Back to Journals » Drug Design, Development and Therapy » Volume 18
Novel Dichloroacetophenone-Based PDHK1 Inhibitors as Potent Anticancer Agents
Authors Wu P, Zhang Z, Zhou Y, Liu Q, Tam KY, Su Z
Received 2 July 2024
Accepted for publication 19 September 2024
Published 18 October 2024 Volume 2024:18 Pages 4661—4679
DOI https://doi.org/10.2147/DDDT.S473437
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Manfred Ogris
Puhua Wu,1,2 Zhicheng Zhang,1 Yan Zhou,2 Quan Liu,2 Kin-Yip Tam,2 Zhenhong Su1
1Hubei Key Laboratory for Kidney Disease Pathogenesis and Intervention, School of Medicine, Hubei Polytechnic University, Huangshi, People’s Republic of China; 2Cancer Centre, Faculty of Health Sciences, University of Macau, Macau, People’s Republic of China
Correspondence: Puhua Wu, Email [email protected]
Background: Pyruvate dehydrogenase kinases (PDHKs), important metabolic and abnormally expressed enzymes in cancer cells, are promising targets for cancer therapy, especially for non-small-cell lung cancer (NSCLC).
Methods: In this study, a new hit, dichloroacetophenone (DAP) analog 9, was postulated to bind to the PDHK1 allosteric pocket, guided by molecular modeling and kinase biochemical experiments. Based on this binding mode, novel DAP analogs were designed and synthesized to confirm the importance of Phe180, Tyr411, and the hydrophobic core at the bottom of the pocket.
Results: This structure–activity relationship (SAR) study led to the discovery of a novel potent hybrid scaffold, dichloroacetophenone biphenylsulfone ether. Dichloroacetophenone biphenylsulfone ether 31 and 32 inhibited PDHK1 with IC50 values of 86 and 140 nM, respectively.
Conclusion: Compound 32 with acceptable in vitro metabolic stability, predicted drug-likeness properties and ADME/T profiles, showed promising therapeutic efficacy in a lung cancer xenograft mouse model.
Keywords: PDHK1, dichloroacetophenone, allosteric pocket, anti-NSCLC activity, tumor xenograft mouse model
Graphical Abstract:

Introduction
Cancer cells predominantly generate ATP through persistent aerobic glycolysis rather than the mitochondrial oxidative phosphorylation (OXPHOS). This phenomenon is known as the “Warburg effect”.1 The disruption of glucose metabolism in cancer may offer new therapeutic opportunities for cancer treatment.2 The pyruvate dehydrogenase complex (PDC) is a pivotal enzyme in cellular glucose metabolism that links pyruvate to the tricarboxylic acid cycle in mitochondrion.3 Dysfunction of PDC is associated with cancer pathobiology. Pyruvate dehydrogenase kinase (PDHK) is a critical negative regulator of the pyruvate dehydrogenase complex (PDC). By catalyzing the phosphorylation of serine residues (S300, S293, and S232) in the E1α subunit of PDC, PDHKs can downregulate the activity of PDC.4 Aberrant elevation of PDHKs is frequently observed in lung cancer, pancreatic ductal adenocarcinoma, and other solid cancers.5–8 Four PDHK isoforms have been reported to be involved in cancer progression.8–11 Among these isoforms, PDHK1 is the most closely associated with cancer malignancy, especially non-small-cell lung cancer (NSCLC).12–15 These results suggest that targeting PDHK1 is a promising strategy for the design of anticancer drugs.
In the past decade, many small molecular PDHK inhibitors have been reported.16,17 Representative PDHK inhibitors are shown in Figure 1. Although these representative inhibitors can inactivate PDHKs at nanomolar concentrations, they cannot be used as single agents for the treatment of cancer owing to their low antiproliferative effects on cancer cells. On the other hand, dichloroacetophenones (DAPs) have gradually emerged as a novel class of promising PDHK inhibitors over the past 6 years.18–20 This type of PDHK inhibitor has displayed more potent anticancer activity than other PDHK inhibitors.
 |
Figure 1 Structures of reported PDHK inhibitors. |
Here, we report the discovery of novel dichloroacetophenones as more potent anti-NSCLC PDHK1 inhibitors than the reported small molecules, guided by molecular modeling and kinase biochemical experiments (Figure 2). In preliminary SAR exploration, different 4′-alkyloxy/aryloxy substituted DAP derivatives and different linkage groups “L” were studied to afford a new PDHK1 inhibitor 9. Based on its binding mode at the allosteric site, a SAR study of the substitution of 4′-phenyloxy was carried out. Further SAR exploration at the 2′-position yielded significantly more potent PDHK1 inhibitors with IC50 values as low as 86 nM. Furthermore, compound 32 demonstrated satisfactory efficacy and safety in a tumor xenograft mouse model.
 |
Figure 2 Design strategy of novel dichloroacetophenones as PDHK1 inhibitors. |
Chemistry
The synthetic routes to the new 4′-alkoxy analogues 1–6 are outlined in Scheme 1. Catalyzed by anhydrous potassium carbonate, the condensation of 4-hydroxy-acetophenone (S1) and the corresponding bromides produced ethers (1a–1e). Subsequently, nitration in 95% nitric acid and acetic anhydride gave intermediates (2a–2e). Intermediates 2a and 2b were converted to compounds 1 and 2 by basic hydrolysis and chlorination with lithium chloride and copper chloride dihydrate in N,N-dimethylformamide, respectively. The chlorination of intermediates 2b–2d with ammonium chloride and 1.3-dichloro-5,5-dimethylhydantoin in acetonitrile produced compounds 3–5, respectively. Compound 6 was prepared from intermediate 2e by treatment with chlorine gas in acetic acid.
 |
Scheme 1 Reagents and conditions: (a) Bromides, K2CO3, CH3CN, 80°C; (b) 95% nitric acid, acetic anhydride, 0°C; (c) aq. NaOH, EtOH; (d) LiCl, CuCl2▪2H2O, DMF, 90°C; (e) NH4Cl, DCDMH, CH3CN, 80°C, 1 h; 35°C, overnight; (f) Cl2, AcOH, r.t. |
As shown in Scheme 2, compounds 7–30 were prepared from 3-nitro-4-fluoro-acetophenone (S2) in two steps. The chlorination of S2 with N-chlorosuccinimide and p-toluenesulfonic acid hydrate in N,N-dimethylformamide gave intermediate 3a. Intermediate 3a was readily converted into compounds 7–30, after treatment with various phenols or amines in the presence of sodium bicarbonate.
 |
Scheme 2 Reagents and conditions: (a) NCS, p-TsOH▪H2O, DMF, 80°C; (b) Corresponding phenols or amines, NaHCO3, DMF, 25–50°C. |
Compounds 31 and 32 were synthesized from commercially available 4-fluoro-2-hydroxy-acetophenone (S3) (Scheme 3). The nitration of S3 in nitric acid and concentrated sulfuric acid produced intermediate 4a. A nucleophilic replacement reaction with the appropriate bromides and chlorination easily converted intermediate 4a to 6a or 6b. In the presence of sodium bicarbonate, 6a and 6b reacted with 4-((4-isopropoxyphenyl)sulfonyl)phenol to furnish compounds 31 and 32, respectively.
 |
Scheme 3 Reagents and conditions: (a) 95% nitric acid, 96% con.H2SO4, 0°C; (b) Bromides, K2CO3, CH3CN, 80°C; (c) NCS, p-TsOH▪H2O, DMF, 80°C; (d) NaHCO3, DMF, 25–50°C. |
Results and Discussion
Preliminary Structure–Activity Relationship Study
According to DAP-64, which inhibited PDHK1 moderately (IN% = 22.3% at 10 μM), we first designed 4′-substituted derivatives to explore the effect of different substitution patterns on PDHK1 inhibition, and to probe the attributes of the binding pocket (Table 1). In the 4′-alkoxy linked series, different functional groups were introduced at the ends of the alkyl chains. Alcohols 1 and 2 showed inhibition rates ranging from 33.2% to 39.1%. Ester 3 (IN% = 45.6%) exhibited the most potent activity among the 4′-alkoxy derivatives tested. Lactam 5 (IN% = 43.8%) showed better PDHK1 inhibitory activity than that of amide 4 (IN% = 32.5%). Amine 6 (IN% = 29.4%) demonstrated the lowest activity because of its positive charge on nitrogen in the assay buffer. These observations suggest that the terminal lipophilic ester and lactam groups improved the PDHK1 inhibitory activity better than the other groups.
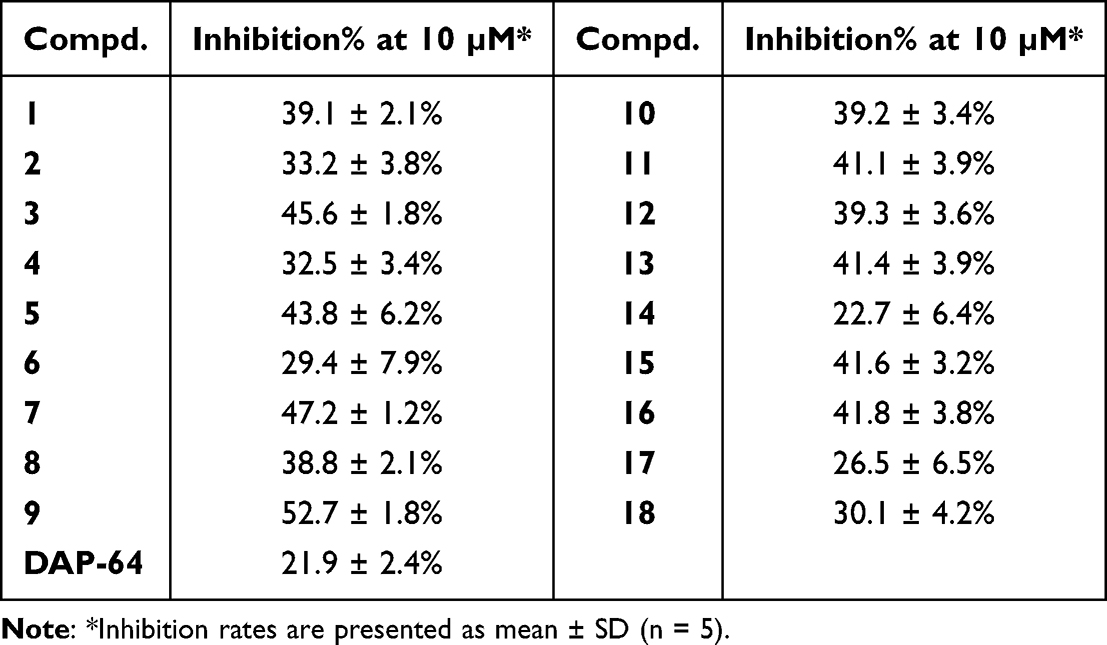 |
Table 1 Inhibitory Effect of Compounds 1–18 on PDHK1 |
In the 4′-phenyloxy series, compound 7 exhibited more potent inhibition of PDHK1 (IN% = 47.2%) than ester 3. Substitution of the linkage oxygen atom with the NH group (8) decreased the anti-PDHK1 activity by approximately 8.4%. The introduction of chlorine at the 4″-position (9, IN% = 52.7%) on the phenyl ring promoted inhibitory activity significantly about 5.5%. Compounds with methoxy 10 and methyl 11 at the 4″-position decreased inhibitory activity by about 11.6–13.5%, relative to compound 9. Other functional groups at the 4″-position, such as cyano (12), amido (13), and acetylamino (14), led to a decrease in inhibitory activity of about 11.3–30%, compared to that of compound 9. By comparing the activity of compounds 14 and 15, it was observed that chlorine at 3″-position improved the anti-PDHK1 activity significantly (by about 18.9%). Polycyclic aryls 16–18 with bulk volume decreased inhibition activity, especially when parallel aryls were located at the 2″,3″-positions (17–18, IN% = 26.5–30.1%), suggesting that the binding pocket is probably a long cylindrical pocket that is unable to accommodate parallel aryls.
New Hit 9 Was Postulated Binding at Allosteric Pocket on PDHK1
For further drug design, the binding site of compound 9 should be validated. Although there are no experimental conditions for us to obtain the direct 3D co-crystal data of PDHK1 and compound 9, we obtained rational binding information using kinase activity assays and molecular modeling.
In the presence of the E2 subunit, AZD7545 showed 86.4% PDHK1 activity inhibition at 10 μM, and hit 9 displayed 51.3% kinase inhibitory activity. When E2 subunit-free inhibitory activity was measured, AZD7545 showed a 4.77% inhibition rate at 10 μM, whereas hit 9 still exhibited 50.1% inhibitory activity (Figure 3A). These observations indicated that compound 9 was different from AZD7545, which inhibited PDHK1 only in the presence of the E2 subunit. On the other hand, the PDHK1 inhibitory percentages of compound 9, VER-246608 and Pfz3, were measured at different ATP concentrations. At an ATP concentration of 1 μM, the inhibition rates of VER-246608, 9, and Pfz3 were 74.9%, 50.3%, and 54.7%, respectively. After the concentration of ATP was increased to 2 μM, the inhibition rate of VER-246608 decreased to 28.8%, whereas the inhibitory activities of compounds 9 and Pfz3 remained at 53.1% and 53.3%, respectively. These observations suggested that the inhibitory activity of 9 and Pfz3 was not affected by ATP concentration, and they were different from VER-246608, which is a known ATP competitive inhibitor of PDHK (Figure 3B). In addition, in silico attempts to dock compound 9 with PDHK1 at the DCA binding site failed to afford refined conformations because of the narrow space in the pocket (Volumecompd.9 = 286.8 Å3 > Volumepocket = 266.9 Å3). It is unlikely that compound 9 occupies the DCA binding site in PDHK1. Taken together, we postulated that compound 9 inhibits PDHK1 in the allosteric pocket for further rational drug design.
 |
Figure 3 (A) Effect of E2 subunit on PDHK1 inhibitory percentages of compound 9, AZD7545 (10 μM). (B) Effect of ATP concentration on PDHK1 inhibitory percentages of VER-246608, compound 9 and Pfz3 (10 μM) (VER-a / 9-a / Pfz3-a: CATP = 1 μM; VER-b/ 9-b / Pfz3-b: CATP = 2 μM). (C) The merged complex of compound 9 with PDHK1 and its binding modes. Data was presented as mean ± SD (n = 5 each group). ns represents P >0.05, **** represents P <0.0001. |
Compound 9 was docked to the allosteric domain in the homology model of PDHK1, which was constructed using hPDHK2 (PDB code 2BU7) as the template. The binding mode showed the formation of a hydrogen bond between the nitro group and Asn100, with a predicted O-N distance of 2.188 Å (Figure 3C). Chlorine at 4″ position formed alkyl interactions with Leu97, Leu101, and Val177, and a special halogen hydrogen bond with Phe180. These affinities made compound 9 more potent than the lipophilic compounds 10 and 11.
Discovery of Compound 30 with Significantly Improved PDHK1 Inhibition Activity
Guided by the binding conformation of compound 9, we designed and synthesized compounds 19–30 to validate the binding properties of the pocket and pursue more potent inhibitors (Table 2). Compared with compound 9, the introduction of a bromo atom at the 4″-position (19) increased the inhibition rate by approximately 5.9% owing to the stronger halogen hydrogen bond interaction between bromine and the hydrogen atom of Phe180 (Figure 4A). At the same position, hydrophobic isopropyl (20) and cyclohexyl (24) induced a decrease in anti-PDHK1 activity of 16.5–19.8%. Compound 21 with an acetyl group at the 4″-position showed a similar inhibitory rate (IN% = 54.1%) as compound 19 by forming two hydrogen bonds with Asn100 and Phe180 (Figure 4B). Compound 25, with a phenyl ring at the 4″-position, showed a 53.2% inhibition rate, whereas compound 26 with coumarin-6-oxyl displayed a 56.3% inhibition rate on PDHK1. Compared with compound 25, 3″-phenyl (27) decreased the inhibition rate to 33.5% owing to the lack of π–π interaction with Phe180. Bis-chloro-substitution at the 3″,4″-positions (22, IN% = 68.5%) resulted in a dramatic increase in the inhibitory rate. The binding mode of compound 22 showed that the two chlorines formed halogen hydrogen bonds with Phe180 and Tyr 411, respectively (Figure 4C). In the absence of chlorine hydrogen bond interactions, 3″,4″-methylenedioxy (23, IN% = 20.3%) induced a significant decrease in anti-PDHK1 activity.
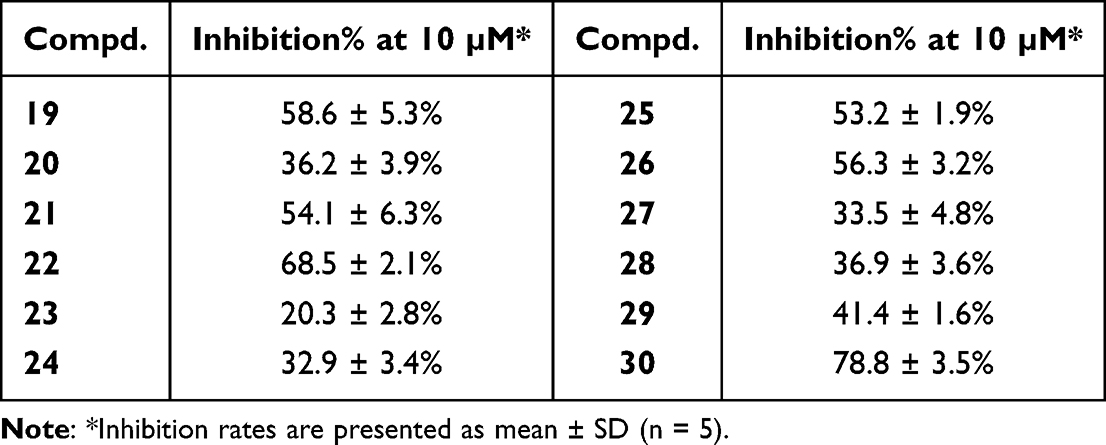 |
Table 2 Inhibitory Effect of Compounds 19–30 on PDHK1 |
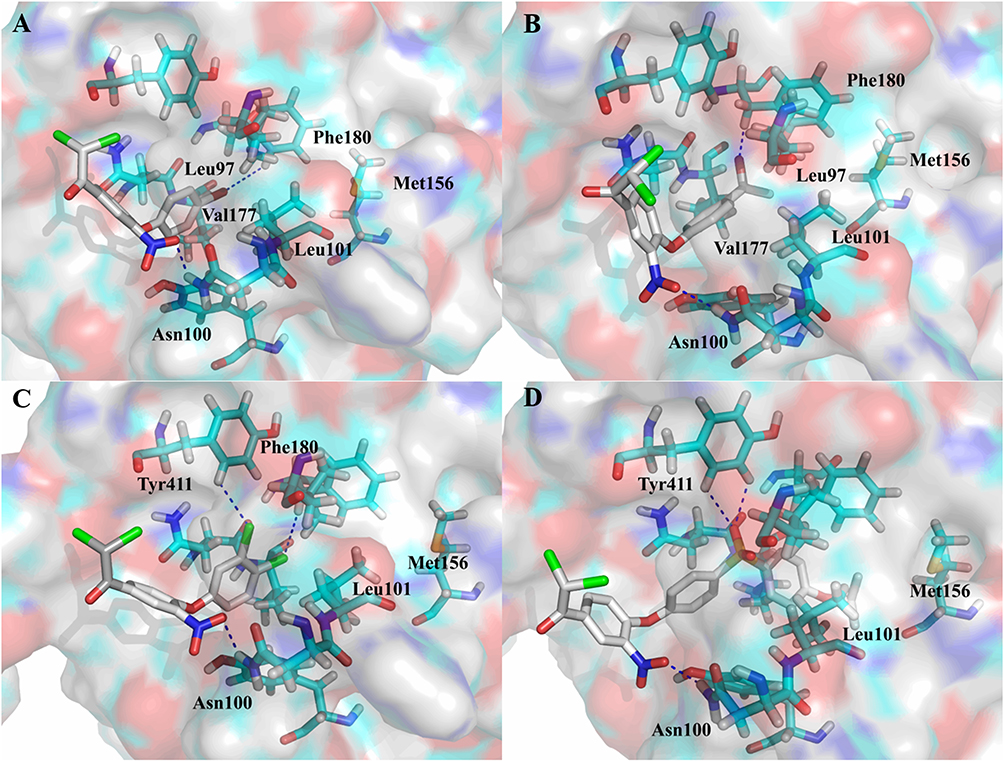 |
Figure 4 Binding modes of compounds with PDHK1 at allosteric pocket. (A) compound 19, (B) compound 21, (C) compound 22, (D) compound 30, blue dash line: hydrogen bond. |
Further analysis of the binding modes of compounds 21 and 22 revealed the existence of a spare space between acetyl/chlorine and Met156, which is the hydrophobic center residue at the bottom of the pocket. Then, 4″-benzoyl (28) was employed as a substitute for the acetyl group to explore deeper areas. However, benzoyl led to a decrease in activity by approximately 17.2% relative to that of compound 21. A possible reason could be that the phenyl ring did not stretch into the hydrophobic bottom of the pocket and weakened the hydrogen bond interaction between carbonyl and Phe180. In contrast, compound 29 with a long benzyloxy at the 4″-position, increased activity by approximately 2.2% relative to compound 10. Hence, to occupy the hydrophobic center at the bottom and retain the hydrogen bond interaction with the residue, a (4-isopropyloxyphenyl)sulfonyl group was designed to link at the 4″-position. This new moiety caused PDHK1 inhibitory activity to increase sharply (30, IN% = 78.8%). The binding pattern of compound 30 showed that the sulfonyl group formed two hydrogen bonds with Tyr411 and the isopropyl group entered the hydrophobic bottom (Figure 4D).
Further Promotion of Potency and Selectivity by Modification at the 2′-Position
Encouraged by the PDHK1 inhibitory rate of compound 30, we measured its inhibitory activity against the other PDHK isoforms (Table 3). Pfz3 and VER-246608 were used as reference inhibitors. Surprisingly, compound 30 displayed weaker inhibitory potency against PDHK2 and PDHK3 than against PDHK1 and PDHK4. To further improve the inhibition and selectivity of PDHK1 and 4, we designed two new compounds (31–32) with acetate ethyl ester and acetamide moieties at the 2′-position by linkages with oxygen atoms. Compared with compound 30, introduction of an acetate ester moiety (31) increased the inhibition rates of PDHK1 and PDHK4 by approximately 5.4% and 16.7%, respectively. Ester 31 (IC50 = 86 nM) exhibited a stronger PDHK1 inhibitory potency than amide 32 (IC50 = 140 nM). However, amide 32 (selectivity = 123 folds) displayed better selectivity for PDHK1 than ester 31 (selectivity = 4.9 folds).
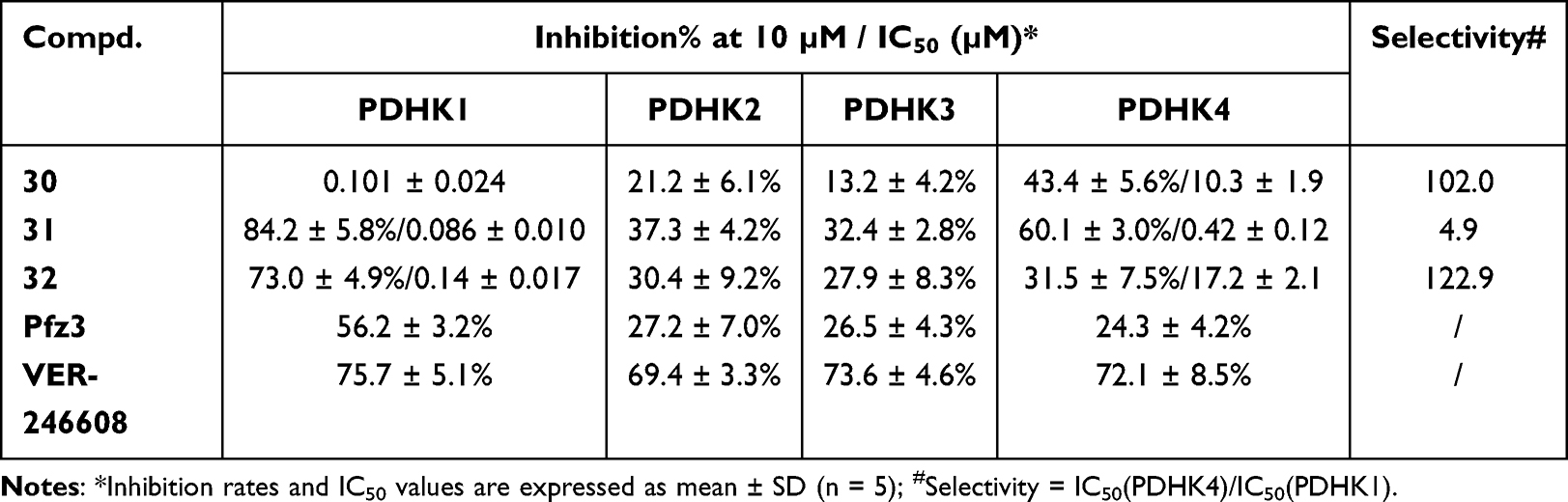 |
Table 3 Inhibitory Effect of Compounds 30–32 on PDHKs and Selectivity |
The binding mode of compound 31 with PDHK1 showed that the acetate moiety formed a hydrogen bond with Asn100, while the nitro group interacted with Tyr411 via another hydrogen bond (Figure 5A). These two hydrogen bonds caused the isopropyl group to enter a crack at the hydrophobic bottom. In contrast, compound 32 displayed different hydrogen bonding modes compared to compounds 30 and 31. The amide moiety made the nitro group of compound 32 form a hydrogen bond with Tyr163 instead of Asn100 (Figure 5B). This change caused isopropyl to move outward and induced a slight decrease in PDHK1 inhibitory activity. These observations suggest the importance and steric/geometric constraints of hydrogen bond interactions from Asn100.
 |
Figure 5 Binding modes of compounds with PDHK1 ((A) compound 31, (B) compound 32; blue dash line: hydrogen bond) and PDHK4 ((C) compound 30, (D) compound 31, (E) compound 32; blue dash line: hydrogen bond) at allosteric pocket. |
The binding modes of compounds 30–32 on PDHK4 are shown in Figure 5C–E. The sulfone group of 31 and Asn154 formed a hydrogen bond, which was not observed in the binding modes of 30 and 32. This strong hydrogen bond may make compound 31 more potent than compounds 30 and 32. Compared to compound 30, the nitro group of compound 32 formed a hydrogen bond with Tyr141 instead of Gln78, and this interaction caused the sulfone moiety to move outward, leading to a decline in its inhibitory activity against PDHK4.
Antiproliferative Potency
Next, we tested the antiproliferative effects of these new PDHK inhibitors on the H1975, H1650, and H1299 cancer cell lines (Table 4). Roughly, compounds 7–32 exhibited better antiproliferative activities against NSCLC cell lines than did compounds 1–6. Thirteen compounds showed a strong inhibition rate (IN% > 85%) in three cancer cell lines at 2.5 μM simultaneously. Compounds 24 and 31–32 exhibited a surprising inhibition rate (IN% > 95%) in all three cell lines. Compound 24, with weak enzymatic activity, showed good anti-NSCLC cellular activity, suggesting that it may inhibit cancer cells through other targets. The IC50 values of compounds 30–32 on cancer cell lines and HEK293 cells were measured for further evaluation (Table 5). Compound 30 displayed potent antiproliferation on these NSCLC cells, with IC50 ranging from 0.33 to 0.88 μM. Compound 32 showed better anti-NSCLC potency compared to compounds 30 and 31. As compounds 30–32 showed both potent anti-NSCLC cytotoxicity and anti-PDHK1 inhibitory activity, it could be inferred that their antiproliferation activity on these NSCLC lines may be mainly caused/promoted by the inhibition of PDHK1. Compound 32 even exhibited a more potent antiproliferative effect on NCI-H1299 cells than DOX. Moreover, its selectivity between cancerous and normal cells is better than that of DOX, which is still in clinical use and highly toxic.
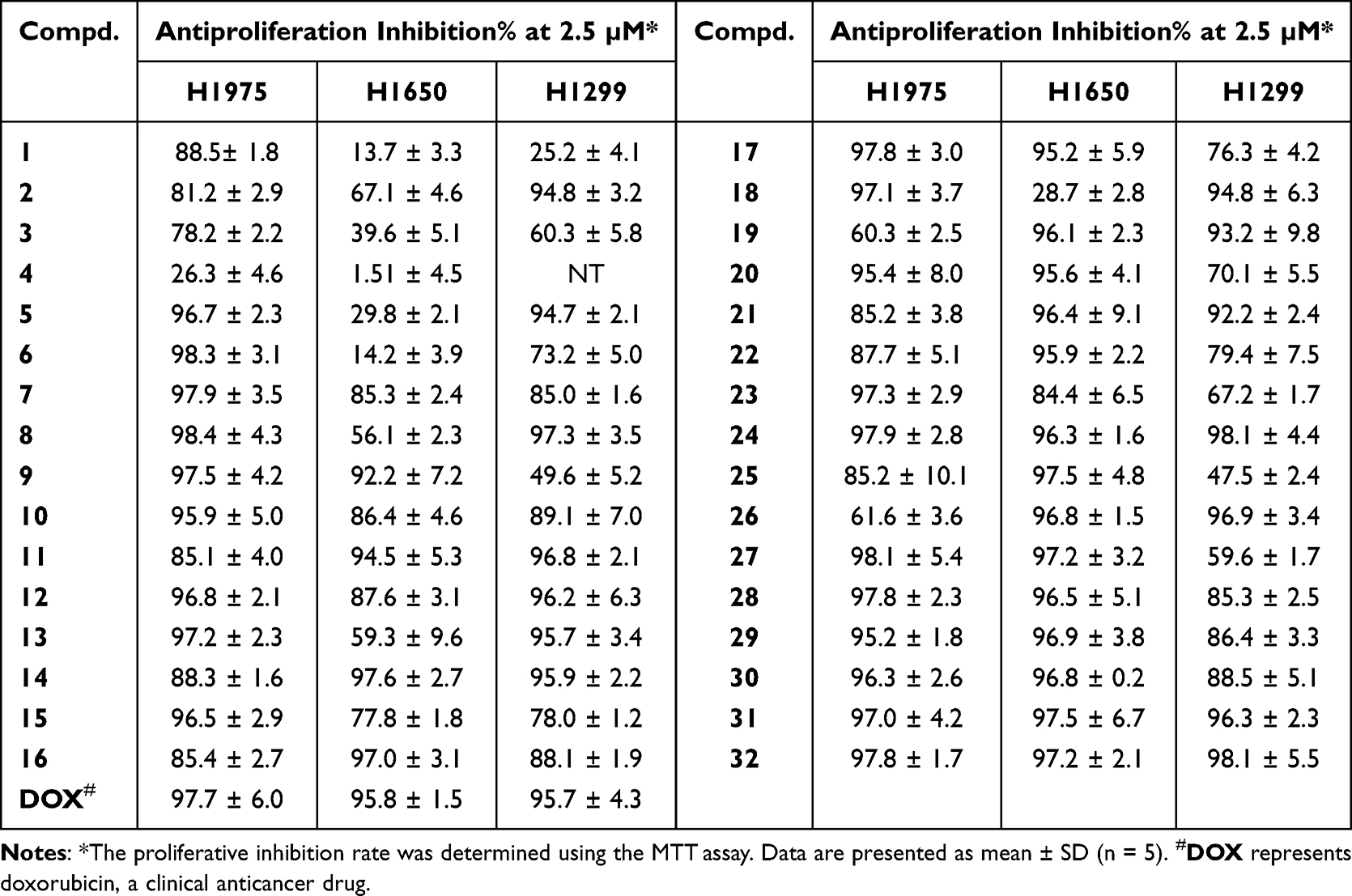 |
Table 4 Inhibitory Effect of Compounds 1–32 on Non-Small-Cell Lung Cancer Cells NCI-H1975, H1650, and H1299 |
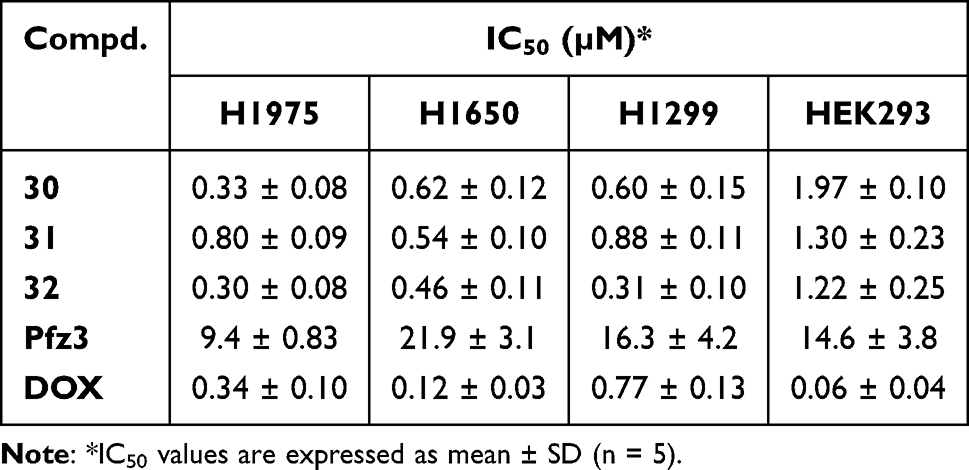 |
Table 5 IC50 Values of DAP Analogs 30–32 on NSCLC Cell Lines and HEK293 |
In vitro Liver Microsome Metabolic Stability Evaluation of Compounds 30–32
To evaluate the druggable potency of compounds 30–32, in vitro liver microsomal metabolic stability tests were performed (Table 6). Compounds 30 and 31 showed poor metabolic stability in the mouse liver system (t1/2 <0.1 h), whereas compound 32 (t1/2 = 0.120 h) showed an equivalent metabolic rate to that of the positive control. In the human liver microsome system, compound 32 exhibited acceptable metabolic stability (t1/2 = 0.778 h, CLint = 1.78 mL/h/mg).
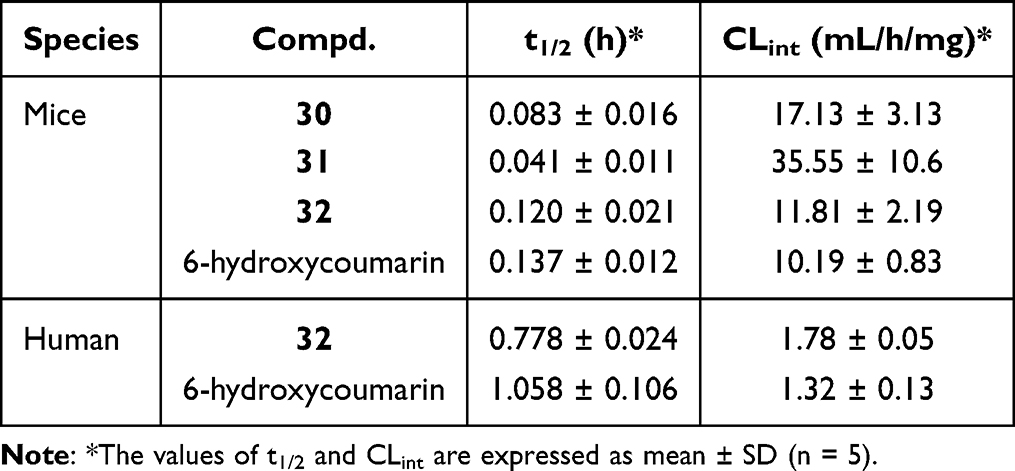 |
Table 6 In vitro Stability Evaluation of Compounds 30–32 in Liver Microsomes |
Predicted Drug-Likeness Properties and ADME/T Profiles of Compound 32
Furthermore, the drug-likeness properties of compound 32 were predicted using Molsoft software (Supplementary Table S1). Compound 32 exhibited acceptable drug-likeness (HBA = 9, HBD = 2, MlogP = 4.11, MlogS = 16.8). In addition, the ADME/T profiles of compound 32 were also predicted (Supplementary Table S2), and its human intestinal absorption (97.1%), MDCK cell permeability (<0.1) and P-glycoprotein inhibition suggest that compound 32 may possess rational bioavailability and excretion. A satisfactory plasma protein binding rate (<99.5%) and low blood-brain barrier penetration (0.2) were also observed. Although the cardiovascular toxicity of compound 32 is ambiguous, acute toxicity can be avoided.
Compound 32 Inhibited Tumor Growth in a Xenograft Mouse Model
Finally, we evaluated the in vivo efficacy of compound 32 in an H1299 xenograft mouse model. Female BALB/c nude mice were intraperitoneally injected with vehicle, compound 32 (6 mg/kg) or DOX (6 mg/kg). Efficacy data revealed that compound 32 exhibited significant antitumor activity during the treatment period, with tumor growth inhibition (TGI) of 66.6%, which is comparable to that of DOX (Figure 6A). After 21 days of administration, DOX-treated mice showed significant body weight loss, whereas the body weight of compound 32-treated mice decreased very little (Figure 6B). No other side-effects were observed after administration of compound 32. Compound 32 may have acceptable toxicity properties; however, further comprehensive toxicity evaluation is required.
 |
Figure 6 In vivo anticancer activity of compound 32 on H1299 xenograft mouse model. Female nude BALB/c mice were administered by intraperitoneal injection with vehicle, compounds 32 and DOX at a dosage of 6 mg/kg, respectively (n = 5 each group). Average tumor volume (A) and average body weight (B) were measured every 3 days. Data are expressed as the mean ± SEM (n = 5). * indicates P <0.05 vs control. |
Conclusion
In summary, a series of novel DAP analogs was designed and synthesized as PDHK inhibitors for the treatment of non-small-cell lung cancer. After 3-round structure–activity relationship studies, compound 32 was discovered to be a novel potent selective PDHK1 inhibitor with good selectivity against other PDHK isoforms (at least 122 folds). Additionally, it showed better anti-proliferative activity than DOX against H1299 cells and excellent selectivity between cancerous and normal cells. More importantly, with acceptable predicted drug-likeness, ADME/T profiles, and in vitro metabolic stability, it significantly suppressed tumor growth in an H1299 tumor xenograft mouse model at a dose of 6 mg/kg. It could be used as a new lead for the development of novel anti-NSCLC drugs in the future.
Methods and Materials
Chemistry
Unless otherwise mentioned, all commercial reagents were used directly without further purification. All the organic solutions were concentrated using an evaporator (EYEL4) below 40°C under reduced pressure. All reactions were monitored by thin layer chromatography (TLC) on 0.1–0.2 mm silica gel plates, which can be visualized under UV light at 254 nm. The chemical structures of the compounds were confirmed by 1H-NMR and 13C-NMR spectroscopy using a Bruker AV-400/600 instrument, with tetramethylsilane (TMS) as the internal standard. High-resolution mass spectra (HRMS) of all compounds were recorded on a Waters Xevo G2-XS QTOF spectrometer (ESI). The purity (>95%) of all biologically evaluated compounds was determined by Agilent 1260 Infinity HPLC on an Agilent ZORBAX SB-C18 column (4.6 mm × 150 mm, 5 μm), using a mixture of acetonitrile, water, and 0.1% trifluoroacetic acid (TFA) at a flow rate of 1 mL/min, and the peaks were determined at 254 nm.
General Procedure for Synthesis of Target Compounds 1–6
The intermediates 2a–2e were easily prepared by condensation and nitration of 4-hydroxy-acetophenone (S1) according to previously reported procedures.21,22
Method A: To a solution of the corresponding intermediates 2a–2b (3.0 mmol) in THF, 1N NaOH was added slowly. The mixture was then stirred overnight at room temperature. To the mixture, 3M HCl was added slowly to adjust the pH to 7. After evaporation, the residue was extracted with DCM and dried over anhydrous Na2SO4 for 3 hours. After filtration, the residue, cupric chloride dihydrate (3.1 g, 18.0 mmol) and lithium chloride (763.2 mg, 18.0 mmol) in DMF (10 mL) was heated to 90°C. After stirring for 3–6 hours, the mixture was cooled to room temperature, diluted with water (50 mL), and extracted with DCM (3 × 30 mL). The organic phase was washed with water (3 × 30 mL) and dried overnight with anhydrous Na2SO4. After evaporation, the residue was purified using silica gel to yield target compounds 1 and 2.
2.2-Dichloro-1-(4-(2-hydroxyethoxy)-3-nitrophenyl)ethan-1-one (1) was obtained as a colorless oil; yield 33%; 1H NMR (400 MHz, CDCl3) δ: 8.66 (d, J = 2.4 hz, 1H), 8.35 (dd, J = 2.4, 8.8 hz, 1H), 7.23 (d, J = 8.8 hz, 1H), 6.53 (s, 1H), 4.35 (dd, J = 4.0, 4.8 hz, 2H), 4.05 (d, J = 3.2 hz, 2H), and 2.32 (brs, 1H) ppm (Supplementary Figure S1). 13C NMR (101 MHz, CDCl3) δ: 183.3, 156.4, 139.5, 135.9, 128.1, 123.7, 114.8, 71.8, 67.7, and 60.7 ppm (Supplementary Figure S2). HRMS m/z calcd for C10H8Cl2NO5 [M-H]− 291.9785, found 291.9827 (Supplementary Figure S3). HPLC analysis: retention time = 12.5 min; purity = 95.4%.
2.2-Dichloro-1-(4-(3-hydroxypropoxy)-3-nitrophenyl)ethan-1-one (2) was obtained as a white solid; yield 28%; m.p.: 68–70 °C; 1H NMR (400 MHz, CDCl3) δ: 8.64 (d, J = 2.4 hz, 1H), 8.34 (dd, J = 2.4, 9.2 hz, 1H), 7.23 (d, J = 8.8 hz, 1H), 6.53 (s, 1H), 4.39 (t, J = 6 hz, 2H), 3.91 (t, J = 5.6 hz, 2H), and 2.14 (quint, J = 6 hz, 1H) ppm (Supplementary Figure S4). 13C NMR (101 MHz, CDCl3) δ: 183.3, 156.6, 139.3, 135.9, 128.0, 123.2, 114.2, 67.8, 67.7, 59.4 and 31.4 ppm (Supplementary Figure S5). HRMS m/z calcd for C11H12Cl2NO5 [M+H]+ 308.0087, found 308.0072 (Supplementary Figure S6). HPLC analysis: retention time = 8.2 min; purity = 97.6%.
Method B: A solution of the corresponding intermediates 2b–2d (2.0 mmol), ammonium chloride (107.0 mg, 2.0 mmol), and 1.3-dichloro-5,5-dimethylhydantoin (788.1 mg, 4.0 mmol) in acetonitrile (10 mL) was stirred for 1 hour at 80°C. After cooling to 35°C, stirring overnight, and evaporating, the mixture was dissolved in EA (30 mL), washed with water (3 × 30 mL), and dried with anhydrous Na2SO4 overnight. After evaporation, the residue was purified using silica gel to yield the target compounds 3–5.
3-(4-(2,2-Dichloroacetyl)-2-nitrophenoxy)propyl acetate (3) was obtained as a light yellow oil; yield 71%; 1H NMR (400 MHz, DMSO‑d6) δ: 8.56 (d, J = 2.4 hz, 1H), 8.31 (dd, J = 2.4, 9.2 hz, 1H), 7.89 (s, 1H), 7.56 (d, J = 9.2 hz, 1H), 4.37 (t, J = 6 hz, 2H), 4.15 (t, J = 6 hz, 2H), 2.07 (quint, J = 6 hz, 1H), and 1.99 (s, 3H) ppm (Supplementary Figure S7). 13C NMR (101 MHz, DMSO‑d6) δ: 183.3, 170.9, 156.3, 139.5, 135.8, 127.9, 123.3, 114.2, 67.7, 66.9, 60.6, 28.3, and 20.9 ppm (Supplementary Figure S8). HRMS m/z calcd for C13H17Cl2N2O6 [M+NH4]+ 367.0458, found 367.0454 (Supplementary Figure S9). HPLC analysis: retention time = 9.3 min; purity = 95.2%.
2-(4-(2,2-Dichloroacetyl)-2-nitrophenoxy)acetamide (4) was obtained as a white solid; yield 61%; m.p.: 160–161°C; 1H NMR (400 MHz, DMSO-d6) δ: 8.60 (d, J = 2.0 hz, 1H), 8.32 (dd, J = 1.6, 8.8 hz, 1H), 7.90 (s, 1H), 7.50 (d, J = 3.2 hz, 2H), 7.42 (d, J = 8.8 hz, 1H), and 4.86 (s, 2H) ppm (Supplementary Figure S10). 13C NMR (101 MHz, DMSO-d6) δ: 184.3, 166.6, 155.6, 139.6, 135.8, 127.2, 124.6, 116.3, 69.2, and 68.1 ppm (Supplementary Figure S11). HRMS m/z calcd for C10H9Cl2N2O5 [M+H]+ 306.9883, found 306.9905 (Supplementary Figure S12). HPLC analysis: retention time = 14.1 min; purity = 98.3%.
(S)-5-((4-(2,2-dichloroacetyl)-2-nitrophenoxy)methyl)pyrrolidin-2-one (5) was obtained as a white solid; yield 57%; m.p.: 176–177°C; 1H NMR (400 MHz, CDCl3) δ: 8.69 (d, J = 2.4 hz, 1H), 8.38 (dd, J = 2.4, 8.8 hz, 1H), 7.22 (d, J = 8.8 hz, 1H), 6.52 (s, 1H), 4.98 (quint, J = 2.8 hz, 1H), 4.79 (dd, J = 3.2, 10 hz, 1H), 4.39 (dd, J = 2.4, 10 hz, 1H), 3.14–3.07 (m, 1H), 2.70–2.62 (m, 1H), 2.49–2.44 (m, 1H) and 2.35–2.28 (m, 1H) ppm (Supplementary Figure S13). 13C NMR (101 MHz, CDCl3) δ: 183.3, 167.5, 155.5, 139.5, 136.3, 128.5, 124.6, 114.5, 68.9, 67.8, 57.2, 29.7, and 19.0 ppm (Supplementary Figure S14). HRMS m/z calcd for C13H13Cl2N2O5 [M+H]+ 347.0196, found 347.0207 (Supplementary Figure S15). HPLC analysis: retention time = 13.7 min; purity = 95.7%.
Method C: Chlorine gas was bubbled slowly into a solution of the corresponding intermediate, 2e (2.0 mmol) in acetic acid (5 mL) at room temperature. The reaction was monitored using high-performance liquid chromatography. After evaporation, the residue was purified using preparative-HPLC to obtain target compound 6.
2.2-Dichloro-1-(4-(2-(dimethylamino)ethoxy)-3-nitrophenyl)ethan-1-one (6) was obtained as a brown oil; yield 62%; 1H NMR (400 MHz, Acetone-d6) δ: 8.63 (d, J = 2.4 hz, 1H), 8.43 (dd, J = 2.4, 9.2 hz, 1H), 7.63 (d, J = 8.8 hz, 1H), 7.61 (s, 1H), 4.91 (t, J = 4.8 hz, 2H), 3.77 (t, J = 4.8 hz, 2H), and 3.04 (s, 6H) ppm (Supplementary Figure S16). 13C NMR (101 MHz, Acetone-d6) δ: 183.6, 155.2, 139.7, 135.5, 126.8, 124.7, 115.3, 68.2, 65.5, 55.5, and 42.9 ppm (Supplementary Figure S17). HRMS m/z calcd for C12H15Cl2N2O4 [M+H]+ 321.0403, found 321.0426 (Supplementary Figure S18). HPLC analysis: retention time = 11.2 min; purity = 97.6%.
General Procedure for Synthesis of Target Compounds 7–30
A solution of the 3-nitro-4-fluoro-acetophenone (S2) (5.5 mmol), p-TsOH monohydrate (1.25 g, 6.6 mmol), and N-chlorosuccinimide (2.9 g, 21.7 mmol) in DMF (10 mL) was heated to 80°C and stirred for 1–2 hours. After cooling to room temperature, the mixture was diluted with water (50 mL) and extracted with DCM (3 × 40 mL). The combined organic layers were washed with water (3 × 30 mL) and dried overnight over anhydrous Na2SO4. After evaporation, the residue was purified using silica gel to yield the intermediate 3a. A solution of the intermediate 3a (1.5 mmol), the corresponding phenols/anilines (1.8 mmol), and NaHCO3 (441.1 mg, 5.25 mmol) in DMF (10 mL) was stirred at room temperature. After stirring for 6–12 hours, the mixture was diluted with water (50 mL) and extracted with DCM (3 × 30 mL). The combined organic layers were washed with water (3 × 30 mL) and dried overnight over anhydrous Na2SO4. After evaporation, the residue was purified using silica gel to yield target compounds 7–30.
2.2-Dichloro-1-(3-nitro-4-phenoxyphenyl)ethan-1-one (7) was obtained as a white solid; yield 62%; m.p.: 74–76°C; 1H NMR (400 MHz, CDCl3) δ: 8.72 (d, J = 2.4 hz, 1H), 8.22 (dd, J = 2.4, 8.8 hz, 1H), 7.49–7.45 (m, 2H), 7.34–7.30 (m, 1H), 7.16–7.13 (m, 2H), 7.00 (d, J = 8.8 hz, 1H), and 6.51 (s, 1H) ppm (Supplementary Figure S19). 13C NMR (101 MHz, CDCl3) δ: 183.4, 156.1, 153.8, 140.3, 135.5, 130.7, 128.2, 126.5, 124.9, 120.8, 118.4, and 67.9 ppm (Supplementary Figure S20). HRMS m/z calcd for C14H9Cl2NO4 [M+H]+ 325.9981, found 325.9983; HRMS m/z calcd for C14H13Cl2N2O4 [M+NH4]+ 343.0247, found 343.0252 (Supplementary Figure S21). HPLC analysis: retention time = 11.6 min; purity = 99.3%.
2.2-Dichloro-1-(3-nitro-4-(phenylamino)phenyl)ethan-1-one (8) was obtained as an orangey yellow solid; yield 71%; m.p.: 129–130°C; 1H NMR (400 MHz, CDCl3) δ: 9.98 (s, 1H), 9.04 (d, J = 2.0 hz, 1H), 8.07 (ddd, J = 0.8, 2.4, 9.2 hz, 1H), 7.51–7.47 (m, 2H), 7.38–7.30 (m, 3H), 7.19 (d, J = 9.2 hz, 1H), and 6.57 (s, 1H) ppm (Supplementary Figure S22). 13C NMR (101 MHz, CDCl3) δ: 183.2, 147.0, 136.9, 136.0, 131.9, 130.3, 130.1, 127.5, 125.5, 119.9, 116.1, and 67.7 ppm (Supplementary Figure S23). HRMS m/z calcd for C14H11Cl2N2O3 [M+H]+ 325.0141, found 325.0140 (Supplementary Figure S24). HPLC analysis: retention time = 15.4 min; purity = 97.3%.
2.2-Dichloro-1-(4-(4-chlorophenoxy)-3-nitrophenyl)ethan-1-one (9) was obtained as a white solid; yield 64%; m.p.: 84–86°C; 1H NMR (400 MHz, CDCl3) δ: 8.72 (d, J = 2.4 hz, 1H), 8.25 (dd, J = 2.0, 8.8 hz, 1H), 7.43 (d, J = 9.2 hz, 2H), 7.09 (d, J = 8.8 hz, 2H), 7.01 (d, J = 9.2 hz, 1H), and 6.51 (s, 1H) ppm (Supplementary Figure S25). 13C NMR (101 MHz, CDCl3) δ: 183.2, 155.3, 152.3, 140.3, 135.5, 131.7, 130.6, 128.1, 125.3, 121.8, 118.4, and 67.7 ppm (Supplementary Figure S26). HRMS m/z calcd for C14H9Cl3NO4 [M+H]+ 359.9592, found 359.9576 (Supplementary Figure S27). HPLC analysis: retention time = 12.6 min; purity = 97.9%.
2.2-Dichloro-1-(4-(4-methoxyphenoxy)-3-nitrophenyl)ethan-1-one (10) was obtained as a light yellow solid; yield 62%; m.p.: 72–74°C; 1H NMR (400 MHz, CDCl3) δ: 8.70 (d, J = 2.4 hz, 1H), 8.20 (dd, J = 2.4, 9.2 hz, 1H), 7.07 (m, 2H), 6.98 (m, 2H), 6.96 (d, J = 8.8 hz, 1H), 6.51 (s, 1H), and 3.85 (s, 3H) ppm (Supplementary Figure S28). 13C NMR (101 MHz, CDCl3) δ: 183.3, 157.8, 156.7, 146.8, 139.7, 135.4, 128.1, 124.4, 121.9, 117.5, 115.5, 67.7, and 55.7 ppm (Supplementary Figure S29). HRMS m/z calcd for C15H12Cl2NO5 [M+H]+ 356.0087, found 356.0067; HRMS m/z calcd for C15H15Cl2N2O5 [M+NH4]+ 373.0353, found 373.0346 (Supplementary Figure S30). HPLC analysis: retention time = 11.6 min; purity = 99.6%.
2.2-Dichloro-1-(3-nitro-4-(p-tolyloxy)phenyl)ethan-1-one (11) was obtained as a white solid; yield, 64%; m.p.: 78–80°C; 1H NMR (400 MHz, CDCl3) δ: 8.70 (d, J = 2.4 hz, 1H), 8.20 (dd, J = 2.4, 8.8 hz, 1H), 7.07 (d, J = 8.4 hz, 2H), 7.03 (d, J = 8.4 hz, 2H), 6.98 (d, J = 8.8 hz, 1H), 6.51 (s, 1H), and 2.40 (s, 3H) ppm (Supplementary Figure S31). 13C NMR (101 MHz, CDCl3) δ: 183.3, 156.4, 151.3, 139.9, 136.2, 135.3, 131.0, 128.0, 124.5, 120.5, 118.0, 67.7, and 20.9 ppm (Supplementary Figure S32). HRMS m/z calcd for C15H12Cl2NO4 [M+H]+ 340.0138, found 340.0134 (Supplementary Figure S33). HPLC analysis: retention time = 12.6 min; purity = 96.1%.
4-(4-(2,2-Dichloroacetyl)-2-nitrophenoxy)benzonitrile (12) was obtained as a white solid; yield 68%; m.p.: 159–162°C; 1H NMR (400 MHz, CDCl3) δ: 8.78 (d, J = 2.4 hz, 1H), 8.35 (dd, J = 2.4, 8.8 hz, 1H), 7.75 (d, J = 8.8 hz, 2H), 7.19 (d, J = 8.8 hz, 2H), 7.17 (d, J = 8.8 hz, 1H) and 6.52 (s, 1H) ppm (Supplementary Figure S34). 13C NMR (101 MHz, CDCl3) δ: 183.1, 158.1, 153.2, 141.3, 135.7, 134.7, 128.3, 127.0, 120.8, 120.0, 117.9, 109.4, and 67.7 ppm (Supplementary Figure S35). HRMS m/z calcd for C15H12Cl2N3O4 [M+NH4]+ 368.0199, found 368.0162 (Supplementary Figure S36). HPLC analysis: retention time = 10.4 min; purity = 97.6%.
4-(4-(2,2-Dichloroacetyl)-2-nitrophenoxy)benzamide (13) was obtained as a white solid; yield, 63%; m.p.: 157–158°C; 1H NMR (400 MHz, DMSO-d6) δ: 8.75 (d, J = 2.0 hz, 1H), 8.31 (dd, J = 2.4, 8.8 hz, 1H), 8.05 (brs, 1H), 8.00 (d, J = 8.8 hz, 2H), 7.92 (s, 1H), 7.43 (brs, 1H), 7.30 (d, J = 8.8 hz, 2H), and 7.28 (d, J = 8.8 hz, 1H) ppm (Supplementary Figure S37). 13C NMR (101 MHz, DMSO-d6) δ: 184.4, 167.4, 156.9, 154.1, 140.9, 136.2, 132.0, 130.6, 127.7, 127.3, 120.7, 119.9, and 69.3 ppm (Supplementary Figure S38). HRMS m/z calcd for C15H11Cl2N2O5 [M+H]+ 369.0040, found 369.0054 (Supplementary Figure S39). HPLC analysis: retention time = 15.4 min; purity = 95.3%.
N-(4-(4-(2,2-dichloroacetyl)-2-nitrophenoxy)phenyl)acetamide (14) was obtained as a white solid; yield 65%; m.p.: 167–169°C; 1H NMR (400 MHz, DMSO-d6) δ: 10.1 (s, 1H), 8.71 (d, J = 2.0 hz, 1H), 8.25 (dd, J = 2.4, 8.8 hz, 1H), 7.89 (s, 1H), 7.70 (d, J = 8.8 hz, 2H), 7.20 (d, J = 9.2 hz, 2H), 7.09 (d, J = 8.8 hz, 1H), and 2.06 (s, 3H) ppm (Supplementary Figure S40). 13C NMR (101 MHz, DMSO-d6) δ: 184.3, 168.9, 155.5, 148.9, 140.2, 137.8, 136.0, 127.6, 126.1, 121.3, 121.2, 118.8, 69.2, and 24.4 ppm (Supplementary Figure S41). HRMS m/z calcd for C16H13Cl2N2O5 [M+H]+ 383.0196, found 383.0267 (Supplementary Figure S42). HPLC analysis: retention time = 7.0 min; purity = 98.8%.
N-(2-chloro-4-(4-(2,2-dichloroacetyl)-2-nitrophenoxy)phenyl)acetamide (15) was obtained as a white solid; yield 79%; m.p.: 142–143°C; 1H NMR (400 MHz, DMSO-d6) δ: 9.60 (s, 1H), 8.73 (d, J = 2.4 hz, 1H), 8.29 (dd, J = 2.4, 9.2 hz, 1H), 7.90 (s, 1H), 7.79 (d, J = 8.8 hz, 1H), 7.49 (d, J = 2.8 hz, 1H), 7.23 (d, J = 8.8 hz, 1H), 7.22 (dd, J = 2.8, 8.8 hz, 1H), and 2.10 (s, 3H) ppm (Supplementary Figure S43). 13C NMR (101 MHz, DMSO-d6) δ: 184.4, 169.3, 154.5, 151.2, 140.6, 136.2, 133.4, 128.2, 127.7, 127.0, 121.6, 120.0, 119.7, 69.2, and 23.7 ppm (Supplementary Figure S44). HRMS m/z calcd for C16H12Cl3N2O5 [M+H]+ 416.9806, found 416.9864 (Supplementary Figure S45). HPLC analysis: retention time = 8.6 min; purity = 98.5%.
1-(4-((6-Bromo-1-chloronaphthalen-2-yl)oxy)-3-nitrophenyl)-2,2-dichloroethan-1-one (16) was obtained as a white solid; yield 76%; m.p.: 149–150°C; 1H NMR (400 MHz, CDCl3) δ: 8.79 (d, J = 2.4 hz, 1H), 8.21 (dd, J = 2.4, 8.8 hz, 1H), 8.17 (d, J = 8.8 hz, 1H), 8.09 (d, J = 2.0 hz, 1H), 7.81 (d, J = 8.8 hz, 1H), 7.76 (dd, J = 2.0, 9.2 hz, 1H), 6.83 (d, J = 8.8 hz, 1H), and 6.51 (s, 1H) ppm (Supplementary Figure S46). 13C NMR (101 MHz, CDCl3) δ: 183.2, 154.7, 146.8, 139.6, 135.5, 133.4, 131.7, 130.4, 130.3, 128.3, 128.2, 126.2, 125.3, 123.7, 121.8, 121.5, 117.3, and 67.7 ppm (Supplementary Figure S47). HRMS m/z calcd for C18H10BrCl3NO4 [M+H]+ 487.8853, found 487.8828 (Supplementary Figure S48). HPLC analysis: retention time = 15.2 min; purity = 98.6%.
2.2-Dichloro-1-(4-(naphthalen-1-yloxy)-3-nitrophenyl)ethan-1-one (17) was obtained as a yellow oil; yield 59%; 1H NMR (400 MHz, CDCl3) δ: 8.77 (d, J = 2.0 hz, 1H), 8.14 (dd, J = 2.4, 9.2 hz, 1H), 7.95 (dd, J = 7.6, 8.0 hz, 2H), 7.84 (d, J = 8.4 hz, 1H), 7.60–7.50 (m, 3H), 7.24 (d, J = 8.8 hz, 1H), 6.85 (d, J = 8.8 hz, 1H), and 6.50 (s, 1H) ppm (Supplementary Figure S49). 13C NMR (101 MHz, CDCl3) δ: 183.2, 156.4, 149.2, 139.8, 135.5, 135.3, 128.2, 128.1, 127.3, 127.2, 126.7, 126.4, 125.7, 124.8, 121.3, 117.7, 116.9, and 67.7 ppm (Supplementary Figure S50). HRMS m/z calcd for C18H12Cl2NO4 [M+H]+ 376.0138, found 376.0139 (Supplementary Figure S51). HPLC analysis: retention time = 13.3 min; purity = 97.9%.
1-(4-(2,2-Dichloroacetyl)-2-nitrophenoxy)anthracene-9,10-dione (18) was obtained as a yellow solid; yield 31%; m.p.: 224–226°C; 1H NMR (400 MHz, CDCl3) δ: 8.87 (d, J = 2.0 hz, 1H), 8.40 (dd, J = 1.2, 7.6 hz, 1H), 8.29 (dd, J = 2.0, 7.6 hz, 1H), 8.17 (dd, J = 2.0, 8.8 hz, 1H), 8.13 (dd, J = 2.0, 7.2 hz, 1H), 7.93 (dd, J = 8.0, 8.0 hz, 1H), 7.79–7.76 (m, 2H), 7.61 (dd, J = 1.2, 8.0 hz, 1H), 6.81 (d, J = 8.8 hz, 1H), and 6.52 (s, 1H) ppm (Supplementary Figure S52). 13C NMR (101 MHz, CDCl3) δ: 183.2, 182.1, 181.0, 156.1, 152.2, 139.6, 136.2, 135.7, 135.5, 134.7, 134.3, 133.9, 132.5, 129.6, 128.5, 127.5, 127.1, 126.5, 125.1, 124.9, 116.6, and 67.7 ppm (Supplementary Figure S53). HRMS m/z calcd for C22H12Cl2NO6 [M+H]+ 456.0036, found 456.0051 (Supplementary Figure S54). HPLC analysis: retention time = 12.2 min; purity = 99.6%.
1-(4-(4-Bromophenoxy)-3-nitrophenyl)-2,2-dichloroethan-1-one (19) was obtained as a white solid; yield 53%; m.p.: 81–82°C; 1H NMR (400 MHz, CDCl3) δ: 8.73 (d, J = 2.4 hz, 1H), 8.25 (dd, J = 2.4, 9.2 hz, 1H), 7.58 (d, J = 8.8 hz, 2H), 7.05–7.02 (m, 3H), and 6.51 (s, 1H) ppm (Supplementary Figure S55). 13C NMR (101 MHz, CDCl3) δ: 183.2, 155.2, 152.9, 140.3, 135.6, 133.7, 128.1, 125.4, 122.2, 119.3, 118.5, and 67.7 ppm (Supplementary Figure S56). HRMS m/z calcd for C14H9BrCl2NO4 [M+H]+ 403.9087, found 403.9165 (Supplementary Figure S57). HPLC analysis: retention time = 13.0 min; purity = 98.6%.
2.2-Dichloro-1-(4-(4-isopropylphenoxy)-3-nitrophenyl)ethan-1-one (20) was obtained as a white solid; yield 31%; m.p.: 76–78°C; 1H NMR (400 MHz, CDCl3) δ: 8.71 (d, J = 2.4 hz, 1H), 8.21 (dd, J = 2.4, 8.8 hz, 1H), 7.31 (d, J = 8.4 hz, 2H), 7.06 (d, J = 8.8 hz, 2H), 7.01 (d, J = 9.2 hz, 1H), 6.51 (s, 1H), 2.97 (hept, J = 6.8 hz, 1H), and 1.27 (d, J = 6.8 hz, 6H) ppm (Supplementary Figure S58). 13C NMR (101 MHz, CDCl3) δ: 183.3, 156.4, 151.4, 147.2, 139.9, 135.3, 128.4, 128.0, 124.5, 120.5, 118.0, 67.7, 33.7, and 24.0 ppm (Supplementary Figure S59). HRMS m/z calcd for C17H16Cl2NO4 [M+H]+ 368.0451, found 368.0401 (Supplementary Figure S60). HPLC analysis: retention time = 14.5 min; purity = 99.1%.
1-(4-(4-Acetylphenoxy)-3-nitrophenyl)-2,2-dichloroethan-1-one (21) was obtained as a white solid; yield 62%; m.p.: 177–179°C; 1H NMR (400 MHz, Acetone-d6) δ: 8.77 (d, J = 2.0 hz, 1H), 8.42 (dd, J = 2.4, 8.8 hz, 1H), 8.15–8.11 (m, 2H), 7.65 (s, 1H), 7.39 (d, J = 8.8 hz, 1H), 7.36–7.32 (m, 2H), and 2.61 (s, 3H) ppm (Supplementary Figure S61). 13C NMR (101 MHz, Acetone-d6) δ: 195.8, 183.6, 158.4, 153.8, 141.4, 135.4, 134.6, 130.9, 127.5, 127.3, 120.8, 119.4, 68.2, and 25.8 ppm (Supplementary Figure S62). HRMS m/z calcd for C16H12Cl2NO5 [M+H]+ 368.0087, found 368.0009 (Supplementary Figure S63). HPLC analysis: retention time = 10.3 min; purity = 99.8%.
2.2-Dichloro-1-(4-(3,4-dichlorophenoxy)-3-nitrophenyl)ethan-1-one (22) was obtained as a white solid; yield 70%; m.p.: 85–87°C; 1H NMR (400 MHz, CDCl3) δ: 8.74 (d, J = 2.0 hz, 1H), 8.30 (dd, J = 2.4, 9.2 hz, 1H), 7.53 (d, J = 8.8 hz, 1H), 7.27 (d, J = 2.8 hz, 1H), 7.08 (d, J = 8.8 hz, 1H), 7.01 (dd, J = 2.8, 8.8 hz, 1H), and 6.51 (s, 1H) ppm (Supplementary Figure S64). 13C NMR (101 MHz, CDCl3) δ: 183.1, 154.5, 152.8, 140.5, 135.7, 134.3, 131.8, 130.1, 128.2, 126.0, 122.3, 119.6, 119.0, and 67.7 ppm (Supplementary Figure S65). HRMS m/z calcd for C14H8Cl4NO4 [M+H]+ 393.9202, found 393.9238 (Supplementary Figure S66). HPLC analysis: retention time = 15.0 min; purity = 98.2%.
1-(4-(Benzo[d][1,3]dioxol-5-yloxy)-3-nitrophenyl)-2,2-dichloroethan-1-one (23) was obtained as a yellow solid; yield, 71%; m.p.: 122–123°C; 1H NMR (400 MHz, CDCl3) δ: 8.70 (d, J = 2.0 hz, 1H), 8.22 (dd, J = 2.0, 8.8 hz, 1H), 7.03 (d, J = 8.8 hz, 1H), 6.85 (d, J = 8.4 hz, 1H), 6.65 (d, J = 2.4 hz, 1H), 6.60 (dd, J = 2.4, 8.4 hz, 1H), 6.51 (s, 1H), and 6.05 (s, 2H) ppm (Supplementary Figure S67). 13C NMR (101 MHz, CDCl3) δ: 183.3, 156.4, 149.0, 147.8, 145.9, 139.8, 135.4, 128.0, 124.6, 117.8, 113.4, 108.8, 102.9, 102.1, and 67.7 ppm (Supplementary Figure S68). HRMS m/z calcd for C15H9Cl2NO6 [M]+ 368.9801, found 368.9865 (Supplementary Figure S69). HPLC analysis: retention time = 13.8 min; purity = 99.8%.
2.2-Dichloro-1-(4-(4-cyclohexylphenoxy)-3-nitrophenyl)ethan-1-one (24) was obtained as a white solid; yield 29%; m.p.: 106–107°C; 1H NMR (400 MHz, CDCl3) δ: 8.70 (d, J = 2.4 hz, 1H), 8.21 (dd, J = 2.4, 9.2 hz, 1H), 7.28 (d, J = 8.4 hz, 2H), 7.05 (d, J = 8.8 hz, 2H), 7.01 (d, J = 9.2 hz, 1H), 6.51 (s, 1H), 2.57–2.51 (m, 1H), 1.91–1.86 (m, 4H), 1.79–1.75 (m, 1H), 1.44–1.39 (m, 4H), and 1.31–1.23 (m, 1H) ppm (Supplementary Figure S70). 13C NMR (101 MHz, CDCl3) δ: 183.3, 156.4, 151.4, 146.4, 139.9, 135.3, 128.8, 128.0, 124.5, 120.4, 118.1, 67.7, 44.0, 34.5, 26.8, and 26.1 ppm (Supplementary Figure S71). HRMS m/z calcd for C20H19Cl2NO4 [M]+ 407.0686, found 407.0758 (Supplementary Figure S72). HPLC analysis: retention time = 17.3 min; purity = 97.9%.
1-(4-([1,1′-Biphenyl]-4-yloxy)-3-nitrophenyl)-2,2-dichloroethan-1-one (25) was obtained as a white solid; yield, 68%; m.p.: 138–139°C; 1H NMR (400 MHz, CDCl3) δ: 8.74 (d, J = 2.4 hz, 1H), 8.25 (dd, J = 2.4, 9.2 hz, 1H), 7.67 (d, J = 8.8 hz, 2H), 7.60–7.58 (m, 2H), 7.49–7.45 (m, 2H), 7.40–7.36 (m, 1H), 7.21 (d, J = 8.8 hz, 2H), 7.10 (d, J = 8.8 hz, 1H), and 6.52 (s, 1H) ppm (Supplementary Figure S73). 13C NMR (101 MHz, CDCl3) δ: 183.3, 155.9, 153.1, 140.2, 139.8, 139.5, 135.4, 129.2, 128.9, 128.1, 127.7, 127.1, 124.9, 120.9, 118.4, and 67.7 ppm (Supplementary Figure S74). HRMS m/z calcd for C20H13Cl2NO4 [M]+ 401.0222, found 401.0154 (Supplementary Figure S75). HPLC analysis: retention time = 14.0 min; purity = 96.9%.
6-(4-(2,2-Dichloroacetyl)-2-nitrophenoxy)-2H-chromen-2-one (26) was obtained as a white solid; yield 80%; m.p.: 159–161°C; 1H NMR (400 MHz, CDCl3) δ: 8.75 (d, J = 2.4 hz, 1H), 8.27 (dd, J = 2.4, 8.8 hz, 1H), 7.67 (d, J = 9.2 hz, 1H), 7.43 (d, J = 8.8 hz, 1H), 7.33 (dd, J = 2.8, 8.8 hz, 1H), 7.28 (d, J = 2.8 hz, 1H), 7.03 (d, J = 8.8 hz, 1H), 6.52 (d, J = 9.6 hz, 1H), and 6.51 (s, 1H) ppm (Supplementary Figure S76). 13C NMR (101 MHz, CDCl3) δ: 183.2, 159.9, 155.2, 151.7, 149.9, 142.2, 140.3, 135.6, 128.2, 125.6, 124.0, 120.1, 119.2, 118.8, 118.4, 118.3, and 67.7 ppm (Supplementary Figure S77). HRMS m/z calcd for C17H10Cl2NO6 [M+H]+ 393.9880, found 393.9928 (Supplementary Figure S78). HPLC analysis: retention time = 10.9 min; purity = 99.8%.
1-(4-([1,1′-Biphenyl]-3-yloxy)-3-nitrophenyl)-2,2-dichloroethan-1-one (27) was obtained as a colorless oil; yield 53%; 1H NMR (400 MHz, CDCl3) δ: 8.74 (d, J = 2.4 hz, 1H), 8.24 (dd, J = 2.4, 9.2 hz, 1H), 7.59–7.53 (m, 4H), 7.46–7.37 (m, 4H), 7.14–7.11 (m, 1H), 7.08 (d, J = 9.2 hz, 1H), and 6.52 (s, 1H) ppm (Supplementary Figure S79). 13C NMR (101 MHz, CDCl3) δ: 183.3, 155.9, 154.1, 144.1, 140.1, 139.5, 135.4, 130.9, 129.0, 128.2, 128.1, 127.1, 125.0, 124.9, 119.2, 119.1, 118.4, and 67.7 ppm (Supplementary Figure S80). HRMS m/z calcd for C20H14Cl2NO4 [M+H]+ 402.0294, found 402.0296 (Supplementary Figure S81). HPLC analysis: retention time = 20.7 min; purity = 95.4%.
1-(4-(4-Benzoylphenoxy)-3-nitrophenyl)-2,2-dichloroethan-1-one (28) was obtained as a white solid; yield, 83%; m.p.: 125–126°C; 1H NMR (400 MHz, CDCl3) δ: 8.77 (d, J = 2.4 hz, 1H), 8.31 (dd, J = 2.4, 8.8 hz, 1H), 7.92 (d, J = 6.8 hz, 2H), 7.82–7.79 (m, 2H), 7.64–7.60 (m, 1H), 7.51 (dd, J = 7.6, 7.6 hz, 2H), 7.22 (d, J = 8.8 hz, 2H), 7.16 (d, J = 8.8 hz, 1H), and 6.53 (s, 1H) ppm (Supplementary Figure S82). 13C NMR (101 MHz, CDCl3) δ: 195.1, 183.2, 157.5, 154.4, 140.9, 137.2, 135.5, 135.1, 132.7, 129.9, 128.5, 128.2, 126.1, 119.9, 119.5, and 67.7 ppm (Supplementary Figure S83). HRMS m/z calcd for C21H14Cl2NO5 [M+H]+ 430.0244, found 430.1468 (Supplementary Figure S84). HPLC analysis: retention time = 14.9 min; purity = 97.3%.
1-(4-(4-(Benzyloxy)phenoxy)-3-nitrophenyl)-2,2-dichloroethan-1-one (29) was obtained as a light yellow solid; yield 57%; m.p.: 125–126°C; 1H NMR (400 MHz, CDCl3) δ: 8.70 (d, J = 2.0 hz, 1H), 8.20 (dd, J = 2.4, 9.2 hz, 1H), 7.46–7.35 (m, 5H), 7.09–7.03 (m, 4H), 6.97 (d, J = 9.2 hz, 1H), 6.51 (s, 1H), and 5.09 (s, 2H) ppm (Supplementary Figure S85). 13C NMR (101 MHz, CDCl3) δ: 183.3, 160.0, 156.7, 147.0, 139.8, 136.5, 135.3, 128.7, 128.2, 128.1, 127.5, 124.4, 121.9, 117.6, 116.5, 70.6, and 67.7 ppm (Supplementary Figure S86). HRMS m/z calcd for C21H17Cl2NO6 [M+H2O]+ 449.0433, found 449.0273 (Supplementary Figure S87). HPLC analysis: retention time = 13.8 min; purity = 99.2%.
2.2-Dichloro-1-(4-(4-((4-isopropoxyphenyl)sulfonyl)phenoxy)-3-nitrophenyl)ethan-1-one (30) was obtained as a white solid; yield 66%; m.p.: 140–141°C; 1H NMR (400 MHz, CDCl3) δ: 8.75 (d, J = 2.0 hz, 1H), 8.31 (dd, J = 2.4, 8.8 hz, 1H), 7.99 (d, J = 8.8 hz, 2H), 7.85 (d, J = 8.8 hz, 2H), 7.18 (d, J = 8.8 hz, 2H), 7.11 (d, J = 8.8 hz, 1H), 6.95 (d, J = 8.8 hz, 2H), 6.52 (s, 1H), 4.62 (hept, J = 6.0 hz, 1H), and 1.35 (d, J = 6.0 hz, 6H) ppm (Supplementary Figure S88). 13C NMR (101 MHz, CDCl3) δ: 183.1, 162.2, 157.9, 153.7, 141.1, 139.7, 135.6, 131.9, 130.1, 129.9, 128.2, 126.6, 120.4, 119.9, 115.9, 70.6, 67.7, and 21.8 ppm (Supplementary Figure S89). HRMS m/z calcd for C23H20Cl2NO7S [M+H]+ 524.0332, found 524.1823 (Supplementary Figure S90). HPLC analysis: retention time = 14.0 min; purity = 99.5%.
General Procedure for Synthesis of Target Compounds 31–32
The intermediates 5a–5b were easily synthesized by nitration and condensation of 4-fluoro-2-hydroxy-acetophenone (S3) according to previously reported procedures.21 Target compounds 31 and 32 can be prepared from the corresponding intermediates 5a–5b by the method mentioned in “General Procedure for Synthesis of Target Compounds 7–30”.
Ethyl 2-(2-(2,2-dichloroacetyl)-5-(4-((4-isopropoxyphenyl)sulfonyl)phenoxy)-4-nitrophenoxy) acetate (31) was obtained as a white solid; yield 82%; mp: 67–68°C; 1H NMR (600 MHz, CDCl3) δ: 8.68 (s, 1H), 7.97 (d, J = 8.4 hz, 2H), 7.84 (d, J = 8.4 hz, 2H), 7.38 (s, 1H), 7.13 (d, J = 9.0 hz, 2H), 6.94 (d, J = 9.0 hz, 2H), 6.42 (s, 1H), 4.64 (s, 2H), 4.62 (hept, J = 6.0 hz, 1H), 4.28 (q, J = 7.2 hz, 2H), 1.34 (d, J = 6.0 hz, 6H), and 1.32 (t, J = 7.2 hz, 3H) ppm (Supplementary Figure S91). 13C NMR (150 MHz, CDCl3) δ: 184.4, 166.0, 162.2, 159.9, 158.3, 154.6, 139.4, 135.8, 131.9, 131.8, 130.1, 129.9, 119.7, 119.2, 115.9, 104.8, 71.0, 70.6, 66.3, 62.6, 21.8, and 14.1 ppm (Supplementary Figure S92). HRMS m/z calcd for C27H26Cl2NO10S [M+H]+ 626.0649, found 626.0649 (Supplementary Figure S93). HPLC analysis: retention time = 15.3 min; purity = 99.1%.
2-(2-(2,2-Dichloroacetyl)-5-(4-((4-isopropoxyphenyl)sulfonyl)phenoxy)-4-nitrophenoxy)acetamide (32) was obtained as a white solid; yield 70%; mp: 142–144°C; 1H NMR (400 MHz, Acetone-d6) δ: 8.63 (s, 1H), 8.00 (d, J = 9.2 hz, 2H), 7.89 (d, J = 9.2 hz, 2H), 7.77 (s, 1H), 7.32 (d, J = 8.8 hz, 2H), 7.18 (brs, 1H), 7.13 (s, 1H), 7.09 (d, J = 8.8 hz, 2H), 6.84 (brs, 1H), 4.89 (s, 2H), 4.75 (hept, J = 6.0 hz, 1H), and 1.31 (d, J = 6.0 hz, 6H) ppm (Supplementary Figure S94). 13C NMR (101 MHz, Acetone-d6) δ: 186.7, 169.7, 163.9, 163.4, 161.0, 155.8, 140.5, 137.4, 134.8, 132.2, 131.8, 131.6, 121.4, 120.5, 117.7, 109.3, 72.9, 72.1, 69.9, and 22.9 ppm (Supplementary Figure S95). HRMS m/z calcd for C25H23Cl2N2O9S [M+H]+ 597.0497, found 597.0516 (Supplementary Figure S96). HPLC analysis: retention time = 16.5 min; purity = 96.7%.
Biological Evaluation
In vitro PDHK1-4 Activity Assay
Recombinant human PDHK1-4 was prepared as previously described.23 PDHK activity was measured using the Kinase-Glo® Luminescent Kinase Kit in 96-well plates. The reaction mixture (50 μL) contained the following components: 20 μL ddH2O, 5 μL assay buffer (250 mm Tris-HCl, 10 mm EDTA, 5 mm EGTA, and 50 mm MgCl2, pH 7.4), 5 μL DTT (10 mm), 5 μL peptide substrate (100 μM RYHGHSMSDP), 5 μL samples at various concentrations in DMSO, 5 μL recombinant PDHK1–4 proteins (10–40 μM), and 5 μL ATP (10–500 μM). After incubation at 30°C for 1 h, 50 μL Kinase-Glo Plus Reagent (Promega, Madison, WI) was added, and the mixture was incubated at 25°C for 10 minutes. Luminescence data were recorded on a SpectraMax® i3 multimode detection platform. The IC50 values were obtained by fitting luminescence values to a dose–response curve with Prism software (Supplementary Figures S97–S102).
Antiproliferative Assays
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays were performed to evaluate cell viability. NCI-H1975, H1650, H1299 (NSCLC cell lines), and HEK293 (human kidney embryo cell) were all bought from Cell Bank (Shanghai, China). Cells were seeded into 96-well plates (about 5,000/well), and incubated for 24 hours. Compounds were added at various concentrations and incubated with the cells for 3 days. Subsequently, 10 μL MTT (5 mg/mL) was added to each well. After incubation for 4 hours, the solution was slowly removed and DMSO (100 μL) was added to each well. Optical density (OD) was recorded at 570 nm using a plate reader. Each concentration was tested in triplicate. Data were analyzed using GraphPad Prism 7.
Molecular Modeling Studies
Homology Models Generation
Sequences of human pyruvate dehydrogenase kinases 1 and 4 (Q15118 and Q16654, respectively) were retrieved from the UniPort database. Discovery Studio 2020 (DS 2020) was used to construct homology models of human pyruvate dehydrogenase kinases 1 and 4, using human pyruvate dehydrogenase kinase 2 (PDB code 2bu7) as the template. Homology models’ generation of human pyruvate dehydrogenase kinase 1 and 4 were performed by “Align sequence to template” and “Building Homology models” protocols. After evaluation with the “Profile-3D” protocol, the optimum homology models of human pyruvate dehydrogenase kinase 1 and 4 were obtained.
Molecular Docking Procedure
All docking calculations were performed using AutoDock Vina (version 1.2.0), which is a classic docking program. Preparation of proteins and compounds were performed by “Dock Prep” and “Minimize Structure” protocols. The parameters used in the docking processes were as follows: center = 41.23, 35.21, and 87.62; size = 20 Å × 20 Å × 20 Å; number of dockings = 100; exhaustiveness of search = 8; maximum energy difference = 3 kcal/mol; and others as default.
In vitro Liver Microsome Stability Evaluation
A 10 mm stock solution of the sample was diluted to a final concentration of 20 μM for the experiments. The compound was mixed with a solution of 0.1 M phosphate buffer, 0.5 mg/mL mice or human liver microsome and 1 mm NADPH. After incubation for 30 minutes at 37°C, the reaction was stopped by adding a two-fold volume of cold acetonitrile containing an internal standard. The mixed solution was stored overnight at −20°C. After centrifugation at 12,000 rpm for 5 minutes, an aliquot of 100 μL of the supernatant was tested using UPLC-MS/MS. 6-Hydroxy-coumarine was used as a positive control. A blank sample without NADPH was used as the negative control.
In vivo Efficacy Studies of the H1299 Xenograft Model in Mice
All animal experiments were approved by the Animal Ethics Committee of Hubei Polytechnic University in accordance with the approved “Animal management regulations” and follow the recommendations concerning laboratory animal welfare. Female BALB/c nude mice aged 5 weeks were purchased from Hubei Bainte Biotechnology Co., Ltd. and maintained under pathogen-free conditions. NCI-H1299 cells were harvested during log phase growth and resuspended in PBS at a density of 1 × 107 cells/mL. Each mouse was inoculated subcutaneously with 1.4 × 106 cells. When the volume of the tumors reached approximately 75 mm3, the mice were randomly divided into three groups (n = 5) and treated intraperitoneally with vehicle control (2% dimethyl sulfoxide, 30% PEG-400, and 68% ddH2O), compound 32, and DOX at a dose of 6 mg/kg. Vehicle and compound 32 were injected once a day for 21 days, whereas DOX was administered once every 3 days. Tumor diameter and body weight were monitored once every 3 days using a digital caliper and an electronic balance, respectively. Tumor volume was calculated using the formula (a × b2)/2, where a is the larger diameter and b is the smaller diameter. Eventually, the mice were euthanized and the tumors were dissected.
Acknowledgments
This work was financially supported by the Talent Introduction Project of the Hubei Polytechnic University (24xjz33R), the Project of the Outstanding Young and Middle-Aged Scientific Innovation Team of Universities in Hubei Province (T2021025), and University of Macau (MYRG2019-00034-FHS).
Disclosure
The authors declare that they have no known competing financial interests or personal relationships that could influence the work reported in this study.
References
1. Semenza GL, Artemov D, Bedi A, et al. ‘The metabolism of tumours’: 70 years later. Novartis Found Symp. 2001;240:251–260; discussion260–254.
2. Jones NP, Schulze A. Targeting cancer metabolism--aiming at a tumour’s sweet-spot. Drug Discov Today. 2012;17:232–241. doi:10.1016/j.drudis.2011.12.017
3. Patel MS, Roche TE. Molecular-biology and biochemistry of pyruvate-dehydrogenase complexes. FASEB J. 1990;4:3224–3233. doi:10.1096/fasebj.4.14.2227213
4. Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans. 2006;34:217–222. doi:10.1042/BST0340217
5. Koukourakis MI, Giatromanolaki A, Bougioukas G, Sivridis E. Lung cancer: a comparative study of metabolism related protein expression in cancer cells and tumor associated stroma. Cancer Biol Ther. 2007;6:1476–1479. doi:10.4161/cbt.6.9.4635
6. Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL. PDK-1 regulates lactate production in hypoxia and is associated with poor prognosis in head and neck squamous cancer. Br J Cancer. 2008;98:1975–1984. doi:10.1038/sj.bjc.6604356
7. Carbone D, De Franco M, Pecoraro C, et al. Structural manipulations of marine natural products inspire a new library of 3-Amino-1,2,4-Triazine PDK inhibitors endowed with antitumor activity in pancreatic ductal adenocarcinoma. Mar Drugs. 2023;21:288. doi:10.3390/md21050288
8. Trinidad AG, Whalley N, Rowlinson R, et al. Pyruvate dehydrogenase kinase 4 exhibits a novel role in the activation of mutant KRAS, regulating cell growth in lung and colorectal tumour cells. Oncogene. 2017;36:6164–6176. doi:10.1038/onc.2017.224
9. McFate T, Mohyeldin A, Lu H, et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem. 2008;283:22700–22708. doi:10.1074/jbc.M801765200
10. Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012;72:560–567. doi:10.1158/0008-5472.CAN-11-1215
11. Lu CW, Lin SC, Chien CW, et al. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am J Pathol. 2011;179:1405–1414. doi:10.1016/j.ajpath.2011.05.050
12. Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Harris AL; Tumor and Angiogenesis Research. Pyruvate dehydrogenase and pyruvate dehydrogenase kinase expression in non small cell lung cancer and tumor-associated stroma. Neoplasia. 2005;7:1–6. doi:10.1593/neo.04373
13. Wang G, Liu X, Xie J, Meng J, Ni X. PDK-1 mediated Hippo-YAP-IRS2 signaling pathway and involved in the apoptosis of non-small cell lung cancer cells. Biosci Rep. 2019;39: BSR20182099.
14. Miao Y, Wang W, Dong Y, et al. Hypoxia induces tumor cell growth and angiogenesis in non-small cell lung carcinoma via the Akt-PDK1-HIF1alpha-YKL-40 pathway. Transl Cancer Res. 2020;9:2904–2918. doi:10.21037/tcr.2020.03.80
15. Liu T, Yin H. PDK1 promotes tumor cell proliferation and migration by enhancing the Warburg effect in non-small cell lung cancer. Oncol Rep. 2017;37:193–200. doi:10.3892/or.2016.5253
16. Zhang SL, Hu X, Zhang W, Yao H, Tam KY. Development of pyruvate dehydrogenase kinase inhibitors in medicinal chemistry with particular emphasis as anticancer agents. Drug Discov Today. 2015;20:1112–1119. doi:10.1016/j.drudis.2015.03.012
17. Stacpoole PW. Therapeutic targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) axis in cancer. J Natl Cancer Inst. 2017;109. doi:10.1093/jnci/djx071
18. Qin L, Tian Y, Yu Z, et al. Targeting PDK1 with dichloroacetophenone to inhibit acute myeloid leukemia (AML) cell growth. Oncotarget. 2016;7:1395–1407. doi:10.18632/oncotarget.6366
19. Zhang SL, Yang Z, Hu XH, Tam Y. Dichloroacetophenones targeting at pyruvate dehydrogenase kinase 1 with improved selectivity and antiproliferative activity: synthesis and structure-activity relationships. Bioorg Med Chem Lett. 2018;28:3441–3445. doi:10.1016/j.bmcl.2018.09.026
20. Xu B, Wang ZP, Liu Q, et al. Synthesis, biological evaluation and structure-activity relationship of novel dichloroacetophenones targeting pyruvate dehydrogenase kinases with potent anticancer activity. Eur J Med Chem. 2021;214:113225. doi:10.1016/j.ejmech.2021.113225
21. Zhong L, Liu Q, Ting YS, et al. Adenine derivatives invert high glucose-induced thioredoxin-interacting protein overexpression. Chem Biol Drug Des. 2018;92:1998–2008. doi:10.1111/cbdd.13371
22. Caldwell ST, Cairns AG, Olson M, et al. Synthesis of an azido-tagged low affinity ratiometric calcium sensor. Tetrahedron. 2015;71:9571–9578. doi:10.1016/j.tet.2015.10.052
23. Zhang SL, Hu X, Zhang W, Tam KY. Unexpected discovery of dichloroacetate derived adenosine triphosphate competitors targeting pyruvate dehydrogenase kinase to inhibit cancer proliferation. J Med Chem. 2016;59:3562–3568. doi:10.1021/acs.jmedchem.5b01828
 © 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2024 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.