, clinically used anticancer drugs are generally limited by toxicity, side effects, and resistance.2 Liver cancer (hepatocellular carcinoma, HCC), with globally rising prevalence remains one of the most common and lethal type of cancer. It ranks as the sixth most frequent cancer type and fourth most common cause of cancer-related death worldwide, as well as being responsible for more than 80% of diagnosed primary liver cancer worldwide.3 Moreover, the therapeutic options remain very limited and restricted on liver resection, transplantation and chemoembolization. Notably, a small proportion of patients are suitable candidates for these options and the relapse rate is high.4 On the other hand, breast cancer is the leading cause of death among cancer-related deaths in women, affecting 2.1 million women every year.5 Despite the recent introduction of several promising novel therapies that have shown considerable therapeutic success including hormone therapy, radiotherapy and chemotherapy, drug resistance remains one of the major challenges in breast cancer treatment.6
Based on the aforementioned aspects, new curative therapeutic strategies are urgently needed that have the advantages of overcoming the anticancer therapy resistance and have superior efficacy, lower toxicity as well as better selectivity. Apart from these therapies, histone deacetylase inhibitors (HDACi)7 are epigenetic drugs, recently discovered, targeting specific parts of cancer cell signalling, particularly epigenome of cancer cells. Studies have shown that the impairment in the balance between the opposing actions of histone deacetylases (HDACs) and histone acetyltransferases (HATs) is responsible for epigenetic dysregulation of gene expression which results in development of many of cancer types. Among HDACs, HDAC2 is the most commonly overexpressed in many cancer types particularly HCC and breast cancers. Interestingly, HDACi can induce growth arrest in transformed cell, cell death and/or inhibition of angiogenesis, activation of the extrinsic and/or intrinsic apoptotic pathways and mitotic cell death.8 Fortunately, normal cells are relatively resistant to HDACi-induced cell death.9 Recently, several HDACi such as vorinostat, belinostat, panobinostat (hydroxamates), romidepsin (cyclic peptides), chidamide (benzamide) have been approved by FDA for treatment of cutaneous T-cell lymphoma, various hematological and solid cancers (Figure 1).10 The benzamide category can selectively inhibit class I HDACs (HDACs 1, 2, 3, and 8) by inducing cell cycle arrest and apoptosis of cancer cell lines, such as entinostat (MS-275) which is in Phase II clinical trial.11,12
 |
Figure 1 Examples of some approved HDACi. |
On the other hand, microtubules are polymers of α- and β-tubulin heterodimers which play the main role in regulating key cellular processes such as cell growth and division, preservation of the cell architecture and motility, spindle formation and intracellular trafficking.13 Consequently, due to these key roles tubulin targeting agents constitute an attractive class of chemotherapeutic drugs and it includes several agents, such as colchicine, combretastatin (CA-4), podophyllotoxin, vincristine (VCR), vinorelbine (VRB) and vinblastine (VBL) (Figure 2).14 CA-4 is a powerful antimitotic compound, possessing potent anticancer activity against a number of human cancer cell lines involving multidrug resistant cancer types.15 It acts as a microtubule destabilizing agent by specifically binding to β-subunit of tubulin at the same site of colchicine and thus strongly inhibits tubulin polymerization.16
 |
Figure 2 Examples on some microtubule targeting agents with affinity for colchicine-binding site. |
While searching for a new anticancer agent, chalcones have shown a successful design of effective antitumor agent. They are considered as one of the privileged scaffolds displaying multifarious applications in a wide variety of scientific domains, such as anticancer,17,18 anti-inflammatory,19 antioxidant,20 antimicrobial, anti-tubercular,21 anti-HIV,22 antimalarial,23 anti-allergic,24 and leishmanicidal activities.25 Notably, different mechanisms of action have been described for chalcones, including the induction of apoptosis, inhibition of cell proliferation, inhibition of angiogenesis, blockade of nuclear factor-kappa B (NF-κB) signaling pathway and reversal of multidrug resistance or a combination of these mechanisms.26,27 Furthermore, chalcones are powerful tubulin assembly inhibitors, with almost similar potency to CA-4.28
Of considerable interest, phthalimide and its derivatives have recently emerged as an important class of compounds which are used as the starting synthone for synthesis of diverse biologically active compounds such as anti-inflammatory,29 antioxidant,30 anti-angiogenic,31 hypoglycemic,32 antitumor,33 anti-tubercular agents34 as well as inhibitor of tumor necrosis factor-α (TNF-α) production.35 Moreover, α-phthalimido-ketones possess liver X receptor antagonistic,36 leukotriene D4 receptor antagonistic,37 anti-inflammatory,38 and angiogenesis inhibitor activities.39
Despite the continuous developments in anticancer drugs, new and more effective compounds are still required for this disease to be under control. These considerations and the above-mentioned findings prompted us to design a novel series of α-phthalimido-substituted chalcones-based hybrid scaffolds by incorporating the two entities in a single molecule via benzamido-link (Scheme 1) aiming at synergizing the anticancer activity and decreasing the human cytotoxicity by demonstrating their potential activity as promising HDAC-tubulin dual inhibitors. The novel hybrids were tested for their in vitro anticancer activity against certain human cancer cell lines. Moreover, the effects of these hybrids as inhibitors of histone deacetylase and tubulin polymerase were studied and supported with evaluation of the cell cycle analysis and apoptotic nature of the most potent compounds. Furthermore, molecular docking studies were also performed to investigate the interaction of inhibitors with the colchicine binding site of tubulin and HDAC2 active site (Figure 3).
 |
Figure 3 Diagram represents the design strategy of novel HDAC/tubulin inhibitors and the reported anticancer compounds CA-4 and entinostat. |
Results and Discussion
Chemistry
The synthesis of the target compounds 7a-j is depicted in Scheme 1. The synthesis of compounds 4 and 5 followed the literature procedures40 using phthalimide as starting material. Reduction of phthalimide with zinc dust in the presence of copper sulphate followed by cyclization gave phthalide, which on reaction with potassium phthalimide in DMF resulted in formation of α-phthalimido-o-toluic acid (4). Treatment of 4 with thionyl chloride gave α-phthalimido-o-toluoyl chloride (5). N-(4-Acetylphenyl)-2-(1,3-dioxoisoindolin-2-yl)benzamide (6) has been synthesized by the reaction of 5 with 4-amino-acetophenone in the presence of triethyl amine (TEA) in 92% yield.
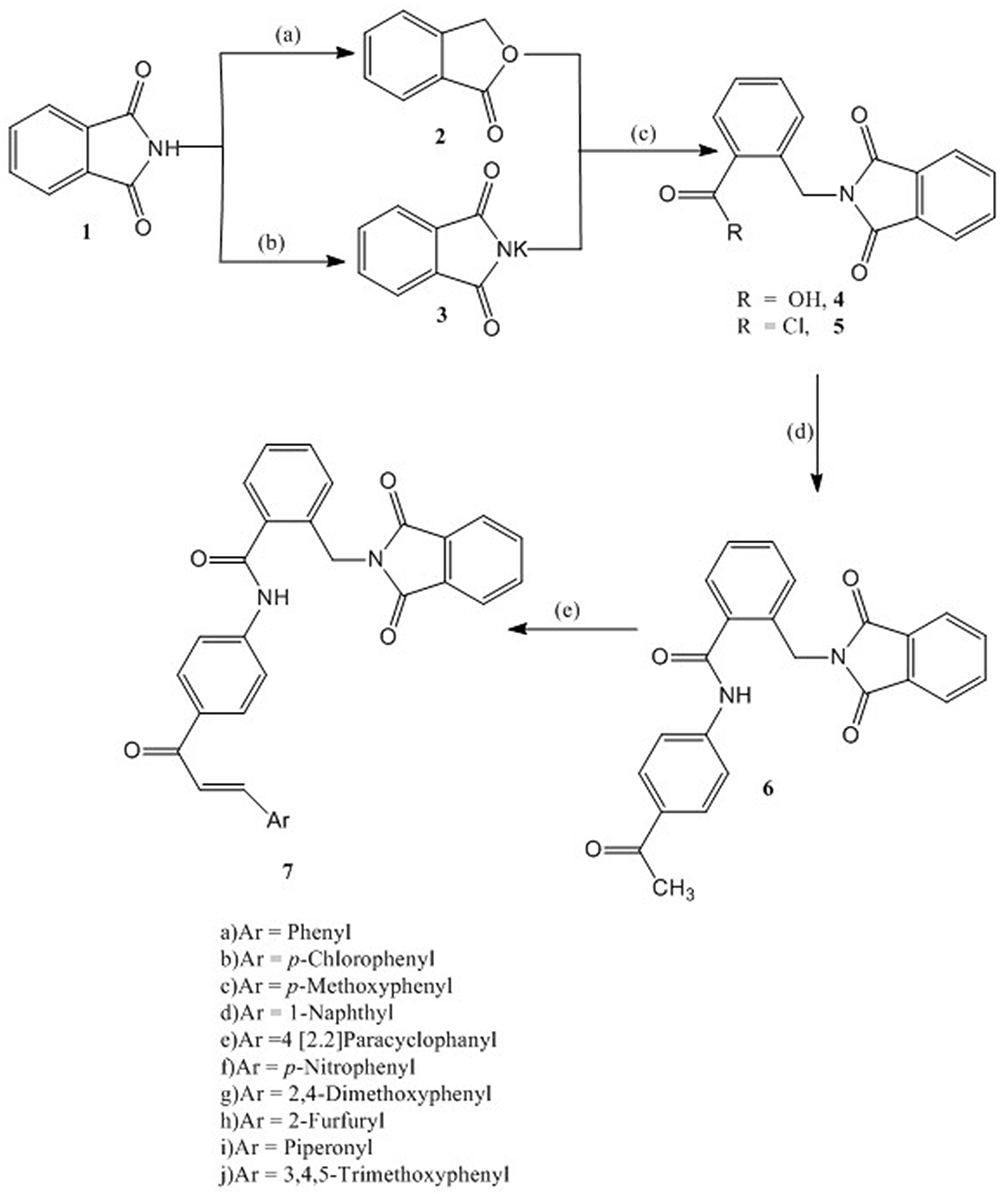 |
Scheme 1 Synthesis of compounds 6 and 7: Reagents and conditions: (A) Zn dust, NaOH, CuSO4; (B) KOH, Acetone; (C) DMF, reflux 6 hr, HCl; SOCl2, reflux, 4 hr; (D) 4-Aminoacetophenone, CH2Cl2, TEA, 4 hr, rt; (E) Aromatic aldehyde, 20% NaOH, EtOH, 8–20 hr, rt. |
The structure of compound 6 was assigned on the basis of satisfactory elemental analysis as well as spectroscopic data. The FT-IR spectrum displayed two bands at 3454 and 3310 cm−1 due to NH, a band at 3097 cm−1 due to Ar-CH, a band at 2926 cm−1 due to Aliph-CH, and three bands at 1766, 1716, 1686 cm−1 due to CO, as well as a band at 1595 cm−1 due to C=N. The 1H NMR Spectrum of compound 6 indicated the presence of three singlets at 10.64, 5.03, 2.56 ppm, arising from NH, CH2 and CH3 resonances, respectively. Moreover, this interpretation is nicely confirmed by MS spectrum, in which the molecular ion peak and the base peak are the same, and appeared at m/z 398. The structure elucidation of compound 6 was also evidenced by X-ray crystallographic analysis of single crystal (Figure 4) (Supplementary Figure S1 and Table S1).
 |
Figure 4 X-ray structural analysis of compound 6. Ellipsoids represent 50% probability levels. |
As illustrated in Scheme 1, compound 6 reacted with substituted aldehydes in the presence of sodium hydroxide under Claisen-Schmidt condensation conditions to form the chalcones 7a-j in 93–71% yield. The chemical structure of the target compounds were elucidated utilizing the spectral data and elemental analyses. The IR spectral data of compounds 7a-j showed intense peaks at 3468–3320 cm−1 (NH) and 1733–1637 cm−1 (CO). Another typical signal of these compounds was found in the 1H NMR spectra at 10.78–10.51 ppm (NH), however the aromatic as well as the olefinic protons appear as complexed multiplets in the region 9.15–6.11 ppm. The methylene protons resonate at 4.90–4.63 ppm. Additionally, the structure of 7a-j is confirmed by some 13C NMR by giving peaks at (CO-CH=CH) at 202.11–187.29 ppm, CO-N-CO at 186.11–169.44 ppm, CO-NH at 169.89–158.23 ppm, and CH2 in compounds 7a-j resonate in the region of the solvent. The EI-MS spectra of 7a-e further support the given structure as they showed correct molecular ion peak.
Biological Evaluation
In vitro Anticancer Activity Screening
The novel synthesized hybrids were investigated for their in vitro anticancer activity against human MCF-7 (breast cancer) and human Hep G2 (hepatocellular liver carcinoma) cell lines by using MTT assay. The CA-4 was used as the reference compound. The results are listed in Table 1 and graphically presented in Figure 5A and B.
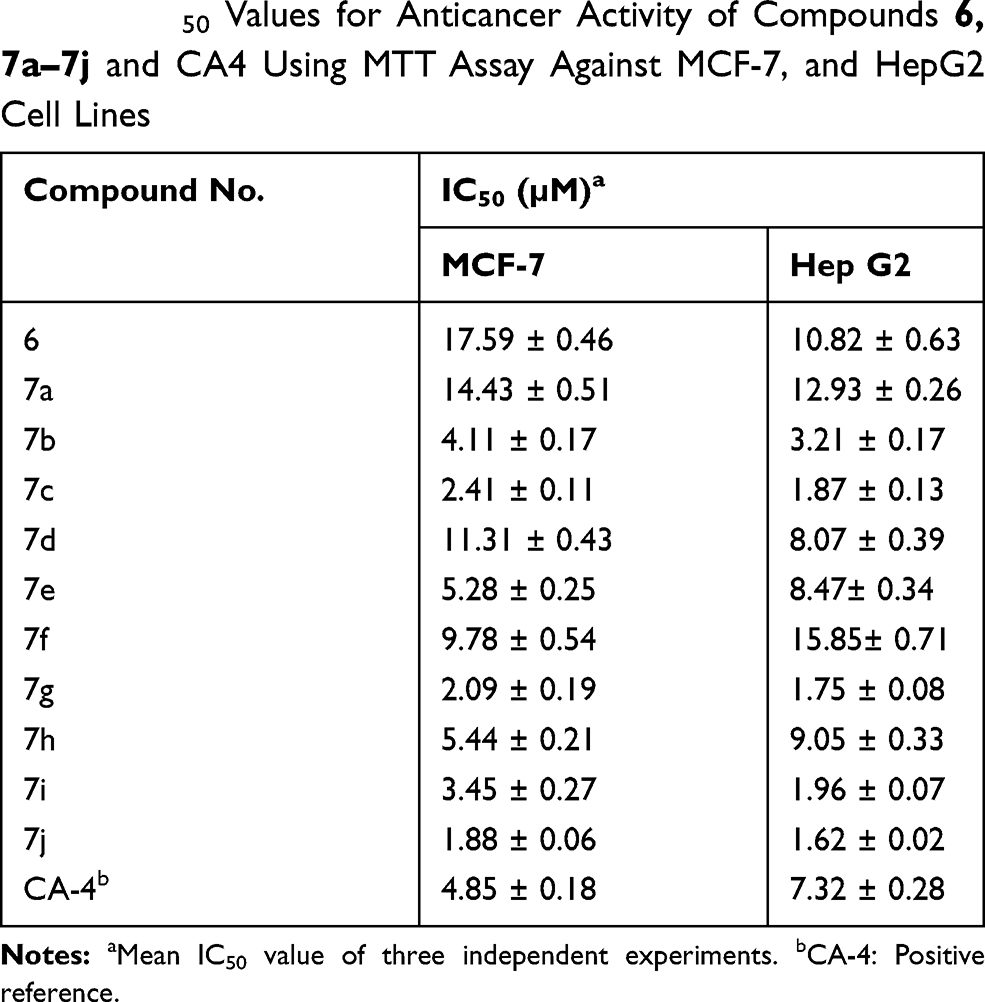 |
Table 1 IC50 Values for Anticancer Activity of Compounds 6, 7a–7j and CA4 Using MTT Assay Against MCF-7, and HepG2 Cell Lines |
 |
Figure 5 (A) IC50 values for anticancer activity of compounds 6, 7a–7j and CA4 using MTT assay against MCF-7 cell line. (B) IC50 values for anticancer activity of compounds 6, 7a–7j and CA-4 using MTT assay against Hep G2 cell line. |
The in vitro screening results revealed that the majority of the synthesized compounds displayed promising anticancer activity against the tested cancer cell lines at low IC50 values. In particular, trimethoxy hybrid 7j was the most active among the synthesized compounds with IC50 values of 1.88 ± 0.06 µM and 1.62 ± 0.02 µM against both MCF-7 and Hep G2 cell lines, respectively; which represents 2.57 and 4.51 fold superior activity than the reference compound. It is noteworthy that, the dimethoxy derivative 7g and the p-methoxy derivative 7c exhibited remarkable anticancer activity of 2.32 and 2.01 times the activity of CA-4 against MCF-7, where the IC50 values were 2.09 ± 0.19 µM and 2.41± 0.11 µM, respectively. While, the anticancer potency of the two hybrids 7g and 7c against Hep G2 was nearly equipotent and nearly 4-fold the reference activity. Introduction of piperonyl group in 7i hybrid increased the anticancer activity by 1.4 and 3.73 fold compared to the reference compound against MCF-7 and Hep G2 cell lines, respectively.
The p-chloro hybrid 7b showed slightly greater anticancer activity compared to CA-4 against MCF-7 cell line. However, this compound achieved IC50 value of 3.21±0.17 µM which represents 2.28 time more potent anticancer activity than CA-4 against Hep G2 cell line. Meanwhile, the [2,2]paracyclophanyl 7e and 2-furyl 7h hybrids experienced nearly equal anticancer activity against both MCF-7 and Hep G2 cell lines, which was slightly weaker than the anticancer activity of CA-4 as indicated by their IC50 values. According to their IC50 values, the p-nitro 7f and 1-naphthyl 7d hybrids had weak anticancer activity compared to CA-4. However, the anticancer activity markedly decreased in the case of the unsubstituted phenyl ring, which in turn confirmed that the substitution in the phenyl ring is critical for potentiating the anticancer activity. Also, the anticancer activity of compound 6 was dramatically decreased which emphasized the importance of the presence of the chalcone-based system. Finally, analyzing the above-mentioned results revealed that the methoxy derivatives experienced the most potent anticancer activity and this activity decreased in this order: tri-OCH3 > di-OCH3 > mono-OCH3. Of considerable interest, modification of the electronic nature of substituents attached to the phenyl ring of the chalcone moiety influenced significantly the anticancer activity. Where, the piperonyl 7i and p-Cl-phenyl 7b derivatives possessed remarkable anticancer activity, the [2,2] paracyclophanyl 7e and 2-furyl 7h hybrids showed a slight decrease in the anticancer activity. Moreover, the anticancer activity was markedly decreased in the p-NO2-phenyl 7f and 1-naphthyl 7d derivatives.
In vitro Enzyme Inhibition
To predict the possible mechanism of action of the synthesized compounds, β-tubulin (TUBb) polymerization inhibitory activity of the most potent synthesized hybrids 7c, 7g and 7j was investigated against the tested cancer cell lines. CA-4 was used as reference compound and the results are presented as IC50 values (in μM concentrations) in Table 2 and Figure 6.
 |
Table 2 IC50 Values for in vitro β-Tubulin Polymerization Inhibitory Activity of 7c, 7g, 7j, and CA-4 Using ELISA Assay |
 |
Figure 6 IC50 values for in vitro β-tubulin polymerization inhibitory activity of 7c, 7g, 7j and CA-4 using ELISA assay. |
The results represented in Table 2 proved that 7j hybrid with IC50 value of 2.32 ± 0.15 μM had the most potent β-tubulin polymerization inhibitory activity among tested and reference compounds. While 7g and 7c hybrids exhibited β-tubulin polymerization inhibition activity comparable to that of CA-4 which cleared by their IC50 values. These results supported that 7j hybrid is a potent inhibitor of tubulin assembly which was in accordance with the in vitro cytotoxic activity of these compounds against MCF-7 and Hep G2 cell line.
To further explore the mechanism of action of the synthesized compounds, compounds 7c and 7g and 7j were tested for in vitro HDAC inhibitory activity, IC50 values for the selected hybrids were determined against HDAC1 and 2 isoforms using entinostat as a reference compound (Table 3, Figure 7).
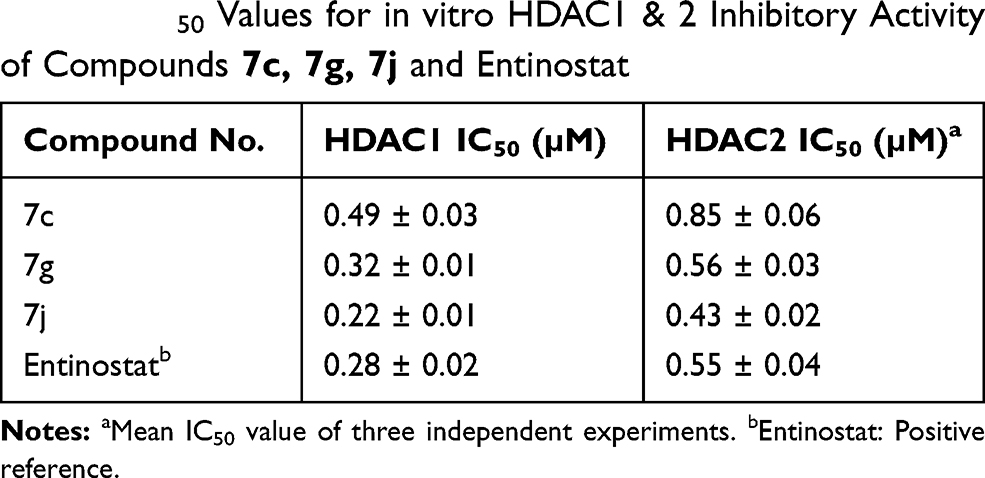 |
Table 3 IC50 Values for in vitro HDAC1 & 2 Inhibitory Activity of Compounds 7c, 7g, 7j and Entinostat |
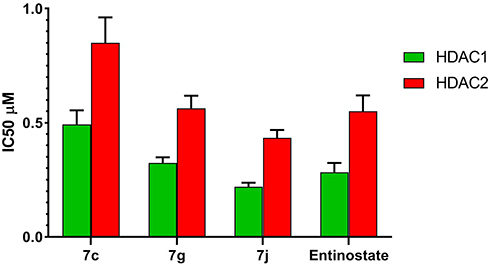 |
Figure 7 IC50 values for in vitro HDAC1 and HDAC2 inhibitory activity of compounds 7c, 7g, 7j and entinostat. |
The results presented in Table 3 reveal that all the selected hybrids possessed potent HDAC1 and HDAC2 inhibitory activity. In particular, 7j hybrid afforded the highest HDAC1and HDAC2 inhibitory activity with IC50 value lower than that of entinostat. Meanwhile, compound 7g exhibited nearly equipotent HDAC1 and HDAC2 inhibitory activity to entinostat which cleared by the proximity of their IC50 values. Moreover, both HDAC1 and HDAC2 inhibitory activities of compound 7c were slightly lower than entinostat IC50 values. In conclusion, the above-mentioned results evidenced that 7g and 7j hybrids are potent inhibitors of both HDAC1 and HDAC2. This was confirmed also by their potent in vitro cytotoxic activity against the tested cancer cell lines.
In vitro Cell Cycle Analysis
In order to further explore the effect of the most promising cytotoxic compounds on the cell cycle, cell cycle distribution using DNA flow cytometry analysis was assessed on 7j and 7c hybrids compared to CA-4 and entinostat (at their IC50 concentration) against MCF-7 cell line. According to the results presented in Tables 4, 7j and 7g hybrids experienced the greatest activity by keeping cells in G2/M and preG1 phases compared to untreated control cell and the reference compounds (Figure 8A and B).
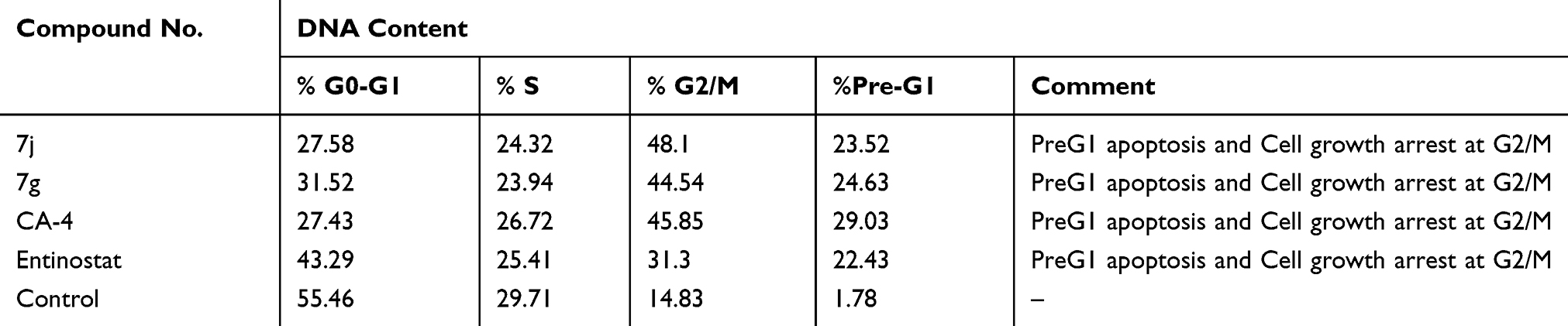 |
Table 4 Cell Cycle Analysis of Compounds 7j, 7g, CA-4 and Entinostat Against MCF-7 Cell Line |
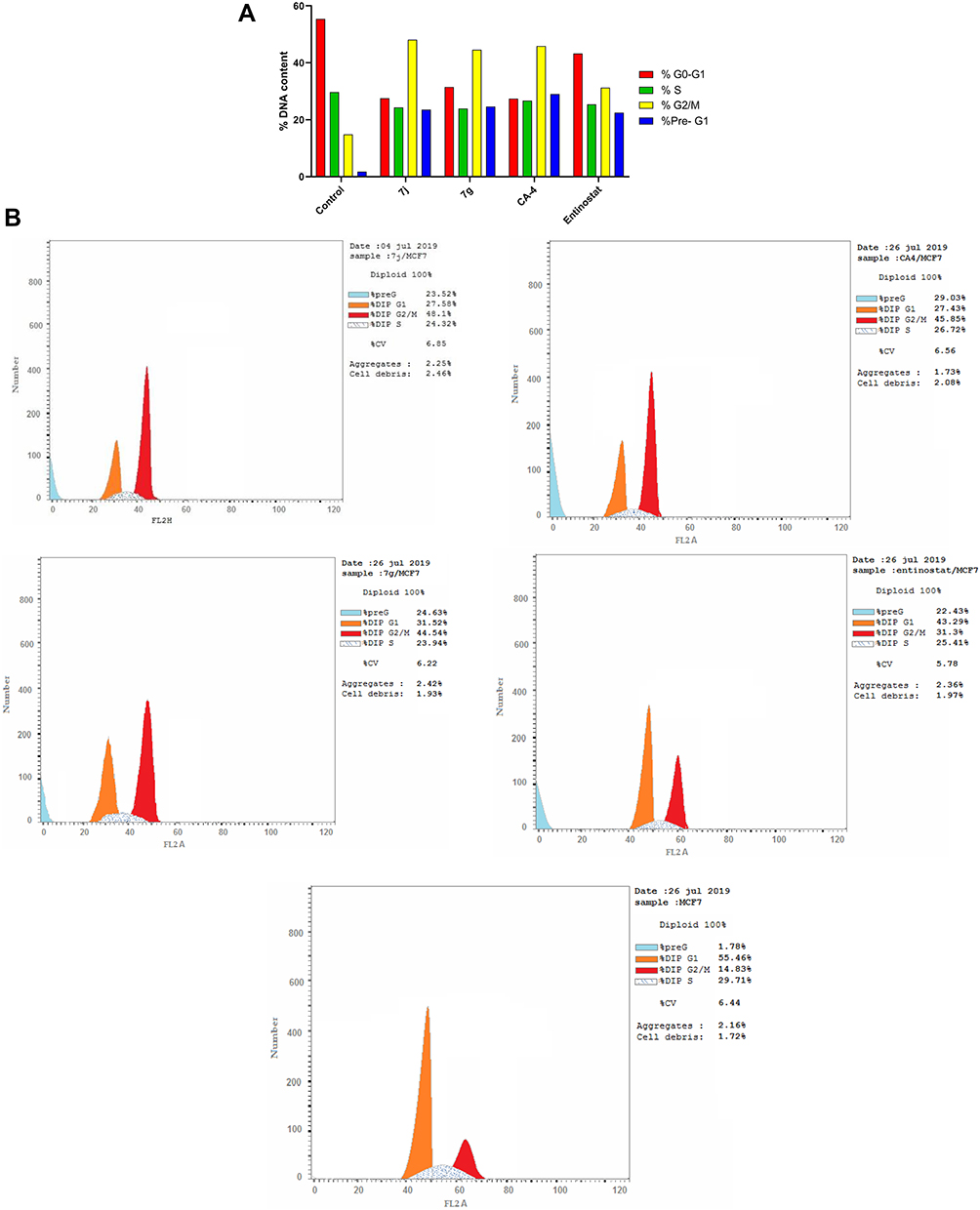 |
Figure 8 (A) Cell cycle analysis in MCF-7 cell line treated with 7j and 7g hybrids at their IC50 (µM). (B) Cell cycle analysis and apoptosis effect in MCF-7 cell line treated with 7j, 7g, CA-4 and entinostat compounds. |
Moreover, the percentage of cells population at G2/M after 7j and 7g treatment was higher than entinostat by 1.53 and 1.42 folds, respectively. Also, 7j hybrid achieved 1.05 fold higher cell accumulation percentage than CA-4 at G2/M phase. While, 7c hybrid increased G2/M cells by nearly equal percentage to CA-4. Interestingly, it is clearly observed that 7j and 7g hybrids exhibited higher apoptosis ability than the untreated control cell as well as the reference compound entinostat at preG1 phase by 1.04 and 1.09 times, respectively.
These data established that the selected hybrids have potent anti-proliferative effects and apoptosis-inducing activity by increasing preG1 apoptosis and developing cell growth arrest at G2/M phase.
It was obvious from cell cycle analysis results that the treatment of MCF-7 with the selected hybrids showed considerable increase in preG1 phase which is a suggestion of apoptosis (Table 5, Figure 9). Consequently, to determine the ability of 7j and 7g hybrids for inducing cell death in MCF-7 cell line via induction of apoptosis, Annexin V-FITC/PI staining assay was carried out for the selected compounds at their IC50 (µM) for 24 hours and analyzed.
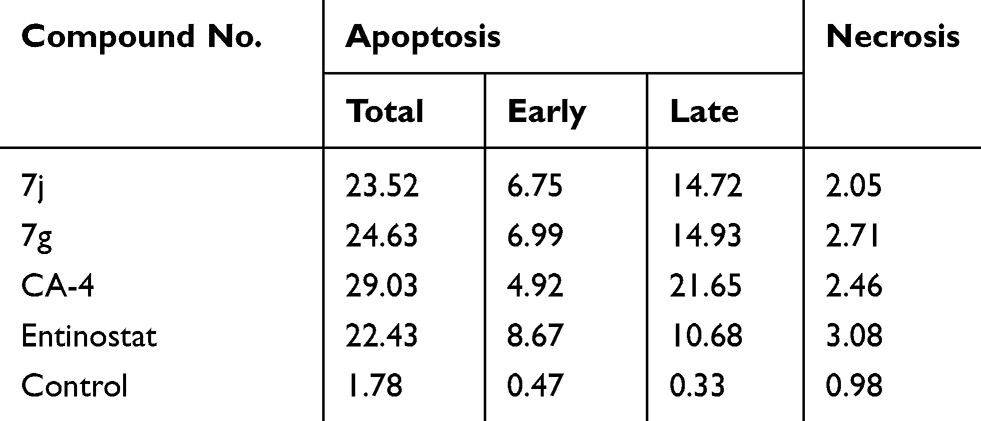 |
Table 5 Results of Apoptotic Assay of 7j, 7g, CA-4 and Entinostat |
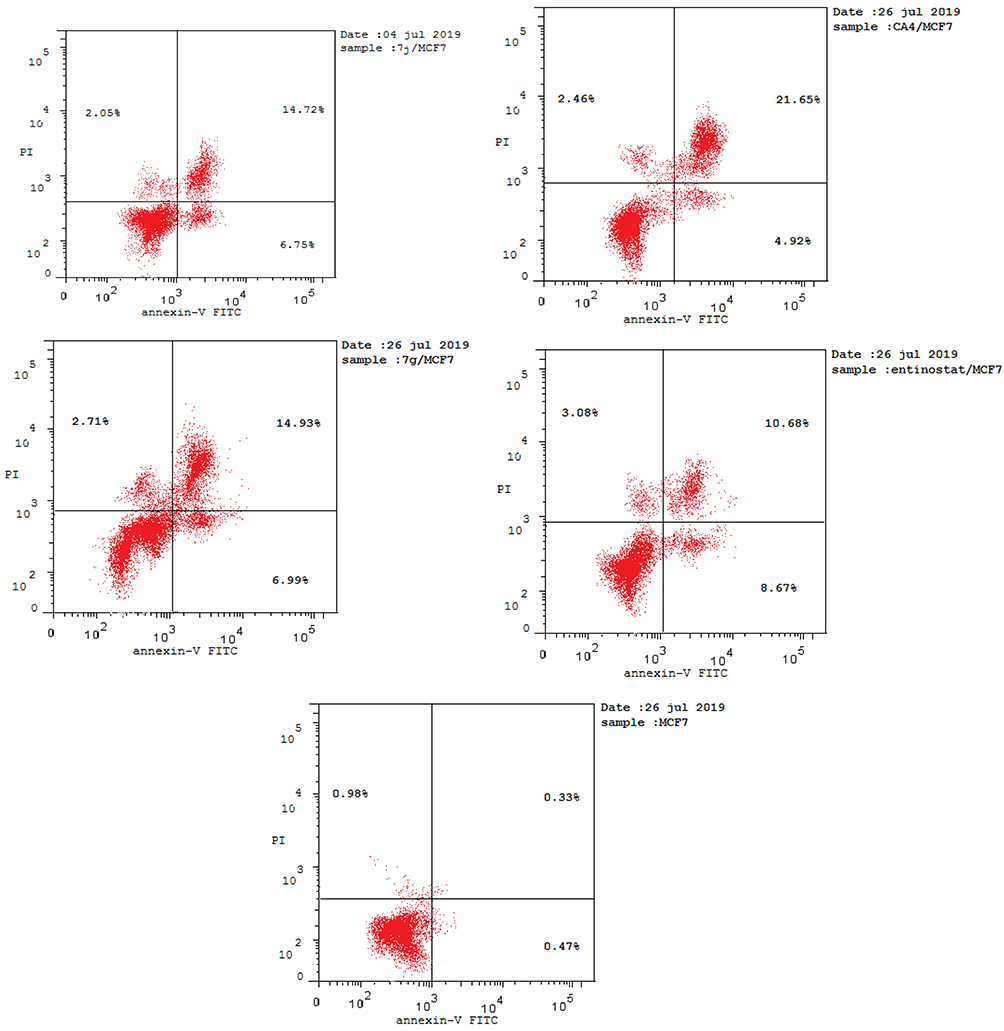 |
Figure 9 Flow cytometric analysis of Annexin V-FITC/PI induced by compounds 7j, 7g, CA-4, entinostat at their IC50 (µM). |
As shown in Table 5, compounds 7j and 7g have the ability for inducing apoptosis at early apoptotic, late apoptotic stages and cell necrosis development, in comparison to untreated controls. It was found that compounds 7j and 7g increased cell populations at the early apoptotic stage by 14.36- and 14.87-fold more than untreated control, respectively. While, 7j and 7g hybrids treatment significantly increased the late apoptotic cells population by 44.60 and 45.24 folds, respectively. Of considerable interest, the selected compounds possessed increasing in both early and late apoptotic cells compared to the reference compounds, CA-4 and entinostat. It is noteworthy that 7j and 7g hybrids displayed significant increase in cell necrosis by 2.09- and 2.76-fold, respectively, compared to the untreated control cell. Moreover, the necrosis percent of the selected hybrids were nearly equal to CA-4 and entinostat. These findings evidenced that the synthesized hybrids cause impairment of cell division with apoptosis-inducing activity and confirmed also that these hybrids can potentially inhibit HDAC and tubulin polymerase enzymes.
Molecular Docking Study
Docking at the Colchicine Binding Site in Tubulin Protein
Docking studies have been carried out to explore the binding ability of the most potent cytotoxic hybrids 7j, 7g and 7c with colchicine binding site of tubulin. MOE program was used for docking the selected hybrids and CA4 into the colchicine binding site using the X-ray crystallographic structure of tubulin-colchicine complex (PDB code: 1SA0) as tubulin protein template. The results of energy binding scores and binding interactions are depicted in Table 6 and Figure 10.
 |
Table 6 Energy Binding Scores (Kcal/mol) for Compounds 7j, 7g, 7c and CA4 with the Colchicine Binding Site |
 |
Figure 10 Docking and binding pattern of compounds 7j, 7g, 7c and CA-4 showing interactions with different amino acid residues found in the active site of colchicine binding site in tubulin protein (PDB code: 1SA0). (A) 3D structure of compound 7j (yellow) (B) 2D structure of compound 7j. (C) 3D structure of compound 7g (yellow) (D) 2D structure of compound 7g. (E) 3D structure of compound 7c (yellow) (F) 2D structure of compound 7c. (G) 3D structure of reference compound CA-4 (yellow) (H) 2D structure of reference compound CA-4. |
The energy binding scores recorded in Table 6 showed that the selected hybrids displayed higher energy binding scores than the reference ligand, consequently, better binding at the colchicine binding site. The highest energy binding score of −9.10 Kcal/mol was observed with the trimethoxy hybrid 7j, followed by dimethoxy 7g and mono-methoxy 7c hybrids, respectively, which justified the cytotoxicity results and the estimated tubulin inhibitory activity. The binding interaction of 7j hybrid with colchicine binding site is shown in Figure 10B, where two hydrogen bonding interactions were formed with GLN 11 and GLY 144 with bond length of 3.30 Å and 2.80 Å, respectively. In addition to π-cation interactions with LYS 254 with bond length of 3.74 Å Moreover, compound 7g achieved two π- hydrogen interactions with GLN 11 and ALA 12 with bond length of 4.23 Å and 3.63 Å, respectively. In addition, another π-π interaction was formed with TYR 224 (bond length: 3.81 Å) Figure 10D. Meanwhile, one hydrogen bonding interaction was formed between NH group of 7c hybrid and ASP 329 (bond length: 2.87 Å) Figure 10F. The reference ligand CA4 showed one hydrogen bonding interaction with SER 140 (3.03 Å) and π-hydrogen interaction with GLY 144 (3.80 Å) Figure 10H.
Docking at the HDAC2 Active Site
In order to investigate the possible binding modes between the synthesized compounds and HDAC active site, the docking studies on the most potent cytotoxic compounds 7j, 7g and 7c were performed using MOE program where the 3D structure of the HDAC active sites is available at RCSB Protein Data Bank, HDAC2 (PDB code: 4LXZ). Entinostat was used as reference compound. The results were expressed as docking energy and binding interactions with the active site of HDAC2 isoform (Table 7 and Figure 11).
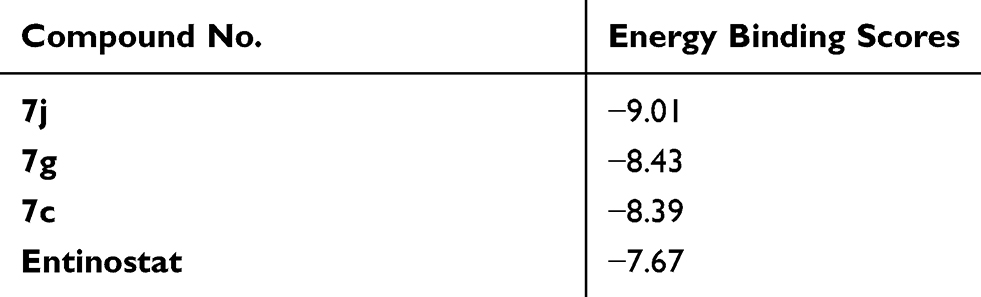 |
Table 7 Energy Binding Scores (Kcal/mol) for Compounds 7j, 7g, 7c and Entinostat at the Active Site of HDAC2 |
 |
Figure 11 Docking and binding pattern of compounds 7j, 7g, 7c and CA-4 showing interactions with different amino acid residues found in HDAC2 active site (PDB code: 4LXZ). (A) 3D structure of compound 7j (yellow) (B) 2D structure of compound 7j. (C) 3D structure of compound 7g (yellow) (D) 2D structure of compound 7g. (E) 3D structure of compound 7c (yellow) (F) 2D structure of compound 7c. (G) 3D structure of entinostat reference compound (yellow) (H) 2D structure of entinostat reference compound. |
Interestingly, the estimated docking energy of the selected compounds confirmed that the selected hybrids possessed higher binding energy than the reference compound which means better binding at the HDAC2 active site. Moreover, the order of binding energy was; tri-OCH3 (7j) > di-OCH3 (7g) > mono-OCH3 (7c), which was consistent with the HDAC inhibition assay. Compound 7j showed an energy binding value of −9.01 Kcal/mol, while compounds 7g and 7c showed energy binding values of −8.43 and −8.39 Kcal/mol, respectively. Compound 7j showed two hydrogen bonding interactions between oxygen of both phthalimido- and chalcone-based moiety with ARG 197, ASN 331 of distances of 2.82 Å and 3.19 Å, respectively Figure 11B. In addition, one hydrogen bonding interaction was formed between oxygen of chalcone nucleus of 7g hybrid and ASN 331 of distance of 3.15 Å. Alsq, π-cation interaction was formed between phthalimido aromatic ring and NH2 group of ARG 311 with distance of 5.00 Å Figure 11D. However, 7c hybrid exhibited one hydrogen bonding interaction with ASN 331 of 2.94 Å Figure 11F. On the other hand, entinostat showed one hydrogen bonding interaction with ASN 331 (3.09 Å) and another π-hydrogen interaction with PRO 344 (4.60 Å) similar to 7g hybrid and 7j hybrid, respectively Figure 11H.
Conclusion
A novel series of α-phthalimido-substituted chalcones-based hybrids as dual HDAC/tubulin inhibitors was synthesized and evaluated for its in vitro anticancer activity by MTT method against MCF-7 and HepG2 human cell lines, using CA-4 as reference compound. Moreover, the in vitro tubulin polymerase and HDAC 1 and 2 inhibitory activity were evaluated for the most potent compounds 7c, 7g and 7j using CA-4 and entinostat as reference compounds. The results revealed that the three substituted methoxy derivatives 7c, 7g and 7j experienced greater anticancer activity than that of CA-4; particularly the trimethoxy derivative 7j which experienced the most potent anticancer activity and potently inhibited β-tubulin polymerase and HDAC 1 and 2 activities. Additionally, the cell cycle analysis proved that these promising hybrids can induce apoptosis and prevent the proliferation of MCF-7 by sequestering higher cell percentage at G2/M and pre G1 phases than untreated control cells, CA-4 and entinostat. The molecular docking studies showed that these potent anticancer hybrids could efficiently bind to colchicine binding site of both tubulin polymerase enzyme and HDAC2 active sites with energy scores higher than the reference ligands. The results of our study proved that α-phthalimido-substituted chalcones-based hybrids may serve as novel promising dual HDAC/tubulin inhibitors in searching for more effective anticancer agents.
Experimental
Chemistry
Melting points were determined on Mel-Temp hot stage apparatus and were uncorrected. Reactions were routinely monitored by thin-layer chromatography on Merck silica gel PF254 plates and spots were visualized with UV light at 254 nm. IR spectra were recorded as KBr disks on a Fourier Transform (FT-IR) Bruker spectrophotometer at Central Lab, Faculty of Science, Minia University, El-Minia, Egypt. 1H NMR and 13C NMR spectra were carried out on a Bruker apparatus operating at 400 MHz and 100 MHz, respectively using TMS as internal reference at the Faculty of Science, Sohag University, Sohag, Egypt. Chemical shifts (δ values) are given in parts per million (ppm) using CDCl3 (7.19) or DMSO-d6 (2.5) as solvents and coupling constants (J) in Hertz. Splitting patterns are designated as follows: s, singlet; bs, broad singlet; d, doublet; t, triplet; m, multiplet. EI-MS: Mass spectrum was carried out on direct probe controller inlet part to single quadruple mass analyzer in (Thermo Scientific Gcms) Model (ISQ LT) using Thermo X-Caslibur Software at the Regonal Center for Mycology and Biotechnology (RCMB) Al-Aazhar University, Naser City, Cairo. Elemental analyses were performed on Perkin–Elmer 2400 CHN Elemental analyzer by the Microanalytical Center, Faculty of Science at the University of Cairo and are within ± 0.4% of theoretical values unless otherwise specified.
Materials
Chemicals and solvents used in the preparation of the target compounds are of commercial grade, and purchased from Alfa Aesar, Cambrian chemicals, Aldrich, Acros Organics, Fluka, Merck, and El-Nasr Pharmaceutical Chemicals Companies. Chemicals were used without purification. Solvents were purified following the .reported
Synthesis of α-Phthalimido-o-Toluoyl Chloride (5)
A mixture of potassium phthalimide, 50 g (0.26 mol) and 34.8 g phthalide (0.26 mol) in 150 mL DMF was refluxed for 5 hours, and then was cooled to r.t. and poured into a mixture of 100 mL glacial acetic acid and 150 mL water. The separated solid was washed with water and 95% ethanol. Recrystallization from acetic acid gave 41.35 g of α-phthalimidi-o-toluic acid (4) (57%), m.p. 264–6°C, Lit.40 266–7°. A suspension of 50 g (0.178 mol) of 4 and 93 g (0.78 mol) thionyl chloride was refluxed for 3 hours. Dry benzene (30 mL) was added to the mixture and distilled with the excess thionyl chloride under reduced pressure. This later process was repeated twice to remove the excess thionyl chloride. The yield of the entitled compound was 47.92 g (90%) as white prisms, m. p. 181–183°C, Lit.40 183.5–184.8°C.
Synthesis of N-(4-Acetylphenyl)-2-(1,3-Dioxoisoindolin-2-Yl)Benzamide (6)
To a magnetically stirred solution of acid chloride 5 (5 g, 0.0167 mol) in abs. CH2Cl2 (20 mL) was added 4-amino-acetophenone (2.25 g, 0.0167 mol) in one portion and 1.686 g (0.0167 mol) TEA. The reaction mixture was stirred for a further 2 hours at r.t. and then treated with a saturated solution of NaCl (20 mL), and extracted with ethyl acetate (30 mL). The organic layer was washed with 1N HCl, NaHCO3 solution and finally with H2O, then dried over anhyd. Na2SO4. The solvent was removed under reduced pressure and the residue was recrystallized from ethyl acetate/n-hexane to give the entitled compound.
White solid, 4.2 g (92%), m.p. 238–240 °C; FTIR (KBr, cm−1) ν: 3454, 3310 (NH), 3097 (Ar-CH), 2926 (Aliph.-CH), 1766, 1716, 1686 (CO), 1595 (C=N). (Supplementary data page 1) 1H NMR (400 MHz, DMSO-d6): δ 10.64 (s, 1H, NH), 7.95 (d, 2H, J= 8.32 Hz, Ar-H)), 7.87 (d, 2H, J= 8.32 Hz, Ar-H), 7.84 (br. S, 4H, Ar-H), 7.61 (d, J= 8.32 Hz, 1H, Ar-H), 7.47 (t, J= 8.32 Hz, 2H, Ar-H), 7.42 (t, J= 8.32 Hz, 2H, Ar-H), 7.32 (d, J= 8.32 Hz, 1H, Ar-H), 5.03 (s, 2H, CH2), 2.56 (s, 3H, CH3) (Supplementary data page 12, 13). MS: m/z (rel. intensity), 399 (M++ 1, 33), 398 (M+, 100), 397 (19), 381 (11), 264 (22), 263 (9), 265 (4), 251 (2), 133 (4), 131 (6), 132 (3), 130 (54), 120 (15), 116 (21), 104 (11), 90 (7), 77 (20), 76 (20), 63 (3) (Supplementary data page 31). Anal. Calcd. for C24H18N2O4 (398.41): C, 72.35; H, 4.55: N, 7.03. Found: C, 72.05; H, 4.75; N, 6.82.
Synthesis of (E)-2-((1,3-Dioxoisoindolin-2-yl)Methyl)-N-(4-(3-(Arylacroyloyl)Phenyl)Benzamides 7
A solution of 20% NaOH (0.5 mL) was added to a well stirred mixture of compound 6 (0.16 g, 0.402 mmol) and different aldehydes (0.402 mmol) in 15 mL absolute ethanol at r.t. Stirring was continued for 8–20 hours. (the reaction was monitored by tlc). The reaction mixture was extracted with ethyl acetate, then treated with brine, 1N HCl, NaHCO3, and washed with H2O. The solvent was removed and the residue was recrystallized from dioxane-ethanol to afford the title compound.
Pale yellow solid, 0.128 g (83%), m.p. 163–165 °C. FTIR (KBr, cm−1) ν: 3343 (NH), 3061 (Ar-CH), 1700, 1653 (CO), 1600, 1523 (C=N) (Supplementary data page 2). 1H NMR (400 MHz, DMSO-d6): δ 10.74 (s, 1H, NH), 8.70–7.39 (m, 17 H, Ar-H, CH=CH), 6.65 (s, 2H, Ar-H), 4.65 (s, 2H, CH2) (Supplementary data page 14, 15, 16); 13C NMR (100 MHz, DMSO-d6): δ 188.24 (COCH=CH), 169.44 (CONCO), 168.38 (CONH), 168.29 (COCH=CH), 143.62 (Ar-C), 138.99 (2Ar-C), 138.02 (Ar-C), 137.19 (Ar-C), 136.07 (Ar-C), 134.11 (Ar-C), 130.19 (Ar-C), 129.16 (2Ar-C), 129.02 (2Ar-C), 127.46 (Ar-C), 127.37 (Ar-C), 126.92 (Ar-C), 123.36 (Ar-C), 122.69 (Ar-C), 119.97 (2Ar-C), 119.81 (2Ar-C), 115.02 (2Ar-C), 113.27 (2Ar-C) (Supplementary data page 42). MS: m/z (rel. intensity) 487 (M++1, 7), 486 (M+, 22), 486 (50), 453 (100), 250 (2), 133 (9), 120 (21), 117 (6), 104 (47), 103 (54), 97 (11), 92 (16), 78 (11), 77 (56), 65 (42), 51 (9) (Supplementary data page 32). Anal. Calcd. for C31H22N2O4 (486.52): C, 76.53; H, 4.56; N, 5.76. Found: C, 76.21; H, 4.72; N, 5.55.
Yellow solid, 0.142 g (92%), m.p. 142–143 °C. FTIR (KBr, cm−1) ν: 3458, 3341 (NH), 3065 (Ar-CH), 2924 (Aliph.-CH), 1702, 1653 (CO), 1601, 1523 (C=N) (Supplementary data page 3). 1H NMR (400 MHz, DMSO-d6): δ 10.75 (s, 1H, NH), 8.22–7.39 (m, 17 H, Ar-H, CH=CH), 6.66 (s, 1H, Ar-H), 4.64 (s, 2H, CH2) (Supplementary data page 17, 18). MS: m/z (rel. intensity) 520 (M+, 9), 512 (3), 510 (19), 492 (7), 469 (7), 452 (10), 410 (6), 393 (7), 387 (100), 364 (18), 358 (44), 340 (17), 226 (7), 208 (7), 201 (14), 178 (12), 160 (4), 133 (20), 113 (25), 104 (34), 101 (51), 91 (37), 70 (29), 54 (34) (Supplementary data page 33). Anal. Calcd. for C31H21ClN2O4 (520.96): C, 71.47; H, 4.06;: N, 5.38. Found: C, 71.80; H, 3.85; N, 5.30.
White solid, 0.115g (74%), m.p. 191–193 °C. FTIR (KBr, cm−1) ν: 3422, 3326 (NH), 3066 (Ar-CH), 1704, 1678, 1654 (CO),1597, 1516 (C=N) (Supplementary data page 4). 1HNMR (400 MHz, DMSO-d6): δ 10.73 (s, 1H, NH), 8.80–7.02 (m, 18H, Ar-H, CH=CH), 4.63 (s, 2H, CH2), 3.85 (s, 3H, OCH3) (Supplementary data page 19); 13C NMR (100 MHz, DMSO-d6): δ 197.02 (CO-CH=CH), 169.43 (CONCO), 168.55 (CONH), 168.45 (COCH=CH), 158.19 (Ar-C), 144.86 (Ar-C), 144.66 (Ar-C), 138.49 (Ar-C), 137.99 (Ar-C), 136.33 (2Ar-C), 136.19 (2Ar-C), 132.19 (Ar-C), 131.07 (Ar-C), 130.81 (Ar-C), 130.05 (2Ar-C), 129.88 (2Ar-C), 128.39 (2Ar-C), 127.07 (Ar-C), 120.35 (Ar-C), 119.22 (Ar-C), 119.71 (2Ar-C), 114.55 (2Ar-C), 55.87 (OCH3) (Supplementary data page 43). MS: m/z (rel. intensity), 516 (M+, 19), 515 (M+-1, 100), 500 (4), 486 (5), 473 (5), 461 (3), 448 (1), 410 (2), 399 (6), 398 (24), 383 (20), 368 (11), 310 (2), 296 (2), 264 (85), 250 (10), 236 (10), 204 (4), 179 (3), 177 (4), 150 (2), 132 (12), 130 (46), 129 (9), 116 (21), 104 (30), 91 (15), 76 (59), 51 (14) (Supplementary data page 34). Anal. Calcd. for C32H24N2O5 (516.54): C, 74.41; H, 4.68; N, 5.42. Found C, 74.20; H, 4.33; N, 5.77.
Pale orange solid, 0.141g (91%), m.p. 141–142 °C. FTIR (KBr, cm−1) ν: 3328, 3320 (NH), 3046 (Ar-CH), 1686, 1639 (CO), 1602, 1584 (C=N) (Supplementary data page 5). 1H NMR (400 MHz, DMSO-d6): δ 10.77 (s, 1H, NH), 9.15–7.42 (m, 20H, Ar-H, CH=CH), 6.69 (s, 1H, Ar-H), 4.65 (s, 2H, CH2) (Supplementary data page 20). MS: m/z (rel. intensity), 537 (M++1, 5) 536 (M+, 18), 526 (36), 512 (10), 491 (12), 461 (2), 434 (7), 413 (10), 411 (100), 409 (23), 402 (16), 383 (15), 367 (3), 455 (2), 279 (2), 258 (30, 175 (3), 152 (18), 126 (3), 105 (2), 79 (4) (Supplementary data page 35). Anal. Calcd. for C35H24N2O4 (536.58): C, 78.34; H, 4.51; N, 5.22. Found C, 78.54; H, 4.51; N, 5.39.
Pale yellow solid, 0.144g (93%), m.p. 153–154 °C. FTIR (KBr, cm−1) ν: 3441 (NH), 3010 (Ar-CH), 2926, 2890 (Aliph.-CH), 1682 (CO), 1626, 1593 (C=N) (Supplementary data page 6). 1H NMR (400 MHz, DMSO-d6): δ 10.53 (s, 1H, NH), 7.04–6.08 (m, 21 H. Ar-H, CH=CH), 4.64 (s, 2H, CH2), 3.17–2.95 (m, 8H, 2 CH2-CH2) (Supplementary data page 21). MS: m/z (rel. intensity), 645 (M++1, 6) 644 (M+, 5), 425 (8), 353 (15), 351 (15), 292 (5), 286 (7), 247 (8), 245 (9), 238 (5), 236 (12), 234 (7), 210 (7), 207 (15), 203 (8), 160 (6), 152 (4), 144 (11). 131 (18), 112 (10), 104 (100), 103 (99), 100 (15), 89 (16), 78 (60), 72 (27), 63 (9), 52 (19) (Supplementary data page 36). Anal. Calcd. for C43H36N2O4 (644.76): C, 80.10; H, 5.63; N, 9.69. Found C, 79.83; H, 5.88; N, 9.98.
Orange solid, 0.139g (90%), m.p. 145–146 °C. FTIR (KBr, cm−1) ν: 3468, 3382 (NH), 2927 (Aliph.-CH), 1637 (CO), 1595, 1517 (C=N) (Supplementary data page 7). 1H NMR (400 MHz, DMSO-d6): δ 10.78 (s, 1H, NH), 8.26–7.53 (m, 16H, Ar-H, CH=CH), 6.68 (s, 2H, Ar-H), 4.62 (s, 2H, CH2) (Supplementary data page 22, 23); 13C NMR (100 MHz, DMSO-d6): δ 187.29 (COCH=CH), 166.22 (CONCO), 158.23 (CONH), 148.11 (Ar-C), 145.01 (Ar-C), 144.34 (Ar-C), 142.12 (Ar-C), 139.01 (Ar-C), 138.72 (Ar-C), 132.32 (2Ar-C), 132.07 (2Ar-C), 130.01 (Ar-C), 128.29 (Ar-C), 128.04 (2Ar-C), 127.12 (Ar-C), 126.14 (2Ar-C), 125.90 (2Ar-C), 125.01 (2Ar-C), 124.72 (2Ar-C), 124.32 (Ar-C), 123.10 (Ar-C), 122.90 (Ar-C) (Supplementary data page 44). MS: m/z (rel. intensity), 532 (M++1,), 531 (M+, 8), 519 (8), 515 (4), 479 (3), 470 (4), 447 (3), 426 (3), 403 (2), 398 (14), 382 (19), 369 (7), 349 (11), 340 (4), 321 (6), 306 (7), 286 (18), 250 (15), 236 (10), 224 (4),205 (4), 193 (2), 175 (5), 167 (7), 150 (3), 132 (6), 77 (42), 76 (91), 65 (100) (Supplementary data page 37). Anal. Calcd. for C31H21N3O6 (531.51): C, 70.05; H, 3.98; N, 7.91. Found C, 70.24; H, 3.74; N, 7.63.
Yellow solid, 0.115 g (74%), m.p. 162–164 °C. FTIR (KBr, cm−1) ν: 3421 (NH), 2933, 2839 (Aliph.-CH), 1733, 1653 (CO), 1596, 1506 (Aliph.-CH) (Supplementary data page 8). 1H NMR (400 MHz, DMSO-d6): δ 10.71 (s, 1H, NH), 8.07–7.41 (m, 13H, Ar-H), 6.65 (m, 4H, Ar-H), 4.63 (s, 2H, CH2), 3.89 (s, 3H, OCH3), 3.85 (s, 3H, OCH3) (Supplementary data page 24, 25). MS: m/z (rel. intensity), 547 (M++1, 4), 546 (M+, 2), 493 (2), 458 (2), 446 (11), 430 (8), 428 (12), 414 (46), 400 (36), 382 (88), 357 (3), 340 (7), 314 (3), 308 (5), 283 (5), 262 (7), 254 (9), 236 (2), 250 (5), 190 (3), 183 (4), 178 (5), 166 (5), 163 (20), 149 (25), 133 (9), 121 (21), 104 (9), 92 (13), 76 (100), 69 (16), 65 (21), 63 (17) (Supplementary data page 38). Anal. Calcd. for C33H26N2O6 (546.57): C, 72.52; H, 4.79; N, 5.13. Found C, 72.21; H, 4.58; N, 5.37.
Pale yellow solid, 0.109g (71%), m.p. 170–172 °C. FTIR (KBr, cm−1) ν: 3411 (NH), 1711, 1647 (CO), 1601, 1530 (Aliph.-CH) (Supplementary data page 9). 1H NMR (400 MHz, DMSO-d6): δ 10.72 (s, 1H, NH), 8.30–6.66 (m, 17H, Ar-H, furyl), 4.90 (s, 2H, CH2) (Supplementary data page 26, 27); 13C NMR (100 MHz, DMSO-d6): δ 202.11 (COCH=CH), 186.11 (CONCO), 169.89 (CONH), 147.33, 146.73, 139.52, 138.18, 138.02, 135.09, 134.11, 130.00, 128.42, 128.22, 126.82, 124.33, 120.87, 119.89, 119.77, 116.82, 115.90, 114.61, 114.11 (Ar-C, furyl-C, CH=CH) (Supplementary data page 45). MS: m/z (rel. intensity), 477 (M++1, 4) 476 (M+, 7), 475 (45), 474 (30), 473 (100), 458 (26), 456 (57), 440 (12), 264 (13), 213 (3), 195 (2), 185 (2), 159 (5), 156 (7), 133 (4), 130 (12), 120 (29), 105 (2), 102 (7), 92 (21), 78 (7), 65 (44) (Supplementary data page 39). Anal. Calcd. for C29H20N2O5 (476.48): C, 73.10; H, 4.23; N, 5.88. Found C, 72.72; H, 4.55; N, 6.09.
Pale yellow solid, 0.114g (74%), m.p. 244–246 °C. FTIR (KBr, cm−1) ν: 3450, 3349, 3241 (NH), 2908 (Aliph.-CH), 1642 (CO), 1588, 1552 (C=N) (Supplementary data page 10). 1H NMR (400 MHz, DMSO-d6): δ 10.51 (s, 1H, NH), 7.91–6.43 (m, 15H, Ar-H, CH=CH), 6.11 (s, 2H, Ar-H), 4.59 (s, 2H, CH2) (Supplementary data page 28, 29); MS. m/z (rel. intensity) 531 (M++1, 2), 530 (M+, 20), 518 (13), 516 (2), 500 (15), 486 (23), 453 (3), 268 (40), 267 (100), 239 (19), 180 (7), 152 (6), 144 (28), 133 (6), 120 (36), 117 (9), 104 (15), 89 (48), 77 (19), 65 (61) (Supplementary data page 40). Anal. Calcd. for C32H22N2O6 (530.53): C, 72.45; H, 4.18; N, 5.28. Found C, 72.31; H, 4.33; N, 5.47.
Yellow solid, 0.181 g (78%), m.p. 187–189 °C. FTIR (KBr, cm−1) ν: 3454 (NH), 2932, 2853 (Aliph.-CH), 1733, 1637 (CO), 1563, 1538 (Aliph.-CH) (Supplementary data page 11). 1H NMR (400 MHz, DMSO-d6): δ 10.69 (s, 1H, NH), 8.57–6.61 (m, 16H, Ar-H, CH=CH), 4.61 (s, 2H, CH2), 3.89 (s, 3H, 1OCH3), 3.85 (s, 6H, 2 OCH3) (Supplementary data page 30). MS: m/z (rel. intensity), 576 (M+, 12), 462 (8), 444 (8), 430 (9), 416 (55), 409 (12), 383 (87), 355 (2), 340 (9), 312 (8). 297 (3), 279 (8), 264 (4). 236 (5), 193 (4). 167 (6), 160 (6), 132 (9), 104 (11), 76 (100), 77 (34) (Supplementary data page 41). Anal. Calcd. for C34H28N2O7 (576.60): C, 70.82; H, 4.89; N, 4.86. Found C, 70.51; H, 5.11; N, 4.65.
Biological Evaluation
In vitro Anti-Proliferative Activity Assay
The anti-proliferative activity of the synthesized hybrids was tested using MTT assay method against MCF-7 and Hep G2. The principle of assay based on the ability of viable cell to cleave tetrazolium ring of MTT reagent yielding purple formazan crystals which are insoluble in aqueous solutions.
The amount of the formazan dye generated by dehydrogenase in cells is directly proportional to the number of living cells.
Cancer cells, (MCF-7) and human hepatocellular carcinoma (Hep G2) were purchased from American type Cell Culture collection (ATCC, Manassas, USA). The tested cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, Invitrogen/Life Technologies) supplemented with 10% fetal bovine serum (FBS, Hyclone) and antibiotics (100 units/mL penicillin and 100 mg/mL streptomycin). After detachment with trypsin, cancer cells were seeded according to the manufacturer’s instructions (1000–2000 cells/well) using 96-well microtiter plates. The cells were incubated in a humidified atmosphere containing 5% CO2 in air at 37°C for 34 hours. The synthesized hybrids 6 and 7a–j at different concentrations (0.1, 1, 10, 25, 100 μM), in addition to CA4, were added to culture media for 72 hours. Media were removed. 200 μL of 5% MTT reagent was added to each well and plates were incubated in CO2 incubator at 37°C for 2 hours. After an incubation period, the formed formazan crystals were dissolved in 200 μL isopropanol/well with continuous shaking using MaxQ 2000 plate shaker (Thermo Fisher Scientific Inc, MI, USA) at room temperature. Absorbance was measured spectrophotometrically at wavelength 570 nm using a Stat FaxR 4200 plate reader (Awareness Technology, Inc., FL, USA).41
Cell viability were calculated as percentage of control and the concentration that inhibits 50% of maximum cell proliferation were determined and expressed as IC50 using Graph Pad Prism 5 software (Graph Pad software Inc, CA, USA).
In vitro Enzyme Inhibitory Assays
To investigate the effect of synthesized hybrids on the enzymatic activity of HDAC and tubulin, MCF-7 line was used and incubated in DMEM supplemented with 10% fetal bovine serum, 1% penicillin-streptomycin and 10 μg/mL insulin (Sigma). All of the other chemicals and reagents were from Sigma, or Invitrogen Cancer cells were seeded at density 1.2–1.8 x 105/well in 100 μL DMEM using 96-well plates. To each well, 100 μL of the synthesized compounds 7c, 7g and 7j were added 18–24 hours prior to HDAC and tubulin inhibition assay.
The assay was done on MCF-7 cell line using TUBb ELISA kit (Cloud-Clone Corp.). Cancer cells were seeded into 96-well microtiter plates and kept in a humidified atmosphere at 37°C for 1 day. After dilution to the specified concentration, 100 μL of the synthesized hybrids 7c, 7g and 7j and CA4 were added to each well, then the plate was incubated for 2 hours at 37°C.
After discarding the supernatant, 100 μL of kit detection reagent A was added to each well, then the plate was re-incubated for 2 hours at 37°C. Kit detection reagent B was added to each well and the plate incubated for 30 minutes at 37°C. After five washing cycles, 90 μL of 3,3ʹ,5,5ʹ-tetramethylbenzidine (TMB) solution was added to each well, then the plate was incubated for 15–25 minutes at 37°C, followed by the addition of 50 μL of stop solution. Different concentrations (0.01, 0.1, 1, 10 μM) of the synthesized hybrids 7c, 7g and 7j and CA4 were incubated with the cells for 72 hours.
Absorbance was measured spectrophotometrically at wavelength 450 nm using a Stat FaxR 4200 plate reader (Awareness Technology, Inc., FL, USA).42 The concentration that inhibits 50% of maximum TUBb activity were determined and expressed as IC50 using Graph Pad Prism 5 software (Graph Pad software Inc, CA, USA ).
The effect of the synthesized hybrids 7c, 7g and 7j on HDAC (1 and 2) were investigated using ELISA assay kit (Mybiosource, Inc.). According to manufacturer’s instructions, MCF-7 cell was seeded into 96-well microtiter plates and kept in a humidified atmosphere at 37°C for 1 day. After dilution to the specified concentration, 100 μL of the synthesized compounds 7c, 7g and 7j and positive control entinostat were added to each well, then the plate incubated for 2 hours at 37°C.
After discarding the supernatant, 100 μL of kit detection reagent A was added to each well, then the plate re-incubated for 2 hours at 37°C. Kit detection reagent B was added to each well and the plate incubated for 30 minutes at 37°C. After five washing cycles, 90 μL of 3,3ʹ,5,5ʹ-tetramethylbenzidine (TMB) solution was added to each well, then the plate was incubated for 15–25 min at 37°C, followed by the addition of 50 μL of stop solution. Different concentrations (0.01, 0.1, 1, 10 μM) of the synthesized hybrids 7c, 7g and 7j and entinostat were incubated with the cells for 72 hours.
Absorbance was measured spectrophotometrically at wavelength 405 nm using a Stat FaxR 4200 plate reader (Awareness Technology, Inc., FL, USA).43 The concentration that inhibits 50% of maximum HDAC activity were determined and expressed as IC50 using Graph Pad Prism 5 software (Graph Pad software Inc, CA).
In vitro Cell Cycle Analysis
MCF-7 cells at density of 2 x 105 were seeded into each well of a 6-well plate. The cells were kept in Dulbecco’s Modified Eagle’s Medium (DMEM), supplemented with 10% fetal bovine serum, and incubated in a humidified atmosphere containing 5% CO2 in air at 37°C for 24 hours. The old medium was replaced with a fresh one containing the synthesized hybrids 7g, 7j in addition to CA-4 and entinostat at their IC50 in DMSO (1% v/v). The cell plates were incubated for 24 hours. The cells were then fixed with 70% ice-cold ethanol after washing twice with cold phosphate buffered saline (PBS). Cells washed with PBS for 30 minutes at 37°C and recovered by centrifugation at 2000 rpm for 5 minutes.
Cells were stained with DNA fluorochrome propidium iodide (PI). The plates were incubated at room temperature in a dark place for 20 minutes. Then the cells were investigated with a FACS Caliber flow cytometer (Becton Dickinson, Heidelberg, Germany).42
MCF-7 cells at a density of 4 x 106/well were incubated with synthesized compounds 7g, 7j in addition to CA-4 and entinostat at their IC50 for 24 hours. Cells were subjected to three washing cycles with ice cold PBS, then suspended in PBS. Cell apoptosis was detected by Annexin V-FITC Apoptosis Detection Kit (BioVisionResearch Products, USA). The cells were stained with PI staining solution, Annexin V-FITC and incubated at room temperature for 15 minutes in a dark place. Cells were investigated by flow cytometry using FACS caliber (Becton Dickinson, Heidelberg, Germany).44
Molecular Docking Study
Molecular modelling studies of the selected derivatives 7c, 7g, 7j, CA-4 and entinostat were performed using MOE software. Ligands were built into the builder interface of the MOE program and their energies were minimized until a root mean square deviation (RMSD) gradient of 0.01 kcal/mol and a root mean square (RMS) distance of 0.1 Å with MMFF94X (Merck molecular force field 94X) force-field and the partial charges were automatically calculated. The X-ray crystallographic structure of tubulin-colchicine complex (PDB code: 1SA0) and HDAC2 active site (PDB code: 4LXZ) were downloaded from protein data bank (www.rcsb.org). The enzymes were prepared, the hydrogens were added then the atoms connection and type were checked with automatic correction. The obtained poses were studied and the poses showing the best ligand-enzyme interactions were selected and stored for energy calculations.
Crystal Structure Determinations
The crystal was mounted in inert oil and transferred to the cold gas stream of a Rigaku-Oxford XtaLAB Synergy diffractometer using mirror-focussed Cu Kα radiation. Absorption corrections were based on multi-scans. The structure was refined anisotropically on F2 using the program S
Crystallographic data for compound 6: C24H18N2O4, Mr= 398.40, Temperature (K) = 100, Crystal habit: colorless plate, Crystal size (mm): 0.15 × 0.10 × 0.04, Crystal system: Monoclinic, Space group: P21/c, Cell dimensions: [a (Å) = 6.5120(3), b (Å) = 33.6736(13), c (Å) = 8.5409(3), β (°) = 95.237(4)], Cell volume (Å3) = 1865.06, Z = 4, Dx (g cm−3) = 1.419, Radiation, wavelength (Å): Cu Kα, 1.54184 Å, μ (mm−1) = 0.80, 2θ(max) (°) =154.8, Reflections collected = 39,381, Independent reflections = 3927, R(int) = 0.031, Transmissions = 0.728–1.000, No. of parameters = 276, Goodness-of-fit on F2 = 1.04, wR2 (all reflections) = 0.092, R1 (F > 4σ(F)) = 0.036, Max. Δρ (e Å−3) = 0.22.
Acknowledgment
The authors are deeply grateful for Professor H. Hopf. Institut fuer Organische Chemie, TU Braunschweig, Germany, for critical reading of the manuscript.
Disclosure
The authors reports no conflicts of interest in this work.
References
1. Petrelli A, Giordano S. From single- to multi-target drugs in cancer therapy: when a specificity becomes an advantage. Curr Med Chem. 2008;15:422–432. doi:10.2174/092986708783503212
2. Ouyang L, Shi Z, Zhao S, et al. Programmed cell death pathways in cancer: a review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45:487–498. doi:10.1111/j.1365-2184.2012.00845.x
3. Fitzmaurice C, Akinyemiju TF, Al Lami FH, et al. Global burden of disease cancer, global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: a systematic analysis for the global burden of disease study. JAMA Oncol. 2017;3:524–548. doi:10.1001/jamaoncol.2016.5688
4. Tang Z-Y. Hepatocellular carcinoma: cause, treatment and metastasis. World J Gastroenterol. 2001;7(4):445–454. doi:10.3748/wjg.v7.i4.445
5. Available from: https://www.who.int/cancer/prevention/diagnosis-screening/breast-cancer/en/.
6. Tang Y, Wang Y, Kiani MF, Wang B. Classification, treatment strategy, and associated drug resistance in breast cancer. Clin Breast Cancer. 2016;16:335–343. doi:10.1016/j.clbc.2016.05.012
7. Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi:10.1038/nrc1779
8. Marks PA, Dokmanovic M. Histone deacetylase inhibitors: discovery and development as anticancer agents. Expert Opin Invest Drugs. 2005;14(12):1497–1511. doi:10.1517/13543784.14.12.1497
9. Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi:10.1038/nrd2133
10. Young JL, Won AJ, Jaewon L, et al. Molecular mechanism of SAHA on regulation of autophagic cell death in tamoxifen-resistant MCF-7 breast cancer cells. Int J Med Sci. 2012;9(10):881–893. doi:10.7150/ijms.5011
11. Rosato RR, Grant S. Histone deacetylase inhibitors in clinical development. Expert Opin Invest Drugs. 2004;13(1):21–38. doi:10.1517/13543784.13.1.21
12. Kumar N, Tomar R, Pandey A, Tomar V, Singh VK, Chandra R. Preclinical evaluation and molecular docking of 1,3-benzodioxole propargyl ether derivatives as novel inhibitor for combating the histone deacetylase enzyme in cancer. Artif Nano Biotech. 2018;46:1288–1299. doi:10.1080/21691401.2017.1369423
13. Downing KH, Nogales E. Tubulin structure: insights into microtubule properties and functions. Curr Opin Struct Biol. 1998;8:785–791. doi:10.1016/S0959-440X(98)80099-7
14. Tomar V, Kumar N, Tomar R, et al. Biological evaluation of noscapine analogues as potent and microtubule-targeted anticancer agents. Sci Rep. 2019;9:19542–19552. doi:10.1038/s41598-019-55839-8
15. Kaur R, Kaur G, Kaur R, Gill RK, Soni R, Bariwal J. Recent developments in tubulin polymerization inhibitors: an overview. Eur J Med Chem. 2014;87:89–124. doi:10.1016/j.ejmech.2014.09.051
16. Sackett DL. Podophyllotoxin, steganacin and combretastatin: natural products that bind at the colchicine site of tubulin. Pharmac Ther. 1993;59:163–228. doi:10.1016/0163-7258(93)90044-E
17. Castaño LF, Cuartas V, Bernal A, et al. New chalcone-sulfonamide hybrids exhibiting anticancer and antituberculosis activity. Eur J Med Chem. 2019;175:50–60. doi:10.1016/j.ejmech.2019.05.013
18. Modzelewska A, Pettit C, Achanta G, Davidson NE, Huang P, Khan SR. Anticancer activities of novel chalcone and bis-chalcone derivatives. Bioorg Med Chem. 2006;14(10):3491–3495. doi:10.1016/j.bmc.2006.01.003
19. Wu J, Li J, Cai Y, et al. Evaluation and discovery of novel synthetic chalcone derivatives as antiinflammatory agents. J Med Chem. 2011;54(23):8110–8123. doi:10.1021/jm200946h
20. Singh N, Kumar N, Rathee G, et al. Privileged scaffold chalcone: synthesis, characterization and its mechanistic interaction studies with BSA employing spectroscopic and chemoinformatics approaches. ACS Omega. 2020;5:2267–2279. doi:10.1021/acsomega.9b03479
21. Chiaradia LD, Mascarello A, Purificação M, et al. Synthetic chalcones as efficient inhibitors of Mycobacterium tuberculosis protein tyrosine phosphatase PtpA. Bioorg Med Chem Lett. 2008;18(23):6227–6230. doi:10.1016/j.bmcl.2008.09.105
22. Cole AL, Hossain S, Cole AM, Phanstiel O. Synthesis and bioevaluation of substituted chalcones, coumaranones and other flavonoids as anti-HIV agents. Bioorg Med Chem. 2016;15(12):2768–2776. doi:10.1016/j.bmc.2016.04.045
23. Syahri J
24. Escribano-Ferrer E, Regué JQ, Garcia-Sala X, Montañés AB, Lamuela-Raventos RM. In vivo anti-inflammatory and antiallergic activity of pure naringenin, naringenin chalcone, and quercetin in mice. Nat Prod. 2019;82:177–182. doi:10.1021/acs.jnatprod.8b00366
25. Espinoza-Hicks JC, Chacón-Vargas KF, Hernández-Rivera JL, et al. Novel prenyloxy chalcones as potential leishmanicidal and trypanocidal agents: design, synthesis and evaluation. Eur J Med Chem. 2019;167:402–413. doi:10.1016/j.ejmech.2019.02.028
26. Sharma V, Kumar V, Kumar P. Heterocyclic chalcone analogues as potential anticancer agents. Anticancer Agents Med Chem. 2013;13(3):422–432. doi:10.2174/1871520611313030006
27. Zuo Y, Yu Y, Wang S, et al. Synthesis and cytotoxicity evaluation of biaryl-based chalcones and their potential in TNFα-induced nuclear factor-kB activation inhibition. Eur J Med Chem. 2012;50:393. doi:10.1016/j.ejmech.2012.02.023
28. Ducki S. Antimitotic chalcones and related compounds as inhibitors of tubulin assembly. Anticancer Agents Med Chem. 2009;9(3):336–347. doi:10.2174/1871520610909030336
29. Alanzi AM, El-Azab AS, Al-Suidan IA, et al. Structure based design of phthalimide derivatives as potential cyclooxygenase-2 (COX-2) inhibitors: anti-inflammatory and analgesic activities. Eur J Med Chem. 2015;92:115–123. doi:10.1016/j.ejmech.2014.12.039
30. Karthik CS, Mallesha L, Mallu P. Investigation of antioxidant properties of phthalimide derivatives. Canad Chem Trans. 2015;3:199–206. doi:10.13179/canchemtrans.2015.03.02.0194
31. Mercurio A, Sharples L, Franchini CC, et al. Phthalimide derivative shows anti-angiogenic activity in a 3D microfluidic model and no teratogenicity in zebrafish embryos. Front Pharmacol. 2019;10:349. doi:10.3389/fphar.2019.00349
32. Mahapatra SP, Ghode P, Jain DK, Chaturvedi SC, Maiti BC, Maity TK. Synthesis and hypoglycemic activity of some phthalimide derivatives. J Pharm Sci & Res. 2010;2:567–578.
33. Belluti S, Orteca G, Rigillo SG, Parenti F, Ferrari E, Imbriano C. Potent anti-cancer properties of phthalimide-based curcumin derivatives on prostate tumor cells. Int J Mol Sci. 2019;20:28–49. doi:10.3390/ijms20010028
34. Santos JL, Yamasaki PR, Chin CM, Takashi CH, Pavan FR, Leite CQF. Synthesis and in vitro anti Mycobacterium tuberculosis activity of a series of phthalimide derivatives. Bioorg Med Chem. 2009;17:3795–3799. doi:10.1016/j.bmc.2009.04.042
35. Kushwaha N, Kaushik D. Recent advances and future prospects of phthalimide derivatives. J App Pharm Sci. 2016;26:159–171. doi:10.7324/JAPS.2016.60330
36. Nomura S, Endo-Umeda K, Aoyama A, Makishima M, Hashimoto Y, Ishikawa M. Styrylphenylphthalimides as novel transrepression-selective Liver X Receptor (LXR) Modulators. ACS Med Chem Lett. 2015;6(8):902–907. doi:10.1021/acsmedchemlett.5b00170
37. Lima ML, de Brito FCF, de Souza SD, et al. Novel phthalimide derivatives, designed as leukotriene D4 receptor antagonists. Bioorg Med Chem Lett. 2002;12:1533–1535. doi:10.1016/S0960-894X(02)00203-2
38. Machado AL, Lima LM, Araujo JX, Fraga CAM, Koatz VLG, Barreiro EJ. Design, synthesis and anti-inflammatory activity of novel phthalimide derivatives, structurally related to thalidomide. Bioorg Med Chem Lett. 2005;15:1169–1172. doi:10.1016/j.bmcl.2004.12.012
39. Noguchi T, Fujimoto H, Sano H, Miyajima A, Miyachi H, Hashimoto Y. Angiogenesis inhibitors derived from thalidomide. Bioorg Med Chem Lett. 2005;15:5509–5513. doi:10.1016/j.bmcl.2005.08.086
40. Bornstein J, Bedell SF, Drummond PE, Kosloski CL. The synthesis of α-amino-o-tolualdehyde diethylacetal and its attempted conversion to pseudoisoindole. J Amer Chem Soc. 1956;78:83–86. doi:10.1021/ja01582a026
41. Jermy BR, Ravinayagam V, Alamoudi WA, et al. Targeted therapeutic effect against the breast cancer cell line MCF-7 with a CuFe2O4/silica/cisplatin nanocomposite formulation. Beilstein J Nanotechnol. 2019;10:2217–2228. doi:10.3762/bjnano.10.214
42. El-Bakhshawangy NM, El-Nassan HB, Kassab AE, Taher AT. Design, synthesis and biological evaluation of chromenopyrimidines as potential cytotoxic agents. Future Med Chem. 2018;10:1465–1481. doi:10.4155/fmc-2017-0324
43. Lu W, Wang F, Zhang T, et al. Search for novel histone deacetylase inhibitors. Part II: design and synthesis of novel isoferulic acid derivatives. Bioorg Med Chem. 2014;22:2707–2713. doi:10.1016/j.bmc.2014.03.019
44. Jamalzadeh L, Ghafoori H, Aghamaali M, Sariri R. Induction of apoptosis in human breast cancer MCF-7 cells by a semisynthetic derivative of artemisinin: a caspase-related mechanism. Iran J Biotech. 2017;15:157–165. doi:10.15171/ijb.1567
45. Sheldrick GM. Crystal structure refinement with SHELXL. Acta Cryst C. 2015;71:3–8. doi:10.1107/S2053229614024218
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.