Back to Journals » Cancer Management and Research » Volume 11
Novel genetic alterations and their impact on target therapy response in head and neck squamous cell carcinoma
Authors Jiang X, Ye J, Dong Z, Hu S, Xiao M ![]()
Received 16 September 2018
Accepted for publication 2 January 2019
Published 8 February 2019 Volume 2019:11 Pages 1321—1336
DOI https://doi.org/10.2147/CMAR.S187780
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Ahmet Emre Eşkazan
Xiaohua Jiang,* Jing Ye,* Zhihuai Dong, Sunhong Hu, Mang Xiao
Department of Otolaryngology Head and Neck Surgery, Sir Run Shaw Hospital, College of Medicine, Zhejiang University, Hangzhou, Zhejiang, China
*These authors contributed equally to this work
Abstract: Head and neck squamous cell carcinoma (HNSCC) is highly variable by tumor site, histologic type, molecular characteristics, and clinical outcome. During recent years, emerging targeted therapies have been focused on driver genes. HNSCC involves several genetic alterations, such as co-occurrence, multiple feedback loops, and cross-talk communications. These different kinds of genetic alterations interact with each other and mediate targeted therapy response. In the current review, it is emphasized that future treatment strategy in HNSCC will not solely be based on “synthetic lethality” approaches directed against overactivated genes. More importantly, biologic, genetic, and epigenetic alterations of HNSCC will be taken into consideration to guide the therapy. The emerging genetic alterations in HNSCC and its effect on targeted therapy response are discussed in detail. Hopefully, novel combination regimens for the treatment of HNSCC can be developed.
Keywords: head and neck malignancy, gene mutations, deletions, amplifications, genetic interactions, target therapy
Introduction
Head and neck squamous cell carcinoma (HNSCC) causes more than 300,000 deaths worldwide each year and its incidence is still growing.1 HNSCC is a fatal heterogeneous disease, with highly variable tumor sites and biologic behaviors.2 The Cancer Genome Atlas (TCGA) has profiled 279 of HNSCC, from the oral cavity (n=172/279, 62%), oropharynx (n=33/279, 12%), and laryngeal sites (n=72/279, 26%).3,4 They generate comprehensive lists of genomic and epigenomic alterations present in diverse tumor samples. The MSK-IMPACT (Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets) assay is a targeted next-generation sequencing (NGS) assay approved through Clinical Laboratory Improvement Amendments.5 There were 151 patients with advanced, treatment-resistant head and neck tumors, including HNSCC, adenoid cystic carcinoma, and other salivary and cutaneous cancers, whose tumors were sequenced.6 Thus, it provided a comprehensive understanding of the molecular pathogenesis of HNSCC progression and treatment response.7
Comprehensive understanding of the biologic, genetic, and epigenetic alterations of HNSCC is necessary to guide the therapy.8 Drug responses are determined largely by pharmacogenetics, cancer microenvironment, and genetic aberrations.9 Identifying molecular aberrations that link to drug sensitivity helps to avoid unnecessary and unhelpful treatment.10 Although associations between genomic alterations and drug response were previously observed in HNSCC, no therapeutically targetable genomic subtypes have been identified. Therefore, surgery, radiation, and chemotherapy still remain to be the standard therapy for the disease.
We reviewed the emerging genetic alterations that represent a novel therapeutic target in HNSCC. These include frequent mutations of several genes (TP53, PIK3CA, CDKN2A, NOTCH1, and MET) as well as copy number gain in EGFR, CCND1, and PIK3CA. Those genomic alterations have been analyzed in relevance of sensitivity to chemotherapy, targeted therapy, and ionizing radiation that could potentially guide the therapy (Table 1). It is essential to determine whether proteins or pathways are involved in the targeted therapy of HNSCCs, aside from gene mutation.11 These genetic alterations may serve well as novel therapeutic candidates and predictive biomarkers for prognosis in HNSCC.
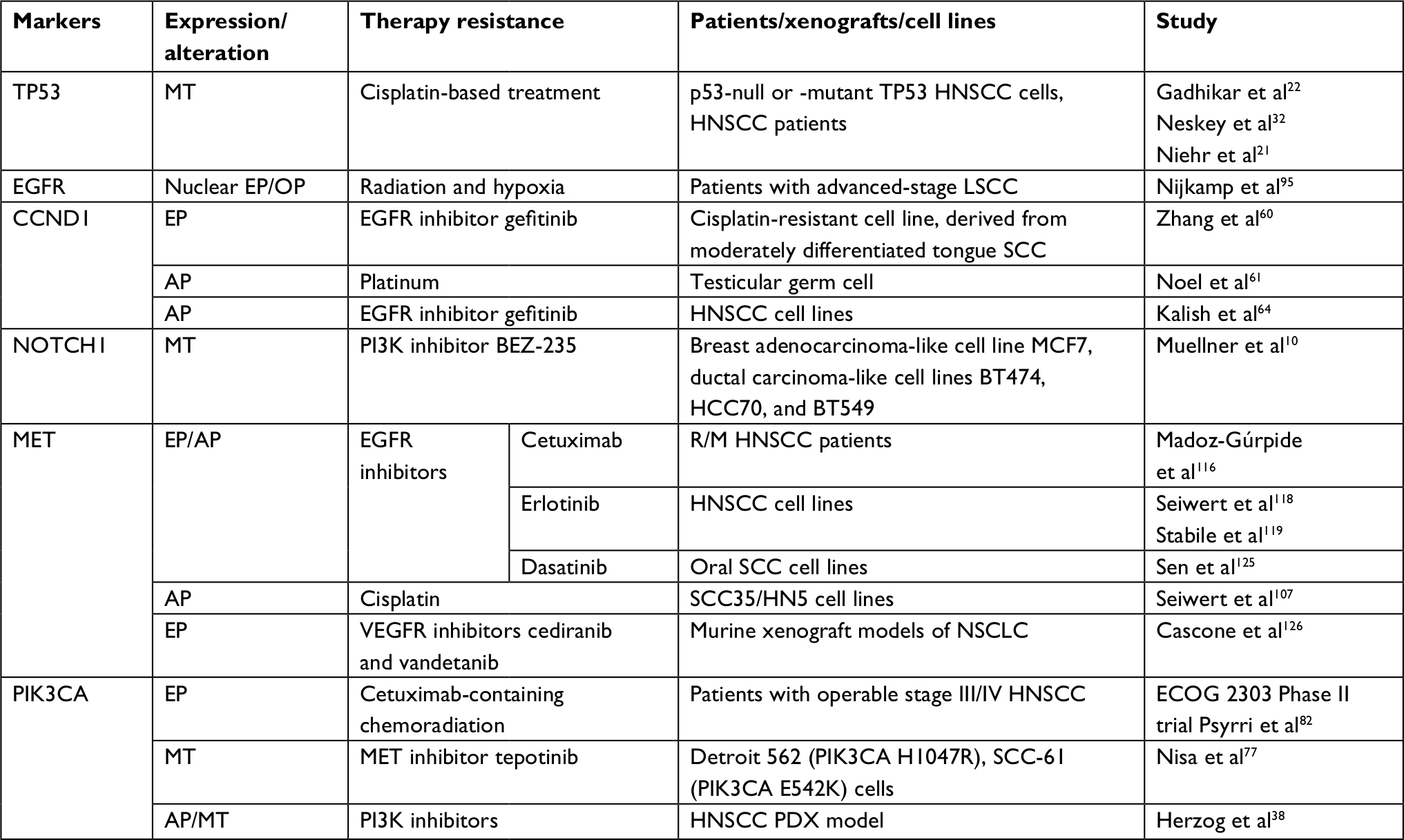 | Table 1 Genomic expression and alterations associated with drug resistance in HNSCC studies Abbreviations: AP, amplification; ECOG, Eastern Cooperative Oncology Group; EP, expression; HNSCC, head and neck squamous cell carcinoma; LSCC, laryngeal squamous cell carcinoma; MT, mutation; NSCLC, non-small-cell lung cancer; OP, OverEP; PI3K, phosphoinositide 3-kinase; R/M, recurrent/metastatic; SCC, squamous carcinoma; VEGFR, vascular endothelial growth factor receptor. |
Based on the recently identified mutations in HNSCCs, the major pathologic pathways implicated in the tumorigenesis of HNSCC include dysregulation of four processes: 1) cellular survival and proliferation (eg, TP53, EGFR, MET, and PIK3CA); 2) cell-cycle control (eg, CDKN2A and CCND1); 3) cellular differentiation (eg, NOTCH1); and 4) adhesion and invasion signaling (eg, FAT1).7 TP53, EGFR, PIK3CA, CDKN2A, CCND1, and MET participate in several common signaling pathways (Figure 1). Alterations of these genes are most frequently seen in alcohol and tobacco-related HNSCC.12 Genetic aberration interactions involve pathway reactivation (downstream re-engagement of original effectors), pathway bypass (recruitment of a parallel pathway converging on the same downstream output), and pathway indifference (development of a cellular state independent of the initial therapeutic target). Gene–drug interactions are analyzed in cBioPortal. Data were from TCGA Head and Neck and Recurrent and Metastatic Head & Neck Cancer (Figure 2).3
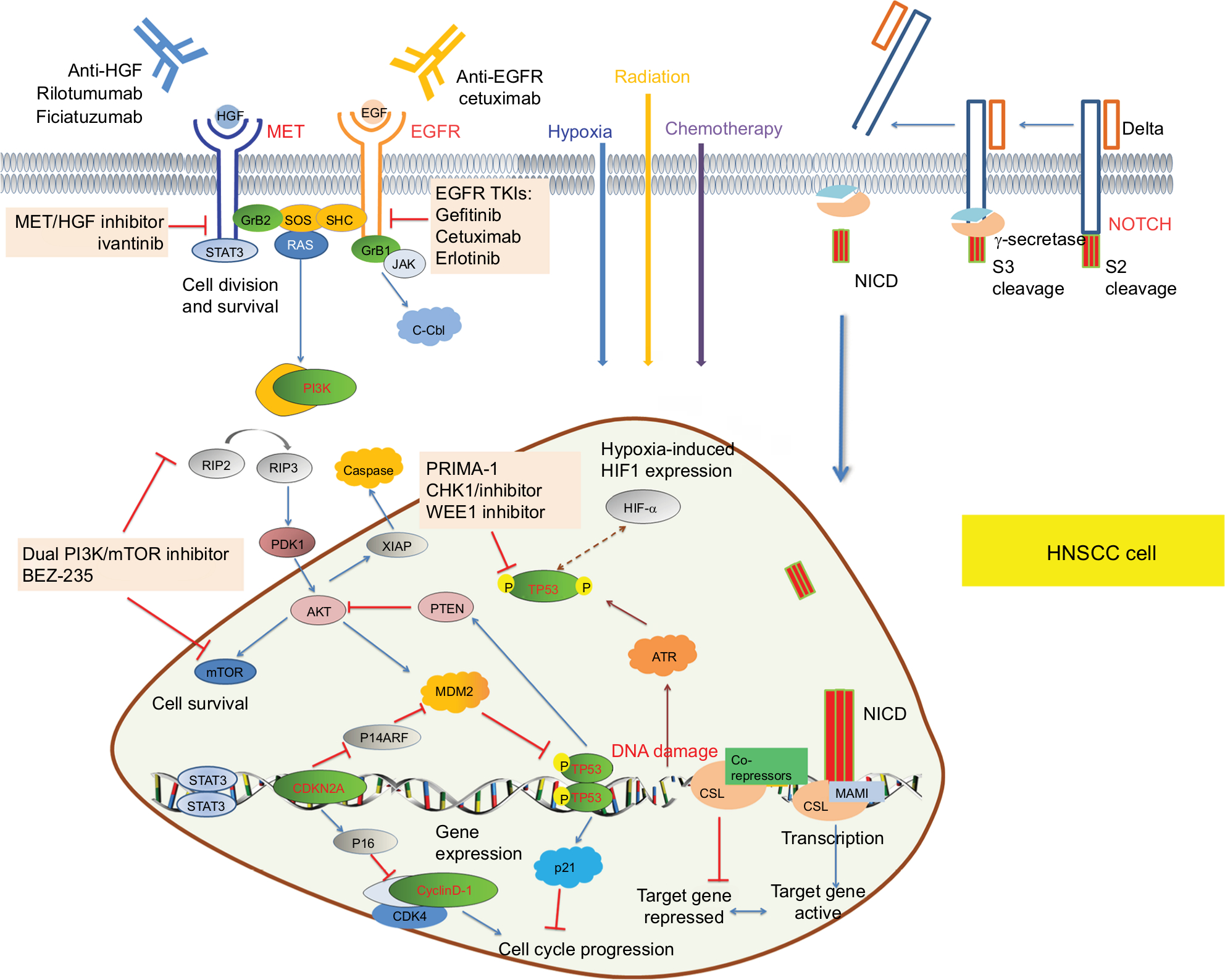 | Figure 1 Schematic diagram of signaling pathways that emerging biomarkers TP53, EGFR, PIK3CA, CDKN2A, CCND1, and MET participated in. Notes: EGFR is a member of the HER family of cell-surface receptor tyrosine kinases. Ligand binding triggers EGFR and activates downstream effectors, thus promoting cell proliferation. Hypoxia induces the expression of EGFR and enhances the phosphorylation of EGFR thereby regulating intrinsic DNA-repair mechanisms, meanwhile EGFR can stabilize HIF-1a. MET is a receptor tyrosine kinase associated with enhanced migration, invasion, and angiogenesis when overexpressed in cancer. PIK3CA encodes p110α, a catalytic subunit of PI3K, which is a heterodimeric kinase with enzymatic activity on lipid and protein substrates. The TP53 has a vital role in the regulation of genes responsible for cell-cycle arrest, senescence, and apoptosis. MDM2 promotes the rapid degradation of the TP53 protein. TP53 missense mutations cause single-amino-acid substitutions that lead to loss of DNA-binding capability. Cyclin D1 is a cell-cycle protein that regulates the key G1-to-S phase transition through formation of complexes with CDKs, such as CDK4 and CDK6. Upon ligand binding, NOTCH receptors undergo a conformational change enabling cleavage and nuclear translocation of the intracellular domain to release the transcriptional repression of downstream target genes. Abbreviations: CDKs, cyclin-dependent kinases; HIF-1a, hypoxia-inducible factor 1a; PI3K, phosphoinositide 3 kinase. |
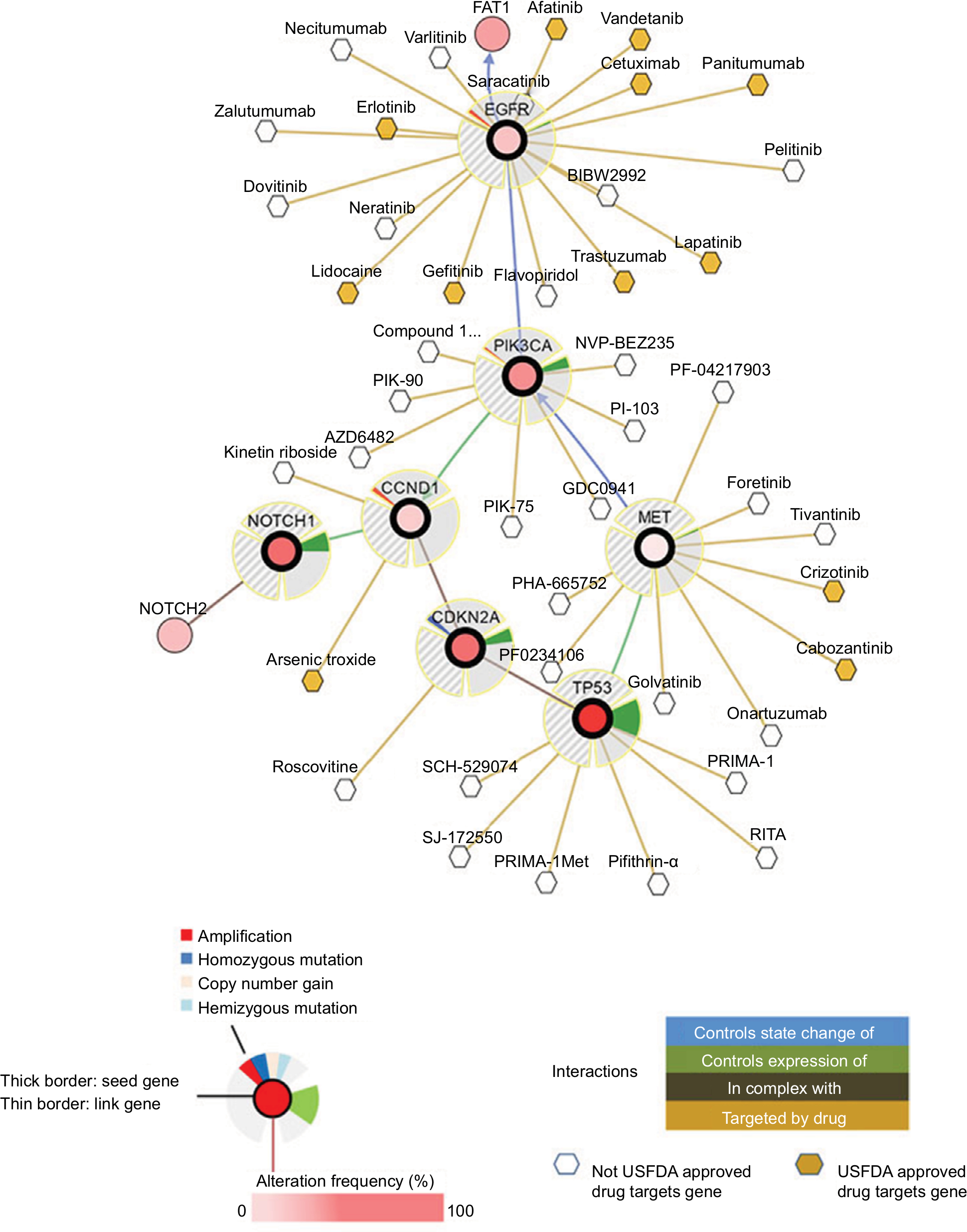 | Figure 2 General interactions of genomic mutations (TP53, CDKN2A, CCND1, NOTCH1, PIK3CA, EGFR, and MET) and current target drugs. Notes: Primary data are from tumor samples with sequencing and CNA data (132 patients/samples) and could be accessed in cBioPortal.6 Detailed description of data mining is (filter neighbors by max 28.8% alteration) found in http://www.cbioportal.org/results/network?Action=Submit&RPPA_SCORE_THRESHOLD=2&Z_SCORE_THRESHOLD=2&cancer_study_list=hnc_mskcc_2016&case_set_id=hnc_mskcc_2016_cnaseq&data_priority=0&gene_list=TP53%2520CDKN2A%2520CCND1%2520NOTCH1%2520PIK3CA%2520EGFR%2520MET&geneset_list=%20&genetic_profile_ids_PROFILE_COPY_NUMBER_ALTERATION=hnc_mskcc_2016_gistic&genetic_profile_ids_PROFILE_MUTATION_EXTENDED=hnc_mskcc_2016_mutations&tab_index=tab_visualize.134,135 Abbreviations: US FDA, US Food and Drug Administration. |
TP53
TP53 mutation
Mutations in the tumor suppressor P53(TP53) gene are present in about 70% of HNSCC.13 Missense mutations in TP53, including those at codons R248, R273, G245, R175, R282, and H179, are the most frequent hotspot mutations in HNSCC.7 Two thousand four hundred ninety-eight samples in seven studies (Head and Neck Squamous Cell Carcinoma [Broad, Science 2011]; Head and Neck Squamous Cell Carcinoma [Johns Hopkins, Science 2011]; Head and Neck Squamous Cell Carcinoma [TCGA, Nature 2015]; Head and Neck Squamous Cell Carcinoma [TCGA, PanCancer Atlas]; Head and Neck Squamous Cell Carcinoma [TCGA, Provisional]; Oral Squamous Cell Carcinoma [MD Anderson, Cancer Discov 2013])4,14,15 showed 62.7% of somatic mutation and 45.2% of missense mutations (http://www.cbioportal.org/). TP53 mutation was markedly higher in metastatic HNSCC.6 TP53 mutation involves varieties of proteins that contribute to tumorigenesis and tumor progression.4,16 It occurs early in carcinoma progression and more frequently in those with greater histologic severity.17,18
Data TCGA Head and Neck and Recurrent and Metastatic Head & Neck Cancer (MSKCC, JAMA Oncol 2016) analyzed by cBioPortal (detailed description of data mining could be found in the figure legends) demonstrated that only TP53 mutation is a predictor for overall survival (OS) rate and disease-free survival rate (Figure 4). Moreover, tumors of the larynx and hypopharynx have the highest TP53 mutation rate (83.5%). Tumors of the tongue and oral cavity have a TP53 mutation rate of 75.6%, and those of the oropharynx (including the tonsils), and base of the tongue have the lowest TP53 mutation rate (28.6%).13 Previous studies have shown that TP53 mutation correlated with resistance to chemotherapy drugs such as cisplatin, doxorubicin, and paclitaxel.19–22 It has been recently further demonstrated that cisplatin resistance was associated with aneuploidy of chromosome 17, increased TP53 copy numbers, and overexpression of mutant variant R248L.21
TP53 mutation has also been indicated for assessment of postoperative radiotherapy.23 If there are no histologically detectable tumors and no TP53 mutations in the surgical margin, patients can be spared postoperative radiotherapy. It is supported by the studies that show the absence of TP53-mutated DNA in surgical margins was significantly associated with local recurrence-free survival.24,25
Classification of TP53 mutation
The value of TP53 mutation in diagnosis is different between subtypes. Some are associated with more aggressive HNSCC phenotypes, whereas others are linked with a more indolent pattern of tumor progression.26
Perrone et al categorized TP53 mutation significance based on the transactivation activity as functional, partially functional, or nonfunctional.27 Loss of function of TP53 (transactivation activities) predicts a significantly low rate of pathologic complete remission and suboptimal response to cisplatin-based neoadjuvant chemotherapy in patients with oral squamous cell carcinoma (OSCC).28 Accumulating evidence suggests that gain of function (GOF) of TP53 mutants as well mediates drug resistance. The underlying mechanisms include apoptotic proteins inhibition and gene regulations.29,30
Another large study classified TP53 mutation as disruptive and nondisruptive, based on alteration of DNA binding.31 The disruptive is defined as any mutation in L2 or L3 loop of the DNA-binding domain or stop codon, resulting in a polarity change within the protein. Disruptive TP53 mutation strongly predicted locoregional recurrence driven by tumor cell radioresistance. The radioresistance is measured by SA-β-gal staining, p21 expression, and release of ROS.31
Evolutionary action (EATP53), a novel computational approach, has been applied to stratify tumor patients with TP53 mutation as high- or low risk. This system was validated both in vivo and in vitro32 (available at http://mammoth.bcm.tmc.edu/EATP53). High-risk mutations promote invasion, metastasis, as well as cisplatin resistance in head and neck cancer cell lines, as they acquired oncogenic GOF properties, while low-risk mutations retained wild-type (WT) TP53 activity.32,33 Different effect of TP53 mutation on cisplatin response has been observed in vitro and in vivo. Mice with HNSCC harboring WT or low-risk mutations responded well to cisplatin treatment. Quite the opposite, TP53 null type or high-risk TP53 mutations failed to show any growth inhibition with cisplatin therapy.34 Similar results were seen in patients with those characters. The high-risk TP53 mutations were associated with decreased OS.32
Targeting TP53 mutation and other coexisting alterations to induce synthetic lethality
Researchers have explored introduction of exogenous WT TP53 into HNSCC cells, or reactivation of some level of WT function in mutant p53-bearing cells, otherwise promotion of mutant TP53 degradation. Of all the compounds that restore WT activity, the only drug directly targeting mutant TP53 that has reached the clinical stage is PRIMA-1(met)/APR-246.35–37 It also induced apoptosis and enhanced the cytotoxicity of standard chemotherapy in HNSCC cells.
In addition to targeting mutant p53 directly, investigators have used strategies targeting mutant TP53-regulated downstream targets and signaling pathways. Mutant TP53 proteins are believed to achieve GOF activity by interacting with other molecular alterations that coexist in HNSCC. Understanding the impact of TP53 mutation on cellular growth and survival signaling pathways can help design more effective therapeutic strategies that target TP53 mutation-bearing HNSCC.
CDKN2A deletions
TP53 and CDKN2A are involved in cell cycle.33 A genomic analysis of human HNSCCs detected that CDKN2A suppresses the oncogenic function of TP53 mutation that promotes malignant progression, and the prognostic value of mutant p53 needs to be considered in the context of CDKN2A.26 The survival of patients with HNSCCs bearing co-occurring high-risk TP53 mutation and CDKN2A homozygous deletions was extremely lower than that of patients with tumors in which high-risk TP53 mutation did not contain CDKN2A homozygous deletions, or that of patients with tumors in which homozygous CDKN2A deletions coexisted with either low-risk TP53 mutation or potential loss-of-function mutations in TP53 (high- and low risk according to the EATP53 classification system).32
PIK3CA activation
Preliminary findings from TCGA Head and Neck Cancer have detected amplifications or putative activating mutations in PIK3CA in 30% of 279 HNSCC tumors, which overlap with WT and mutant TP53 subsets.38 The aforementioned studies indicate that increase in phosphoinositide 3-kinase (PI3K)/Akt activation has been linked to frequent alterations in TP53 via multiple mechanisms.10,38–40 Comparing half-maximal inhibitory concentration in a wider panel of HNSCC cell lines suggests that TP53 status contributes to the sensitivity of PI3K/mTOR inhibitor.
In human xenograft, PI3K/mTOR inhibition has been proven to enhance WT TP53 expression, enhance the TP53 DNA-damage response, and further delay tumor regrowth with radiation;38 thus, the combination of PI3K/TP53 events seems to be necessary for PI3K inhibitor sensitivity. The dual PI3K/PLK inhibitor rigosertib has such potent antiproliferative effects on patient tumor xenografts, which carried combination of a PIK3CA-activating event driven by amplification or mutation and a TP53-inactivating event driven by either human papillomavirus (HPV)-negative or nonsense TP53 mutation.41
CDKN2A
CDKN2A mutations
Global genomic analyses identified the tumor suppressor CDKN2A as the second most commonly altered gene in HNSCC.13 CDKN2A (also known as P16, INK4, p16INK4A, and MTS1) is allelic to chromosome 9p21 and encodes a CDK4/CDK6 kinase inhibitor that constrains cells from progressing through the G1 restriction point. It is thought to be involved in early stages of HNSCC development. It is affected in up to 80% of HNSCC – often deleted, hypermethylated, or, much rarely, mutated.42 CDKN2A mutation is considered as “noncoding mutations”, “inactivation”, or “loss of function”.43 It is associated with worse oversurvival in patients with recurrent and metastatic HNSCC (MSKCC, JAMA Oncol 2016) (Figure 4).13 Most of the mutations in CDKN2A were found in exon 2 of the gene.42 However, these are likely insufficient to drive tumorigenesis by CDKN2A mutations themselves.44 For example, CDKN2A mutations in benign epithelial lesions have low potential to transform into malignancy.44 Therapeutic targeting of CDKN2A presents the challenge of restoring tumor suppressor activity or inhibiting downstream targets that have been rendered overactive.
CDKN2A mutation reflects p16INK4a function
p16INK4a immunohistochemical expression has been proposed as a surrogate marker for viral oncogene activity in HNSCC.45 The overexpression of p16INK4a is an outstanding surrogate for HPV positivity extensively documented in OSCC.46,47 Numerous studies have reported a considerably improved survival (death risk reduced by 40%–80%) or lower locoregional recurrences (reduced by 60%–70%) and better response to chemo- and radiotherapy in HPV-positive patients with p16INK4a expression compared to HPV-negative patients, when treated with standard multimodality treatments.48–53
HPV-positive HNSCC rarely has CDKN2A mutation and thus has the p16INK4a function;26,54 those patients are significantly less likely to develop a locoregional recurrence.43 On the other hand, up to 90% of HPV-negative HNSCCs have loss of p16INK4a function as a result of CDKN2A mutation, promoter methylation, or gene/chromosome 9p21 deletion.45 But the recent study found that CDKN2A mutation still reflects the p16INK4a expression in HPV-negative HNSCC. Loss of CDKN2A function as a result of CDKN2A mutation induced p16INK4a mRNA transcription strongly.45 Hence, p16INK4a overexpression is associated with CDKN2A mutation in a subset of cases. In those cases, the value of p16INK4a for inducing HPV infection was not reliable.
CCND1
CCND1 amplification
CCND1 encodes cyclin D1 on chromosome 11q13, and regulates the key G1-to-S phase transition through formation of complexes with cyclin-dependent kinases (CDKs), such as CDK4 and CDK6.55 Mutations, amplification, and overexpression of this gene, which alters cell-cycle progression, are observed frequently in a variety of tumors and may contribute to tumorigenesis. CCND1 is amplified in as many as 30%–40% of head and neck cancers with cyclin D1 overexpression.56,57 Comparative studies have either shown no prognostic advantage of protein expression over gene amplification or have favored cyclin D1 gene amplification as a better marker of poor prognosis. CCND1 amplification had a statistically significant association with recurrence, distant metastasis, platinum and cisplatin resistance, and EGFR-inhibitor resistance.58–61 Allele A may be a risk factor for upper aerodigestive tract cancer, especially in nonalcoholics. Patients of recurrent and metastatic head and neck cancer with CCND1 alterations have poor OS rate (Figure 4). CCND1 may be a valuable marker for predicting regional response to radiotherapy in metastatic HNSCC and might assist when deciding on appropriate primary therapy.62 Kinetin riboside, an inhibitor of CCND2 transactivation, rapidly suppressed cyclin D1,63 which is a promising CCND1 inhibitor.
CCND1 amplification confers to EGFR inhibitor resistance
Maintenance of cyclin D1 levels is critical to the resistance HNSCC cells display to gefitinib.54 Sensitivity of the six HNSCC cell lines (FaDu, Detroit 562, SCC 9, SCC 15, SCC 25, and CAL 27) to gefitinib was related to cyclin D1 overexpression. These cell lines that overexpressed cyclin D1 continued to proliferate when treated with up to lethal level of gefitinib, but cell numbers remained static or decreased after gefitinib treatment in the non-overexpressed cyclin D1 cell lines.65 Furthermore, the most resistant cell line displayed the highest level of CCND1 amplification and cyclin D1 overexpression. Cyclin D1 upregulation also combines with cortactin overexpression to promote resistance to gefitinib.66 Cortactin overexpression attenuated ligand-induced downregulation of EGFR, leading to sustained signaling.66
A study in vitro explored the relationship between deregulated cyclin D1 expression and sensitivity to gefitinib to determine whether this frequently occurring oncogenic change affected the cellular response to gefitinib. Three of six cell lines displayed cyclin D1 amplification and/or overexpression, and these cell lines were resistant to gefitinib.65 No preclinical studies have yet addressed whether cyclin D1 expression is maintained in CCND1-amplified cancers after EGFR inhibition, and this is an issue warranting investigation.
NOTCH1
NOTCH1 mutations
The NOTCH pathway consists of four receptors (NOTCH1–4) and two families of ligands: the Jagged (JAG1 and JAG2) and the Delta-like (DLL1, DLL3, and DLL4) proteins. NOTCH1 is a transmembrane receptor and controls cell differentiation and embryonic development. Upon ligand binding, functions of NOTCH1 in oral SCC are considered tumor-suppressive. In tongue cancer xenografts, the presence of NOTCH1 caused a dramatic tumor reduction compared to tumors in which this gene was absent.67 Higher expression and activity of NOTCH1 have previously been associated with radio- and chemoresistance.68
Point mutations affecting this gene occurred in 11% of the HNSCC tumors.13 It is considered to represent “loss of function”.13,69,70 Missense mutations in NOTCH1 are considered tumor-suppressive in OSCC. Notably, OSCC patients with NOTCH pathway mutations (AR, ARNT, EP300, CREBBP, JAK2, JAK, NOTCH1, NOTCH2, NOTCH3, and PARP1) were 3.3 times more likely to die with recurrent disease compared to those who did not have these alterations.71 The NOTCH1 mutation status recently served as biomarker for the identification of HNSCC with higher sensitivity to radio- and chemotherapy, as significant association of NOTCH1 mutations with improved localregional control and increased overall survival was observed after chemoradiotherapy.70 Using of gamma-secretase inhibitors supposed to be an attractive strategy for improvement of chemoradiotherapy in malignancies harboring wild-type NOTCH1. For HNSCC it is should be carefully evaluated given evidence of an increased frequency of squamous cell carcinoma in vivo with genetic reductional γ-secretase activity.133
Analysis of recurrent and metastatic head and neck cancer suggests a similar trend toward better 5-year (60 months) OS in patients with alterations in NOTCH1, but after 5 years the OS in patients with alterations was worse; however, this was not statistically significant (Figure 4).
Activated NOTCH1 contributes to resistance of PI3K inhibitors
Active domain of NOTCH1 conferred resistance to the dual PI3K/mTOR inhibitor BEZ-235 in breast adenocarcinoma-like cell line MCF7 and the ductal carcinoma-like cell lines BT474, HCC70, and BT549.10 They showed resistance to BEZ-235 treatment upon expression of ICN1 (intracellular active domain of NOTCH1).72 Furthermore, in NOTCH1 mutant breast cells that lack the extracellular domain, BEZ-235 sensitivity could also be restored by inhibiting γ-secretase, indicating that naturally cleaved NOTCH1 also confers resistance to PI3K inhibition. A significant correlation was detected between low expression of negative regulator of NOTCH (NUMB) and resistance to PI3K/mTOR inhibition in cell lines derived from various tumor types, including melanoma and hepatocellular carcinoma. These results suggest that uncoupling proliferation from the PI3K pathway via NOTCH1 activation may be a general phenomenon across cancer cell lines.
PIK3CA
PIK3CA mutations
The PI3K pathway (PIK3CA, PTEN, AKT, and STK11) was the most frequently somatically mutated oncogenic pathway in HNSCC tumors.73–75
PIK3CA alterations encoding the key catalytic subunit of PI3K pathway were subsequently found to be prevalent. Data from the querying 2,498 samples showed PIK3CA alterations around 29%, including copy number gain (high polysomy or amplification) or mutations13 (Figure 3).4,14,15,67 However, PIK3CA alteration has no significant prognoses value for recurrent and metastatic head and neck cancer analyzed in cBioPortal (Figure 4).
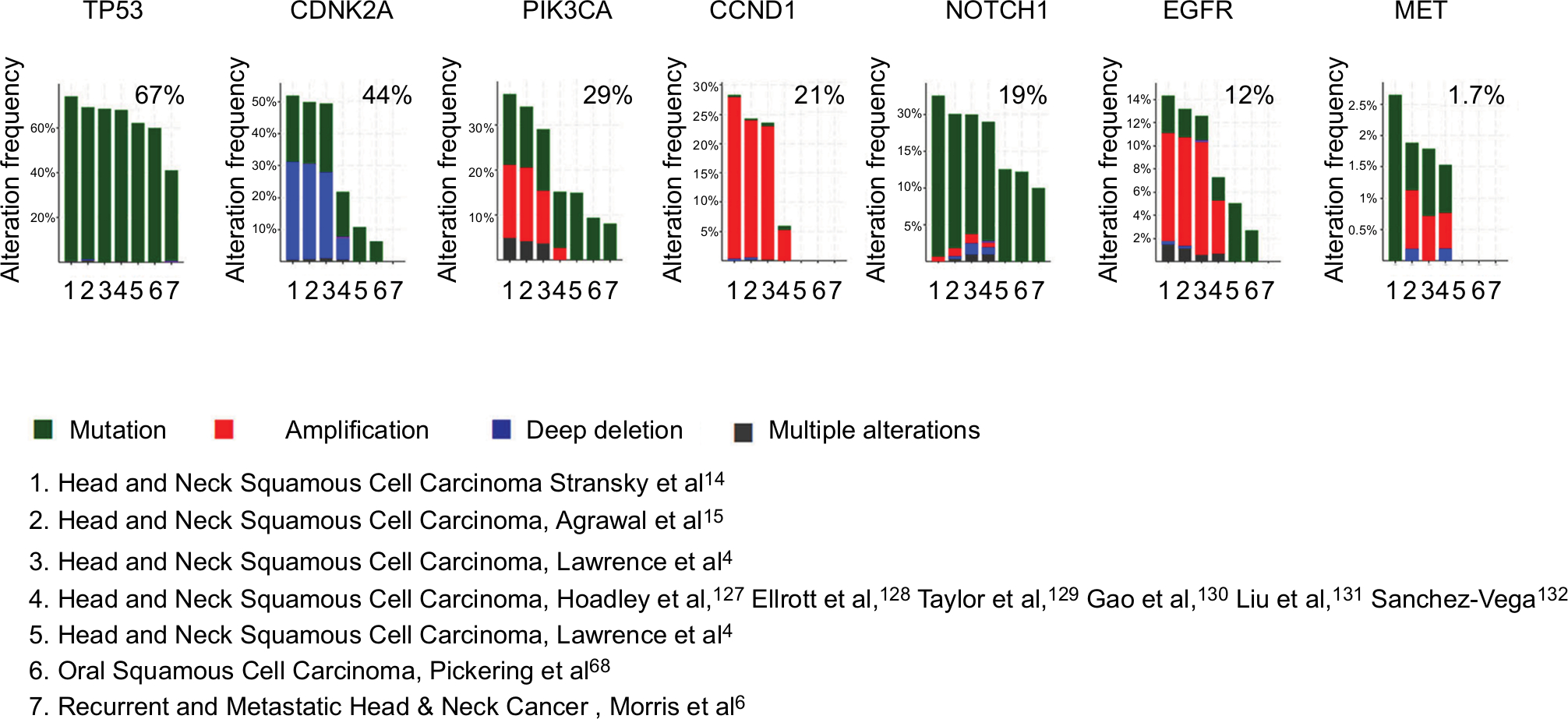 | Figure 3 Alteration profiles of the emerging biomarkers from querying 1,627 patients/1,629 samples in 7 studies analyzed in cBioPortal. Notes: Data from 11,000 cases and all TCGA tumor types from TCGA. Detailed description for data mining could be found in http://www.cbioportal.org/results/cancerTypesSummary?Action=Submit&RPPA_SCORE_THRESHOLD=2&Z_SCORE_THRESHOLD=2&cancer_study_list=hnc_mskcc_2016%2Chnsc_broad%2Chnsc_jhu%2Chnsc_tcga_pub%2Chnsc_tcga_pan_can_atlas_2018%2Chnsc_tcga%2Chnsc_mdanderson_2013&case_set_id=all&data_priority=0&gene_list=TP53%2520CDKN2A%2520CCND1%2520NOTCH1%2520PIK3CA%2520EGFR%2520MET&geneset_list=%20&tab_index=tab_visualize.134,135 Abbreviation: TCGA, The Cancer Genome Atlas. |
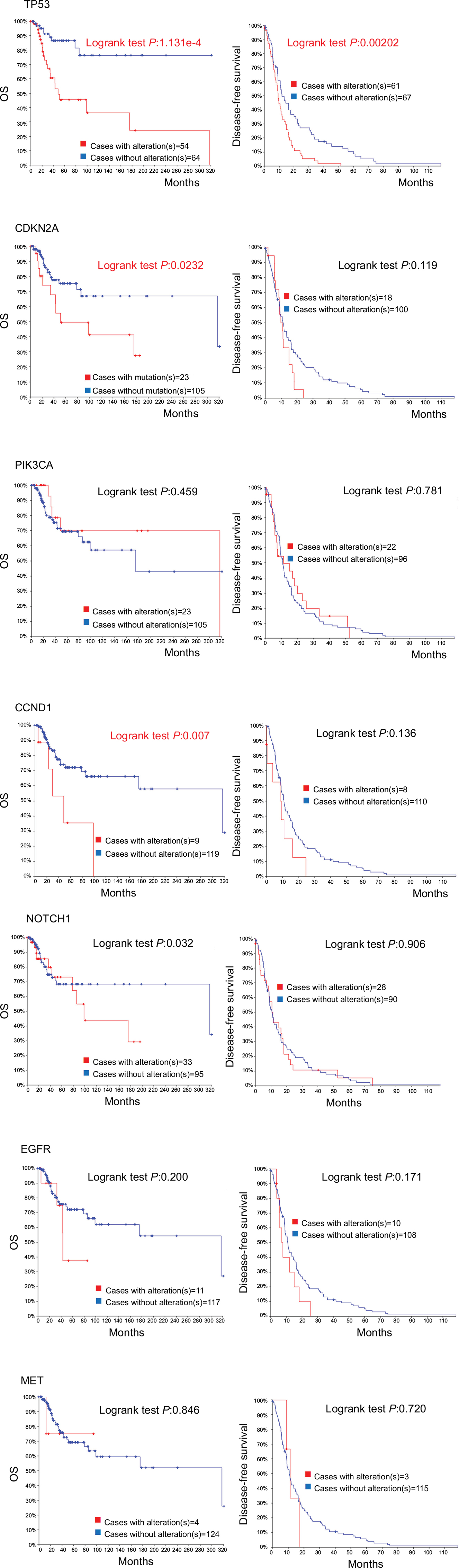 | Figure 4 Recurrent and metastatic head and neck cancer samples with sequencing and CNA data (132 patients/samples) analyzed in cBioPortal. Notes: OS/recurrence-free survival, for cases with/without alteration(s) in query gene. Survival probabilities were calculated with the Kaplan–Meier method, according to the original article.6 Detailed description of data mining could be found in http://www.cbioportal.org/results/survival?Action=Submit&RPPA_SCORE_THRESHOLD=2&Z_SCORE_THRESHOLD=2&cancer_study_list=hnc_mskcc_2016&case_set_id=hnc_mskcc_2016_cnaseq&data_priority=0&gene_list=TP53&geneset_list=%20&genetic_profile_ids_PROFILE_COPY_NUMBER_ALTERATION=hnc_mskcc_2016_gistic&genetic_profile_ids_PROFILE_MUTATION_EXTENDED=hnc_mskcc_2016_mutations&tab_index=tab_visualize.134,135 Abbreviation: OS, overall survival. |
PIK3CA mutations seem to favor advanced HNSCC, which harbor multiple PI3K-pathway mutations.38 Lui et al detected that 100% HNSCC (10/10 cases) with concurrent PI3K mutations were advanced (Stage IV).75 PIK3CA copy number gain (high polysomy or amplification) but not PIK3CA mutation was detected to be associated with significantly lower disease-free survival, resulting from an NGS.77 The most frequent mutations occur in exons 9 and 20, with hotspots at H1047R (eight mutations total), E542K (three mutations), and E545K/G (four mutations).78 H1047R was more potent than E545K at inducing resistance in PI3K pathway activation.10 PIK3CA canonical and novel mutations increase survival. These canonical hotspot mutations showed significantly enhanced HNSCC growth compared to overexpression of WT PIK3CA.74 This observation implies that the PI3K pathway-mutated HNSCC tumors have “oncogenic” advantage even with genomic instability, which can be partly explained by PIK3CA “driver” mutations’ growth-promoting activity.
Although there is not enough evidence associated with oversurvival and recurrence rates, PIK3CA alterations are providing a potential approach to treat a substantial subset of patients in advanced stage.
Targeted PIK3CA activation
There are currently a variety of different inhibitors that are being studied for targeting the PI3K pathway.74,79 PIK3CA activation either caused by mutation or copy number gain is oncogenic and is thus a commonly activating point mutation with targeted agents.73,74 Targeting the PIK3CA activation has emerged as one of the most promising therapeutic targets in HNSCC.75 They are already in clinical development for other cancer types.10
HNSCC cell lines containing hotspots endogenous PIK3CA (H1047R) mutations demonstrated increased sensitivity to PI3K pathway inhibition by the mTOR/PI3K inhibitor BEZ-235 compared to the representative HNSCC cells with WT PIK3CA.80 Furthermore, they were more sensitive to the combination of BEZ-235 plus cetuximab compared to cetuximab alone.80 PIK3CA mutations are more common in HPV-positive HNSCC and are associated with activation of mTOR.4,74,81 Patient-derived HPV-positive OPSCC tumorgrafts in vivo with PIK3CA (E542K) mutation were also sensitive to a dual mTOR/PI3K inhibitor (BEZ-235), in contrast to PIK3CA WT tumorgrafts.76,80 These results suggest that mTOR/PI3K inhibitors may have activity against PIK3CA mutant HNSCC, particularly in HPV-positive HNSCC.
Activated PI3K/Akt confers resistance to MET inhibitors
PIK3CA and MET may play an important role in oncogenesis of certain specific subtypes of head and neck cancer. In several cancers, gene copy numbers of MET and PIK3CA have been found to be prognostic and predictive for therapy response.82 The MET receptor tyrosine kinase represents a promising target in cancer. PIK3CA activating mutations are common in several tumor types and can potentially confer resistance to anti-receptor tyrosine kinase therapy.78 Hotspots E545K and H1047R confer resistance to MET inhibition in MET-driven models. Hence, resistance to MET inhibition could be synergistically overcome by co-targeting PI3K. Combination of PI3K/MET inhibitors led to more-than-additive effects.
Combined MET/PI3K inhibition leads to enhanced antitumor activity in MET-expressing HNSCC harboring endogenous PIK3CA (H1047R).78 PIK3CA mutations can lead to resistance to MET inhibition, supporting future clinical evaluation of combinations of PI3K and MET inhibitors in common scenarios of malignant neoplasms featuring aberrant MET expression and PIK3CA mutations. In conclusion, PI3K signaling pathway was a potential target for treatment optimization across all risk groups.
Activated PI3K/Akt causes resistance of EGFR inhibitors
Activated PI3K/Akt and RAS/MAPK/ERK pathways are associated with resistance to cetuximab-containing chemoradiation (weekly cetuximab, paclitaxel, and carboplatin followed by chemoradiation with the same regimen) in operable stage III/IV HNSCC, implicated by Eastern Cooperative Oncology Group (ECOG) 2303 Phase II trial.82 Inferior OS and/or PFS were observed in patients harboring activated PI3K/Akt and RAS/MAPK/ERK and maintained significance in multivariable analysis.83 Preclinical studies suggest that PIK3CA and RAS mutations may predict intrinsic resistance to cetuximab, which may be prevented by combination of cetuximab and PI3K and/or mTOR inhibitors in HNSCC patients.84
EGFR
EGFR plays an important role in the emergence and progression of epithelial malignancies and is the most frequently activated receptor tyrosine kinase in HNSCC.85 The first molecular targeting approach to demonstrate a survival advantage for HNSCC patients has emerged in the context of EGFR biology. TCGA data have detected EGFR alteration in 14.34% cases including 65.0% with EGFR amplification. High-level EGFR amplifications in ~10% of HNSCC, and polysomy are present in even more.13 Primary somatic mutations in EGFR are uncommon in HNSCC.86 Most of the previous studies found that EGFR overexpression was associated with radioresistance, and worse locoregional and disease-free survival.87–90 However, EGFR alteration also has no significant prognoses value in metastatic head and neck cancer analyzed in cBioPortal (Figure 4). And because there has been no consistently identifiable alteration in the EGFR that correlates to sensitivity to EGFR inhibitors, EGFR analysis has not been incorporated into the pathologic evaluation of HNSCC.85
EGFR amplification is highly associated with resistance to chemotherapy and radiation in HNSCC.22,91 Hypoxia induces radioresistance, directly as DNA damage is maximized in the presence of oxygen and indirectly by promoting genetic instability92 (Figure 1). Hypoxia induces the expression of EGFR and enhances the phosphorylation of EGFR thereby regulating intrinsic DNA-repair mechanisms, meanwhile EGFR can stabilize hypoxia-inducible factor 1a.93,94 These radioresistance mechanisms play a role in HNSCC because EGFR is overexpressed in most of these tumors. That is why EGFR inhibition has subsequently been explored as a potential therapeutic adjunct to radiotherapy in HNSCC.95,96
Targeting EGFR is lack of response evaluation
Anti-EGFR agents are largely divided into two classes of drugs: monoclonal antibodies and tyrosine kinase inhibitors (TKIs). EGFR inhibitors have been used to treat both lung and head and neck cancers with squamous cell histology.97 These tumors often show high EGFR expression and/or increased gene copy number. The efficacy of cetuximab is demonstrated in the recurrent/metastatic (R/M) HNSCC settings of clinical trials: ECOG, Erbitux in First-line Treatment of Recurrent or Metastatic Head and Neck Cancer (EXTREME) trial. Recent developments included US Food and Drug Administration approval of pembrolizumab and cetuximab. Cetuximab has conferred a survival advantage when combined with platinum-based combination chemotherapy in the first-line R/M setting.98 However, only a fraction of HNSCC are sensitive to the cetuximab, 5-year survival of HNSCC is not improved by the cetuximab, which is currently <40%.82,99
The use of EGFR as a biomarker for response to EGFR inhibitor is limited by the semiquantitative nature of and subjectivity of immunohistochemical scoring.100–102 It might be due to inaccurate assays as well as the nature of increased gene copy number being a less predictive biomarker compared to activating mutations.55 Several studies explored the biology, genomics, and patterns of response to EGFR inhibitors to inform identification of potential biomarkers that have shown promise in preclinical studies and clinical trials.85,97,101,103–105 But no biomarkers are known to predict response to the treatment.
Vermorken et al demonstrated the superiority of incorporating cetuximab, in combination with cisplatin/carboplatin and fluorouracil in patients with recurrent/metastatic HNSCC.97 Genomic studies suggest that EGFR alteration is more common in HPV-negative tumors compared to HPV-positive tumors.74 Clinical data further contradict the HPV positivity as a biomarker for EGFR-inhibitor resistance.97 Development of EGFR inhibitors could be more relevant in treating HPV-negative tumors.81,106,107
MET
MET alterations as oncogenic driver of HNSCC
Hepatocyte growth factor (HGF) receptor (MET) is functionally novel in HNSCC with prominent overexpression, and increased copy number and mutations.95,108 It regulates cancer cell plasticity through reversible reprogramming of epithelial–mesenchymal transition (EMT).109,110 It is a process considered a critical step in the progression of HNSCC. Accumulating evidence suggests that EMT and its ligand HGF favor the distant dissemination of single carcinoma cells from the site of the primary tumor.109,111
MET/HGF pathway activation driven by MET overexpression correlated with worse outcome in R/M HNSCC patients for progression-free survival and OS.112 Although MET alterations are relatively low from querying 2,498 samples in 7 studies in TCGA head and neck carcinoma (1.3%) (Figure 3), MET is proposed as an escape mechanism mediating drug resistance.109,110,113 MET copy number gain or somatic mutations drive metastatic spread of HNSCC108,114 and are significantly associated with shorter disease-specific survival.99 Therefore, MET may be a substantial ontogenetic driver of certain specific subtypes of HNSCC, which should be further explored.
Hence, multiple clinical scenarios targeting the MET in HNSCC have been undertaken because of significant preclinical work demonstrating a relationship between MET/HGF signaling and cancer cell survival (eg, tivantinib, cabozantinib, and crizotinib).114 HGF/c-MET pathway mediates vascular endothelial growth factor receptor (VEGFR) inhibitor resistance and vascular remodeling in non-small-cell lung cancer (NSCLC). Forced HGF expression in NSCLC reduced tumor sensitivity to VEGFR TKIs and produced tumors with increased blood vessels. Dual VEGFR/c-MET signaling inhibition could prevent the vascular morphology alterations.115 These preclinical results indicated that the level of MET expression is a determinant of outcome.112
MET mutations associated with resistance to EGFR inhibitor
The cross-talk of MET and its ligand with EGFR, such as coactivation of alternative kinases, may contribute to EGFR inhibitor resistance.82,112 For example, MET mutations are present in up to 20% of patients with resistance to EGFR inhibitors, like cetuximab.116 In head and neck cancer cell lines MET was shown to mediate resistance to dasatinib and erlotinib.117,118 HGF/MET pathway activation is associated with poorer outcome in R/M HNSCC patients. Therefore, patients treated with EGFR inhibitors have a worse prognosis.119,120 Experimental studies in vitro and in vivo of R/M HNSCC documented a synthetic lethal effect of dual blockade using c-MET (SU11274, PF2341066, and PF04217903) and EGFR TKIs (cetuximab, erlotinib, and gefitinib).118,121,122
Conclusion
Precision medicine in oncology seeks to match each patient with the most effective and appropriate targeted therapies.6 NGS data-guided therapy is promising, in revealing novel molecular alterations, which might be used as drug targets.6
Since the last decade, concepts of cross-talk, co-occurrence, and interactions have been intensively introduced into biochemical research. Various etiologic factors are implicated in the genesis of distinct molecular subsets of HNSCC. Apparently, signaling pathways and transcription factors are not regulated by mutations of a specific gene. Multiple gene mutations, along with the corresponding protein dysfunction or molecular pathway dysregulations, are causative in the process of carcinogenesis or drug resistance.
HPV-positive and -negative HNSCCs are quite distinct clinically and biologically. However, they both can have deregulations within the PI3K pathway.123,124,81 Our review paid less attention on HPV-positive HNSCC, because studies of the HPV-positive HNSCC are scarce. The TCGA cohort comprised 85% HPV-negative HNSCC.3 HPV-negative HNSCC featured novel focal deletions in tumor suppressor genes (eg, NOTCH1 and CDKN2A).13 Also, mutations of TP53, CDKN2A, PIK3CA, and NOTCH genes are enriched. Combination therapy is required. The major challenge is to identify and make good use of the novel genetic interactions, and develop more accurate model systems and innovative clinical trials.
Acknowledgments
We sincerely thank Prof Dr Weidong Han for his suggestions on the theme and language modification.
This study is sponsored by grants from Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (grant no. 2019336033); Zhejiang Provincial Natural Science Foundation of China (grant no. LY15H160028); Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (grant no. 2014KYB129).
Disclosure
The authors report no conflicts of interests in this work.
References
Brauswetter D, Dános K, Gurbi B, et al. Copy number gain of PIK3CA and Met is associated with poor prognosis in head and neck squamous cell carcinoma. Virchows Arch. 2016;468(5):579–587. | ||
Walter V, Yin X, Wilkerson MD, et al. Molecular subtypes in head and neck cancer exhibit distinct patterns of chromosomal gain and loss of canonical cancer genes. PLoS One. 2013;8(2):e56823. | ||
[No authors listed] TCGA releases head and neck cancer data. Cancer Discov. 2015;5(4):340–341. | ||
Lawrence MS, Sougnez C, Lichtenstein L; Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–582. | ||
Hyman DM, Solit DB, Arcila ME, et al. Precision medicine at Memorial Sloan Kettering Cancer Center: clinical next-generation sequencing enabling next-generation targeted therapy trials. Drug Discov Today. 2015;20(12):1422–1428. | ||
Morris LG, Chandramohan R, West L. The molecular landscape of recurrent and metastatic head and neck cancers: insights from a precision oncology sequencing platform. JAMA Oncol. 2017;3(2):244–255. | ||
Zhou G, Liu Z, Myers JN. TP53 mutations in head and neck squamous cell carcinoma and their impact on disease progression and treatment response. J Cell Biochem. 2016;117(12):2682–2692. | ||
Weiss J, Hayes DN. Classifying squamous cell carcinoma of the head and neck: prognosis, prediction and implications for therapy. Expert Rev Anticancer Ther. 2014;14(2):229–236. | ||
Konieczkowski DJ, Johannessen CM, Garraway LA. A convergence-based framework for cancer drug resistance. Cancer Cell. 2018;33(5):801–815. | ||
Muellner MK, Uras IZ, Gapp BV, et al. A chemical-genetic screen reveals a mechanism of resistance to PI3K inhibitors in cancer. Nat Chem Biol. 2011;7(11):787–793. | ||
Zhou G, Liu Z, Myers JN. TP53 mutations in head and neck squamous cell carcinoma and their impact on disease progression and treatment response. J Cell Biochem. 2016;117(12):2682–2692. | ||
Perri F, Ionna F, Scarpati G. Translational research: a future strategy for managing squamous cell carcinoma of the head and neck? Anticancer Agents Med Chem. 2018; 18(9):1220–1227. | ||
Cancer Genome Atlas Network. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–582. | ||
Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. | ||
Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in Notch1. Science. 2011;333(6046):1154–1157. | ||
Omura G, Ando M, Ebihara Y, et al. The prognostic value of TP53 mutations in hypopharyngeal squamous cell carcinoma. BMC Cancer. 2017;17(1):898. | ||
Shahnavaz SA, Regezi JA, Bradley G, Dubé ID, Jordan RC. P53 gene mutations in sequential oral epithelial dysplasias and squamous cell carcinomas. J Pathol. 2000;190(4):417–422. | ||
Manterola L, Aguirre P, Larrea E, et al. Mutational profiling can identify laryngeal dysplasia at risk of progression to invasive carcinoma. Sci Rep. 2018;8(1):6613. | ||
Zhao D, Tahaney WM, Mazumdar A, Savage MI, Brown PH. Molecularly targeted therapies for p53-mutant cancers. Cell Mol Life Sci. 2017;74(22):4171–4187. | ||
Ock CY, Son B, Keam B, et al. Identification of genomic mutations associated with clinical outcomes of induction chemotherapy in patients with head and neck squamous cell carcinoma. J Cancer Res Clin Oncol. 2016;142(4):873–883. | ||
Niehr F, Eder T, Pilz T, et al. Multilayered OMICs-based analysis of a head and neck cancer model of cisplatin resistance reveals intratumoral heterogeneity and treatment-induced clonal selection. Clin Cancer Res. 2018;24(1):158–168. | ||
Gadhikar MA, Sciuto MR, Alves MV, et al. Chk1/2 inhibition overcomes the cisplatin resistance of head and neck cancer cells secondary to the loss of functional p53. Mol Cancer Ther. 2013;12(9):1860–1873. | ||
Strojan P, Karner K, Smid L, et al. Concomitant chemoradiotherapy with mitomycin C and cisplatin in advanced unresectable carcinoma of the head and neck: phase I-II clinical study. Int J Radiat Oncol Biol Phys. 2008;72(2):365–372. | ||
van Houten VM, Leemans CR, Kummer JA, et al. Molecular diagnosis of surgical margins and local recurrence in head and neck cancer patients: a prospective study. Clin Cancer Res. 2004;10(11):3614–3620. | ||
van Houten VM, Leemans CR, Kummer JA, et al. Molecular diagnosis of surgical margins and local recurrence in head and neck cancer patients: a prospective study. Clin Cancer Res. 2004;10(11):3614–3620. | ||
Li Z, Gonzalez CL, Wang B, et al. Cdkn2a suppresses metastasis in squamous cell carcinomas induced by the gain-of-function mutant p53(R172H). J Pathol. 2016;240(2):224–234. | ||
Zhou G, Liu Z, Myers JN. TP53 mutations in head and neck squamous cell carcinoma and their impact on disease progression and treatment response. J Cell Biochem. 2016;117(12):2682–2692. | ||
Perrone F, Bossi P, Cortelazzi B, et al. TP53 mutations and pathologic complete response to neoadjuvant cisplatin and fluorouracil chemotherapy in resected oral cavity squamous cell carcinoma. J Clin Oncol. 2010;28(5):761–766. | ||
Hanel W, Marchenko N, Xu S, Yu SX, Weng W, Moll U. Two hot spot mutant p53 mouse models display differential gain of function in tumorigenesis. Cell Death Differ. 2013;20(7):898–909. | ||
Song H, Hollstein M, Xu Y. P53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol. 2007;9(5):573–580. | ||
Skinner HD, Sandulache VC, Ow TJ, et al. TP53 disruptive mutations lead to head and neck cancer treatment failure through inhibition of radiation-induced senescence. Clin Cancer Res. 2012;18(1):290–300. | ||
Neskey DM, Osman AA, Ow TJ, et al. Evolutionary action score of TP53 identifies high-risk mutations associated with decreased survival and increased distant metastases in head and neck cancer. Cancer Res. 2015;75(7):1527–1536. | ||
Osman AA, Monroe MM, Ortega Alves MV, et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Mol Cancer Ther. 2015;14(2):608–619. | ||
Osman AA, Neskey DM, Katsonis P, et al. Evolutionary action score of TP53 coding variants is predictive of platinum response in head and neck cancer patients. Cancer Res. 2015;75(7):1205–1215. | ||
Lambert JM, Moshfegh A, Hainaut P, Wiman KG, Bykov VJ. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene. 2010;29(9):1329–1338. | ||
Rieber M, Strasberg-Rieber M. Hypoxia, Mn-SOD and H(2)O(2) regulate p53 reactivation and PRIMA-1 toxicity irrespective of p53 status in human breast cancer cells. Biochem Pharmacol. 2012;84(12):1563–1570. | ||
Zandi R, Selivanova G, Christensen CL, Gerds TA, Willumsen BM, Poulsen HS. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin Cancer Res. 2011;17(9):2830–2841. | ||
Herzog A, Bian Y, vander Broek R, et al. PI3K/mTOR inhibitor PF-04691502 antitumor activity is enhanced with induction of wild-type TP53 in human xenograft and murine knockout models of head and neck cancer. Clin Cancer Res. 2013;19(14):3808–3819. | ||
Friedman J, Nottingham L, Duggal P, et al. Deficient TP53 expression, function, and cisplatin sensitivity are restored by quinacrine in head and neck cancer. Clin Cancer Res. 2007;13(22 Pt 1):6568–6578. | ||
Singh B, Reddy PG, Goberdhan A, et al. P53 regulates cell survival by inhibiting PIK3CA in squamous cell carcinomas. Genes Dev. 2002;16(8):984–993. | ||
Anderson RT, Keysar SB, Bowles DW, et al. The dual pathway inhibitor rigosertib is effective in direct patient tumor xenografts of head and neck squamous cell carcinomas. Mol Cancer Ther. 2013;12(10):1994–2005. | ||
Todorova TA, Jordanov SH, Stancheva GS, et al. Mutational status of CDKN2A and TP53 genes in laryngeal squamous cell carcinoma. Pathol Oncol Res. 2015;21(2):413–421. | ||
Ben-Dayan MM, Ow TJ, Belbin TJ, et al. Nonpromoter methylation of the CDKN2A gene with active transcription is associated with improved locoregional control in laryngeal squamous cell carcinoma. Cancer Med. 2017;6(2):397–407. | ||
Loyo M, Li RJ, Bettegowda C, et al. Lessons learned from next-generation sequencing in head and neck cancer. Head Neck. 2013;35(3):454–463. | ||
Larque AB, Conde L, Hakim S, et al. P16(INK (4)a) overexpression is associated with CDKN2A mutation and worse prognosis in HPV-negative laryngeal squamous cell carcinomas. Virchows Arch. 2015;466(4):375–382. | ||
Posner MR, Lorch JH, Goloubeva O, et al. Survival and human papillomavirus in Oropharynx cancer in Tax 324: a subset analysis from an international phase III trial. Ann Oncol. 2011;22(5):1071–1077. | ||
Rischin D, Young RJ, Fisher R, et al. Prognostic significance of p16INK4a and human papillomavirus in patients with oropharyngeal cancer treated on TROG 02.02 phase III trial. J Clin Oncol. 2010;28(27):4142–4148. | ||
Chung CH, Parker JS, Karaca G, et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell. 2004;5(5):489–500. | ||
Jou A, Hess J. Epidemiology and molecular biology of head and neck cancer. Oncol Res Treat. 2017;40(6):328–332. | ||
Huang SH, Perez-Ordonez B, Weinreb I, et al. Natural course of distant metastases following radiotherapy or chemoradiotherapy in HPV-related oropharyngeal cancer. Oral Oncol. 2013;49(1):79–85. | ||
Rieke DT, Keilholz U. Systemic treatment in HPV-induced recurrent or metastatic HNSCC. Recent Results Cancer Res. 2017;206:149–160. | ||
López F, Sampedro T, Llorente JL, Hermsen M, Álvarez-Marcos C. Alterations of p14 ARF, p15 INK4b, and p16 INK4a genes in primary laryngeal squamous cell carcinoma. Pathol Oncol Res. 2017;23(1):63–71. | ||
Sinha P, Thorstad WT, Nussenbaum B, et al. Distant metastasis in p16-positive oropharyngeal squamous cell carcinoma: a critical analysis of patterns and outcomes. Oral Oncol. 2014;50(1):45–51. | ||
Todorova TA, Jordanov SH, Stancheva GS, et al. Mutational status of CDKN2A and TP53 genes in laryngeal squamous cell carcinoma. Pathol Oncol Res. 2015;21(2):413–421. | ||
Kang H, Kiess A, Chung CH. Emerging biomarkers in head and neck cancer in the era of genomics. Nat Rev Clin Oncol. 2015;12(1):11–26. | ||
Hunt JL, Barnes L, Lewis JS, et al. Molecular diagnostic alterations in squamous cell carcinoma of the head and neck and potential diagnostic applications. Eur Arch Otorhinolaryngol. 2014;271(2):211–223. | ||
Namazie A, Alavi S, Olopade OI, et al. Cyclin D1 amplification and p16(MTS1/CDK4I) deletion correlate with poor prognosis in head and neck tumors. Laryngoscope. 2002;112(3):472–481. | ||
Namazie A, Alavi S, Olopade OI, et al. Cyclin D1 amplification and p16(MTS1/CDK4I) deletion correlate with poor prognosis in head and neck tumors. Laryngoscope. 2002;112(3):472–481. | ||
Maahs GS, Machado DC, Jeckel-Neto EA, Michaelses VS. Cyclin D1 expression and cervical metastases in squamous cell carcinoma of the mouth. Braz J Otorhinolaryngol. 2007;73(1):87–94. | ||
Zhang P, Zhang Z, Zhou X, Qiu W, Chen F, Chen W. Identification of genes associated with cisplatin resistance in human oral squamous cell carcinoma cell line. BMC Cancer. 2006;6:224. | ||
Noel EE, Yeste-Velasco M, Mao X, et al. The association of CCND1 overexpression and cisplatin resistance in testicular germ cell tumors and other cancers. Am J Pathol. 2010;176(6):2607–2615. | ||
Mielcarek-Kuchta D, Olofsson J, Golusinski W. P53, Ki67 and cyclin D1 as prognosticators of lymph node metastases in laryngeal carcinoma. Eur Arch Otorhinolaryngol. 2003;260(10):549–554. | ||
Tiedemann RE, Mao X, Shi CX, et al. Identification of kinetin riboside as a repressor of CCND1 and CCND2 with preclinical antimyeloma activity. J Clin Invest. 2008;118(5):1750–1764. | ||
Kalish LH, Kwong RA, Cole IE, Gallagher RM, Sutherland RL, Musgrove EA. Deregulated cyclin D1 expression is associated with decreased efficacy of the selective epidermal growth factor receptor tyrosine kinase inhibitor gefitinib in head and neck squamous cell carcinoma cell lines. Clin Cancer Res. 2004;10(22):7764–7774. | ||
Timpson P, Wilson AS, Lehrbach GM, Sutherland RL, Musgrove EA, Daly RJ. Aberrant expression of cortactin in head and neck squamous cell carcinoma cells is associated with enhanced cell proliferation and resistance to the epidermal growth factor receptor inhibitor gefitinib. Cancer Res. 2007;67(19):9304–9314. | ||
Kalish LH, Kwong RA, Cole IE, Gallagher RM, Sutherland RL, Musgrove EA. Deregulated cyclin D1 expression is associated with decreased efficacy of the selective epidermal growth factor receptor tyrosine kinase inhibitor gefitinib in head and neck squamous cell carcinoma cell lines. Clin Cancer Res. 2004;10(22):7764–7774. | ||
Pickering CR, Zhang J, Yoo SY, et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013;3(7):770–781. | ||
Vermezovic J, Adamowicz M, Santarpia L, et al. Notch is a direct negative regulator of the DNA-damage response. Nat Struct Mol Biol. 2015;22(5):417–424. | ||
Tinhofer I, Budach V, Saki M, et al. Targeted next-generation sequencing of locally advanced squamous cell carcinomas of the head and neck reveals druggable targets for improving adjuvant chemoradiation. Eur J Cancer. 2016;57:78–86. | ||
Tinhofer I, Stenzinger A, Eder T, et al. Targeted next-generation sequencing identifies molecular subgroups in squamous cell carcinoma of the head and neck with distinct outcome after concurrent chemoradiation. Ann Oncol. 2016;27(12):2262–2268. | ||
Vettore AL, Ramnarayanan K, Poore G, et al. Mutational landscapes of tongue carcinoma reveal recurrent mutations in genes of therapeutic and prognostic relevance. Genome Med. 2015;7:98. | ||
Cortelazzi B, Verderio P, Ciniselli CM, et al. Receptor tyrosine kinase profiles and human papillomavirus status in oropharyngeal squamous cell carcinoma. J Oral Pathol Med. 2015;44(9):734–745. | ||
Sewell A, Brown B, Biktasova A, et al. Reverse-phase protein array profiling of oropharyngeal cancer and significance of PIK3CA mutations in HPV-associated head and neck cancer. Clin Cancer Res. 2014;20(9):2300–2311. | ||
Feldman R, Gatalica Z, Knezetic J, et al. Molecular profiling of head and neck squamous cell carcinoma. Head Neck. 2016;38 (Suppl 1): E1625–E1638. | ||
Lui VW, Hedberg ML, Li H, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013;3(7):761–769. | ||
Chau NG, Li YY, Jo VY, et al. Incorporation of next-generation sequencing into routine clinical care to direct treatment of head and neck squamous cell carcinoma. Clin Cancer Res. 2016;22(12):2939–2949. | ||
Nisa L, Häfliger P, Poliaková M, et al. PIK3CA hotspot mutations differentially impact responses to Met targeting in MET-driven and non-driven preclinical cancer models. Mol Cancer. 2017;16(1):93. | ||
Fokas E, Im JH, Hill S, et al. Dual inhibition of the PI3K/mTOR pathway increases tumor radiosensitivity by normalizing tumor vasculature. Cancer Res. 2012;72(1):239–248. | ||
Soulières D, Faivre S, Mesía R, et al. Buparlisib and paclitaxel in patients with platinum-pretreated recurrent or metastatic squamous cell carcinoma of the head and neck (BERIL-1): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Oncol. 2017;18(3):323–335. | ||
Seiwert TY, Zuo Z, Keck MK, et al. Integrative and comparative genomic analysis of HPV-positive and HPV-negative head and neck squamous cell carcinomas. Clin Cancer Res. 2015;21(3):632–641. | ||
Arnold L, Enders J, Thomas SM. Activated HGF-c-Met axis in head and neck cancer. Cancers. 2017;9(12):E169:169. | ||
Psyrri A, Lee JW, Pectasides E, et al. Prognostic biomarkers in phase II trial of cetuximab-containing induction and chemoradiation in resectable HNSCC: Eastern Cooperative Oncology Group E2303. Clin Cancer Res. 2014;20(11):3023–3032. | ||
Wang Z, Martin D, Molinolo AA, et al. mTOR co-targeting in cetuximab resistance in head and neck cancers harboring PIK3CA and RAS mutations. J Natl Cancer Inst. 2014;106(9). | ||
Misiukiewicz K, Dang RP, Parides M, et al. Endothelial growth factor receptor inhibitors in recurrent metastatic cancer of the head and neck. Head Neck. 2016;38(Suppl 1):E2221–E2228. | ||
Martin D, Abba MC, Molinolo AA, et al. The head and neck cancer cell oncogenome: a platform for the development of precision molecular therapies. Oncotarget. 2014;5(19):8906–8923. | ||
Ahmad P, Sana J, Slavik M, Slampa P, Smilek P, Slaby O. MicroRNAs involvement in radioresistance of head and neck cancer. Dis Markers. 2017;2017:1–8. | ||
Flezar MS, Kirbis IS, Popović KS, Strojan P. Radiosensitivity of squamous cell carcinoma metastases to the neck assessed by immunocytochemical profiling of fine-needle aspiration biopsy cell specimens: a pilot study. Radiother Oncol. 2009;93(3):575–580. | ||
Metheetrairut C, Slack FJ. MicroRNAs in the ionizing radiation response and in radiotherapy. Curr Opin Genet Dev. 2013;23(1):12–19. | ||
Summerer I, Niyazi M, Unger K, et al. Changes in circulating microRNAs after radiochemotherapy in head and neck cancer patients. Radiat Oncol. 2013;8:296. | ||
Brand TM, Iida M, Luthar N, Starr MM, Huppert EJ, Wheeler DL. Nuclear EGFR as a molecular target in cancer. Radiother Oncol. 2013;108(3):370–377. | ||
Toulany M, Kasten-Pisula U, Brammer I, et al. Blockage of epidermal growth factor receptor-phosphatidylinositol 3-kinase-Akt signaling increases radiosensitivity of K-ras mutated human tumor cells in vitro by affecting DNA repair. Clin Cancer Res. 2006;12(13):4119–4126. | ||
Janssens GO, Rademakers SE, Terhaard CH, et al. Accelerated radiotherapy with carbogen and nicotinamide for laryngeal cancer: results of a phase III randomized trial. J Clin Oncol. 2012;30(15):1777–1783. | ||
Swinson DE, O’Byrne KJ. Interactions between hypoxia and epidermal growth factor receptor in non-small-cell lung cancer. Clin Lung Cancer. 2006;7(4):250–256. | ||
Moreira J, Tobias A, O’Brien MP, Agulnik M. Targeted therapy in head and neck cancer: an update on current clinical developments in epidermal growth factor receptor-targeted therapy and immunotherapies. Drugs. 2017;77(8):843–857. | ||
Nijkamp MM, Span PN, Terhaard CH, et al. Epidermal growth factor receptor expression in laryngeal cancer predicts the effect of hypoxia modification as an additive to accelerated radiotherapy in a randomised controlled trial. Eur J Cancer. 2013;49(15):3202–3209. | ||
Juergens RA, Bratman SV, Tsao MS, et al. Biology and patterns of response to EGFR-inhibition in squamous cell cancers of the lung and head & neck. Cancer Treat Rev. 2017;54:43–57. | ||
Vermorken JB, Mesia R, Rivera F, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359(11):1116–1127. | ||
Brauswetter D, Dános K, Gurbi B, et al. Copy number gain of PIK3CA and Met is associated with poor prognosis in head and neck squamous cell carcinoma. Virchows Arch. 2016;468(5):579–587. | ||
Milik SN, Lasheen DS, Serya RAT, Abouzid KAM. How to train your inhibitor: design strategies to overcome resistance to epidermal growth factor receptor inhibitors. Eur J Med Chem. 2017;142:131–151. | ||
Kontić M, Milovanović J, Čolović Z, et al. Epidermal growth factor receptor (EGFR) expression in patients with laryngeal squamous cell carcinoma. Eur Arch Otorhinolaryngol. 2015;272(2):401–405. | ||
Marimuthu A, Chavan S, Sathe G, et al. Identification of head and neck squamous cell carcinoma biomarker candidates through proteomic analysis of cancer cell secretome. Biochim Biophys Acta. 2013;1834(11):2308–2316. | ||
Ansell A, Jedlinski A, Johansson AC, Roberg K. Epidermal growth factor is a potential biomarker for poor cetuximab response in tongue cancer cells. J Oral Pathol Med. 2016;45(1):9–16. | ||
Zanotti L, Paderno A, Piazza C, et al. Epidermal growth factor receptor detection in serum and saliva as a diagnostic and prognostic tool in oral cancer. Laryngoscope. 2017;127(11):E408–E414. | ||
van Dijk LK, Boerman OC, Kaanders JH, Bussink J. Epidermal growth factor receptor imaging in human head and neck cancer xenografts. Acta Oncol. 2015;54(9):1263–1267. | ||
Beck TN, Georgopoulos R, Shagisultanova EI, et al. EGFR and Rb1 as dual biomarkers in HPV-negative head and neck cancer. Mol Cancer Ther. 2016;15(10):2486–2497. | ||
Romanitan M, Näsman A, Munck-Wikland E, Dalianis T, Ramqvist T. EGFR and phosphorylated EGFR in relation to HPV and clinical outcome in tonsillar cancer. Anticancer Res. 2013;33(4):1575–1583. | ||
Seiwert TY, Jagadeeswaran R, Faoro L, et al. The Met receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009;69(7):3021–3031. | ||
Nieto MA, Huang RY, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21–45. | ||
Tsai JH, Yang J. Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 2013;27(20):2192–2206. | ||
Tam WL, Weinberg RA. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat Med. 2013;19(11):1438–1449. | ||
Szturz P, Raymond E, Faivre S. c-MET-mediated resistance to EGFR inhibitors in head and neck cancer: how to move from bench to bedside. Oral Oncol. 2016;59:e12–e14. | ||
Citron F, Armenia J, Franchin G, et al. An integrated approach identifies mediators of local recurrence in head and neck squamous carcinoma. Clin Cancer Res. 2017;23(14):3769–3780. | ||
Szturz P, Raymond E, Abitbol C, Albert S, de Gramont A, Faivre S. Understanding c-MET signalling in squamous cell carcinoma of the head & neck. Crit Rev Oncol Hematol. 2017;111:39–51. | ||
Garufi A, Trisciuoglio D, Porru M, et al. A fluorescent curcumin-based Zn(II)-complex reactivates mutant (R175H and R273H) p53 in cancer cells. J Exp Clin Cancer Res. 2013;32:72. | ||
Cañadas I, Rojo F, Arumí-Uría M, Rovira A, Albanell J, Arriola E. C-Met as a new therapeutic target for the development of novel anticancer drugs. Clin Transl Oncol. 2010;12(4):253–260. | ||
Madoz-Gúrpide J, Zazo S, Chamizo C, et al. Activation of Met pathway predicts poor outcome to cetuximab in patients with recurrent or metastatic head and neck cancer. J Transl Med. 2015;13:282. | ||
Xu H, Stabile LP, Gubish CT, Gooding WE, Grandis JR, Siegfried JM. Dual blockade of EGFR and c-Met abrogates redundant signaling and proliferation in head and neck carcinoma cells. Clin Cancer Res. 2011;17(13):4425–4438. | ||
Seiwert TY, Jagadeeswaran R, Faoro L, et al. The Met receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009;69(7):3021–3031. | ||
Stabile LP, He G, Lui VW, et al. c-Src activation mediates erlotinib resistance in head and neck cancer by stimulating c-Met. Clin Cancer Res. 2013;19(2):380–392. | ||
Mooren JJ, Gültekin SE, Straetmans JM, et al. P16(INK4A) immunostaining is a strong indicator for high-risk-HPV-associated oropharyngeal carcinomas and dysplasias, but is unreliable to predict low-risk-HPV-infection in head and neck papillomas and laryngeal dysplasias. Int J Cancer. 2014;134(9):2108–2117. | ||
Walline HM, Carey TE, Goudsmit CM, et al. High-risk HPV, biomarkers, and outcome in matched cohorts of head and neck cancer patients positive and negative for HIV. Mol Cancer Res. 2017;15(2):179–188. | ||
Ohnami S, Ohshima K, Nagashima T, et al. Comprehensive characterization of genes associated with the TP53 signal transduction pathway in various tumors. Mol Cell Biochem. 2017;431(1–2):75–85. | ||
Vainshtein JM, Spector ME, McHugh JB, et al. Refining risk stratification for locoregional failure after chemoradiotherapy in human papillomavirus-associated oropharyngeal cancer. Oral Oncol. 2014;50(5):513–519. | ||
Kanyilmaz G, Ekinci O, Muge A, Celik S, Ozturk F. HPV-Associated p16INK4A Expression and Response to Therapy and Survival in Selected Head and Neck Cancers. Asian Pacific Journal of Cancer Prevention. 2015;16(1):253-258. | ||
Sen B, Peng S, Saigal B, Williams MD, Johnson FM. Distinct Interactions Between c-Src and c-Met in Mediating Resistance to c-Src Inhibition in Head and Neck Cancer. Clin Cancer Res. 2011;17(3):514–524. | ||
Cascone T, Xu L, Lin HY, et al. The HGF/c-MET Pathway Is a Driver and Biomarker of VEGFR-inhibitor Resistance and Vascular Remodeling in Non-Small Cell Lung Cancer. Clin Cancer Res. 2017;23(18):489–5501. | ||
Hoadley KA, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018 Apr 5;173(2):291–304. | ||
Ellrott K, Bailey MH, Saksena G, et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell systems. 2018 Mar 28;6(3):271–281. | ||
Taylor AM, Shih J, Ha G, et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018 Apr 9;33(4):676–689. | ||
Gao Q, Liang WW, Foltz SM, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell reports. 2018 Apr 3;23(1):227–338. | ||
Liu J, Lichtenberg T, Hoadley KA, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018 Apr 5;173(2):400–416. | ||
Sanchez-Vega F, Mina M, Armenia J, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018 Apr 5;173(2):321–337. | ||
Li T, Wen H, Brayton C, et al. Epidermal growth factor receptor and notch pathways participate in the tumor suppressor function of gamma-secretase. J Biol Chem. 2007;282(44):32264–32273. | ||
Gao J, Aksoy BA, Dogrusoz U, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):l1. | ||
Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.