Back to Journals » Drug Design, Development and Therapy » Volume 10
Novel glyoxalase-I inhibitors possessing a “zinc-binding feature” as potential anticancer agents
Authors Al-Balas Q ![]() , Hassan M, Al-Shar’i N
, Hassan M, Al-Shar’i N ![]() , Mhaidat N, Almaaytah A, Al-Mahasneh F, Isawi I
, Mhaidat N, Almaaytah A, Al-Mahasneh F, Isawi I
Received 20 April 2016
Accepted for publication 23 May 2016
Published 17 August 2016 Volume 2016:10 Pages 2623—2629
DOI https://doi.org/10.2147/DDDT.S110997
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Wei Duan
Qosay A Al-Balas,1 Mohammad A Hassan,1 Nizar A Al-Shar’i,1 Nizar M Mhaidat,2 Ammar M Almaaytah,3 Fatima M Al-Mahasneh,1 Israa H Isawi1
1Department of Medicinal Chemistry and Pharmacognosy, 2Department of Clinical Pharmacy, 3Department of Pharmaceutical Technology, Faculty of Pharmacy, Jordan University of Science and Technology, Irbid, Jordan
Background: The glyoxalase system including two thiol-dependent enzymes, glyoxalase I (Glo-I) and glyoxalase II, plays an important role in a ubiquitous metabolic pathway involved in cellular detoxification of cytotoxic 2-oxoaldehydes. Tumor cells have high glycolytic activity, leading to increased cellular levels of these toxic metabolites. The increased activity of the detoxification system in cancerous cells makes this pathway a viable target for developing novel anticancer agents. In this study, we examined the potential utility of non-glutathione-based inhibitors of the Glo-I enzyme as novel anticancer drugs.
Methods: Computer-aided drug design techniques, such as customized pharmacophoric features, virtual screening, and flexible docking, were used to achieve the project goals. Retrieved hits were extensively filtered and subsequently docked into the active site of the enzyme. The biological activities of retrieved hits were assessed using an in vitro assay against Glo-I.
Results: Since Glo-I is a zinc metalloenzyme, a customized Zn-binding pharmacophoric feature was used to search for selective inhibitors via virtual screening of a small-molecule database. Seven hits were selected, purchased, and biologically evaluated. Three of the seven hits inhibited Glo-I activity, the most effective of which exerted 76.4% inhibition at a concentration of 25 µM.
Conclusion: We successfully identified a potential Glo-I inhibitor that can serve as a lead compound for further optimization. Moreover, our in silico and experimental results were highly correlated. Hence, the docking protocol adopted in this study may be efficiently employed in future optimization steps.
Keywords: cancer, glyoxalase-I, zinc-binding feature, flexible docking, Discovery Studio 3.5, ketol
Introduction
Cancer is a rebound system of growth that originates in the human body. The cancer cells are capable of avoiding apoptosis, a programmed suicide mechanism, which confers them the ability of limitless cell expansion.1 Cancer is the leading cause of mortality worldwide; it was accountable for 8.2 million deaths in 2012 according to the World Health Organization statistics, and its incidence is expected to increase to 13.0 million by 2030. Consequently, immense research attention has been focused on successful treatment of the disease. Unfortunately, due to the lack of selectivity for cancerous cells, no optimal chemotherapeutic agents have been identified to date. Researchers are therefore centralizing their efforts on discovering valid targets and designing new therapeutic inhibitors.2–5
Glyoxalase system enzymes have been approved as valid targets for cancer treatment.6–8 These enzymes present an attractive target for cancer therapy owing to their overexpression in malignant cells and involvement in increasing resistance to antitumor agents. The glyoxalase system comprising two enzymes, glyoxalase-1 (Glo-I) and glyoxalase-2 (Glo-II), is responsible for the detoxification of metabolic byproducts produced by cells, particularly reactive aldehydes, such as methylglyoxal (MG). Glo-I catalyzes the first step, which is isomerization of the toxic metabolite, and the detoxification process is subsequently accomplished via hydrolysis of the Glo-I product, S-D-lactoylglutathione, to generate nontoxic bystanders that are safely excreted outside the body (Figure 1).9–13
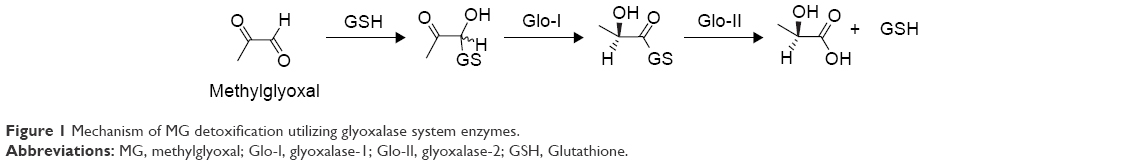 | Figure 1 Mechanism of MG detoxification utilizing glyoxalase system enzymes. |
Tumor cells are metabolically more active than normal cells, resulting in increased cellular levels of toxic MG and S-D-lactoylglutathione metabolites. Tumor cells respond by augmenting the activity of the detoxifying glyoxalase system to minimize the intracellular concentrations of toxic metabolites. This phenomenon has been reported in breast carcinoma by Rulli et al14 and human superficial or invasive bladder cancer by Mearini et al,15 who showed that activities of both Glo-I and Glo-II are significantly increased in tumors, compared to those in normal cells. This finding rationalizes that inhibition of the glyoxalase system (Glo-I and/or Glo-II) may be detrimental for cancer cells, potentially leading to their self-destruction.14,16–22 Structurally, Glo-I is a homodimeric enzyme composed of two identical polypeptide chains, each comprising 183 amino acids. In addition, the enzyme possesses a structural zinc metal. Zinc is an essential cofactor present within the Glo-I active site that plays an important structural and catalytic role in the isomerization of reactive and toxic aldehydes via stabilizing the enediolate intermediate by lowering the free activation energy of proton transfer.23 The Glo-I active site is located at the interface of the two polypeptide chains, and the zinc atom displays octahedral coordination with adjacent amino acids from both chains.24,25
Glo-I, an established pivotal anticancer target, has been the focus of research by several groups. For example, More et al26–32 designed a number of Glo-I transition-state competitive inhibitors incorporating different zinc-binding groups (ZBGs). Takasawa et al33–35 additionally identified natural flavonoids, such as Glo-I inhibitors, and showed that coplanar hydroxyl and ketone groups are important for zinc chelation (Figure 2).
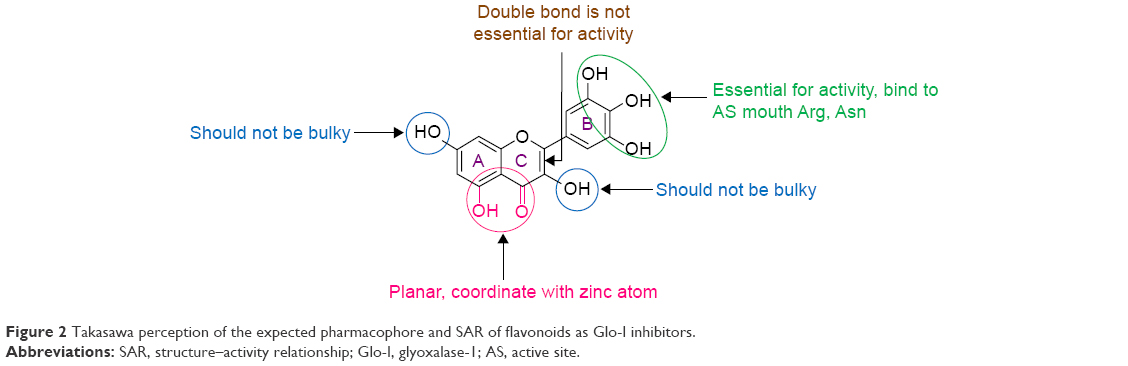 | Figure 2 Takasawa perception of the expected pharmacophore and SAR of flavonoids as Glo-I inhibitors. |
Previously, we performed active site analysis of Glo-I to determine the pivotal features essential for optimal inhibitor binding with the active site.28 Three features were determined as critical: a zinc-binding region in the inhibitor possessing a ZBG to chelate the Zn2+ ion, the presence of a negatively ionizable group in the inhibitor for binding to the positively ionized active site mouth, and a deep hydrophobic pocket that could be filled with a hydrophobic group.28
In this study, we mined the commercially available AldrichCPR database for extraction of potential inhibitors of Glo-I by selecting a ketol group (present in myricetin) as the ZBG and carboxylic acid to complement the positively ionized active site mouth. Final selection of potential inhibitors was achieved by performing flexible docking to estimate binding energy, along with establishing a good perception of the binding modes of these inhibitors within the active site. The biological inhibitory activities of the selected hits against Glo-I were evaluated via in vitro assay.
Materials and methods
Computational materials
Discovery Studio (DS) 3.5 from Biovia® Software Inc http://www.3ds.com/products-services/biovia/ (San Diego, CA, USA) was used for active site analysis, extraction of potential inhibitors from commercial databases (AldrichCPR), and flexible docking. The crystal structure of Glo-I (accession code: 1QIN) was retrieved from the Protein Data Bank (PDB) as our structural model.25
Glo-I active site analysis
The crystal structure of Glo-I, obtained from PDB, was prepared using the Prepare Protein Protocol, in which missing loops and alternate conformations were corrected. The active site was determined from the cocrystallized ligand bound within, along with the zinc atom. The Glo-I enzyme active site is mapped as three main areas: a highly positive active site mouth containing arginine and lysine, a hydrophobic pocket deep within the active site, and zinc atom coordinated between these two areas.28
Virtual screening of the commercial database
The commercially available AldrichCPR database (containing ~250,000 compounds) from Sigma-Aldrich Co. (St Louis, MO, USA) was downloaded and converted into a three-dimensional database using the Build 3D Database protocol in DS. The database was searched for potential inhibitors of Glo-I based on the active site and the function of the ketol group, which can chelate zinc atom.34
The search query was based on two steps: extraction of all compounds containing a ketol group with the potential to chelate zinc atoms, followed by selection of carboxyl-containing compounds from hits retrieved from the first step which were expected to bind to the mouth of the active site. These steps were performed using the Ligand Pharmacophore Mapping protocol within DS, which aligns and identifies the ligands that map to a pharmacophore. Moreover, with application of Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) profiling and Lipinski’s and Veber’s rules, hits were reduced to obtain drug-like compounds and facilitate the process of flexible docking, which is computationally expensive.
Docking
Final selection of the anticipated inhibitors was based on docking studies in parallel with visual inspection. The protein active site was determined using the Edit and Define Active Site Tool available in DS. The active site was defined by a sphere of 11 Å radius encompassing the three main areas. Other parameters for docking were left at default values.
Flexible Docking protocol in DS 3.5 was used to facilitate selection of potential Glo-I enzyme inhibitors. Selection of the proposed inhibitors from the docked set was based on their score, in addition to orientation within the active site and potential chelation between the inhibitor and zinc atom (considered the main criterion of selection).
This type of docking protocol is distinct from other docking programs in that it allows side chains of the active site residues to move with high flexibility. Despite being computationally demanding, the flexible docking procedure results in better mimicking of real inhibitor–enzyme binding.36
Glo-I in vitro assay
Bulk solvents and chemicals were purchased from Sigma-Aldrich Co. and Acros (Thermo Fisher Scientific, New Jersey, US) via their local agents and utilized with no further purification or distillation.
Biological activities of the selected compounds were evaluated by measuring their in vitro inhibitory activities against human recombinant Glo-I following the protocol provided by the manufacturer (R&D Systems, Inc., Minneapolis, MN, USA). The enzyme was reconstituted in our laboratory by dissolving to a concentration of 0.5 mg/mL in sterile, deionized water, stored at −70°C, and thawed for use on the day of the test. Test compounds were dissolved in dimethyl sulfoxide to prepare 10 mM stock solution, and absorbance was measured at λmax of 240 nm for 200 seconds at 25°C.
The assay buffer was prepared using 0.5 M sodium phosphate dibasic and 0.5 M sodium phosphate monobasic. The substrate mixture contained 100 mM glutathione (freshly prepared) and 100 mM MG. The two solutions were combined and added to assay buffer, and the mixture was allowed to stand at room temperature for 15 minutes. The blank was prepared by mixing assay buffer with substrate mixture, and test compounds were prepared by diluting with assay buffer followed by the addition of human recombinant Glo-I into a cuvette at a concentration of 50 μM. All tests were performed in triplicate, and the mean was calculated.
Results and discussion
In silico design
The Glo-I enzyme has been investigated previously by our research group, and three main criteria that contribute to the binding of potential inhibitors have been established. In the present study, the crystal structure of Glo-I complexed with a substrate analog (PDB accession code: 1QIN) was employed for active site analysis. The zinc atom localized at the bottom of the active site is considered the main determinant of specific binding of potential inhibitors. The reason behind this assumption is that the zinc atom plays a structural role in the catalytic mechanism of aldehyde detoxification. Structurally, Zn2+ has octahedral geometry, coordinating with four amino acids in addition to the inhibitor (Figure 3).25
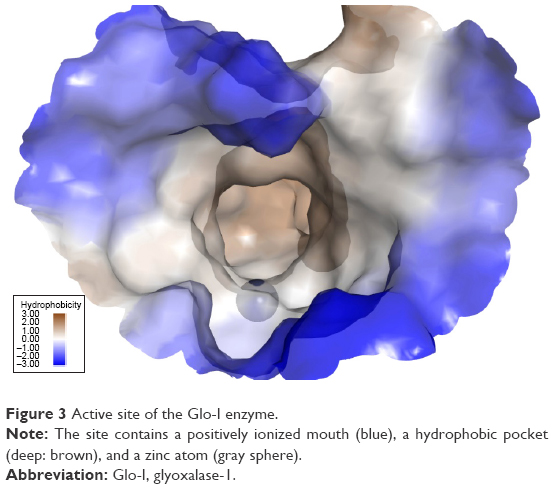 | Figure 3 Active site of the Glo-I enzyme. |
The second important criterion for selecting Glo-I inhibitors is the presence of positively ionizable amino acids, such as arginine and lysine, at the mouth of the active site. The nature of this area has guided the design process to preferably select ligands carrying negatively ionizable groups, such as carboxylic acid. Finally, the hydrophobic pocket located deep within the active site of the enzyme has been utilized in the design process via incorporation of a hydrophobic feature in the final pharmacophore that fits into the pocket.
The next step in the selection of suitable inhibitors of Glo-I enzyme was to identify compounds bearing the correct binding features. Many functional groups are known to chelate the zinc atom.34,37,38
We selected the ketol group as a guide, in view of its presence in many natural compounds and lack of association with visible toxicity that is evident with thiol or hydroxamate groups. A ketol-mapping feature was incorporated into our final pharmacophore (Figure 4). Several naturally occurring compounds, such as flavonoids, possess such a group. Myricetin, containing a ketol group for chelating the zinc atom, is an established inhibitor of Glo-I, and therefore used as a positive control in our enzyme assay. Based on this hypothesis, a query root pharmacophore was built, and the AldrichCPR database was virtually screened to extract the compounds displaying this feature. Following the first search step, a refining step was conducted on retrieved hits to further identify compounds possessing carboxylic acid groups, resulting in a collection of compounds possessing both ketol and carboxylic acid groups. The third feature for consideration is the presence of a hydrophobic moiety, which was investigated manually due to the difficulty of assigning specific groups for the search, since numerous candidate chemical groups are capable of forming hydrophobic interactions.
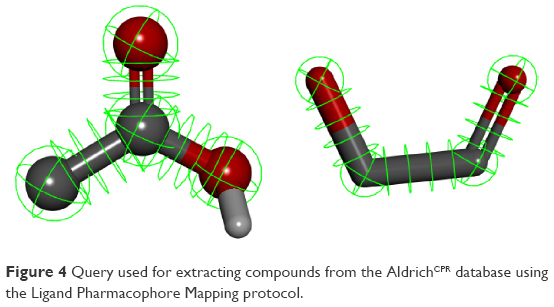 | Figure 4 Query used for extracting compounds from the AldrichCPR database using the Ligand Pharmacophore Mapping protocol. |
To reduce the number of returned hits and enhance the chances of finding potential inhibitors of Glo-I, Lipinski’s and Veber’s rules for orally available drug-like compounds (within DS) were implemented. Additional restrictions for the selection criteria were imposed prior to docking by filtering compounds that had passed the previous filtration step using the ADMET profile filter. The resulting set of filtered compounds were considered effective drug candidates, as they complied with the known rules of safe oral compounds.
Successful candidates that passed the harsh filtration criteria were subjected to a semifinal ranking procedure based on docking. Flexible docking is used as a predictor of binding energy and the binding mode (orientation) of the final filtered set of compounds. Final selection of the proposed inhibitors for biological evaluation was performed by visual inspection of the chemical structure. Three main features were required for binding (ZBG, COOH, hydrophobic) in addition to their docking score and binding orientation.
Seven compounds were ultimately selected from the AldrichCPR commercial database (Table 1). All seven compounds possessed a ZBG, carboxylic acid forming a salt bridge at the mouth of the active site, and a hydrophobic group next to the ZBG.
 | Table 1 Chemical structures of retrieved hits from AldrichCPR, their in vitro enzyme assay results, and corresponding in silico docking scores |
The compounds were assayed (at a concentration of 50 μM) using the method described in the “Materials and methods” section.
As shown in Table 1, compounds 1, 2, and 3 showed high inhibitory activities, compared to the other four candidates, with compound 2 exhibiting the highest inhibition efficiency (76.4%) at a concentration of 25 μM. Compounds 1–3 with high structural similarity (apart from the hydrophobic region with different patterns of substitution) were selected to investigate the importance of hydrophobic interactions. Compound 1 contained an unsubstituted benzene ring, compound 2 had a chlorine atom on the benzene ring as electron-withdrawing group, and compound 3 contained an isopropyl group as electron-donating group. This variation was in favor of the presence of a chlorine atom at the para position of the benzene ring, which produced a compound at least twice as active as the isopropyl analog and three times as active as the unsubstituted compound. This could be explained by the fact that the hydrophobic pocket is capable of accommodating more than the benzene ring and electrostatic forces are important, as the Cl atom has better activity than the isopropyl group, although both groups are considered classical bioisosteres. The most active hit was selected as a lead compound for further derivatization and optimization of pharmacokinetic and pharmacodynamic profiles. Ongoing research in our laboratory has focused on generating a robust structure–activity relationship that can be utilized in further development and optimization.
In silico docking of the proposed compounds was well correlated with experimental data obtained from the in vitro enzyme assay. Highest scores were obtained with the first three compounds (1, 2, 3) in docking studies, with significant differences in activity, compared with the inactive compounds. These results have enhanced credibility and confidence in our parametrization of the flexible docking protocol, which may be effectively used in the future to screen other commercial databases for further potential Glo-I inhibitors. Compound 2 (the most active) is depicted in Figure 5 docked within the active site of the enzyme in which the ketol group clearly chelates the zinc atom, the carboxylic acid group forms a salt bridge with the positively ionized mouth, and the hydrophobic pocket is filled with a para-chlorobenzene ring.
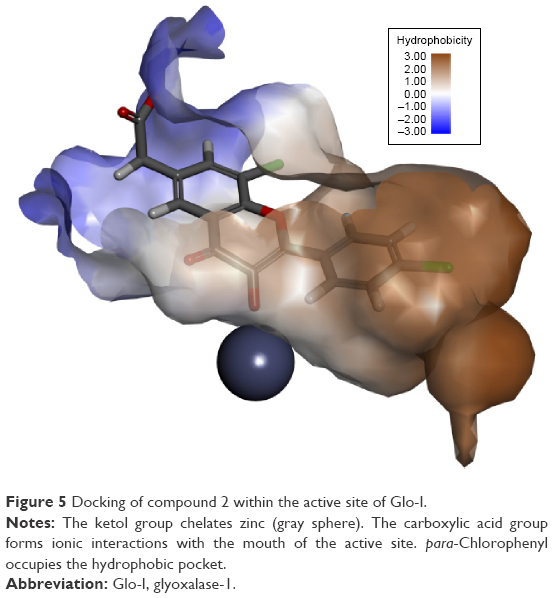 | Figure 5 Docking of compound 2 within the active site of Glo-I. |
Conclusion
In this research, computer-aided drug design techniques were implemented to identify novel Glo-I inhibitors. Thorough investigation of the active site of the target enzyme revealed three areas crucial for effective binding that were used to construct the pharmacophore. The electrostatic criteria of the three main areas of the active site were fulfilled, that is, a zinc-binding region with a ketol group, a positively ionized active site mouth that can bind carboxylic acid, and finally, a hydrophobic pocket with a lipophilic moiety.
Seven compounds were selected based on evaluation of Glo-I inhibitory activity in vitro. The computer-aided drug design techniques used facilitated the rapid and successful identification of a new class of Glo-I inhibitors. The most effective compound exerted 76.4% inhibition at a concentration of 25 μM. We observed a clear correlation between the in vitro results obtained with the selected compounds and their in silico docking scores.
Acknowledgment
This work was supported by the Deanship of Research, Jordan University of Science and Technology, The Hashemite Kingdom of Jordan.
Disclosure
The authors report no conflicts of interest in this work.
References
National Cancer Institute. Available from: http://www.cancer.gov/. Accessed October 28, 2015. | ||
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61(2):69–90. | ||
World Health Organization (WHO). The Global Burden of Disease: 2004 Update. Geneva: WHO. Available from: http://www.who.int/topics/global_burden_of_disease/en/. Accessed October 28, 2015. | ||
Aziz NM, Rowland JH. Trends and advances in cancer survivorship research: challenge and opportunity. Semin Radiat Oncol. 2003;13(3):248–266. | ||
Petrelli NJ, Winer E, Brahmer J, et al. Clinical Cancer Advances 2009: major research advances in cancer treatment, prevention, and screening – a report from the American Society of Clinical Oncology. J Clin Oncol. 2009;27(35):6052–6069. | ||
Davidson SD, Milanesa DM, Mallouh C, Choudhury MS, Tazaki H, Konno S. A possible regulatory role of glyoxalase I in cell viability of human prostate cancer. Urol Res. 2002;30(2):116–121. | ||
Rabbani N, Thornalley PJ. Glyoxalase in diabetes, obesity and related disorders. Semin Cell Dev Biol. 2011;22(3):309–317. | ||
Thornalley PJ, Rabbani N. Glyoxalase in tumourigenesis and multidrug resistance. Semin Cell Dev Biol. 2011;22(3):318–325. | ||
Xue M, Rabbani N, Thornalley PJ. Glyoxalase in ageing. Semin Cell Dev Biol. 2011;22(3):293–301. | ||
Sakamoto H, Mashima T, Kizaki A, et al. Glyoxalase I is involved in resistance of human leukemia cells to antitumor agent-induced apoptosis. Blood. 2000;95(10):3214–3218. | ||
National Toxicology Program. Final report on carcinogens background document for formaldehyde. Rep Carcinog Backg Doc. 2010;10-5981:i-512. | ||
Chaplen FW. Incidence and potential implications of the toxic metabolite methylglyoxal in cell culture: a review. Cytotechnology. 1998;26(3):173–183. | ||
Suttisansanee U, Honek JF. Bacterial glyoxalase enzymes. Semin Cell Dev Biol. 2011;22(3):285–292. | ||
Rulli A, Carli L, Romani R, et al. Expression of glyoxalase I and II in normal and breast cancer tissues. Breast Cancer Res Treat. 2001;66(1):67–72. | ||
Mearini E, Romani R, Mearini L, et al. Differing expression of enzymes of the glyoxalase system in superficial and invasive bladder carcinomas. Eur J Cancer. 2002;38(14):1946–1950. | ||
Feierberg I, Luzhkov V, Åqvist J. Computer simulation of primary kinetic isotope effects in the proposed rate-limiting step of the glyoxalase I catalyzed reaction. J Biol Chem. 2000;275(30):22657–22662. | ||
Thornalley P. Protecting the genome: defence against nucleotide glycation and emerging role of glyoxalase I overexpression in multidrug resistance in cancer chemotherapy. Biochem Soc Trans. 2003;31(6):1372–1377. | ||
Thornalley P. Glyoxalase I-structure, function and a critical role in the enzymatic defence against glycation. Biochem Soc Trans. 2003;31(6):1343–1348. | ||
Clelland J, Allen R, Thornalley P. Inhibition of growth of human leukaemia 60 cells by S-2-hydroxyacylglutathiones and monoethyl ester derivatives. Biochem Pharmacol. 1992;44(10):1953–1959. | ||
Edwards L, Adesida A, Thornalley P. Inhibition of human leukaemia 60 cell growth by S-d-lactoylglutathione in vitro. Mediation by metabolism to N-d-lactoylcysteine and induction of apoptosis. Leuk Res. 1996;20(1):17–26. | ||
Edwards L, Thornalley P. Prevention of S-d-lactoylglutathione-induced inhibition of human leukaemia 60 cell growth by uridine. Leuk Res. 1994;18(9):717–722. | ||
Thornalley P, Tisdale M. Inhibition of proliferation of human promyelocytic leukaemia HL60 cells by S-D-lactoylglutathione in vitro. Leuk Res. 1988;12(11–12):897–904. | ||
Feierberg, I. (2003). Computational Studies of Enzymatic Enolization Reactions and Inhibitor Binding to a Malarial Protease. Department of Cell and Molecular Biology. Uppsala, Acta Universitatis Upsaliensis. Doctor of Philosophy: 54. | ||
Cameron AD, Olin B, Ridderström M, Mannervik B, Jones TA. Crystal structure of human glyoxalase I – evidence for gene duplication and 3D domain swapping. EMBO J. 1997;16(12):3386–3395. | ||
Cameron AD, Ridderström M, Olin B, Kavarana MJ, Creighton DJ, Mannervik B. Reaction mechanism of glyoxalase I explored by an X-ray crystallographic analysis of the human enzyme in complex with a transition state analogue. Biochemistry. 1999;38(41):13480–13490. | ||
More SS, Vince R. A metabolically stable tight-binding transition-state inhibitor of glyoxalase-I. Bioorg Med Chem Lett. 2006;16(23):6039–6042. | ||
Vince R, Daluge S, Wadd WB. Inhibition of glyoxalase I by S-substituted glutathiones. J Med Chem. 1971;14(5):402–404. | ||
Al-Balas Q, Hassan M, Al-Oudat B, Alzoubi H, Mhaidat N, Almaaytah A. Generation of the first structure-based pharmacophore model containing a selective “zinc binding group” feature to identify potential glyoxalase-1 inhibitors. Molecules. 2012;17(12):13740–13758. | ||
More SS, Vince R. Design, synthesis, and binding studies of bidentate Zn-chelating peptidic inhibitors of glyoxalase-I. Bioorg Med Chem Lett. 2007;17(13):3793–3797. | ||
More SS, Vince R. Inhibition of glyoxalase I: the first low-nanomolar tight-binding inhibitors. J Med Chem. 2009;52(15):4650–4656. | ||
Suzuki T, Miyata N. Rational design of non-hydroxamate histone deacetylase inhibitors. Mini Rev Med Chem. 2006;6(5):515–526. | ||
Maingot L, Leroux F, Landry V, et al. New non-hydroxamic ADAMTS-5 inhibitors based on the 1, 2, 4-triazole-3-thiol scaffold. Bioorg Med Chem Lett. 2010;20(21):6213–6216. | ||
Takasawa R, Saeki K, Tao A, et al. Delphinidin, a dietary anthocyanidin in berry fruits, inhibits human glyoxalase I. Bioorg Med Chem. 2010;18(19):7029–7033. | ||
Takasawa R, Takahashi S, Saeki K, Sunaga S, Yoshimori A, Tanuma S-I. Structure–activity relationship of human GLO I inhibitory natural flavonoids and their growth inhibitory effects. Bioorg Med Chem. 2008;16(7):3969–3975. | ||
Takasawa R, Tao A, Saeki K, et al. Discovery of a new type inhibitor of human glyoxalase I by myricetin-based 4-point pharmacophore. Bioorg Med Chem. 2011;21(14):4337–4342. | ||
Koska JR, Spassov VZ, Maynard AJ, et al. Fully automated molecular mechanics based induced fit protein–ligand docking method. J Chem Inf Model. 2008;48(10):1965–1973. | ||
Madsen AS, Kristensen HM, Lanz G, Olsen CA. The effect of various zinc binding groups on inhibition of histone deacetylases 1–11. ChemMedChem. 2014;9(3):614–626. | ||
Kawai K, Nagata N. Metal–ligand interactions: an analysis of zinc binding groups using the Protein Data Bank. Eur J Med Chem. 2012;51:271–276. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.