")
Back to Journals » Journal of Experimental Pharmacology » Volume 13
Novel Experimental Drugs for Treatment of Multiple Myeloma
Authors Offidani M , Corvatta L, Morè S, Olivieri A
Received 18 December 2020
Accepted for publication 23 February 2021
Published 9 March 2021 Volume 2021:13 Pages 245—264
DOI https://doi.org/10.2147/JEP.S265288
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Bal Lokeshwar
Massimo Offidani,1 Laura Corvatta,2 Sonia Morè,1 Attilio Olivieri1
1Clinica di Ematologia Azienda Ospedaliero-Universitaria Ospedali Riuniti di Ancona, Ancona, Italy; 2U.O.C. Medicina, Ospedale Profili, Fabriano, Italy
Correspondence: Massimo Offidani Email [email protected]
Abstract: Multiple myeloma (MM) is the second most frequent hematological malignancy characterized by bone marrow aberrant plasma cells proliferation leading to a genetic complex and heterogeneous disease, with a median survival ranging from two to more than 10 years. By using new drugs such as proteasome inhibitors (PIs), immunomodulatory drugs (IMiDs), monoclonal antibodies (mAbs) in different combinations and high-dose therapy followed by auto-transplantation, there has been an amazing improvement in the outcome of this disease in recent years. Despite this, MM is still considered an incurable disease, characterized by remission periods alternated with relapse/progression episodes finally leading to resistant disease. In particular, patients who become refractory to PIs, IMiDs and mAbs have a very poor outcome. Moreover, to overcome resistant residual disease, a large combination of drugs will be increasingly used in early lines of therapy; this further reduces the therapeutic options at each relapse. This natural history means that MM always needs new drugs/strategies to overcome the incoming resistance. New combinations of naked mAbs are becoming the therapy of choice for patients refractory to lenalidomide and/or PI; conjugated mAbs will be useful in triple- and more-refractory patients; CAR-T cells and bispecific mAbs have shown relevant results in very advanced stages of disease. In this review, we reported the results of these new therapies and explored their potential applications. Personalized and precision medicine seem to be the new frontier of cancer treatment. Although no single or few factors have been identified as disease drivers in MM, recurrent gene mutations were recognized and specific compounds targeting these alterations were developed and studied. Therefore, we reviewed these targeted drugs to try to understand what the best therapeutic strategy in MM is.
Keywords: multiple myeloma, immunotherapy, CAR T cell therapy, monoclonal antibodies, targeted therapy
Introduction
Multiple myeloma (MM) represents the second most frequent hematological malignancy1 and its incidence and prevalence are growing concurrently with the increasing half-life being a typical cancer of advanced age. It is characterized by bone marrow aberrant plasma cells proliferation that commonly produces a high amount of monoclonal immunoglobulin leading to organ damage, such as anemia, bone disease, renal impairment and hypercalcemia.2
MM is a very heterogeneous disease as per biological, clinical, and prognostic points of view with a median survival ranging from two to more than 10 years;3,4 it is considered an incurable disease characterized by remission periods alternated with relapse/progression episodes, finally leading to resistant disease.
In the past two decades hematologists have seen extremely significant changes in the management of this complex hematologic disease due to refinement of transplant strategy and supportive care as well as the introduction of novel agents as immunomodulatory drugs (IMiDs) thalidomide, lenalidomide and pomalidomide and proteasome inhibitors (PIs) bortezomib, carfilzomib and ixazomib. These two classes of drugs revolutionized the natural history of MM because of their strong synergistic action.
IMiDs exert anti-myeloma properties through several mechanisms since they induce direct cytotoxic effects including growth arrest, activation of caspase-8 and apoptosis, and cereblon (CRBN)-dependent degradation of ikaros (IKZF1) and aiolos (IKZF3), followed by IRF4 downregulation.5 Moreover, IMiDs disrupt the stromal and myeloma cells interaction, modulating cytokine and growth factor secretion and, remarkably, they upregulate T, NK and NKT cells while downregulate regulatory T cells.6 Alternatively, the main effect of PIs is the direct inhibition of the NF-kB pathway, essential for proliferation of MM cells.7 Bortezomib is further able to directly induce apoptosis via c-Jun-NH2-terminal kinase (JNK) and p53,8 to inhibit production of cytokines as IL-6, IGF-1 and VEGF in bone marrow stromal cells, to modify bone turnover and osteoclast activity and to inhibit VLA-4 expression, resulting in overcoming cell adhesion-mediated drug resistance.9 Triplet combinations in which bortezomib plus dexamethasone (VD) is a backbone for adding IMiDs (VTD, VRD) have represented, until now, suggested upfront regimens in transplant eligible (TE) MM patients.10,11
Currently, continuous therapies with lenalidomide plus dexamethasone (Rd) or VRD regimen, recently approved by the EMA, or fixed-duration of VMP cycles are the standard of care for older and nontransplant eligible patients (NTE).10,11 Despite no clear evidence of a cure, by using these treatments the overall survival (OS) of MM was significantly prolonged and it continues to improve over the years, as demonstrated by recent data showing a median OS of 6.8 years in a cohort of 3783 newly diagnosed MM (NDMM) patients treated between 2004 and 2018.12
Despite this improvement in first-line therapy, almost all patients eventually relapse and the outcome progressively worsens with each disease progression. Patients who become refractory to PIs and IMiDs have a very poor outcome with a median OS of 13 months.13 Furthermore, the large combination of drugs used in early lines of therapy further reduces the therapeutic options at each subsequent disease progression.
In this paper we reviewed data provided by studies on innovative drugs for MM treatment with the aim to update the results in this difficult-to-treat group of refractory MM patients.
“Old” Generation Immunotherapies
Monoclonal Antibodies
Based on the success of monoclonal antibody (mAb) therapy in the treatment of other hematologic malignancies.14,15 This approach has been explored in MM by identifying specific cellular targets. Elotuzumab, that binds SLAMF7 (signaling lymphocytic activation molecule F7) (Figure 1), represents the first mAb evaluated in clinical trials, to show antitumor activity and to be introduced in the treatment of MM.16
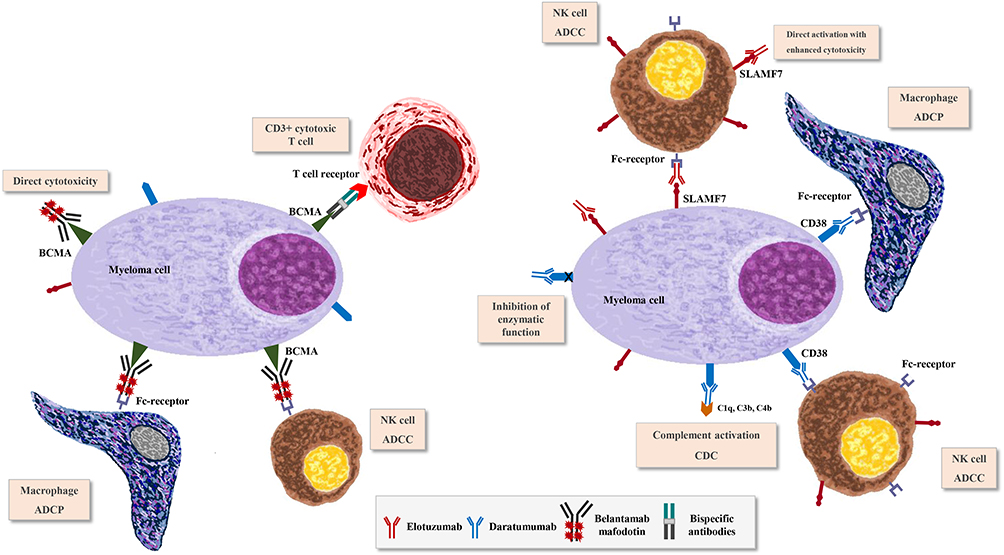 |
Figure 1 Mechanisms of action of: naked monoclonal antibodies as elotuzumab and daratumumab; antibody–drug conjugate as belantamab mafodotin and bispecific antibodies. |
It is currently approved in combination with lenalidomide and dexamethasone (Elo-Rd) or pomalidomide and dexamethasone (Elo-Pd) for treatment of relapsed/refractory MM (RRMM) based on the results of Phase III ELOQUENT-217 and ELOQUENT-318 trials.
In the first study, Elo-Rd significantly improved PFS vs Rd (median 19.4 months vs 14.9 months; HR=0.71; p=0.0004) after a four-year follow-up.19
Remarkably, final analysis of study demonstrated a significant OS benefit for Elo-Rd vs Rd since median OS was 48.3 vs 39.6 months, respectively (HR=0.82; p=0.04), making ELOQUENT-2 the first trial showing an OS benefit with an antibody-based triplet regimen in RRMM.20
In more heavily pretreated patients enrolled in ELOQUENT-3 trial, the addition of elotuzumab to pomalidomide plus dexamethasone (Elo-Pd) induced a 46% reduction in progression or death, with median PFS being 10.3 vs 4.7 months in patients receiving Elo-Pd and Pd, respectively, after a median follow-up of 9.1 months. In the same setting, ongoing studies are evaluating quadruplets as elotuzumab, bortezomib, pomalidomide, dexamethasone (Elo-PVd) (NCT02718833), elotuzumab, carfilzomib, lenalidomide, dexamethasone (Elo-KRd) (NCT02343042) or selinexor, pomalidomide, elotuzumab, dexamethasone (SPEd) within STOMP study (NCT02343042). Results with elotuzumab-containing regimens in NDMM patients seem to be less significant. In the final analysis of the phase III ELOQUENT-1 trial, comparing Elo-Rd vs Rd in NDMM NTE, the addition of elotuzumab did not show a statistically significant improvement in PFS, the primary endpoint of study.21 In NDMM TE, preliminary data from the phase III GMMG-HD6 trial show that adding elotuzumab to VRD did not result in increased ≥ VGPR rates after induction.22
Daratumumab is the first fully human IgG1κ mAb targeting CD38 to be explored in MM after it showed antimyeloma activity as single agent.23,24
It elicits cell death through complement-dependent cytotoxicity (CDC), antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP), induction of apoptosis and modulation of CD38 enzyme activities (Figure 1).25
In the RRMM setting, daratumumab has been approved in combination with lenalidomide and dexamethasone (DRd) or bortezomib and dexamethasone (DVd) in patients who have received at least one prior line of therapy, based on the results from the phase III POLLUX26 and CASTOR27 trials. More recently, using Pd instead of Rd as backbone, the phase III APOLLO trial28 compared DPd vs Pd with the mAb administered subcutaneously. After a median follow-up of 16.9 months, the study met its primary endpoint of improved PFS since in patients receiving DPd median PFS was 12.4 vs 6.9 in those treated with Pd (HR=0.63; p=0.0018). In the phase III CANDOR trial29 triplet combination daratumumab, carfilzomib, dexamethasone (DKd) showed a significant benefit in term of PFS compared with Kd since median PFS was 28.6 months vs 15.2 months (HR=0.59) after a median follow-up of 27.8 months. Moving into the front-line MM setting, in early 2000 regulatory agencies approved combination daratumumab, bortezomib, thalidomide, dexamethasone (D-VTd) as induction therapy in TE patients based on the results of phase III CASSIOPEIA trial.30 In the phase II randomized GRIFFIN study31 the addition of daratumumab to VRd led to deeper response after post-transplant consolidation with sCR and MRD negativity at 10−5 being 42% and 50% in D-VRd group vs 32% and 20% in VRd group, respectively. Phase III PERSEUS trial comparing D-VRd (with subcutaneous daratumumab) vs VRd has completed enrollment and results are yet to be published. In NTE patients, daratumumab has been explored in combinations with standard-of-care regimens (VMP and Rd) so D-VMP and D-Rd have been recently approved after results of phase III ALCYONE32 and MAIA33 trials. An overview of clinical trials with daratumumab in NDMM and RRMM patients is shown in Table 1.
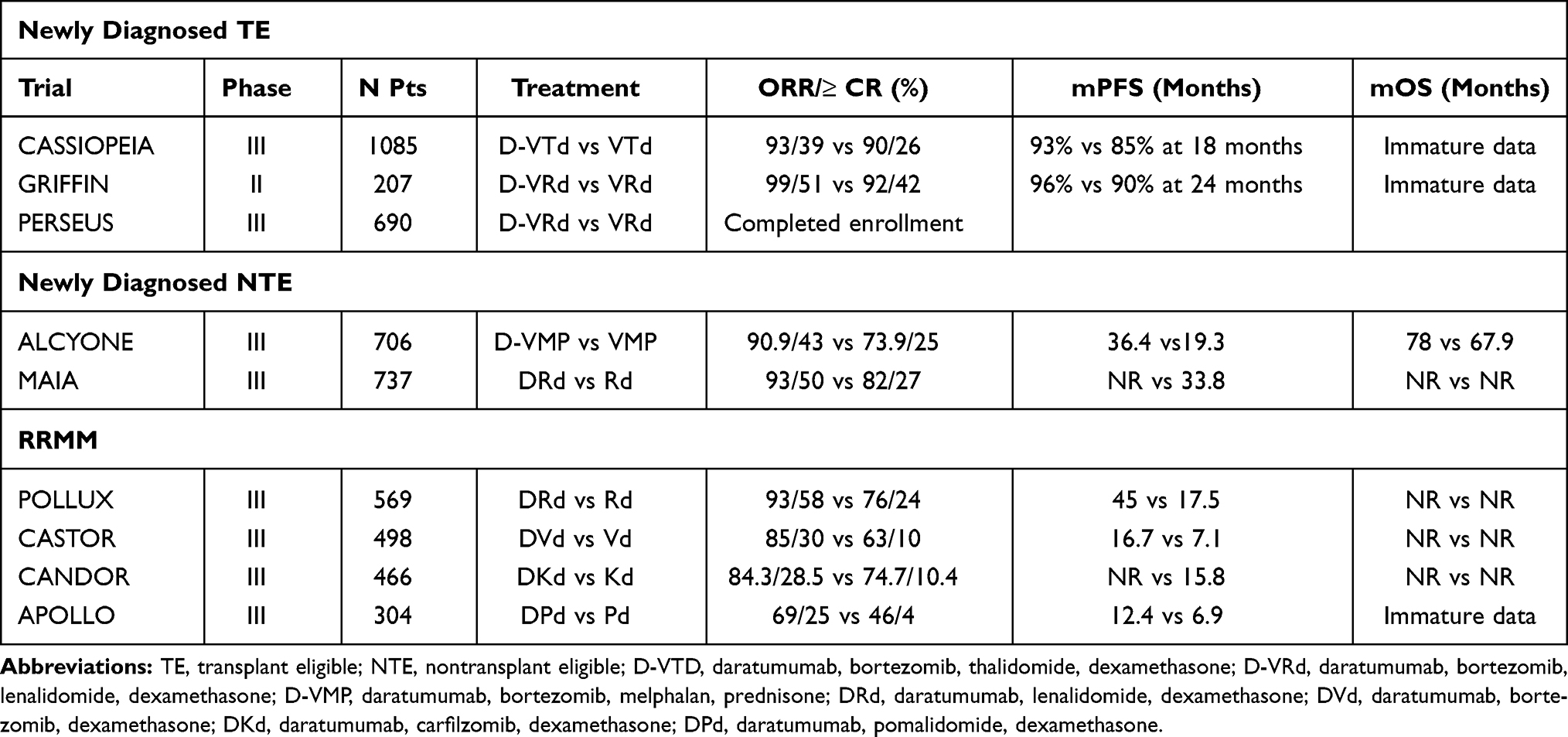 |
Table 1 Main Clinical Trials with Daratumumab in NDMM and RRMM |
Despite being generally well tolerated, intravenous formulation of daratumumab is characterized by a long infusion time (seven to eight hours for the first infusion) so the phase III COLUMBA trial34 explored the noninferiority for ORR of a co-formulation of daratumumab (DARA SC) (1800 mg flat dose) with recombinant human hyaluronidase (rHuPH20), needing a median administration duration of five minutes, vs conventional daratumumab. The study met the primary endpoint since ORR was 41% in the SC group vs 37% in the IV group with a significant reduction of infusion-related reaction (IRR) rate being 12.7% vs 34.5% in the SC and IV arm, respectively.
The phase II PLEIADES study35 assessed daratumumab SC in combination with standard care as VRD (D-VRD in TE patients), VMP (D-VMP in NTE) and Rd (DRd in RRMM), showing response rates comparable with those obtained in phase III trials using daratumumab IV and low IRRs.
Isatuximab is a chimeric IgG1κ mAb binding a specific epitope on CD38 and, unlike daratumumab, is capable of inducing direct apoptosis in MM cells, also in those harboring p53 mutations.36 Moreover, isatuximab was found to inhibit CD38 enzymatic activity more effectively than daratumumab whereas it is less able to induce CDC.37 Isatuximab exerts antitumor activity indirectly by eliminating CD38 immunosuppressive regulatory T cells and restoring immune effectors against MM.38 Remarkably, in vitro and in vivo studies demonstrated enhanced antimyeloma activity combining isatuximab with pomalidomide or PIs such as bortezomib or carfilzomib.39 Triplet isatuximab, pomalidomide, and dexamethasone has been explored in the phase III ICARIA trial40 which compared Isa-Pd vs Pd in patients previously treated with ≥2 lines of therapy. After a median follow-up of 11.6 months, median PFS was 11.5 months in Isa-Pd and 6.5 months in the Pd group (HR=0.59; p=0.001). This benefit was documented also in older (≥75 year-olds) and frail patients, among whom 8.3% vs 16.7% receiving Isa-Pd and Pd, respectively, discontinued treatment due to adverse events.41 As for OS final analysis, a longer follow-up is required but in patients treated with Isa-Pd a trend in OS improvement was seen at the last analysis (HR=0.68).39 Based on results from the ICARIA trial, the FDA and EMEA approved triplet Isa-Pd in RRMM patients who have received at least two prior lines of therapy. In the phase III IKEMA study,42 isatuximab has been evaluated in combination with carfilzomib and dexamethasone (Isa-Kd) and compared with doublet Kd. At the last update of trial, after a median follow-up of 20.7 months, PFS was statistically longer in Isa-Kd vs Kd group (not reached vs 19.15 months; HR=0.53; p<0.0007). Interestingly, using mass spectrometry analysis to measure monoclonal component without isatuximab interference, CR rate was reached for 45.8% of patients vs 39.7% achieved with immunofixation assay.43 In the near future, this triplet could become a new standard for patients with RRMM.
Several ongoing studies are assessing isatuximab in NDMM patients. The Phase III GMMG HD7 trial (NCT03617731) is evaluating the benefit of adding isatuximab to VRD (Isa-VRD) induction and to lenalidomide maintenance in TE patients whereas the same quadruplet (Isa-VRD) is compared to VRD in the ongoing Phase III IMROZ (NCT03319667), enrolling NTE patients. The Phase II GMMG-CONCEPT trial (NCT03104842) investigated combination isatuximab, carfilzomib, lenalidomide and dexamethasone (Isa-KRd) in high risk NDMM, defined by del(17p) or t(4;14) or t(14;16) or >3 copies 1q21 plus ISS stage 2 or 3, in both TE and NTE patients. Preliminary results of 46 TE patients showed ORR of 100%, ≥VGPR of 90%, CR/sCR of 46% and MRD negativity of 60% after six induction cycles of Isa-KRd.44 The Phase III IsKia trial by European Myeloma Network (NCT 04483739) will explore the efficacy in term of MRD negativity of Isa-KRd vs KRd as induction and post-ASCT consolidation. Finally, in a phase Ib study of Isa-VCD or Isa-VRD in NTE patients, ORR and ≥VGPR were 93% and 67% vs 100% and 42%, respectively.45
Other mAbs targeting CD38 are under evaluation starting from MOR202/TJ202, an investigational human mAb derived from MorphoSys’s HuCAL antibody technology that can be administered safely within 30 min. In a phase 1/2a study46 including RRMM patients with at least two previous lines of therapy, it showed activity either as single agent or in combinations with dexamethasone, lenalidomide and dexamethasone (Rd) (ORR=65%) or pomalidomide and dexamethasone (Pd) (ORR=48%). A Phase III trial (NCT03952091) is assessing MOR202 plus Rd vs Rd in RRMM.
Another receptor exerting a role in the dissemination of MM cells out of the bone marrow is the chemokine CXCR4 and ulocuplumab represents the first-in-class fully mAb targeting CXCR4, showing activity in RRMM in a phase Ib/II study.47 Combined with lenalidomide and dexamethasone, ulocuplumab showed high ORR (55%) with a benefit even in patients previous treated with IMiDs.
Although introduction of mAbs has been a turning point, a key and emergent issue regarding daratumumab refractoriness that is related to several mechanisms starting from decreased CD38 expression, whose role in acquired resistance toward mAb targeting CD38 has not been completely clarified. Other resistance mechanisms include overexpression of complement inhibitory protein as CD55 and CD59; bone marrow stromal cells (BMSC)-mediated suppression of ADCC by upregulating protein survivin in MM cells (cell adhesion-mediated immune resistance, CAM-IR); Fc-gamma-receptor polymorphisms, affecting ADCC and ADCP; reduction of NK cells, the most important mediators of daratumumab-mediated ADCC; production of neutralizing antibodies.48 Several studies are evaluating strategies that may overcome resistance to CD38 mAbs, such as adding pembrolizumab, a humanized PD-1 mAb, to daratumumab49 or combining daratumumab with azacitidine.50 Moreover, recent data support the possibility of treating patients refractory to daratumumab with isatuximab, due to MM cells “re-sensitization” after withdrawal daratumumab.51 However, the multicenter retrospective MAMMOTH study52 demonstrated that patients who become refractory to CD38 and those “penta-refractory” (refractory to CD38 mAb, two PIs and two IMiDs) had median OS of 8.6 and 5.6 months, respectively, so this poor outcome requires development and introduction in clinical practice of innovative therapeutic options.
New Generation Immunotherapies
In the last years, the early use of mAbs in MM therapeutic armamentarium led to a new refractory MM patient population with a few therapeutic chances. The new frontier of immunotherapy in MM is the use of a smart technique that can boost the immune system against MM cells.53 B-cell maturation antigen (BCMA) is the most used target because of its important functions in the proliferation, survival, and tumor progression in MM.54
BCMA is a receptor for two different soluble growth factors in the BM microenvironment: BAFF whose affinity for BAFF-R is much higher than the affinity for BCMA, and APRIL, which has a great selectivity for BCMA. BAFF and APRIL ligation to their receptors induces the activation of NF-kB pathways and the upregulation of antiapoptotic proteins, maintaining a crucial role in survival of neoplastic plasma cells (PCs).55 BCMA is highly expressed in neoplastic PCs and it is absent on naïve and memory B lymphocytes, T cells and other nonlymphoid organs.
Anti-BCMA antibodies are able to induce clinical response through multiple mechanisms: firstly they can induce a direct apoptosis of neoplastic PCs that express BCMA; secondly they can induce an ADCC response involving the effector cells of immune system toward an immunogenic cell death; thirdly, they can active the ADCP response; fourthly, they can kill the neoplastic sPC by the BCMA receptor signalling inhibition.56
Bispecific Monoclonal Antibodies
Bispecific monoclonal antibodies consist in a Fc domain, a Fab region (including a variable domain and a constant domain) and two binding sites, one for CD3 on T cells and the other for the specific target on cancer cells. The convergence of cytotoxic T lymphocytes and tumor cells due to BsAb binding activates cytotoxic T cells and promotes killing of tumor cells (Figure 1). Among bispecific antibodies, bispecific T cell engagers (BiTE) differ for the presence of two single chain variable fragments connected by a linker and for the lack of the Fc domain, characteristic that gives them a short half-life requiring frequent or continuous dosing.56
AMG-420 and AMG-701
AMG-420 is the first BiTE targeting BCMA studied in the setting of RRMM in a phase 1 study57 including 42 patients with a median of five prior lines of therapy (range: 2–14). Patients received AMG-420 at doses ranging from 0.2 to 800 µg/d for up to 10 cycles with each six-week cycles including four weeks of continuous intravenous infusion followed by two weeks off treatment. ORR was 31% but at the maximum tolerated dose (MTD), found to be 400 µg/d, ORR was 70% (7/10 patients) with five patients achieving MRD negativity at level of 10−4. The most common grade 3–4 adverse events were infections occurring in 33% of patients, whereas CRS was 2% and neurotoxicity was uncommon. Logistical challenges for patients and health-care system due to the need of continuous infusion, led the sponsor to stop AMG-420 trials and to developed a new anti-BCMA BiTe molecule with an extended half-life (AMG-701). An ongoing Phase I study is evaluating DLTs and safety of AMG-701 monotherapy, or in combination with pomalidomide, with or without dexamethasone, in RRMM patients after three or more lines of therapy that must include IMIDs, PIs and anti-CD38 mAbs (NCT03287908). At the 2020 ASH Meeting, Harrison et al58 reported preliminary data of 85 patients with a median of six prior therapies (range: 2–25) who received weekly AMG-701 at escalated doses. ORR was 36% at doses of 3–18 mg with 24% achieving at least VGPR, median time to response was one month. The most common grades of adverse events were CRS (61%), hematological toxicity (anemia 43%, neutropenia 23%, thrombocytopenia 20%) and diarrhoea (31%).
Teclistamab
Teclistamab is a humanized IgG-4 bispecific BCMA⨰CD3 antibody that induces T cell mediated cytotoxicity against BCMA-expressing MM cells. A phase 1 study59 evaluated teclistamab in RRMM patients either with intravenous administration or with subcutaneous one. Seventy-eight patients received a weekly dose of teclistamab and their median number of previous therapies was six (80% triple-class refractory and 41% penta-drug refractory). The best responses were observed at 270 µg/kg dose with ORR of 67% and 50% of patients obtaining at least VGPR. The most common AEs were grade 1–2 CRS (62%), whereas grade 3–4 hematologic toxicities consisted of neutropenia (38%) and thrombocytopenia (24%). During the last ASH meeting, Garfall et al60 presented results of subcutaneous teclistamab administered to 33 patients at dose of 1500 µg/kg. ORR was 73%, being 70% and 75% in triple-class and penta-drug refractory patients. As for the toxicity, no significant differences were observed in comparison with intravenous administration.
CC-93269
This humanized bispecific antibody is an immunoglobulin G1 based, with bivalent BCMA and monovalent CD3 binding in a 2+1 format. In a dose escalation phase 1 study,61 19 patients with a median of six prior lines of therapy (range: 3–12) were enrolled. All patients had been exposed to lenalidomide and bortezomib, 95% to daratumumab, 84% to pomalidomide and 84% to carfilzomib. Grade 3.4 adverse events included neutropenia (52%), infections (26%), thrombocytopenia (21%). With regard to CRS, they occurred in 89% of patients, mainly during first or second doses, and majority of them were of grade 1–2. Among patients receiving a dose ≥6 mg, ORR was 83%, at least VGPR 58% with 75% of patients achieving MRD negativity at level of 10−5.
Talquetamab (JNJ-64407564)
Several studies are exploring bispecific mAbs with antigen target different from BCMA. Talquetamab is a BiTE binding GPRC5D and CD3. GPRC5D is an orphan receptor whose transcript is highly expressed on MM cells but less expressed in other cells, encouraging its use as therapeutic target.62 In preclinical models, talquetamab induced cell killing of primary MM cells and inhibited tumor formation and growth in MM mouse models. Chari et al,63 reported initial results of a phase 1 study enrolling 157 in the part 1, aiming to identify RP2D (intravenous and subcutaneous), and 19 patients in the part 2 exploring safety and activity of RP2D. Among 157 patients who had received a median of six prior therapies (range: 2–20), 82% were triple-class refractory and 33% penta-drug refractory. ORR was 69% at the RP2D of 405 mg/kg subcutaneously with a median time to response of one month (0.2–3). Using this dose, most common grade 3–4 adverse events were neutropenia (42%) and lymphopenia (16%). CRS occurred in 68% of patients but none were higher than grade 2.
Cevostamab (BFCR4350A)
Cevostamab is a humanized immunoglobulin G-based T cell-engaging bispecific antibody that targets the most membrane-proximal domain of FcRH5 on MM cells and CD3 on T cells. Fc receptor-homolog 5 (FcRH5) is a type I membrane protein expressed on B cells and PC with a near 100% prevalence. At the last ASH Meeting Cohen et al64 reported the initial results from a dose-escalation phase 1 trial. Fifty-three patients, with a median of six prior lines of therapy, 72% triple-refractory and 45% penta-refractory, received BFCR4350A by IV infusion every 21 days. ORR was 61% for ≥3.6 mg first dose followed by 20 mg target dose and 6/15 patients were in response for >6 months at data cutoff. The most common adverse events were CRS (76%), all grade 1–2, except for one patient with grade 3 CRS. Treatment-related grade 3–4 toxicities included thrombocytopenia (25%) and neutropenia (15%), In Table 2 we summarized ongoing clinical trials with bispecific antibodies.
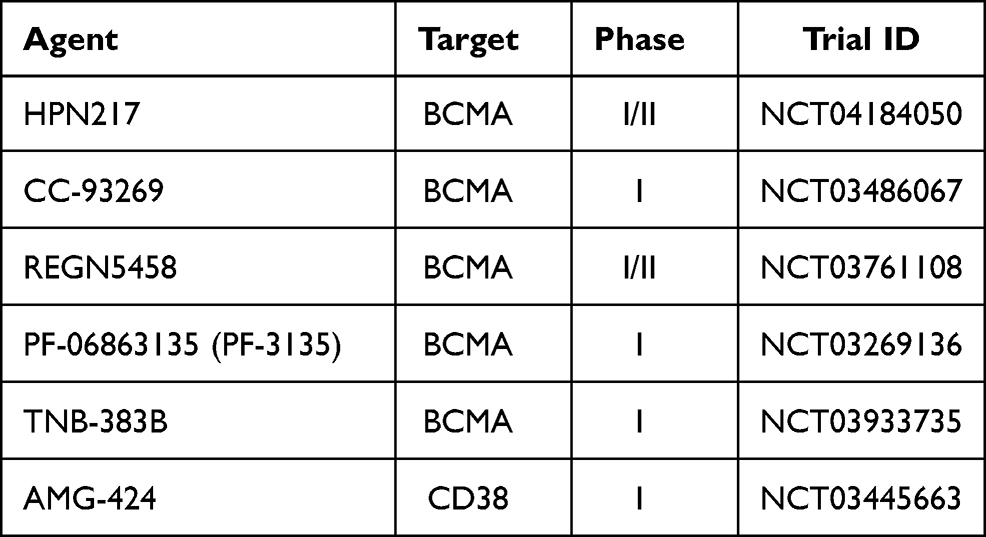 |
Table 2 Ongoing Trials with Bispecific Antibodies |
Antibody-Drug Conjugates (ADCs)
ADCs represent a new class of target therapy in the management of MM. They consist in three components: a mAb directed against a tumor-specific antigen, a cytotoxic payload and a linker that connects the targeting part to the cancer-killing molecule. Among the optimal strategies to create an ADC, the choice of the antibody is crucial: the preferred one should be a human antibody because of longer half-life (IgG is the best type), better cancer cell specificity and lower immunogenicity. Then, it is necessary to pay attention to the choice of the killing molecule: the predominant payloads used are microtubule inhibitors, which bind to tubulin and cause a G2/M arrest and apoptosis. Auristatins include monomethyl auristatin E and F. DNA damaging agents are also used as toxic payloads, ie calicheamicin. The ideal number of payloads attached to a single antibody should be three to four molecules. The third part is the linker: it should have the same half-like as the antibody and it should not release the payload prematurely in order to avoid off-target toxicity.56
Belantamab Mafodotin (GSK2857916, Belamaf)
Belamaf is a humanized IgG1 ADC that binds specifically to BCMA (Figure 1). It can induce cell death by multiple mechanism: the first one is the caspase-dependent mechanism, through which ADC is internalized into the cell after binding it. The second one is the ADCC and macrophage phagocytosis through retention of IgG effectors by the antibody, that promote death in dividing and nondividing cancer cells. The third one is the ICD (immunogenic cell death), through the expression of antigens specific to dying tumor cells.65
The phase 1 study (DREAMM-1)66 included a part 1, a dose-escalation phase establishing the recommended dose of belamaf, and a part 2 in which this dose was used. Overall, 35 patients were enrolled and 40% of them had received more than five prior lines of therapy. Dose of intravenous 3.4 mg/kg administered in a one-hour infusion every three weeks was selected as the recommended dose. Sixty percent of patients had a confirmed response of PR or better (6% sCR, 9% CR). ORR was 71.4% in patients without prior daratumumab exposure and 42.9% with prior daratumumab exposure. ORR was 70% in patients who had received ≤3 prior therapies and 50% in >5 prior therapies, but ORR was not different between high and standard cytogenetic risk patients. Median time to response was 1.2 months and responses deepened over time. Median PFS was 12 months (7.9 months in IMIDs and PI refractoriness, 6.8 months in the prior daratumumab group) and median DOR was 14.3 months. The most common hematologic grade 3–4 adverse event was thrombocytopenia (35%). All grades cornel events occurred in 69% of patients, most commonly blurred vision (51%), dry eye (37%) and photophobia (29%), whose median duration was 35 days. Treatment-related serious AEs were experienced in 20% of patients, most commonly infusion-related reactions (6%).
The phase 2 study (DREAMM-2)67 enrolled 196 RRMM patients (97 in the 2.5 mg/kg cohort and 99 in the 3.4 mg/kg cohort). ORR was 31% in 2.5 mg/kg and 34% in 3.4 mg/kg cohort: there were no differences between patients with ≤4 and >4 prior lines of therapy, neither between high and standard cytogenetic risk but ORR was lower in patients with extramedullary MM. The most common grade 3–4 adverse event was keratopathy (27% and 21% in the two cohorts, respectively), thrombocytopenia (20% and 33%) and anemia (25% and 40%). Ocular events included keratopathy (microcyst-like epithelial changes [MECs]: an eye exam finding with/without symptoms), best-corrected visual acuity (BCVA) changes, and symptoms (blurred vision and dry eye). The majority of patients did not experience a clinically meaningful BCVA decline, and events rarely led to treatment discontinuation. The first keratopathy (MEC) event or clinically meaningful BCVA decline recovered in the majority of patients with events.68 Despite ocular toxicity, patients showed a general improvement in fatigue during the treatment period and their quality of life did not worsen over time.69
After a median follow-up of 13 months, IRRs were 21%, none of them grade 4–5, mostly occurring during the first cycle with a median onset and duration of one day (1–3). They were resolved in 90% of patients, without a discontinuation of the drug. The 2.5 mg/kg dose was selected as the recommended dose for future studies on the basis of its similar antimyeloma activity with a more favourable safety profile.70
The combination of belamaf, pomalidomide, and dexamethasone was assessed in a phase I study (Algonquin study)71 enrolling 37 RRMM patients who had received a median of three prior lines of therapy, 35% triplet-refractory. In part 1 of the study MTD was established to be belamaf 2.5 mg/kg every four weeks and at this dose ORR was 100%, at least VGPR 100% and CR 57%. With a median PFS not reached after a median follow-up of 13 months. Main grade 3–4 adverse events were keratopathy (51%), neutropenia (40%), thrombocytopenia (32%) whereas severe IRR occurred in 5.4% of patients.
In Table 3 we summarized an ongoing clinical trial with belamaf and in Table 4 ongoing studies with other antibody-drug conjugates in MM patients.
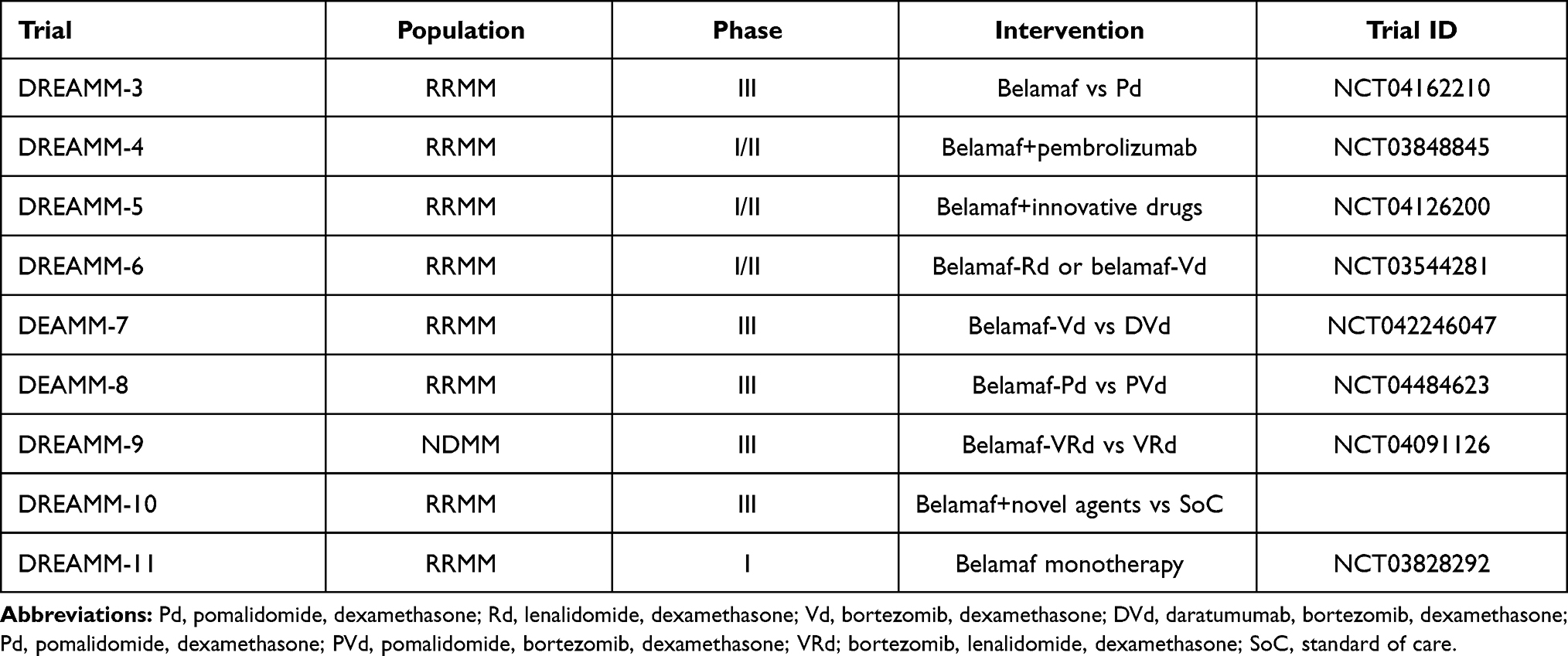 |
Table 3 Ongoing Clinical Trials with Belantamab Mafodotin |
 |
Table 4 Ongoing Trials with Antibody–Drug Conjugates |
CAR-T Cell Therapies
CAR-T cell therapy represents the new frontier in the treatment of hematologic malignancies and it consists in genetically modified T cell to induce cytotoxic ability by targeting specific tumor antigens.72 Recently, three CAR-T therapies against CD19 antigen, namely tisagenlecleucel, axicabtagene ciloleucel and brexucabtagene autoleucel have been approved by FDA for the treatment of B-ALL, respectively, large B-cell lymphoma and mantle cell lymphoma. Briefly, chimeric antigen receptor (CAR) is characterized by an extracellular target-binding domain, a hinge region, a transmembrane domain anchoring the CAR to the cell membrane, an intracellular domain as CD3ζ activation domain and a costimulatory domain (CD28 or 4-1BB) that transmit activation signals. According to number of costimulatory domains, CARs can be classified into first (CD3ζ only), second (CD3ζ plus one costimulatory domain) or third generation (CD3ζ plus combined costimulatory domain).73 An ideal target for CARs is a surface antigen that is uniformly expressed on tumor cells but not present on normal cells to minimized toxicity.74 In MM cells this requirement has been identified in the BCMA, a member of the TNF receptor superfamily, above described. Manufacturing of CAR-T cells is a very complex process and the first step is the collection of CD3 positive T cells by nonmobilized leukapheresis (Figure 2). T cells are expanded manifold in culture and activated using beads coated with anti-CD3 or anti-CD28 mAbs or cell based artificial antigen presenting cells.75 Subsequently, in a process during up to four weeks, T cells are transduced with a vector either lentiviral or retroviral carrying the gene encoding a receptor to an antigen present on plasma cells. Before infusion, eligible patients underwent lymphodepleting chemotherapy to enhance engraftment and persistence of CAR-T cells that, once they encounter antigens, proliferate and kill tumor cells.76
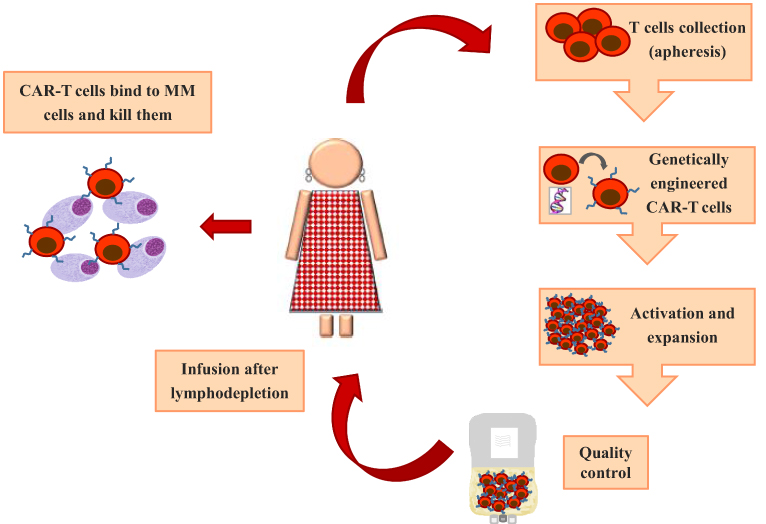 |
Figure 2 CAR-T cell therapy in multiple myeloma. |
Idecabtagene Vicleucel (Ide-Cel)
The most advanced CAR-T cells product against BCMA for treatment of advanced MM is idecabtagene vicleucel (ide-cel, bb2121), a lentiviral vector-based and including a 4-1BB costimulatory domain, which was explored in a phase I study (CRB-401),77 enrolling 33 RRMM patients with a median of five prior lines of therapy, who received 50 to 800⨰106 CAR-T cells/kg body weight. ORR and CR were 85% and 45%, respectively, but high quality responses (≥ VGPR) were observed only with doses of at least 150⨰106 CAR-T cells. Median PFS was 11.3 months and, regarding safety profile, the primary endpoint of the study, grade 3–4 neutropenia and thrombocytopenia were the most common adverse events occurring in 85% and 45% of patients, respectively. Cytokine release syndrome (CRS) developed in 76% of patients but it was of grade 1–2 in 70% of them. CRS is characterized by fever, tachypnea, headache, tachycardia, hypotension, rash, hypoxia and it is caused by release of cytokines from cells. In the recent update of this study,78 among 38 patients receiving 450⨰106 CAR-T cells 89.5% and 37% obtained ORR and at least CR, respectively, with median PFS and OS of nine and 34.2 months. Ide-cel has been subsequently evaluated in the phase II pivotal KarMMa study79 enrolling 128 heavily pretreated patients (six prior antimyeloma regimens, 84% triple-refractory) who received doses of CAR-T cells ranging from 150 to 450⨰106. Overall, ORR and CR/sCR rates were 73% and 33%, respectively, with a median PFS of 8.8 months. Remarkably, patients who received the highest target dose of 450⨰106 CAR-T cells had the best results, 82% being ORR, 65% at least VGPR, 48% at least VGPR plus MRD negativity with a median PFS of 12.1 months. The benefit was observed in all subgroups of patients including those with high-risk cytogenetics, extramedullary disease and penta-refractory (refractory to two PIs, two IMiDs and CD38 mAb). Grade 3 or higher CRS and neurotoxicity were observed in less than 6% of patients. Neurotoxicity related to CAR-T cell therapy is now named ICANS (immune effector cell associated neurotoxicity syndrome) and it consists of several symptoms or signs beginning with disorientation to time or place, aphasia, depressed level of consciousness, seizure, motor weakness and cerebral edema. Cytopenias were common, with grade ≥3 neutropenia and thrombocytopenia occurring in 89% and 52% of patients. Of note, efficacy outcomes in patients aged ≥65 and ≥70 years were comparable without differences in safety profile.80
Several clinical studies using ide-cel are ongoing: Phase II KarMMa-2 study (NCT03601078) is evaluating ide-cel in early and late relapsed MM; Phase III KarMMa-3 trial (NCT03651128) is comparing ide-cel with standard regimens in RRMM; Phase I KarMMa-4 (NCT04196491) is a study of ide-cel in patients with high-risk newly diagnosed MM.
Ciltacabtagene Autoleucel (Cilta-Cel, JNJ-4528)
LCAR-B38M, currently known as JNJ-4528, is a CAR-T cell construct characterized by two BCMA-targeting single-domain antibodies, CD3ζ activation domain and 4-1BB costimulatory domain. It was explored in China for the first time in the LEGEND-2 phase I study81 including 57 patients with a median of three prior therapies (range: 1–9). This therapy was very active with an ORR of 88%, median PFS of 19.9 months and median OS of 36.1 months. CRS occurred in 90% of patients, being grade 3 in 7% of them. The same CAR-T cell therapy has been evaluated in the CARTITUDE-1 study82 which, in the 1b portion, showed ORR of 100%, ≥VGPR of 97% and sCR of 86% in 29 patients who had been treated with a median of five prior lines of therapy (range: 3–18). Patients received a single infusion of cilta-cel at a target dose of 0.75⨰106 CAR-T cells/kg. Median time to response was one months and PFS at nine months resulted to be 86%. Safety profile was consistent with LEGEND-2 since CRS were mostly grade 1–2, neurotoxicity (ICANS) occurred infrequently and grade 3–4 cytopenias resolved after 60 days. In the updated data from the phase 1b portion along with initial phase 2 of CARTITUDE-1 study,83 97 patients with a median of six prior therapies, 42% penta-refractory, received cilta-cel. ORR was 97% with 93% of patients achieving at least VGPR and 67% sCR. Among evaluable patients, 93% achieved MRD negativity at level of 10−5. PFS and OS at 12 months were 76.6% and 88.5%, respectively. Therefore, preliminary data suggest that CAR-T cell therapy can induce deep and maybe durable responses in heavily pretreated MM population. The Phase II CARTITUDE-2 study (NCT04133636) is exploring cilta-cel in early and late MM relapse whereas the phase III CARTITUDE-4 is comparing cilta-cel with triplets as daratumumab, pomalidomide and dexamethasone (DPd) or pomalidomide, bortezomib, dexamethasone (PVd).
Orvacabtagene Autoleucel (Orva-Cel, JCAR-H125)
Orva-cel is a BCMA CAR-T cell construct characterized by a fully human binder and a manufacturing process enriching for central memory T cell phenotype. The phase I/II EVOLVE study84 showed promising results in heavily pretreated RRMM as reported at the last ASCO meeting. Sixty-two patients with a median of six prior lines of therapy (range: 3–18, 94% triple-refractory and 48% penta-refractory) received CAR-T cells dose ranging from 300 to 600⨰106. ORR was 92% with 68% of patients achieving at least VGPR and 36% CR/sCR. Remarkably, 100% of patients treated with 600⨰106 obtained a MRD negativity at level of 10−5 at three months. The safety profile was encouraging, since grade 3–4 CRS was 3% and neurologic toxicity 2%. Most common grade 3–4 side effects were neutropenia (90%) and thrombocytopenia (47%).
Other CAR-T Cell Products
Several other BCMA-targeted CAR-T cell products are under investigation in phase I/II studies, starting from bb21217, constructed on the basis of bb2121 CAR structure with an extra domain of bb007 to enrich for T cells displaying a memory-like phenotype to allow a longer persistence. A phase I CRB-402 study85 evaluated bb21217 in 62 patients with a median of six prior lines of therapy (range: 3–17; 64% triple-refractory) who received CAR-T cells dose ranging from 150 to 450⨰106. Overall, ORR was 68% with 54% achieving at least VGPR and 29% CR/sCR. Median duration of response was 11.9 months across different target dose levels. With regards to toxicities, cytopenias were common and not dose related, infections occurred in 26% of patients whereas grade 3–4 neurotoxicity was rare (4%) as was severe CRS (4%). CT053 is another second-generation CAR-T product incorporating a fully human anti-BCMA scFv, less immunogenic, a 4-1BB co-stimulatory motif and a CD3ζ domain. This product, manufactured in nearly 8–10 days, was at first assessed in a phase I study in China86 and after a follow-up of 24 months, among 24 patients enrolled in one year, ORR was 87% including 79% with CR/sCR and median PFS 18.8 months. In the USA, the phase I trial Ib/II Lummicar-2 study,87 using CT053, has enrolled 20 patients to date. Preliminary data presented at the last ASH meeting showed the achievement of ORR in 94% of patients with 11/12 evaluable patients obtaining MRD negativity at level of 10−5. Very interesting is the phase I/II PRIME study88 assessing P-BCMA-101, a novel anti-BCMA CAR-T cell product manufactured with transposons instead of viral vectors to increase efficacy and minimize toxicity. Bispecific CAR-T cells targeting BCMA and another tumor antigen are under development and preliminary data from the first-in-human study evaluating GC012F, a dual BCMA/CD19 targeted CAR-T cells with a platform enabling 24–36-h manufacturing, are very encouraging.89
Finally, the phase I Universal study90 is evaluating allogenic CAR-T cells therapy, allowing a standard quality product in a short time.
Emerging Drugs
Selinexor
Exporting-1 (XPO-1) is the major regulator of intracellular oncoprotein transport and it is overexpressed in MM plasma cells.91 It induces nuclear retention of tumor suppression protein and suppresses oncoprotein expression (Figure 3).92 Selinexor is an oral, potent XPO-1 inhibitor that has been investigated in clinical trials as monotherapy and in combinations. The STORM trial is a phase II study93 in which 122 heavily pretreated MM patients (seven median prior lines of therapy; range: 3–18), most of them (68%) penta-refractory, were treated with selinexor and dexamethasone. In this difficult-to-treat population ORR was 26%, PFS 3.7 months and OS 8.6 months. Hematological and gastrointestinal toxicities were of concern in this trial. Another phase I/II trial termed STOMP,94 evaluated the combination of selinexor, bortezomib and dexamethasone in patients with less advanced disease. This regimen allowed an ORR of 63% and a PFS of nine months. This study was preparatory to the phase III BOSTON trial95 that compared standard of care bortezomib-dexamethasone (Vd) with selinexor-bortezomib-dexamethasone (SVd) in 402 patients treated with one to three prior lines of therapy. Triplet therapy containing selinexor was significantly better in terms of PFS (13.9 vs 9.5; HR=0.70; p=0.0075) although the announced high rate of gastrointestinal toxicity was confirmed. This trial was updated at the last ASH meeting, with regards to particular subgroups of patients such as elderly or frail patients,96 high-risk cytogenetic patients,97 prior PI or lenalidomide exposure98 and >1 prior therapy.99
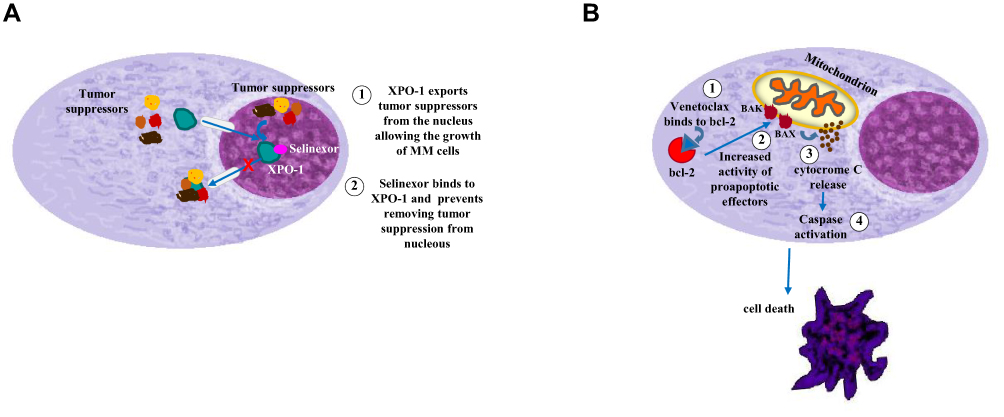 |
Figure 3 Mechanism of action of selinexor (A) and venetoclax (B). |
Triplet therapy containing selinexor performed better than Vd alone in any of these prespecified subgroups in terms of response, PFS and TTNT although adverse events were sometimes more frequent.
An update of the STOMP trial was also presented at the 2020 ASH meeting, particularly selinexor plus dexamethasone in combination with carfilzomib100 or pomalidomide.101 In the first study,100 the MTD for triplet combination (SKd) was once-weekly selinexor 80 mg, carfilzomib 56 mg/m2 and dexamethasone 40 mg. In patients treated with a median of three prior lines of therapy (range: 1–8), ORR was 75% (CR 20.8%) in the total 24 patients and 57% in patients pretreated with daratumumab. Median follow-up is too short for PFS evaluation. Besides the well-known gastrointestinal toxicity, thrombocytopenia was of concern with this combination, requiring TPO receptor antagonist to maintain the drug schedule. Regarding the combination with pomalidomide (SPd),101 MTD was achieved at pomalidomide 4 mg days 1–21 and selinexor 60 mg once weekly. Characteristics of patients were similar to the population treated with SKd, with a median of three prior lines of therapy (range: 1–10). Overall, ORR was 60% at the recommended phase 2 dose (RP2D), with a PFS of 12.2 months for all patients and not reached for RP2D patients. Main toxicities were hematologic and gastrointestinal.
Selinexor is a convenient oral drug that broadens the therapeutic armamentarium for triple refractory RRMM patients although its suboptimal gastrointestinal tolerability may be a limitation.
Melfuflen
Melphalan-flufenamide (melflufen) is a first-in-class peptide-drug conjugate targeting intracellular aminopeptidases.102 Melflufen, being highly lipophilic, passively diffuses across cellular membrane, binds to aminopeptidases and releases melphalan which remains inside the cells being hydrophilic. It penetrates into the nucleus and induces DNA damage leading to cellular apoptosis.103
After the phase I/II trial (O-12-M1)104 established melflufen 40 mg on day one in 28-day cycles as the MTD in combination with dexamethasone 40 mg weekly, phase 2 HORIZON (OP-106) trial105 included 157 patients with RRMM who had received ≥2 prior lines of therapy (median of five prior lines), and were triple-class refractory defined as refractory to one IMiD, one PI and anti-CD3 mAb. This trial included patients who had extramedullary disease (EMD) (35%) and/or cytogenetic high-risk features (38%). Patients received melflufen 40 mg of on day one of each 28-day cycle along with 40 mg of dexamethasone weekly (20 mg for patients aged ≥75 years). Treatment was administered until progression or unacceptable toxicity. ORR was the primary endpoint, while secondary objectives included progression-free survival (PFS), overall survival (OS) and safety. The ORR in the ITT population (n=157) was 29% while that in the triple-class refractory group (n=119) was 26% and 24% in the EMD group (n=55). The median PFS in ITT population was 4.2 months, the median OS was 11.6 months. Median PFS and OS were 2.9 months and 6.5 in patients with EMD. Grade 3/4 adverse events were mainly thrombocytopenia, neutropenia and pneumonia.
This trial was recently updated at the last ASH meeting regarding subgroups such as the elderly (n=25; age ≥75 years)106 high-risk (n=59),107 EMD (n=55)108 and alkylators refractory (n=92)109 patients. All these populations had results consistent with that of the ITT population one ORR rate, median PFS and OS as well as safety profile.
The phase III OCEAN trial (OP-103) (NCT03151811) is randomizing patients with RRMM who had received two to four prior lines of therapy and who were refractory to lenalidomide in the last line of therapy to receive melflufen 40 mg on day one every 28 days plus dexamethasone 40 mg (or 20 mg if aged >75 years) days 1, 8, 15, 22 or pomalidomide 4 mg on days 1–21 plus dexamethasone 40 mg (or 20 mg if aged >75 years) days 1, 8, 15, 22. The trial is ongoing and estimated enrollment is 450 patients until March 2021.
ANCHOR is the name of a phase I/II trial110 evaluating activity and safety of two triplet therapies including melflufen and dexamethasone plus bortezomib or daratumumab in patients with RRMM treated with one to four prior lines of therapy and who were refractory to lenalidomide and/or bortezomib (in the daratumumab arm). Most patients were exposed to alkylating agents. In the dose-escalation phase, no DLT was achieved. Interim analysis in the daratumumab arm included 33 patients (median two prior lines of therapy, range: 1–4) showed an ORR of 73% and a PFS of 12.9 months after a median follow-up of 18.9 months. Main grade 3–4 treatment related adverse events were thrombocytopenia (73%), neutropenia (67%), and pneumonia (6%). In the bortezomib arm only six patients were enrolled at data cutoff and DLT was not reported. ORR was encouraging at 67% and grade 3–4 side effects were consistent with the single drug profiles.
Considering these promising results coupled with manageable toxicity profile, the phase III LIGHTHOUSE trial (NCT04649060) was planned with the intent to randomize 170 patients with three or more prior lines of therapy, refractory to IMiDs and PIs to receive melflufen-daratumumab vs daratumumab.
Triple refractory MM patients have a very poor outcome.52 Although several new drugs will be approved in the near future, melflufen, either as a single agent or in association, will have its own specific room in this setting due to its peculiar mechanism of action, its predictable and limited toxicity and its convenient monthly schedule. Efficacy in extramedullary disease could be of particular interest, although its safety and activity were demonstrated in all RRMM subgroups of patients including those exposed/refractory to alkylating agents and refractory to mAbs who will be an increasingly growing wave of patients.
CELMoDs
Cererebron E3 ligase modulators (CELMoDs) are the next-generation IMiDs that degrade Ikaros and Aiolos in a novel and more potent mechanism compared to lenalidomide and pomalidomide.111 Moreover, stimulation of immune system is even more powerful. Iberdomide (CC-220) is the first CELMoD to be evaluated in clinical trials. In a phase Ib/II dose escalation study,112 iberdomide plus dexamethasone yielded an ORR of 31% in 59 patients with RRMM treated with a median of five prior lines of therapy (range: 2–12). Similar results were obtained in the subgroups refractory to lenalidomide and to pomalidomide and daratumumab. The most common adverse events were neutropenia and infections. In a more recent phase I/II trial,113 iberdomide was studied in association with bortezomib-dexamethasone (IberVd) (n=23) and daratumumab-dexamethasone (IberDd) (n=27). Patients had received at least one prior regimen containing lenalidomide or pomalidomide and a PI in the first cohort while they had to be refractory to IMiDs and PI in at least their two prior lines of therapy in the second cohort. MTD of iberdomide was not reached in both cohorts. Overall, main grade 3–4 adverse events were neutropenia (66%) and infections (18.5%) in IberDd group and thrombocytopenia (26%) and infections (13%) in IberVd group. ORR was 42% in the daratumumab cohort and 60% in the bortezomib one. Interestingly, both regimens were active in the daratumumab and bortezomib refractory patients.
Another CELMoD currently on study is CC-92480.114 It is a novel and possibly more powerful, cereblon ligase modulator compared to iberdomide. This agent, due to its rapid and maximal degradation of target proteins Ikaros and Aiolos, exerts potent antiproliferative and tumoricidal activity in MM cell lines, including those lenalidomide and pomalidomide resistant. Moreover, CC-92480 enhanced direct antimyeloma and immune-stimulatory activities in preclinical models and showed marked synergistic effects with dexamethasone, proteasome inhibitors and mAbs. In the phase I CC-92480 MM-01 trial,115 76 patients treated with a median of six prior lines of therapy (range: 2–13), 50% triple-refractory and 36% with extramedullary disease, were enrolled to receive escalated dose of CC-92480 plus dexamethasone. MTD was achieved at 1.6 mg, so 1 mg for 14–21 days on 28 days cycle was chosen for the recommended phase 2 dose. ORR in the ITT population was 20% but, in the expansion phase at the MTD, it increased to 54.5% (27% ≥VGPR) in 11 patients most of them triple-refractory and/or with extramedullary disease. Main adverse events were neutropenia, thrombocytopenia, pneumonia and febrile neutropenia. The expansion phase of this study is ongoing and phase I/II studies combining CC-92480 with standard treatments are ongoing.
Ibrutinib
Ibrutinib is an oral Bruton's tyrosine kinase (BTK) inhibitor approved for the B cell malignancies such as LLC, MCL and WM116 playing a central role in survival and proliferation of B cell lineage. BTK is overexpressed in MM cell favoring proliferation and migration of plasma cells, bone destruction, and dexamethasone resistance. Moreover, ibrutinib enhances activity of PIs and IMiDs.117 In a phase I/II study,118 92 patients prior treated with a median of four lines of therapy underwent increasing doses of ibrutinib and dexamethasone. MTD was reached at 840 mg/daily of ibrutinib. Clinical benefit rate, the primary endpoint of the study, was 28% with DOR of 11 months. PFS was 4.5 months. Main toxicity was hematological whereas nonhematological adverse events (infections and syncope) were rare. Therefore, another phase I/II trial119 was conducted with ibrutinib in association with carfilzomib and dexamethasone in 59 patients, previously treated with a median of three lines of therapy and most of them double refractory. MTD was ibrutinib 840 mg/daily, carfilzomib 36 mg/m2 twice weekly and dexamethasone 20 mg. ORR was 71% and PFS was 7.4 months (very similar to those in high-risk patients: 67% and 7.7 months). No unexpected side effects were seen with this association.
Anti-PD1
Immune checkpoints are a potential target for MM therapy since they are overexpressed in MM plasma cells conferring drug resistance and immune reaction imbalance. After promising phase I/II studies120,121 demonstrated synergism of action with IMiDs, two randomized trials, comparing Rd vs Rd-pembrolizumab (KEYNOTE 185)122 in first-line therapy and Pd vs Pd-pembrolizumab (KEYNOTE 183)123 in relapsed-refractory MM, concluded in an interim unplanned analysis that led to halt the studies, that both triple therapies had an unfavorable risk–benefit profile, due to an inferior PFS and an excess of attributable mortality in the pembrolizumab arm. The future of this therapeutic strategy is uncertain although it has not been abandoned.
Emerging Targeted Drugs
Advances in MM biology knowledge paved the way to transition from patient- and drug characteristic-guided therapy to biomarker-driven therapy, ie from personalized to precision medicine.
MM is a very heterogeneous disease in which no single, or few disease, drivers were identified. Moreover, intraclonal and clonal evolution of disease complicate the scenario.124 Nevertheless, recurrent gene mutations such as those of MYK, KRAS, NRAS and BRAF activating MAP kinase pathway, and of TP53 were recognized.125 Recently, specific compounds targeting these alterations were developed and studied in preclinical and clinical trials.
Inhibition of MAPK pathway by vemurafenib and trametinib, MEK1/2 inhibitors, showed potent reduction of cancer cell growth in vitro. In the phase I VE-Basket study126 nine patients with very advanced MM and BRAFV600 mutation were treated with vemurafenib monotherapy. ORR was 33% and PFS was 4.6 months. Main adverse events were alopecia, skin disorders and infections. Another trial127 explored trametinib monotherapy or with standard therapy in 40 real-life heavily pretreated patients with MM carrying the BRAF mutation. Ten percent of patients receiving trametinib monotherapy had a response whereas 57% of them responded with trametinib in association. Skin rash, diarrhea, and cardiac toxicity were the main side effects leading to not negligible drug discontinuation.
Seeing the poor effectiveness of these compounds in monotherapy, My-DRUG Umbrella trial (NCT03732703) is assessing several drugs targeting: RAF/RAS mutations (cobimetinib), IDH activating mutations (enasidenib), CDK pathway activating alterations (abemaciclib, dinaciclib), FGRF3 activating alterations (erdafitinib), in combination with dexamethasone and with ixazomib-pomalidomide-dexamethasone in high-risk RRMM patients based on the MMRF CoMMpass study that recognized 12 different molecular types of MM by genomic sequencing. Results of this trial are highly awaited. A phase I study128 with cobimetinib single drug or associated with venetoclax and with atezolizumab is ongoing but response was seen only in patients with t(11;14) in the venetoclax cohort.
Deletion of chromosomal 17p (del17p) is a well-known marker of high-risk MM although the population of del17 MM is heterogeneous due to variability in cytogenetic assay cutoff. The TP53 gene, affecting tumor suppression function of the proteins, is included in 17p chromosome and its aberration or deletion led to inactivation of its anti-oncogenic function. Biallelic inactivation of TP53 is considered one of the worst adverse prognostic factors in MM since it provided genomic instability resulting in tumor progression and drug resistance. Consequently, TP53 is now considered as a suitable target for MM therapy particularly inhibition of MDM2 cascade by nutlin compounds.129 Idasanutlin is a NDM2 antagonist that activates TP53, blocks proliferation and induces apoptosis. A Phase I/II study in combination with ixazomib and dexamethasone is ongoing (NCT02633059). MCL-1 promotes cell survival by its antiapoptotic action. Hyperexpression of MCL-1 confers resistance of proapoptotic drugs such as bortezomib and venetoclax. Studies targeting MCL-1 with AZD5991 (NCT03218683) and AMG176 (NCT02675452) are ongoing and combination studies are in project.
Venetoclax
Anti-BCL-2 targeted therapy was the first in class biomarker-guided therapy (Figure 3). As in other hematological malignancies, overexpression of BCL-2 protein inhibits apoptosis so representing one of the most frequent mechanism of resistance also in MM. In particular, MM harboring t(11;14) translocation is associated with increase dependency upon BCL-2 for plasma cell survival.130 Venetoclax is a selective, potent, oral BCL-2 inhibitor recently approved for treatment of CLL and AML.131 In MM, phase 1b studies with venetoclax monotherapy,132 venetoclax plus dexamethasone133 and venetoclax plus bortezomib and dexamethasone134 yielded promising results leading to the phase III BELLINI trial135 that compared bortezomib plus dexamethasone to triplet combination venetoclax, bortezomib, dexamethasone in 291 patients, treated with one to three prior lines of therapy and not refractory to PIs. Patients receiving triplet therapy containing venetoclax had a significantly longer PFS, the primary endpoint of the trial, compared with patients receiving doublet (median 22.4 vs 11.5 months; HR=0.63; p=0.010). However, a substantial proportion of early deaths due to infection occurring in patients treated with venetoclax led to a significantly worse OS (HR=2.03; p=0.034). Remarkably, impressive prolonged PFS was observed in patients with t(11;14) or high BCL-2 expression (median PFS not reached vs 9.9 months; HR=0.30; p<0.001) without significantly differences in term of toxicity, particularly in early mortality as happened in the ITT population. This result confirms that venetoclax is a selective drug to use in patients with t(11;14) or high BCL-2 expression as indicated by development drug plan that includes a Phase III trial (CANOVA; NCT03539744) comparing venetoclax plus dexamethasone vs pomalidomide plus dexamethasone in patients with RRMM with the above-mentioned characteristics.
Conclusions
MM is a complex disease difficult to render chronic or to cure despite the paramount improvements achieved so far with the introduction of chemoimmunotherapy. Patients refractory to IMiDs and/or PIs benefit now from combinations containing anti-CD38 mAbs and from CELMoDs in the near future. Triple-refractory or penta-refractory patients benefit now from belantamab-mafodotin or selinexor and from CAR-T cells and or BiTE in the near future. Targeted therapy with venetoclax is a reality whereas the possibilities of using other compounds are unknown yet.
However, this scenario is in a constant evolution based on the results that gradually come from ongoing trials that informed us on the MRD, survival parameters, toxicity, and feasibility of these complex and expensive therapies. Really, many questions remain open: firstly, how to achieve the best sequencing of therapy in MM, particularly regarding retreatment with anti-CD38 mAbs and, secondly, how to position conjugated mAbs, BiTE and CAR-T cells, which are in competition for the same target named BCMA? Moreover, could high-risk disease benefit from combinations of conventional drugs and compounds that target specific genetic lesions? Will it be possible to modulate these therapies on the basis of MRD or other markers, avoiding continuous therapy?
The future of MM therapy is a fascinating challenge to achieve a cure and a good quality of life for all our patients.
Disclosure
Dr Massimo Offidani reports grants Amgen, BMS, Celgene, Janssen, GSK, Sanofi, and Takeda, during the conduct of the study. Dr Laura Corvatta reports grants from Janssen, Celgene, and Amgen, during the conduct of the study. Dr Sonia Morè reports grants from Amgen, during the conduct of the study. The authors report no other conflicts of interest in this work.
References
1. National Cancer Institute. Cancer stat facts: myeloma. Available from: https://seer.cancer.gov/statfacts/html/mulmy.html. Accessed Dec 01, 2020.
2. Anderson KC. Progress and paradigms in multiple myeloma. Clin Cancer Res. 2016;22:5419–5427. doi:10.1158/1078-0432.CCR-16-0625
3. Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–2520. doi:10.1182/blood-2007-10-116129
4. Kumar SK, Dispenzieri A, Lacy MQ, et al. Continued improvement in survival in multiple myeloma: changes in early mortality and outcomes in older patients. Leukemia. 2014;28:1122–1128. doi:10.1038/leu.2013.313
5. Kronke J, Udeshi ND, Narla A, et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343:301–305. doi:10.1126/science.1244851
6. Davies F, Baz R. Lenalidomide mode of action: linking bench and clinical findings. Blood Rev. 2010;24(Suppl 1):S13–S19. doi:10.1016/S0268-960X(10)70004-7
7. Chauhan D, Hideshima T, Mitsiades C, et al. Proteasome inhibitor therapy in multiple myeloma. Mol Cancer Ther. 2005;4:686–692. doi:10.1158/1535-7163.MCT-04-0338
8. Hideshima T, Mitsiades C, Akiyama M, et al. Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood. 2003;101:1530–1534. doi:10.1182/blood-2002-08-2543
9. Ito S. Proteasome inhibitors for the treatment of multiple myeloma. Cancers. 2020;12:265. doi:10.3390/cancers12020265
10. Moreau P, San Miguel J, Sonneveld P, et al. Multiple myeloma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2017;28(Suppl. 4):iv52–iv61. doi:10.1093/annonc/mdx096
11. Mikhael J, Ismaila N, Cheung MC, et al. Treatment of multiple myeloma: ASCO and CCO joint clinical practice guidelines. J Clin Oncol. 2019;37:1228–1263. doi:10.1200/JCO.18.02096
12. Nandakumar B, Kapoor P, Binder M, et al. Continued improvement in survival of patients with newly diagnosed multipole myeloma. Blood. 2020;136(Suppl. 1):abstract 2280. doi:10.1182/blood-2020-140388
13. Kumar SK, Dimopoulos MA, Kastritis E, et al. Natural history of relapsed myeloma, refractory to immunomodulatory drugs and proteasome inhibitors: a multicenter IMWG study. Leukemia. 2017;31:2443–2448. doi:10.1038/leu.2017.138
14. Coiffier B, Thieblemont C, Van Den Neste E, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood. 2010;116:2040–2045. doi:10.1182/blood-2010-03-276246
15. Connors JM, Jurczak W, Strauss DJ, et al. Brentuximab vedotin with chemotherapy for stage III or IV Hodgkin’s lymphoma. N Engl J Med. 2018;378:331–344. doi:10.1056/NEJMoa1708984
16. Offidani M, Corvatta L. A review discussing elotuzumab and its use in the second-line plus treatment of multiple myeloma. Future Oncol. 2018;14:319–329. doi:10.2217/fon-2017-0371
17. Lonial S, Dimopoulos MA, Palumbo A, et al. Elotuzumab therapy for relapsed or refractory multiple myeloma. N Engl J Med. 2015;373:621–631. doi:10.1056/NEJMoa1505654
18. Dimopoulos MA, Dytfeld D, Grosicki S, et al. Elotuzumab plus pomalidomide and dexamethasone for multiple myeloma. N Engl J Med. 2018;379:1811–1822. doi:10.1056/NEJMoa1805762
19. Dimopoulos MA, Lonial S, Betts KA, et al. Elotuzumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: extended 4-year follow-up and analysis of relative progression-free survival from the randomized ELOQUENT-2 trial. Cancer. 2018;124:4032–4043. doi:10.1002/cncr.31680
20. Dimopoulos MA, Lonial S, White D, et al. Elotuzumab, lenalidomide and dexamethasone in RRMM: final overall survival results from the Phase 3 randomized ELOQUENT-2 study. Blood Cancer J. 2020;10:91. doi:10.1038/s41408-020-00357-4
21. Bristol-Myers Squibb. Bristol-Myers Squibb reports primary results of ELOQUENT-1 study evaluating Empliciti (elotuzumab) plus Revlimid (lenalidomide) and dexamethasone in patients with newly diagnosed, untreated multiple myeloma. Available from: https://news.bms.com/news/corporate-financial/2020/Bristol-Myers-Squibb-Reports-Primary-Results-of-ELOQUENT-1-Study-Evaluating-Empliciti-elotuzumab-Plus-Revlimid-lenalidomide-and-Dexamethasone-in-Patients-with-Newly-Diagnosed-Untreated-Multiple-Myeloma/default.aspx. Accessed Dec 01, 2020.
22. Goldschmidt H, Mai EK, Salwender HJ, et al. Bortezomib, lenalidomide and dexamethasone with or without elotuzumab as induction therapy for newly diagnosed, transplant-eligible multiple myeloma. Hemasphere. 2020;4(S1):abstract S 203.
23. Lokhorst HM, Plesner T, Laubach JP, et al. Targeting CD38 with daratumumab monotherapy in multiple myeloma. New Engl J Med. 2015;373:1207–1219. doi:10.1056/NEJMoa1506348
24. Lonial S, Weiss BM, Usmani SZ, et al. Daratumumab monotherapy in patients with treatment-refractory multiple myeloma (SIRIUS): an open-label, randomised, phase 2 trial. Lancet. 2016;387:1551–1560. doi:10.1016/S0140-6736(15)01120-4
25. Nooka AK, Kaufman JL, Hofmeister CC, et al. Daratumumab in multiple myeloma. Cancer. 2019;125:2364–2382. doi:10.1002/cncr.32065
26. Dimopoulos MA, Oriol A, Nahi H, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. New Engl J Med. 2016;375:1319–1331. doi:10.1056/NEJMoa1607751
27. Palumbo A, Chanan-Khan A, Weisel K, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. New Engl J Med. 2016;375:754–766. doi:10.1056/NEJMoa1606038
28. Dimopoulos MA, Terpos E, Boccadoro M, et al. Apollo: phase 3 randomized study of subcutaneous daratumumab plus pomalidomide and dexamethasone (D-Pd) versus pomalidomide and dexamethasone (Pd) alone in patients with relapsed/refractory multiple myeloma. Blood. 2020;136(Suppl 1):abstract 412. doi:10.1182/blood-2020-135874
29. Dimopoulos MA, Quach H, Mateos M-V, et al. Carfilzomib, dexamethasone and daratumumab versus carfilzomib and dexamethasone in relapsed or refractory multiple myeloma: updated efficacy and safety results of the phase 3 Candor study. Blood. 2020;136(Suppl 1):abstract 2325. doi:10.1182/blood-2020-137602
30. Moreau P, Attal M, Hulin C, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet. 2019;394:29–38. doi:10.1016/S0140-6736(19)31240-1
31. Voorhees PM, Kaufman JL, Laubach J, et al. Daratumumab, lenalidomide, bortezomib, and dexamethasone for transplant-eligible newly diagnosed multiple myeloma: the GRIFFIN trial. Blood. 2020;136:936–945. doi:10.1182/blood.2020005288
32. Mateos MV, Dimopoulos MA, Cavo M, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2018;378:518–528. doi:10.1056/NEJMoa1714678
33. Kumar SK, Facon T, Usmani SZ, et al. Updated analysis of daratumumab plus lenalidomide and dexamethasone (D-Rd) versus lenalidomide and dexamethasone (Rd) in patients with transplant-ineligible newly diagnosed multiple myeloma: the phase 3 Maia study. Blood. 2020;136(Suppl 1):abstract 2276. doi:10.1182/blood-2020-134847
34. Mateos MV, Nahi H, Legiec W, et al. Subcutaneous versus intravenous daratumumab in patients with relapsed or refractory multiple myeloma (COLUMBA): a multicentre, open-label, non-inferiority, randomised, phase 3 trial. Lancet Haematol. 2020;7:e370–e380. doi:10.1016/S2352-3026(20)30070-3
35. Chari A, Rodriguez-Otero P, McCarthy H, et al. Subcutaneous daratumumab plus standard treatment regimens in patients with multiple myeloma across lines of therapy (PLEIADES): and open-label Phase II study. Br J Haematol. 2020
36. Van de Domnk NW, Usmani SZ. CD38 antibodies in multiple myeloma: mechanisms of action and modes of resistance. Front Immunol. 2018;9:2134. doi:10.3389/fimmu.2018.02134
37. Van Bueren JL, Jakobs D, Kaldenhoven N, et al. Direct in vitro comparison of daratumumab with surrogate analogs of CD38 antibodies MOR03087, SAR650884 and Ab79. Blood. 2014;124(21):abstract 3474. doi:10.1182/blood.V124.21.3474.3474
38. Feng X, Zhang L, Acharya C, et al. Targeting CD38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res. 2017;23:4290–4300. doi:10.1158/1078-0432.CCR-16-3192
39. Martin TG, Corzo K, Chiron M, et al. Therapeutic opportunities with pharmacological inhibition of CD38 with isatuximab. Cells. 2019;8:1522. doi:10.3390/cells8121522
40. Attal M, Richardson PG, Rajkumar SV, et al. Isatuximab plus pomalidomide and low-dose dexamethasone versus pomalidomide and low-dose dexamethasone in patients with relapsed and refractory multiple myeloma (ICARIA-MM): a randomised, multicentre, open-label, phase 3 study. Lancet. 2019;394:2096–2107. doi:10.1016/S0140-6736(19)32556-5
41. Schjesvold F, Bringhen S, Richardson PG, et al. Isatuximab plus pomalidomide and dexamethasone in frail patients with relapsed/refractory multiple myeloma: icaria-MM subgroup analysis. Blood. 2020;136(Suppl 1):abstract 1411.
42. Moreau P, Dimopoulos MA, Mikhael J, et al. Isatuximab plus carfilzomib and dexamethasone vs carfilzomib and dexamethasone in relapsed/ refractory multiple myeloma (IKEMA): interim analysis of a phase 3, randomized, open-label study. Blood. 2020;136(Suppl 1):abstract 2316. doi:10.1182/blood.2020008150
43. Martin T, Mikhael J, Hajek R, et al. Depth of response and response kinetics of isatuximab plus carfilzomib and dexamethasone in relapsed/refractory multiple myeloma: IKEMA interim analysis. Blood. 2020;136(Suppl 1):abstract 414. doi:10.1182/blood-2020-137681
44. Weisel KC, Asemissen AM, Besemer B, et al. Depth of response to isatuximab, carfilzomib, lenalidomide and dexamethasone (Isa-KRd) in front-line treatment of high-risk multiple myeloma: interim analysis of the GMMG-CONCEPT trial. J Clin Oncol. 2020;38(Suppl.):abstract 8505. doi:10.1200/JCO.2020.38.15_suppl.8508
45. Ocio EM, Bringhen S, Oliva S, et al. Updates from a phase Ib study of isatuximab, bortezomib, and dexamethasone plus cyclophosphamide or lenalidomide in transplant ineligible newly diagnosed multiple myeloma. Blood. 2020;136(Suppl 1):abstract 2288. doi:10.1182/blood-2020-136569
46. Raab MS, Engelhardt M, Blank A, et al. MOR202, a novel anti-CD38 monoclonal antibody, in patients with relapsed or refractory multiple myeloma: a first-in-human, multicenter, phase 1-2a trial. Lancet Hematol. 2020;7:e381–394. doi:10.1016/S2352-3026(19)30249-2
47. Ghobrial IM, Liu C-J, Redd R, et al. A phase Ib/II trial of the first-in-class anti-CXCR4 antibody ulocuplumab in combination with lenalidomide or bortezomib plus dexamethasone in relapsed multiple myeloma. Clin Cancer Res. 2020;26:344–353. doi:10.1158/1078-0432.CCR-19-0647
48. Franssen LE, Stege CAM, Zweegman S, et al. Resistance mechanisms towards CD38-directed antibody therapy in multiple myeloma. J Clin Med. 2020;9:1195. doi:10.3390/jcm9041195
49. Paul B, Symanowski J, Osipoff P, et al. A phase 2 trial of daratumumab and pembrolizumab in refractory multiple myeloma. Blood. 2020;136(Suppl 1):abstract 1399. doi:10.1182/blood-2020-141623
50. Kambhampati S, Wong SW, Martin T, et al. Phase II study of daratumumab in combination with azacitidine and dexamethasone in relapsed/refractory multiple myeloma previously treated with daratumumab: darazadex. Blood. 2020;136(Suppl 1):abstract 2326. doi:10.1182/blood-2020-138869
51. Perez De Acha O, Idler BI, Walker ZJ, et al. Myeloma drug sensitivity testing to optimize retreatment with anti-CD38 monoclonal antibodies in daratumumab-refractory patients. Blood. 2020;136(Suppl 1):abstract 1387. doi:10.1182/blood-2020-137819
52. Gandhi UH, Cornell RF, Lakshman A, et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia. 2019;33:2266–2275. doi:10.1038/s41375-019-0435-7
53. Dalla Palma B, Marchica V, Catarozzo MT, Giuliani N, Accardi F. Monoclonal and bispecific anti-BCMA antibodies in multiple myeloma. J Clin Med. 2020;9:3022. doi:10.3390/jcm9093022
54. Tai Y-T, Li XF, Breitkreutz I, et al. Role of B-cell–activating factor in adhesion and growth of human multiple myeloma cells in the bone marrow microenvironment. Cancer Res. 2006;66:6675–6682. doi:10.1158/0008-5472.CAN-06-0190
55. Moreaux J, Legou E, Jourdan E, et al. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood. 2004;103:3148–3157. doi:10.1182/blood-2003-06-1984
56. Lancman G, Richter J, Chari A. Bispecific, trispecific, and other novel immune treatments in myeloma. Hematology. 2020;2020:264–271. doi:10.1182/hematology.2020000110
57. Topp MS, Duell J, Zugmaier G, et al. Anti-B-cell maturation antigen BiTE molecule AMG 420 induces responses in multiple myeloma. J Clin Oncol. 2020;38:775–778. doi:10.1200/JCO.19.02657
58. Harrison JS, Minnema MC, Lee HC, et al. A phase 1 first in Human (FIH) study of AMG 701, an anti-B-Cell maturation antigen (BCMA) half-life extended (HLE) BiTE® (bispecific T-cell engager) molecule, in relapsed/refractory (RR) multiple myeloma (MM). Blood. 2020;136(Suppl 1):abstract 181. doi:10.1182/blood-2020-134063
59. Usmani SZ, Mateos M-V, Nahi H, et al. Phase I study of teclistamab, a humanized B-cell maturation antigen (BCMA) x CD3 bispecific antibody, in relapsed/ refractory multiple myeloma (R/R MM). J Clin Oncol. 2020;38(Suppl.):abstract 100. doi:10.1200/JCO.2020.38.15_suppl.100
60. Garfall AL, Usmani SZ, Mateos M-V, et al. Updated phase 1 results of teclistamab, a B-Cell Maturation Antigen (BCMA) x CD3 bispecific antibody, in relapsed and/or refractory multiple myeloma (RRMM). Blood. 2020;136(Suppl 1):abstract 27. doi:10.1182/blood-2020-138831
61. Costa LJ, Wong SW, Bermudez A, et al. First clinical study of the B-Cell Maturation Antigen (BCMA) 2+1 cell engager CC-93269 in patients with relapsed/refractory multiple myeloma: interim results of a phase 1 multicenter trial. Blood. 2019;134(Suppl. 1):abstract 143. doi:10.1182/blood-2019-122895
62. Atamaniuk J, Gleiss A, Porpaczy E, et al. Overexpression of G protein coupled receptor 5D in the bone marrow is associated with poor prognosis in patients with multiple myeloma. Eur J Clin Invest. 2012;42:953–960. doi:10.1111/j.1365-2362.2012.02679.x
63. Chari A, Berdeja JG, Oriol A, et al. A phase 1, first-in-human study of Talquetamab, a G Protein-Coupled Receptor Family C Group 5 Member D (GPRC5D) x CD3 bispecific antibody, in patients with relapsed and/or refractory multiple myeloma (RRMM). Blood. 2020;136(Suppl 1):abstract 290. doi:10.1182/blood-2020-133873
64. Cohen AD, Harrison SJ, Krishnan A, et al. Initial clinical activity and safety of BFCR4350A, a FcRH5/CD3 T-Cell-Engaging bispecific antibody, in relapsed/ refractory multiple myeloma. Blood. 2020;136(Suppl 1):abstract 292. doi:10.1182/blood-2020-136985
65. Sheikh S, Lebel E, Trudel S. Belantamab mafodotin in the treatment of relapsed or refractory multiple myeloma. Fut Oncol. 2020;16:2783–2798. doi:10.2217/fon-2020-0521
66. Trudel S, Lendvai N, Popat R, et al. Antibody–drug conjugate, GSK2857916, in relapsed/refractory multiple myeloma: an update on safety and efficacy from dose expansion phase I study. Blood Cancer J. 2019;9:37. doi:10.1038/s41408-019-0196-6
67. Lonial S, Lee HC, Badros A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): a two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020;21:207–221. doi:10.1016/S1470-2045(19)30788-0
68. Lonial S, Nooka A, Thulasi P, et al. Recovery of ocular events with longer-term follow-up in the DREAMMM-2 Study of single-agent belantamab mafodotin (Belamaf) in patients with relapsed or refractory multiple myeloma (RRMM). Blood. 2020;136(Suppl. 1):abstract 3224. doi:10.1182/blood-2020-140078
69. Popat R, Lonial S, Voorhees PM, et al. DREAMM-2: single-agent belantamab mafodotin (Belamaf) effects on patient-reported outcome (PRO) measures in patients with relapsed/refractory multiple myeloma (RRMM). Blood. 2020;136(Suppl. 1):abstract 2278.
70. Nooka A, Lee HC, Badros AZ, et al. Infusion-related reactions (IRRs) in the DREAMM-2 study of single-agent belantamab mafodotin (Belamaf) in patients with relapsed/refractory multiple myeloma (RRMM). Blood. 2020;(Suppl. 1):abstract 3221.
71. Trudel S, McCurdy A, Sutherland HJ, et al. Part 1 results of a dose finding study of belantamab mafodotin (GSK2857916) in combination with pomalidomide and dexamethasone for the treatment of relapsed/refractory multiple myeloma. Blood. 2020;(Suppl. 1):abstract 725.
72. Sadelain M, Brentjens R, Riviere I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013;3:338–398. doi:10.1158/2159-8290.CD-12-0548
73. Guedan S, Calderon H, Posey AD, Maus MV. Engineering and design of chimeric antigen receptors. Mol Ther Methods Clin Dev. 2018;12:145–156. doi:10.1016/j.omtm.2018.12.009
74. Mikkilinemi L, Kochenderfer JN. Chimeric antigen receptor T-cell therapies for multiple myeloma. Blood. 2017;130:2594–2602. doi:10.1182/blood-2017-06-793869
75. Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2016;4:92–101. doi:10.1016/j.omtm.2016.12.006
76. Sha UA, Mailankody S. Emerging immunotherapies in multiple myeloma. BMJ. 2020;370:m3176. doi:10.1136/bmj.m3176
77. Raje N, Berdeja J, Lin Y, et al. Anti-BCMA CAR T-cell therapy bb2121 in relapsed or refractory multiple myeloma. N Engl J Med. 2019;380:1726–1737. doi:10.1056/NEJMoa1817226
78. Lin Y, Raje NS, Berdeja JG, et al. Idecabtagene vicleucel (ide-cel, bb2121), a BCMA-directed CAR T cell therapy, in patients with relapsed and refractory multiple myeloma: updated results from phase 1 CRB-401 study. Blood. 2020;(Suppl. 1):abstract 131. doi:10.1182/blood.2020006245
79. Munshi NC, Anderson LD, Shah N, et al. Idecabtagene vicleucel (ide-cel, bb2121), a BCMA-targeted CAR T cell therapy, in patients with relapsed and refractory multiple myeloma: initial KarMMa results. J Clin Oncol. 2020;38(Suppl.):abstract 8503. doi:10.1200/JCO.2020.38.15_suppl.8503
80. Berdeja J, Raje NS, Siegel DS, et al. Efficacy and safety of idecabtagene vicleucel (ide-cel, bb2121) in elderly patients with relapsed and refractory multiple myeloma: karMMa subgroup analysis. Blood. 2020;(Suppl. 1):abstract 1367. doi:10.1182/blood.2020007522
81. Wang B-Y, Zhao W-H, Liu J, et al. Long-term follow-up of a phase 1, first-in-human open-label study of LCAR-B38M, a structurally differentiated chimeric antigen receptor T (CAR-T) cell therapy targeting B-Cell maturation antigen (BCMA) in patients with relapsed/refractory multiple myeloma. Blood. 2019;134(Suppl. 1):abstract 579. doi:10.1182/blood-2019-124953
82. Berdeja JG, Madduri D, Usmani SZ, et al. Update of CARTITUDE-1: a phase 1b/2 study of JNJ-4528, a B-cell maturation antigen (BCMA)-directed ccAR-T cell therapy, in relapsed/ refractory multiple myeloma. J Clin Oncol. 2020;38(Suppl.):abstract 8505. doi:10.1200/JCO.2020.38.15_suppl.8505
83. Madduri D, Berdeja JG, Usmani SZ, et al. CARTITUDE-1: phase 1b/2 study of ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T cell therapy, in relapsed/ refractory multiple myeloma. Blood. 2020;(Suppl. 1):abstract 177.
84. Mailankody S, Jakuboviak A, Htut M, et al. Orvacabtagene autoleucel (orva-cel), a B-cell maturation antigen (BCMA)-directed CAR T cell therapy for patients with relapsed/refractory multiple myeloma: update of the phase 1/2 EVOLVE study (NCT03430011). J Clin Oncol. 2020;38(Suppl.):abstract 8504. doi:10.1200/JCO.2020.38.15_suppl.8504
85. Alsina M, Shah N, Raje NS, et al. Updated results from the phase I CRB-402 study of anti-BCMA CAR-T cell therapy bb21217 in patients with relapsed and refractory multiple myeloma: correlation of expansion and duration of response with T cell phenotypes. Blood. 2020;136(Suppl. 1):abstract 130. doi:10.1182/blood-2020-140410
86. Hao S, Jin J, Jiang S, et al. Two-years follow-up of investigator-initiated phase 1 trials of the safety and efficacy of fully human anti-bcma CAR T cells (CT053) in relapsed/ refractory multiple myeloma. Blood. 2020;136(Suppl. 1):abstract 132. doi:10.1182/blood-2020-140156
87. Kumar SK, Baz RC, Orlowski RZ, et al. Results from Lummicar-2: a phase 1b/2 study of fully human B-cell maturation antigen-specific CAR T cells (CT053) in patients with relapsed and/or refractory multiple myeloma. Blood. 2020;136(Suppl. 1):abstract 133. doi:10.1182/blood-2020-139802
88. Costello VCL, Cohen AD, Patel KK, et al. Phase 1/2 study of the safety and response of P-BCMA-101 CAR-T cells in patients with relapsed/refractory multiple myeloma (PRIME) with novel therapeutic strategies. Blood. 2020;136(Suppl. 1):abstract 134.
89. Jiang H, Dong B, Gao L, et al. Clinical results of a multicenter study of the first-in-human dual BCMA and CD19 targeted novel platform fast CAR-T cell therapy for patients with relapsed/refractory multiple myeloma. Blood. 2020;136(Suppl. 1):abstract 178. doi:10.1182/blood-2020-138614
90. Mailankody S, Matous JV, Liedtke M, et al. Universal: an allogeneic first-in-human study of the anti-BCMA ALLO-715 and the anti-CD52 ALLO-647 in relapsed/ refractory multiple myeloma. Blood. 2020;136(Suppl. 1):abstract 129. doi:10.1182/blood-2020-140641
91. Hill R, Cautain B, De Pedro N, et al. Targeting nucleocytoplasmic transport in cancer therapy. Oncotarget. 2014;5:11–28. doi:10.18632/oncotarget.1457
92. Gravina GL, Senapedis W, McCauley D, et al. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J Hematol Oncol. 2014;7:85. doi:10.1186/s13045-014-0085-1
93. Chari A, Vogl DT, Gavriatopoulou M, et al. Oral selinexor-dexamethasone for triple-class refractory multiple myeloma. N Engl J Med. 2019;381:727–738. doi:10.1056/NEJMoa1903455
94. Bahlis NJ, Sutherland H, White D, et al. Selinexor plus low-dose bortezomib and dexamethasone for patients with relapsed or refractory multiple myeloma. Blood. 2018;132:2546–2554. doi:10.1182/blood-2018-06-858852
95. Grosicki S, Simonova M, Spicka I, et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): a randomized, open-label, phase 3 trial. Lancet. 2020;396:1563–1573. doi:10.1016/S0140-6736(20)32292-3
96. Auner HW, Gavriatopoulou M, Delimpasi S, et al. Once weekly selinexor, bortezomib, and dexamethasone versus twice weekly bortezomib and dexamethasone in relapsed or refractory multiple myeloma: age and frailty subgroup analyses from the phase 3 Boston study. Blood. 2020;136(Suppl 1):abstract 3215.
97. Richard S, Chari A, Delimpasi S, et al. Once weekly selinexor, bortezomib, and dexamethasone versus twice weekly bortezomib and dexamethasone in relapsed or refractory multiple myeloma: high-risk cytogenetic risk planned subgroup analyses from the phase 3 Boston study. Blood. 2020;136(Suppl 1):abstract 1385. doi:10.1182/blood-2020-137154
98. Mateos M-V, Gavriatopoulou M, Facon T, et al. Effect of prior treatment with proteasome inhibitors on the efficacy and safety of once weekly selinexor, bortezomib, and dexamethasone in comparison with twice weekly bortezomib and dexamethasone in relapsed or refractory multiple myeloma: subgroup analysis from the Boston study. Blood. 2020;136(Suppl 1):abstract 2297.
99. Mateos M-V, Jagannath S, Delimpasi S, et al. Effect of prior therapies on the safety and efficacy of once weekly selinexor, bortezomib, and dexamethasone compared with twice weekly bortezomib and dexamethasone in relapsed or refractory multiple myeloma: subgroup analysis from the Boston study. Blood. 2020;136(Suppl 1):abstract 3245.
100. Gasparetto C, Lipe B, Tuchman SA, et al. Selinexor in combination with carfilzomib and dexamethasone, all once weekly (SKd), for patients with relapsed/refractory multiple myeloma. Blood. 2020;136(Suppl 1):abstract 1366. doi:10.1182/blood-2020-137183
101. Chen CI, Bahlis NJ, Gasparetto C, et al. Selinexor in combination with pomalidomide and dexamethasone (SPd) for treatment of patients with relapsed refractory multiple myeloma. Blood. 2020;136(Suppl 1):abstract 726. doi:10.1182/blood-2020-139858
102. Chauhan D, Ray A, Viktorsson K, et al. In vitro and in vivo antitumor activity of a novel alkylating agent, melphalan-flufenamide, against multiple myeloma cells. Clin Cancer Res. 2013;19:3019–3031. doi:10.1158/1078-0432.CCR-12-3752
103. Oriol A, Larocca A, Leleu X, et al. Melflufen for relapsed and refractory multiple myeloma. Expert Opin Investig Drugs. 2020;29:1069–1078. doi:10.1080/13543784.2020.1808884
104. Richardson P, Bringhen S, Voorhees P, et al. Melflufen plus dexamethasone in relapsed and refractory multiple myeloma (0-12-M1): a multicenter, international, open-label, phase 1-2 study. Lancet Hematol. 2020;7:e395–e407. doi:10.1016/S2352-3026(20)30044-2
105. Richardson PG, Oriol A, Larocca A, et al. HORIZON (OP-106): melflufen plus dexamethasone in relapsed/refractory multiple myeloma refractory to pomalidomide and/or anti-CD38 monoclonal antibody-primary and subgroup analysis. Hemasphere. 2020;4(S1):abstract EP 945.
106. Larocca A, Richardson PG, Oriol A, et al. HORIZON (OP-106): melflufen plus dexamethasone in patients with relapsed/refractory multiple myeloma-age subgroup analysis of elderly patients. Blood. 2020;136(Suppl 1):abstract 2293. doi:10.1182/blood-2020-136856
107. Mateos M-V, Oriol A, Larocca A, et al. HORIZON (OP-106): melflufen plus dexamethasone in patients with relapsed/refractory multiple myeloma with high-risk cytogenetics-subgroup analysis. Blood. 2020;136(Suppl 1):abstract 3237. doi:10.1182/blood-2020-136002
108. Richardson PG, Mateos M-V, Oriol A, et al. HORIZON (OP-106): melflufen plus dexamethasone in 55 patients with relapsed/refractory multiple myeloma with extramedullary disease (EMD)-subgroup analysis. Blood. 2020;136(Suppl 1):abstract 3214. doi:10.1182/blood-2020-137118
109. Rodriguez-Otero P, Mateos M-V, Oriol A, et al. HORIZON (OP-106): melflufen plus dexamethasone in patients with relapsed/refractory multiple myeloma exposed to prior alkylator therapy-subgroup analysis. Blood. 2020;136(Suppl 1):abstract 2321. doi:10.1182/blood-2020-136466
110. Ocio EM, Efebera YA, Hajek R, et al. ANCHOR (OP-104): melflufen plus dexamethasone and daratumumab or bortezomib in relapsed/refractory multiple myeloma refractory to an IMiD and/or a proteasome inhibitor-updated efficacy and safety. Blood. 2020;136(Suppl 1):abstract 417. doi:10.1182/blood-2020-135991
111. Bjorklund CC, Kang J, Amatangelo M, et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia. 2020;34:1197–1201. doi:10.1038/s41375-019-0620-8
112. Lonial S, Van de Donk N, Popat R, et al. A phase 1b/2a study of the CELMoD Iberdomide (CC-220) in combination with dexamethasone in patients with relapsed/refractory multiple myeloma. Clin Lymphoma Myeloma Leuk. 2019;19:abstract e52–e53. doi:10.1016/j.clml.2019.09.080
113. Van de Donk N, Popat R, Larsen J, et al. First results of iberdomide (IBER; CC-220) in combination with dexamethasone and daratumumab or bortezomib in patients with relapsed/refractory multiple myeloma. Blood. 2020;136(Suppl 1):abstract 724. doi:10.1182/blood-2020-137743
114. Hansen JD, Correa M, Nagy MA, et al. Discovery of CRBN E3 ligase modulator CC-92480 for the treatment of relapsed and refractory multiple myeloma. J Med Chem. 2020;63:6648–6676. doi:10.1021/acs.jmedchem.9b01928
115. Richardson PG, Vangsted AJ, Ramasamy K, et al. First-in-human phase 1 study of the novel CELMoD agent CC-92480 combined with dexamethasone in patients with relapsed/refractory multiple myeloma (RRMM). J Clin Oncol. 2020;38(Suppl.):abstract 8500. doi:10.1200/JCO.2020.38.15_suppl.8500
116. Imbruvica (Ibrutinib) capsules, for oral use (package insert). Sunnyvale, CA: Pharmacyclis LLC; 2019.
117. Tai YT, Chang BY, Kong SY, et al. Bruton tyrosine kinase inhibition is a novel therapeutic strategy targeting tumor in the bone marrow microenvironment in multiple myeloma. Blood. 2012;120:1877–1887. doi:10.1182/blood-2011-12-396853
118. Richardson PG, Bensinger WI, Huff CA, et al. Ibrutinib alone or with dexamethasone for relapsed or relapsed and refractory multiple myeloma: phase 2 trial results. Br J Haematol. 2018;180:821–830. doi:10.1111/bjh.15058
119. Chari A, Corbnell RF, Gasparetto C, et al. Final analysis of a phase 1/2b study of ibrutinib combined with carfilzomib/dexamethasone in patients with relapsed/refractory multiple myeloma. Hematol Oncol. 2020;38:353–362. doi:10.1002/hon.2723
120. Mateos M-V, Orlowski RZ, Ocio EM, et al. Pembrolizumab combined with lenalidomide and low-dose dexamethasone for relapsed or refractory multiple myeloma: phase I KEYNOTE-023 study. Br J Haematol. 2019;186:e117–e121. doi:10.1111/bjh.15946
121. Badros A, Hyjek E, Ma N, et al. Pembrolizumab, pomalidomide and low-dose dexamethasone for relapsed/refractory multiple myeloma. Blood. 2017;130:1189–1197. doi:10.1182/blood-2017-03-775122
122. Usmani SZ, Schjesvold F, Oriol A, et al. Pembrolizumab plus lenalidomide and dexamethasone for patients with treatment-naïve multiple myeloma (KEYNOTE-185): a randomized, open-label, phase 3 trial. Lancet Hematol. 2019;6:e448–e458. doi:10.1016/S2352-3026(19)30109-7
123. Mateos M-V, Blacklock H, Schjesvold F, et al. Pembrolizumab plus pomalidomide and dexamethasone for patients with relapsed or refractory multiple myeloma (KEYNOTE-183): a randomized, open-label, phase 3 trial. Lancet Hematol. 2019;6:e459–e469.
124. Neuse CJ, Lomas OC, Schliemann C, et al. Genome instability in multiple myeloma. Leukemia. 2020;34:2887–2897. doi:10.1038/s41375-020-0921-y
125. Walker BA, Boyle EM, Wardell CP, et al. Mutational spectrum, copy number changes, and outcome; results of a sequencing study of patients with newly diagnosed myeloma. J Clin Oncol. 2015;33:3911–3920. doi:10.1200/JCO.2014.59.1503
126. Raje N, Chau I, Hyman DM, et al. Vemurafenib in patients with relapsed refractory multiple myeloma harboring BRAFV600 mutations: a cohort of the histology-independent VE-BASKET Study. JCO Precis Oncol. 2019;3:
127. Heuck CJ, Jethava Y, Khan R, et al. Inhibiting MEK in MAPK pathway-activated myeloma. Leukemia. 2016;30:976–980. doi:10.1038/leu.2015.208
128. Schjesvold F, Ribrag V, Rodriguez-Otero P, et al. Safety and preliminary efficacy results from a phase Ib/II study of cobimetinib as a single agent and in combination with venetoclax with or without atezolizumab in patients with relapsed/refractory multiple myeloma. Blood. 2020;136(Suppl 1):abstract 295. doi:10.1182/blood-2020-135845
129. Flynt E, Bisht K, Sridharan V, et al. Prognosis, biology, and targeting of TP53 dysregulation in multiple myeloma. Cells. 2020;9:287. doi:10.3390/cells9020287
130. Touzeau C, Dousset C, Le Gouill S, et al. The bcl-2 specific BH3 mimetic ABT-199: a promising targeted therapy for t(11;14) multiple myeloma. Leukemia. 2014;28:210–212. doi:10.1038/leu.2013.216
131. Touzeau C, Maciag P, Amiot M, Moreau P. Targeting bcl-2 for the treatment of multiple myeloma. Leukemia. 2018;32:1899–1907. doi:10.1038/s41375-018-0223-9
132. Kumar S, Kaufman JL, Gasparetto C, et al. Efficacy of venetoclax as targeted therapy for relapsed/refractory t(11;14) multiple myeloma. Blood. 2017;130:2401–2409. doi:10.1182/blood-2017-06-788786
133. Kaufman JL, Gasparetto C, Mikhael J, et al. Phase 1 study of venetoclax in combination with dexamethasone as targeted therapy for t(11;14) relapsed/refractory multiple myeloma. Blood. 2017;130:3131.
134. Moreau P, Chanan-Khan A, Roberts AW, et al. Promising efficacy and acceptable safety of venetoclax plus bortezomib and dexamethasone in relapsed/refractory MM. Blood. 2017;130:2392–2400. doi:10.1182/blood-2017-06-788323
135. Kumar SK, Harrison SJ, Cavo M, et al. Venetoclax or placebo in combination with bortezomib and dexamethasone in patients with relapsed or refractory multiple myeloma (BELLINI): a randomized, double-blind, multicenter, phase 3 trial. Lancet Oncol. 2020;21:1630–1642. doi:10.1016/S1470-2045(20)30525-8
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.