")
Back to Journals » International Journal of Nanomedicine » Volume 9 » Issue 1
Novel delivery system for T-oligo using a nanocomplex formed with an alpha helical peptide for melanoma therapy
Authors Uppada S, Erickson T, Wojdyla L, Moravec D, Song Z, Cheng J, Puri N
Received 29 September 2013
Accepted for publication 6 November 2013
Published 17 December 2013 Volume 2014:9(1) Pages 43—53
DOI https://doi.org/10.2147/IJN.S55133
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Srijayaprakash B Uppada,1,* Terrianne Erickson,1 Luke Wojdyla,1 David N Moravec,1 Ziyuan Song,2 Jianjun Cheng,2 Neelu Puri1,*
1Department of Biomedical Sciences, University of Illinois College of Medicine at Rockford, Rockford, 2Department of Materials Science and Engineering, University of Illinois at Urbana-Champaign, Urbana, IL, USA
*These authors contributed equally to this work
Abstract: Oligonucleotides homologous to 3'-telomere overhang (T-oligos) trigger inherent telomere-based DNA damage responses mediated by p53 and/or ATM and induce senescence or apoptosis in various cancerous cells. However, T-oligo has limited stability in vivo due to serum and intracellular nucleases. To develop T-oligo as an innovative, effective therapeutic drug and to understand its mechanism of action, we investigated the antitumor effects of T-oligo or T-oligo complexed with a novel cationic alpha helical peptide, PVBLG-8 (PVBLG), in a p53 null melanoma cell line both in vitro and in vivo. The uptake of T-oligo by MM-AN cells was confirmed by immunofluorescence, and fluorescence-activated cell sorting analysis indicated that the T-oligo-PVBLG nanocomplex increased uptake by 15-fold. In vitro results showed a 3-fold increase in MM-AN cell growth inhibition by the T-oligo-PVBLG nanocomplex compared with T-oligo alone. Treatment of preformed tumors in immunodeficient mice with the T-oligo-PVBLG nanocomplex resulted in a 3-fold reduction in tumor volume compared with T-oligo alone. This reduction in tumor volume was associated with decreased vascular endothelial growth factor expression and induction of thrombospondin-1 expression and apoptosis. Moreover, T-oligo treatment downregulated procaspase-3 and procaspase-7 and increased catalytic activity of caspase-3 by 4-fold in MM-AN cells. Furthermore, T-oligo induced a 10-fold increase of senescence and upregulated the melanoma tumor-associated antigens MART-1, tyrosinase, and thrombospondin-1 in MM-AN cells, which are currently being targeted for melanoma immunotherapy. Interestingly, siRNA-mediated knockdown of p73 (4–10-fold) abolished this upregulation of tumor-associated antigens. In summary, we suggest a key role of p73 in mediating the anticancer effects of T-oligo and introduce a novel nanoparticle, the T-oligo-PVBLG nanocomplex, as an effective anticancer therapeutic.
Keywords: T-oligo, melanoma, senescence, angiogenesis, apoptosis
Introduction
Metastatic melanoma is the most fatal skin cancer, for which there is no effective chemotherapy.1–4 Currently, therapies targeting melanoma and approved by the US Food and Drug Administration, such as BRAF inhibitors, have limited efficacy due to the development of drug resistance.5 Hence, more effective alternative therapies need to be developed for melanoma. In this study, we explored the therapeutic potential of a nanocomplex composed of a recently discovered positively charged helical peptide (PVBLG)6 and an eleven-base oligonucleotide identical to the 3′ telomere overhang (T-oligo)7–9 in melanoma cells and tumor xenografts.
T-oligo mimics the exposure of the 3′ telomere overhang and induces DNA damage response (DDR) signals in several cancers.8–13 T-oligo mediates DDRs by activating multiple p53 dependent/independent DDR pathways which modulate several downstream effectors.12,14 Recent evidence by our group suggests that T-oligo interacts with the shelterin complex, thereby disrupting telomere integrity and inducing DDRs.12 Our earlier studies indicate that T-oligo treatment reduces tumorigenicity and metastasis, and induces differentiation of melanoma tumors.8 Furthermore, we have also shown that T-oligo reduces tumorigenicity by induction of senescence and reduced vascularity in lung cancer.15 However, to be clinically effective, T-oligo requires stabilization for intravenous delivery in cancer patients since serum and intracellular nucleases could degrade its phosphodiester backbone.16
Recently, Gabrielson et al developed a highly efficient gene and siRNA delivery system using PVBLG, a cationic helical polypeptide that is significantly more effective and less toxic than liposomal formulations or other cationic polymer vehicles.17 The helical structure of PVBLG allows for high stability across a wide range of pH, temperatures, and salt concentrations in the presence of denaturing agents.6 Moreover, it has been shown to be highly effective in delivering plasmid DNA and siRNA to a variety of cell lines.6 Therefore, T-oligo was stabilized via complex formation with PVBLG. PVBLG self-assembles with oligonucleotides such as T-oligo, neutralizing their negative charges and stabilizing them for delivery in vivo.
The aim of the present study was to investigate the anticancer effects of T-oligo and test the efficacy of a novel T-oligo-PVBLG (TOP) nanocomplex. We found that the TOP complex enhanced the therapeutic potential of T-oligo in malignant melanoma in vitro and in vivo. We propose that the TOP complex could be used as a novel therapeutic approach for melanoma.
Materials and methods
Oligonucleotides
Oligonucleotides homologous (pGTTAGGGTTAG) (T-oligo) and complementary to the 3′ overhang sequence (pCTAACCCTAAC) (C-oligo) were obtained from Midland Certified Reagent Co, Midland, TX, USA.
Synthesis of PVBLG and preparation of TOP nanoparticles
PVBLG was prepared by Jianjun Cheng at the University of Illinois, via ring-opening polymerization of N-carboxyanhydrides as described previously.18 A range of PVBLG concentrations (0.025–0.05 mg/mL) were then complexed with T-oligo (20 μM) in HEPES buffer to form nanoparticles.
Particle size and zeta potential analysis
T-oligo and PVBLG solutions were prepared separately in DNAse/RNAse-free distilled water (Invitrogen, Grand Island, NY, USA) and complexed at various weight ratios. The TOP complex was incubated at room temperature for 20 minutes. Particle size and zeta potential was measured using a NICOMP 380 dynamic light scattering instrument (Particle Sizing Systems, Inc, Santa Barbara, CA, USA). PVBLG or T-oligo alone was used as the control.
Antibodies
Antibodies for vascular endothelial growth factor (VEGF, sc-152), thrombospondin (TSP)-1 (sc-12312) CD31 (PECAM-1, sc-1506), and tyrosinase-related protein (TRP-1, sc 10443) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). β-actin (A5441) antibody was sourced from Sigma-Aldrich (St Louis, MO, USA). MART-1/Melan-A (clone M2-7C10) monoclonal antibody was sourced from Signet (Dedham, MA, USA). Tyrosinase (clone T311) monoclonal antibody (Novacastra™) was purchased from Leica Biosystems (Buffalo Grove, IL, USA). p73 antibody was obtained from EMD Millipore (Billerica, MA, USA), cleaved caspase-3 (clone 5 A1E, rabbit monoclonal antibody #9664) and cleaved caspase-7 (clone D6H1, rabbit monoclonal antibody #8438) were purchased from Cell Signaling Technology (Danvers, MA, USA). All antibodies were used according to the manufacturers’ instructions.
Cell lines and cell culture
MU and MM-AN cell lines were obtained by explant culture8 and maintained at 37°C in Minimum Essential Medium (Catalog Number MT-10-010-CM, Thermo Fisher Scientific, Pittsburg, PA, USA) supplemented with 10% (v/v) fetal bovine serum (Catalog Number S11150, Atlanta Biologicals, Lawrenceville, GA, USA) and 1% (v/v) antibiotic/antimycotic (Catalog Number 15240, Invitrogen).
Senescence and clonogenicity experiments
T-oligo-induced senescence in MU melanoma cells was evaluated by treating the cells with 40 μM of T-oligo or C-oligo for one week in 35 mm2 dishes, after which the cells were fixed with 4% paraformaldehyde in phosphate-buffered saline for 5 minutes and washed twice with phosphate-buffered saline. The cells were then stained with senescence-associated β-galactosidase (SA-β-gal)7 for 16 hours at 37°C. The cell phenotypes were examined, and images were captured at 200× magnification to determine senescent morphology. For clonogenicity experiments, MU and MM-AN cells were plated at 25,000 cells/60 mm dish and treated for one week with 40 μM of T-oligo or C-oligo. Cells were then trypsinized, collected, and seeded at 3,000 cells per dish and allowed to grow for 10 days in growth medium. Cells were then fixed in 100% ethanol and stained with 1% methylene blue for 10 minutes. Colonies were counted using Kodak ROI analysis software (Eastman Kodak Company, Rochester, NY, USA). All experiments were performed in triplicate. The paired Student’s t-test was used to evaluate the differences between T-oligo and C-oligo groups. Significance was established at α=0.05.
Transfection of p73 siRNA and tumor-associated antigen expression in vitro
Cells were cultured in Minimum Essential Medium supplemented with 5% fetal bovine serum and transfected with control siRNA or p73 siRNA (four pooled siRNA duplexes, Dharmacon, Pittsburgh, PA, USA) using Oligofectamine™ transfection reagent (Catalog Number 12252-011, Life Technologies, Grand Island, NY, USA) for 12 hours. Mock transfection was used as a negative control in parallel using signal silence control siRNA (Catalog Number 6568S, Cell Signaling Technology). The cells were then treated with 40 μM T-oligo or diluent. Cells were collected after 72–96 hours, cell lysates were made, and immunoblotting was performed with the lysates for p73 and the tumor-associated antigens (TAAs) MART-1, tyrosinase, and TRP-1.
Immunoblotting for procaspases and evaluation of caspase activity
MM-AN cells were grown in Minimum Essential Medium containing 5% fetal bovine serum and treated with diluent, 40 μM of T-oligo or C-oligo at various time points, after which cells were harvested and protein lysates were immunoblotted for procaspase-3 and procaspase-7. These lysates were further used to determine caspase-3 activity using the CaspACE assay system (Catalog Number G7351, Promega, Madison, WI, USA), as described previously.19 Quadruplicate cultures of MM-AN cells were treated with diluent, or 40 μM of T-oligo or C-oligo, for 72 or 96 hours. Cells were then collected by centrifugation, washed, and lysed by repeated freeze/thaw cycles. The lysates were then clarified by centrifugation and the supernatant was used for protein estimation and for the caspase assay. This assay uses Ac-DEVD-pNA as a substrate and colorimetrically measures the release of free pNA (p-nitroaniline). Statistical significance was determined using the paired Student’s t-test, with significance established at α=0.05.
MTT cell viability assay
To study the effects of T-oligo with or without PVBLG on cell viability, MM-AN cells were treated with T-oligo or the TOP complex, and cell viability was determined by measuring 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric dye reduction (Sigma-Aldrich, Catalog Number TOX1) according to the manufacturer’s instructions. Five thousand cells per well were plated in replicates of six in a 96-well plate in Minimum Essential Medium supplemented with 10% fetal bovine serum. After 24 hours, the cells were treated with T-oligo (20 μM) with or without PVBLG (0.025, 0.05 mg/mL) at concentrations found to be nontoxic and maximally effective for 4 hours, after which an MTT assay was performed as described previously.20 At 96 hours, the MTT reagent was added, cells were incubated for 4 hours at 37°C, after which solubilization solution was added and absorbance was measured at 570 nm. Background absorbance at 690 nm was subtracted from the 570 nm measurement and percentage of cell viability was determined relative to the control. Each experiment was performed in replicates of six for each treatment condition and repeated three times. Statistical significance was obtained using analysis of variance test with α at 0.05, and paired Student’s t-tests were used to determine the statistical significance of differences between two individual groups.
Intracellular uptake of FITC-labeled T-oligo
The uptake of fluorescein isothiocyanate (FITC)-labeled T-oligo (Midland Certified Reagent Co) or the TOP complex (40 μM FITC-T-oligo and 0.025 mg/mL PVBLG) by MM-AN cells was assessed using fluorescence-activated cell sorting (FACS) and fluorescence microscopy. For FACS analysis, 1.5×105 MM-AN cells in Minimum Essential Medium were plated in 35×10 mm cell culture dishes. After 24 hours, the cells were treated with 40 μM T-oligo with or without PVBLG (0.025 mg/mL) in Minimum Essential Medium with 2% calf serum for 4 hours. Cells were then washed twice with Hank’s Balanced Salt Solution (Invitrogen, Carlsbad, CA, USA), trypsinized, pelleted, counted, and fixed with 4% paraformaldehyde. Next, 1×106 cells were analyzed on a Becton Dickinson FACS device (Immunocytometric Systems, San Jose, CA, USA) and the data were analyzed using Cell Quest analysis software. For microscopy, 10,000 MM-AN cells were plated in a chamber slide. After 24 hours, cells were treated as described above and images were taken with a fluorescence microscope (IX 71, Olympus America Inc, Center Valley, PA, USA).
Animals and tumorigenicity experiment
Five-week-old male nu/nu (nude) mice were purchased from Taconic (Hudson, NY, USA) and housed in the pathogen-free animal facility at the University of Illinois College of Medicine at Rockford. First, 2.5×106 MM-AN cells were injected into the flanks of the mice (n=10). After 24 hours, the mice were treated with daily intravenous injections of diluent or 52 nmol of T-oligo with or without 0.025 mg/mL PVBLG. After 3 weeks, the mice were euthanized by CO2 inhalation and the tumors were evaluated. Analysis of tumor growth was performed using the paired Student’s t-test to compare mice treated with T-oligo with or without PVBLG. Significance was established at α=0.05.
The protocol was approved by the Institutional Animal Care and Use Committee of the University of Illinois College of Medicine at Rockford and animal studies were performed at a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care and the United States Department of Agriculture. Care was taken to ensure that the animals did not suffer discomfort, distress, or pain.
Immunohistochemistry
Tumors resected from mice were formalin-fixed, paraffin-embedded, and sectioned at the Rockford Memorial Hospital pathology laboratory (Rockford, IL, USA). Immunostaining procedures were performed as described previously.8,15 Briefly, sections were rehydrated and then rinsed in phosphate-buffered saline twice for 5 minutes. Antigen retrieval was performed by incubating rehydrated sections in a 0.05% trypsin, 1% calcium chloride solution for 10 minutes at 37°C. Tumor sections were then stained using the Mouse on Mouse (TOM™) immunohistochemistry kit (Vector Laboratories, Burlingame, CA, USA). Once the desired staining intensity was obtained, the slides were counterstained using hematoxylin (Sigma-Aldrich) for 15 seconds, dehydrated in increasing concentrations of ethanol and xylene, and mounted using Permount (Fisher, Rockford, IL, USA). Photomicrographs were obtained with an Olympus BH-2 microscope (Olympus America Inc) at 200× magnification. Immunohistochemistry was performed on at least three separate tumors with similar results, and the most representative section is shown.
TUNEL analysis
Apoptosis in mice tumors was detected by terminal transferase dUTP nick end labeling (TUNEL) assay. Tissues were fixed in 4% paraformaldehyde for 30 minutes, permeabilized with 0.1% Triton X-100 for 2 minutes, and a TUNEL assay was performed with a cell death detection kit (Roche Diagnostics, Indianapolis, IN, USA) as per the manufacturer’s instructions.
Statistical analysis
Statistical analyses were carried out using Statistical Package for the Social Sciences version 17 software (SPSS Inc, Chicago, IL, USA). A one-way analysis of variance with α at 0.05 and paired Student’s t-tests were used to determine statistical significance.
Results
T-oligo induces senescence and decreases clonogenic capacity in melanoma cells
Senescence has been shown to be a key restraining factor in cancer formation and progression.21 Earlier studies have demonstrated that T-oligo induces senescence in lung and breast cancer; however, senescence has not been demonstrated in melanoma with T-oligo treatment.11,15 Therefore, we investigated the ability of T-oligo to induce senescence in melanoma cells. MU cells showed a 10-fold increase in SA-β-gal staining after treatment with T-oligo compared with cells treated with C-oligo (Figure 1A and B). Further, they showed an enlarged, dendritic-like morphology similar to a senescent phenotype7 (Figure 1A). Furthermore, T-oligo-induced senescence was indicated by a 3.7-fold and 4.4-fold reduction in the clonogenic capacity of MU and MM-AN cells, respectively, as compared with C-oligo treatment (Figure 1C).
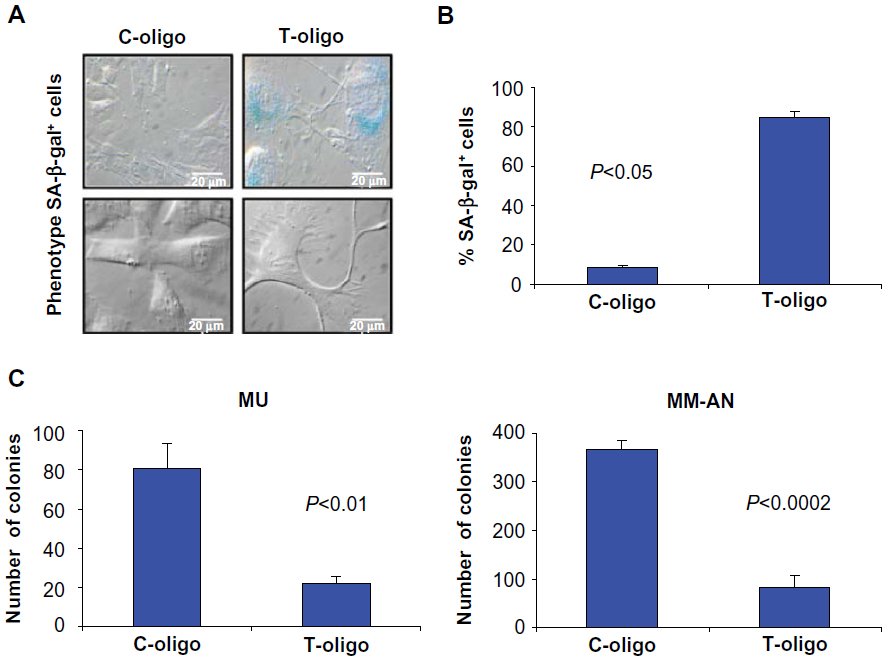 | Figure 1 Effect of T-oligo on senescence and clonogenicity in melanoma cells. (A) MU melanoma cells were treated with T-oligo or C-oligo for 1 week. Cells were then stained with SA-β-gal and observed under an Olympus BH2 microscope. T-oligo-induced senescence was indicated by SA-β-gal staining (blue) and changes in cell morphology similar to a senescent phenotype. (B) T-oligo treatment resulted in a 10-fold increase in SA-β-gal staining in melanoma cells as quantified by cell counting after trypan blue exclusion. (C) MU and MM-AN melanoma cells were treated with 40 μM T-oligo or C-oligo for 1 week. Cells were trypsinized and plated at 3,000 cells per dish and grown for 10 days. Cells were fixed in 100% ethanol, stained with 1% methylene blue for 10 minutes, and counted using Kodak ROI analysis software. T-oligo treatment decreased clonogenic capacity by 3.7-fold and 4.4-fold in MU and MM-AN melanoma cells, respectively. Experiments were carried out in triplicate and the most representative results are shown. |
T-oligo increases expression of TAAs in vitro
In melanoma cells, the expression of TAAs, such as MART-1, enhances targeting by specific CD8+ and CD4+ T cells.22 We have previously demonstrated upregulation of TAAs in p53 null and p73-expressing MM-AN cells after T-oligo treatment.8 In this study, we investigated the expression of TAAs in these cells after T-oligo treatment with/without knockdown of p73. Western blot analysis confirmed p73 knockdown (4–10-fold) at 48–96 hours (Figure 2A). T-oligo treatment resulted in upregulation of TRP-1 (1.5-fold) and tyrosinase (2.8-fold) at 72 hours, and MART-1 (4.5-fold) at 96 hours (Figure 2B and C) that was abolished by p73 knockdown, indicating a role for p73 in the modulation of TAAs in melanoma.
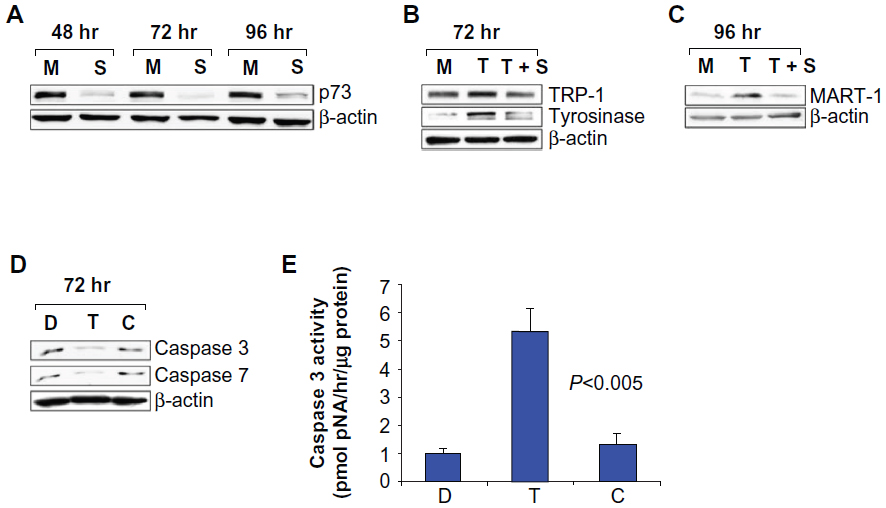 | Figure 2 Knockdown of p73 expression by siRNA downregulates T-oligo-induced tumor-associated antigens. MM-AN melanoma cells were transfected with p73-specific siRNA duplexes or mock siRNA for 12 hours, treated with diluent or 40 μM T-oligo, and analyzed by Western blotting at 48–96 hours. (A) siRNA-mediated knockdown of p73 (4–10-fold) at 48–96 hours was confirmed in MM-AN cells. (B and C) p73 knockdown inhibited T-oligo-induced expression of TRP-1 (1.5-fold), tyrosinase (2.8-fold), and MART-1 (4.5-fold) at 72–96 hours. (D) Immunoblots indicate a decrease in procaspase-3 (2-fold) and procaspase-7 (2-fold) after 72 hours of T-oligo treatment. Blots are representative of at least three independent experiments and β-actin was used to demonstrate even loading of samples. (E) Treatment with T-oligo increased caspase-3 activity in MM-AN cells by 4-fold at 72 hours as determined by caspase colorimetric assay. |
T-oligo induces apoptosis by activation of caspases
Earlier studies suggest that p73 mediates T-oligo-induced apoptosis in a p53 null cell line;8 however, caspase activation has not been studied in these cells. Therefore, we explored the modulation of procaspase-3 and procaspase-7 in MM-AN cells after T-oligo treatment. MM-AN cells displayed downregulation of procaspase-3 (2-fold) and procaspase-7 (2-fold) at 72 hours upon exposure to T-oligo (Figure 2D). Caspase-3 catalytic activity was found to be upregulated 4-fold in cells treated with T-oligo compared with cells treated with diluent or C-oligo (Figure 2E).
Particle size, zeta potential analysis, and enhanced cellular uptake of T-oligo by PVBLG
Dynamic light scattering was performed to analyze the particle size of the TOP complex. The mean particle diameter was observed to be 147.3 nm at a DNA to polymer weight ratio of 15:1 (Figure 3), which was similar to other DNA to polymer weight ratios (1:1–15:1, data not shown). Neither PVBLG nor T-oligo formed detectable particles when measured independently. In addition, the zeta potential of the TOP complex was found to be −25 mV ±3.13 mV, thereby indicating that PVBLG combined with T-oligo forms a stable nanocomplex.
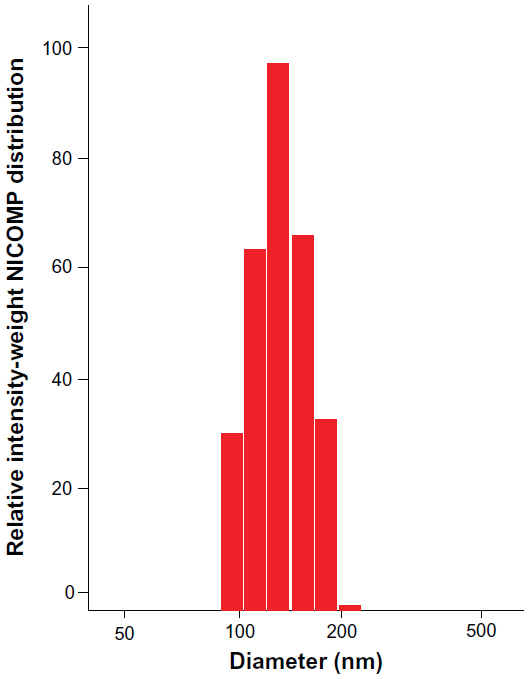 | Figure 3 Particle size analysis of the TOP complex. T-oligo and PVBLG solutions were prepared at a DNA to polymer weight ratio of 15:1 in DNAse/RNAse-free distilled water. T-oligo and PVBLG solutions were then mixed and incubated at room temperature for 20 minutes. The particle size was analyzed by dynamic light scattering. The results obtained are shown in NICOMP (dynamic light scattering) distribution, indicating that the TOP complex had a mean diameter of 147.3 nm. PVBLG or T-oligo alone was used as the control. |
FACS analysis of MM-AN melanoma cells treated with the TOP complex showed a greater than 15-fold increase in fluorescence as compared with T-oligo alone (Figure 4A). Uptake was verified using immunofluorescence microscopy studies which also showed enhanced cellular uptake of T-oligo after complexing it with PVBLG (Figure 4B). These results indicate that PVBLG stabilizes and enhances the uptake of T-oligo by MM-AN melanoma cells.
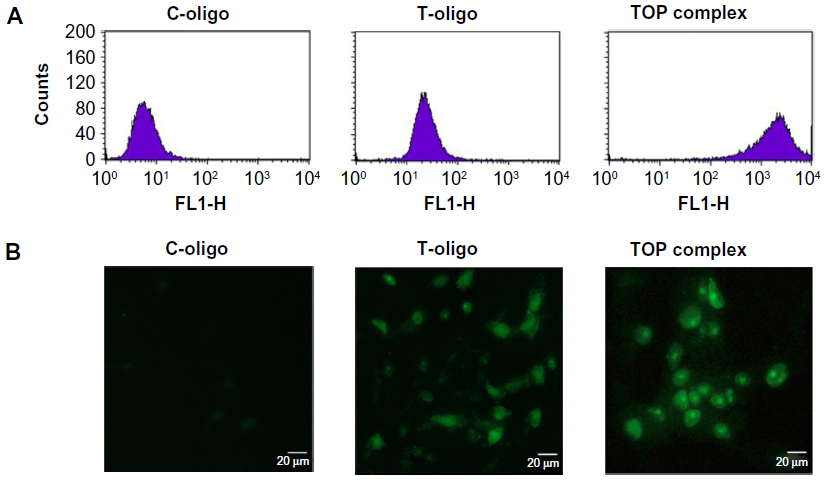 | Figure 4 Uptake studies of T-oligo and TOP complex in melanoma cells using fluorescence-activated cell sorting analysis and fluorescence microscopy. (A) 1.5×105 MM-AN cells in Minimum Essential Medium were plated in 35 mm2 cell culture dishes. After 24 hours, the cells were treated with 40 μM FITC-T-oligo or the TOP complex (40 μM FITC-T-oligo and 0.025 mg/mL PVBLG) in Minimum Essential Medium with 2% calf serum for 4 hours. Cells were then washed twice with Hank’s Balanced Salt Solution, trypsinized, pelleted, counted, and fixed with 4% paraformaldehyde and analyzed on a Becton Dickinson fluorescent-activated cell sorter. Data analysis was performed using Cell Quest software. (B) 10,000 MM-AN cells were plated in chamber slides, treated with T-oligo or the TOP complex as described above, and observed under a fluorescent microscope (Olympus IX71) at 200× magnification. |
PVBLG enhances efficacy of T-oligo in vitro
The effect of PVBLG on T-oligo efficacy in MM-AN cells was measured in vitro by cell counting after trypan blue exclusion. T-oligo and increasing concentrations of PVBLG were determined to be effective and nontoxic (0.025–0.05 mg/mL, n=6, P<0.001) and enhanced the growth inhibitory effect of T-oligo in a dose-dependent manner by 2.3–4.5-fold relative to T-oligo treatment alone (Figure 5).
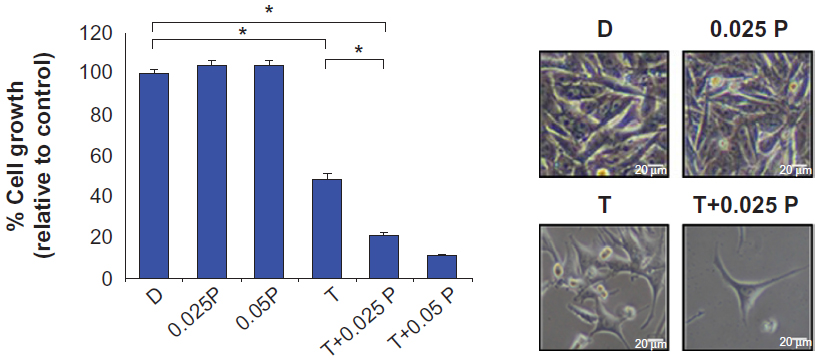 | Figure 5 Effect of T-oligo with or without PVBLG on growth and viability of MM-AN cells. First, 5,000 MM-AN cells/well were plated (n=6) in Minimum Essential Medium supplemented with 10% fetal bovine serum. After 24 hours, the cells were treated for 96 hours with 20 μM T-oligo with or without PVBLG at concentrations found to be nontoxic (0.025–0.05 mg/mL). Cell viability was determined using the MTT colorimetric dye reduction assay and percent cell viability was determined relative to the control. T-oligo inhibited growth by 51% while T-oligo with PVBLG (0.025–0.05 mg/mL) inhibited growth by 78%–89%. PVBLG alone had no effect at the tested concentrations. *P<0.001. |
PVBLG enhances efficacy of T-oligo in an immunodeficient mouse model
Oligonucleotides are quickly degraded by nucleases in serum as well as within cells. Therefore, to increase its therapeutic efficacy in vivo, an attempt was made to stabilize T-oligo. Recent studies by Gabrielson et al indicated that PVBLG enhanced the delivery of oligonucleotides and siRNA.17 Therefore, the TOP complex was further studied on melanoma tumors in immunodeficient mice. Different concentrations of PVBLG were tested on mice (0.01–0.1 mg/mL) and no toxicity was observed, and based on these studies and in vitro experiments, a concentration of 0.025 mg/mL PVBLG was selected. The T-oligo concentration used in this study were comparable with concentrations used in earlier investigations.8 Our results indicate a 3-fold and 9-fold reduction in tumor volume in T-oligo-treated and TOP complex treated mice respectively, as compared with mice treated with diluent (Figure 6). These results indicate that PVBLG enhances the therapeutic efficacy of T-oligo by 3-fold.
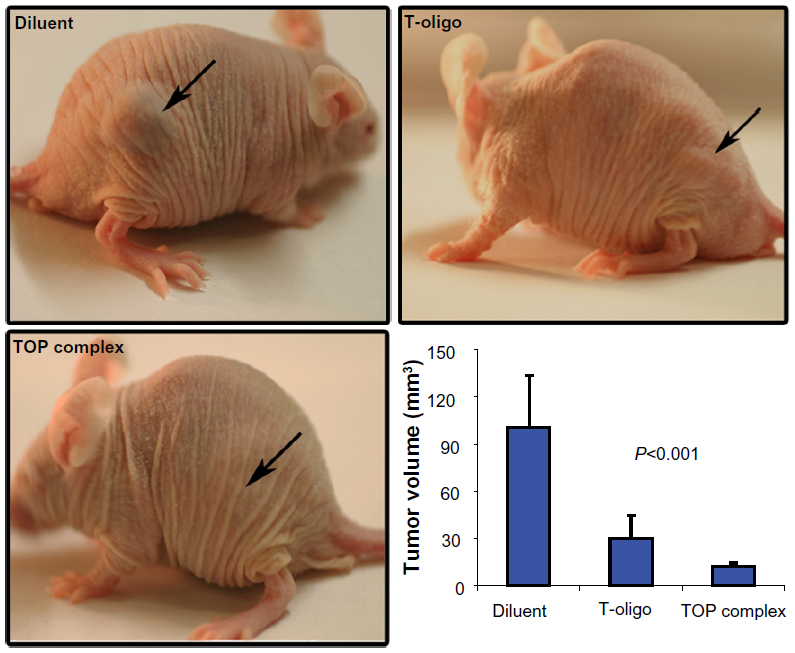 | Figure 6 Increased reduction of MM-AN melanoma xenograft tumors by TOP complex in nu/nu mice. First, 2.5×106 MM-AN cells were injected into the flank of nu/nu mice (n=10). After 24 hours, the mice were treated daily with diluent, T-oligo (52 nmol), TOP complex (52 nmol T-oligo and 0.025 mg/mL PVBLG) intravenously for 3 weeks. Mice treated with T-oligo demonstrated reduced tumors, while most TOP complex treated mice showed no visible tumors. The graph shows a 3-fold and 9-fold reduction in tumor volume after treatment with T-oligo or the TOP complex, respectively, as compared with diluent (P<0.001). |
T-oligo treatment inhibits angiogenesis and induces apoptosis in vivo
Increased expression of VEGF and angiogenesis are essential for tumor progression.23 TSP-1 plays a major role in inhibiting VEGF expression and angiogenesis and in inducing apoptosis.24,25 Moreover, levels of VEGF and TSP-1 were shown to correlate with prognosis in cancer patients.26 Therefore, we investigated the modulation of TSP-1, VEGF expression, and induction of apoptosis in melanoma tumor xenografts of immunodeficient mice treated intravenously with T-oligo or the TOP complex. Immunohistochemistry analysis displayed an increase in TSP-1 expression and a decrease in VEGF expression in the TOP complex treated tumors as compared with tumors treated using T-oligo alone (Figure 7). Moreover, there was an enhanced induction of apoptosis in the TOP complex treated tumors as compared with those treated with T-oligo alone (Figure 7).
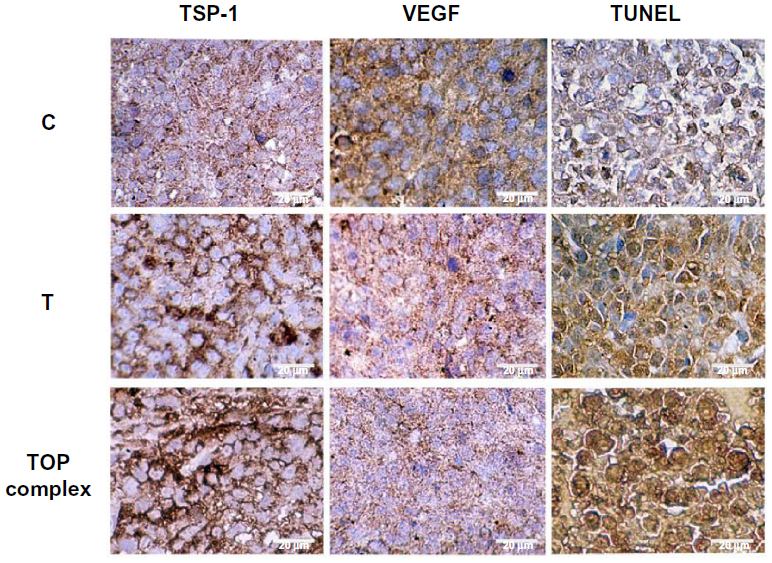 | Figure 7 Enhanced antitumor effects of the TOP complex in xenograft tumors of MM-AN melanoma cells. First, 2.5×106 MM-AN cells were injected into the flank of each immunodeficient mouse (n=10). After 24 hours, the mice were treated daily with diluent, T-oligo (52 nmol), or TOP complex (52 nmol T-oligo and 0.025 mg/mL PVBLG) intravenously for 3 weeks. Xenograft tumors were resected on day 21 and subjected to immunohistochemistry for VEGF and TSP-1. Apoptosis was evaluated by TUNEL assay, which was increased in tumors treated with the TOP complex compared with T-oligo. Immunohistochemical staining showed enhanced expression of TSP-1 and decreased expression of VEGF in mice treated with the TOP complex as compared with T-oligo alone. |
Discussion
Targeting the telomere and telomerase with anticancer oligonucleotides is an attractive approach in melanoma therapy.27 However, there is a severe hindrance in their delivery in vivo due to their degradation by serum and intracellular nucleases.28 Stabilization of oligonucleotides with nanoparticles is a potential solution since several nanoparticles have been developed for enhanced gene delivery.29,30 Recently, siRNA delivery using cationic lipid nanoparticles for melanoma therapy has been demonstrated to be effective;31 however, cationic liposomes require further modification to curb their cytotoxicity.32 PVBLG-8 (PVBLG), a recently discovered positively charged helical peptide,6 has been used as a highly efficient gene and siRNA delivery system.17 PVBLG is significantly more effective and less toxic than liposomal formulations or other cationic polymer delivery systems, and can also be used for targeted gene delivery as a supramolecular assembly system.33 Moreover, our earlier studies demonstrate that PVBLG protects DNA and siRNA oligonucleotides from nucleolytic degradation.17,34 The present study substantiates the therapeutic potential of the anticancer effects of T-oligo in melanoma cells and demonstrates enhanced uptake and efficacy of T-oligo with PVBLG nanoparticles (TOP complex) both in vitro and in vivo.
While treatment with T-oligo demonstrates potent antitumorigenic effects in several cancers, it has no cytotoxicity on their normal counterparts.8,11,15,35 In the present study, T-oligo treatment increased SA-β-gal expression, induced a morphology similar to that of senescent cells, and decreased clonogenic capacity in melanoma cells, suggesting that T-oligo-induced senescence inhibited their growth in vitro. The TAp73 tumor suppressor isoform of p73 mediates apoptosis irrespective of p53 status.36,37 Earlier studies show that p73 siRNA blocked apoptosis in MM-AN melanoma cells.8 Since apoptosis is linked with activation of caspases,38 we suggest a role of p73-mediated activation of caspase-3 and caspase-7 after T-oligo treatment. Additionally, T-oligo-induced expression of TAAs, which are known to slow cellular proliferation, was also abolished following p73 knockdown. Based on these results, we hypothesize that T-oligo mediates its anticancer effects through p73 (TAp73). Presently, studies are being carried out in our laboratory to understand further the p73-mediated effects of T-oligo in melanoma tumors in vivo.
To be effective in patients, T-oligo must be stabilized to avoid degradation by nucleases.39 Phosphorothioate-modified oligonucleotides have been used to increase stability against nucleolytic degradation;40 however, they release toxic products due to 3′-exonuclease-mediated degradation.41 Moreover, they exhibit low binding capacity to complementary sequences and show nonspecific binding to proteins,42 leading to toxic effects.43 To increase the efficacy of gene delivery, various viral and nonviral delivery strategies are being developed; however, viral approaches are hindered by side effects such as immunogenicity and toxicity, whereas nonviral delivery systems have limited transfection efficiency and biodegradability issues.43 To enhance the delivery and efficacy of T-oligo without toxicity, we have formed a nanocomplex using PVBLG, which has been shown to be efficient in delivering siRNA, genes, and oligos, with low toxicity.6 PVBLG facilitates delivery of oligonucleotides by disrupting the cell membrane irrespective of pH conditions, allowing oligonucleotides to escape endocytic vesicles, an otherwise limiting factor in their delivery. In addition, PVBLG proved to be 50% more effective than the commonly used commercial transfectant Lipofectamine® 2000.44 We hypothesize that PVBLG self-assembles with the negatively charged T-oligo, forming a stable nanocomplex thereby protecting it from nuclease degradation.
In the present study, we investigated the effect of the TOP complex on tumor growth, angiogenesis, and apoptosis. Inhibition of angiogenesis is an important approach to cancer therapy in which VEGF is a key mediator23 and TSP-1 is an inherent inhibitor.45 In several cancers, tumor progression depends on an “angiogenic switch” that results in a change of phenotype from avascular to vascular and is regulated by a balance of the proangiogenic factor VEGF and the antiangiogenic factor TSP-1.46–48 Upregulation of VEGF results in increased angiogenesis in melanoma and other cancers,49,50 thereby promoting tumor progression, while its inhibition increases progression-free survival.51,52 Moreover, VEGF has also been suggested to be a key predictive marker of patient prognosis and response to therapy in melanoma and other cancers.52,53 Currently, bevacizumab, a VEGF-specific antibody that inhibits angiogenesis,54 has been demonstrated to be an effective cancer therapy,55–57 thus substantiating that inhibition of angiogenesis by blocking VEGF is an effective approach to inhibit tumorigenicity in several cancers.58 TSP-1, a matricellular protein,59 has been shown to inhibit angiogenesis in human prostate cancer xenografts60 and liver metastasis in renal and colon carcinoma,61 and also modulates apoptosis, affecting tumor progression.62 Moreover, in a preclinical study, exogenous administration of TSP-1 inhibited angiogenesis and metastasis in human melanoma xenografts;63 however, in contrast with the above studies, other investigations have demonstrated that TSP-1 promotes metastasis in breast cancer.64 In our in vivo studies, we observed that treatment with the TOP complex was more effective in reducing melanoma tumor size as compared with T-oligo alone. Accordingly, both T-oligo and the TOP complex reduced VEGF and concomitantly increased TSP-1 expression, thereby reducing vasculogenesis and enhancing apoptosis, thereby substantiating their antitumorigenic role in melanoma. These results indicate that T-oligo reduces tumorigenicity through multiple anticancer approaches, and its stability and therapeutic potential can be enhanced by complexing it with PVBLG. Therefore, we propose that the TOP complex may be an effective therapeutic agent for the treatment of melanoma and possibly other cancers.
In conclusion, we suggest that T-oligo induces anticancer responses in melanoma cells through p73. We further demonstrate that the TOP complex is more efficient than T-oligo in inhibiting tumor growth, induction of apoptosis, and inhibition of angiogenesis. Our in vivo studies provide strong support for the enhanced therapeutic efficacy of the TOP complex in the treatment of melanoma, which may be extended to the treatment of other cancers in further investigations. Currently, studies are being carried out in our laboratory to improve the efficacy of the TOP complex by using ligands such as folic acid or mannose which have recently been developed into a supramolecular self-assembly system for oligonucleotide delivery.33
Acknowledgments
This research was funded by the University of Illinois at Chicago Cancer Center 2010 Pilot Grant Program. The authors are grateful to Nathan Gabrielson for his help with the technology and synthesis of PVBLG and Hayat Onyuksel for kindly helping with the particle size and zeta potential analysis.
Disclosure
The authors report no conflicts of interest in this work.
References
Khan KH, Goody RB, Hameed H, Jalil A, Coyle VM, McAleer JJ. Metastatic melanoma: a regional review and future directions. Tumori. 2012;98:575–580. | |
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. | |
Viola JR, Rafael DF, Wagner E, Besch R, Ogris M. Gene therapy for advanced melanoma: selective targeting and therapeutic nucleic acids. J Drug Deliv. 2013;2013:897348. | |
Haluska FG, Tsao H, Wu H, Haluska FS, Lazar A, Goel V. Genetic alterations in signaling pathways in melanoma. Clin Cancer Res. 2006;12:2301s–2307s. | |
Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. | |
Lu H, Wang J, Bai Y, et al. Ionic polypeptides with unusual helical stability. Nat Commun. 2011;2:206. | |
Li GZ, Eller MS, Firoozabadi R, Gilchrest BA. Evidence that exposure of the telomere 3′ overhang sequence induces senescence. Proc Natl Acad Sci U S A. 2003;100:527–531. | |
Puri N, Eller MS, Byers HR, Dykstra S, Kubera J, Gilchrest BA. Telomere-based DNA damage responses: a new approach to melanoma. FASEB J. 2004;18:1373–1381. | |
Rankin AM, Sarkar S, Faller DV. Mechanism of T-oligo-induced cell cycle arrest in Mia-PaCa pancreatic cancer cells. J Cell Physiol. 2012;227:2586–2594. | |
Li GZ, Eller MS, Hanna K, Gilchrest BA. Signaling pathway requirements for induction of senescence by telomere homolog oligonucleotides. Exp Cell Res. 2004;301:189–200. | |
Yaar M, Eller MS, Panova I, et al. Telomeric DNA induces apoptosis and senescence of human breast carcinoma cells. Breast Cancer Res. 2007;9:R13. | |
Pitman RT, Wojdyla L, Puri N. Mechanism of DNA damage responses induced by exposure to an oligonucleotide homologous to the telomere overhang in melanoma. Oncotarget. 2013;4:761–771. | |
Gnanasekar M, Thirugnanam S, Zheng G, Chen A, Ramaswamy K. T-oligo induces apoptosis in advanced prostate cancer cells. Oligonucleotides. 2009;19:287–292. | |
Sarkar S, Faller DV. T-oligos inhibit growth and induce apoptosis in human ovarian cancer cells. Oligonucleotides. 2011;21:47–53. | |
Puri N, Pitman RT, Mulnix RE, et al. Non-small cell lung cancer is susceptible to induction of DNA damage responses and inhibition of angiogenesis by telomere overhang oligonucleotides. Cancer Lett. September 4, 2013. [Epub ahead of print.] | |
Wickstrom E. Oligodeoxynucleotide stability in subcellular extracts and culture media. J Biochem Biophys Methods. 1986;13:97–102. | |
Gabrielson NP, Lu H, Yin L, Kim KH, Cheng J. A cell-penetrating helical polymer for siRNA delivery to mammalian cells. Mol Ther. 2012;20:1599–1609. | |
Lu H, Bai Y, Wang J, et al. Ring-opening polymerization of gamma-(4-vinylbenzyl)-(L)-glutamate N-carboxyanhydride for the synthesis of functional polypeptides. Macromolecules. 2011;44:6237–6240. | |
Eller MS, Puri N, Hadshiew IM, Venna SS, Gilchrest BA. Induction of apoptosis by telomere 3′ overhang-specific DNA. Exp Cell Res. 2002;276:185–193. | |
Puri N, Salgia R. Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J Carcinog. 2008;7:9. | |
Kang TW, Yevsa T, Woller N, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547–551. | |
Butterfield LH, Comin-Anduix B, Vujanovic L, et al. Adenovirus MART-1-engineered autologous dendritic cell vaccine for metastatic melanoma. J Immunother. 2008;31:294–309. | |
Cavallaro U, Christofori G. Molecular mechanisms of tumor angiogenesis and tumor progression. J Neurooncol. 2000;50:63–70. | |
Kaur S, Martin-Manso G, Pendrak ML, Garfield SH, Isenberg JS, Roberts DD. Thrombospondin-1 inhibits VEGF receptor-2 signaling by disrupting its association with CD47. J Biol Chem. 2010;285:38923–38932. | |
Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat Med. 2000;6:41–48. | |
Fleitas T, Martinez-Sales V, Vila V, et al. VEGF and TSP1 levels correlate with prognosis in advanced non-small cell lung cancer. Clin Transl Oncol. 2013;15:897–902. | |
Parkinson EK, Minty F. Anticancer therapy targeting telomeres and telomerase: current status. BioDrugs. 2007;21:375–385. | |
Eder PS, DeVine RJ, Dagle JM, Walder JA. Substrate specificity and kinetics of degradation of antisense oligonucleotides by a 3′ exonuclease in plasma. Antisense Res Dev. 1991;1:141–151. | |
Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. | |
Ali HM, Urbinati G, Raouane M, Massaad-Massade L. Significance and applications of nanoparticles in siRNA delivery for cancer therapy. Expert Rev Clin Pharmacol. 2012;5:403–412. | |
Chen Y, Bathula SR, Yang Q, Huang L. Targeted nanoparticles deliver siRNA to melanoma. J Invest Dermatol. 2010;130:2790–2798. | |
Nguyen LT, Atobe K, Barichello JM, Ishida T, Kiwada H. Complex formation with plasmid DNA increases the cytotoxicity of cationic liposomes. Biol Pharm Bull. 2007;30:751–757. | |
Yin L, Song Z, Qu Q, et al. Supramolecular self-assembled nanoparticles mediate oral delivery of therapeutic TNF-alpha siRNA against systemic inflammation. Angew Chem Int Ed Engl. 2013;52:5757–5761. | |
Yin L, Song Z, Kim KH, Zheng N, Gabrielson NP, Cheng J. Non-viral gene delivery via membrane-penetrating, mannose-targeting supramolecular self-assembled nanocomplexes. Adv Mater. 2013;25:3063–3070. | |
Rankin AM, Forman L, Sarkar S, Faller DV. Enhanced cytotoxicity from deoxyguanosine-enriched T-oligo in prostate cancer cells. Nucleic Acid Ther. 2013;23:311–321. | |
Terrinoni A, Ranalli M, Cadot B, et al. p73-alpha is capable of inducing scotin and ER stress. Oncogene. 2004;23:3721–3725. | |
Ramadan S, Terrinoni A, Catani MV, et al. p73 induces apoptosis by different mechanisms. Biochem Biophys Res Commun. 2005;331:713–717. | |
Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. 2004;5:897–907. | |
Dias N, Stein CA. Antisense oligonucleotides: basic concepts and mechanisms. Mol Cancer Ther. 2002;1:347–355. | |
Wojcik M, Cieslak M, Stec WJ, Goding JW, Koziolkiewicz M. Nucleotide pyrophosphatase/phosphodiesterase 1 is responsible for degradation of antisense phosphorothioate oligonucleotides. Oligonucleotides. 2007;17:134–145. | |
Brown DA, Kang SH, Gryaznov SM, et al. Effect of phosphorothioate modification of oligodeoxynucleotides on specific protein binding. J Biol Chem. 1994;269:26801–26805. | |
Levin AA. A review of the issues in the pharmacokinetics and toxicology of phosphorothioate antisense oligonucleotides. Biochim Biophys Acta. 1999;1489:69–84. | |
Elsabahy M, Nazarali A, Foldvari M. Non-viral nucleic acid delivery: key challenges and future directions. Curr Drug Deliv. 2011;8:235–244. | |
Yen J, Zhang Y, Gabrielson NP, et al. Cationic, helical polypeptide-based gene delivery for IMR-90 fibroblasts and human embryonic stem cells. Biomater Sci. 2013;1:719–727. | |
Lawler J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J Cell Mol Med. 2002;6:1–12. | |
Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev. 2004;56:549–580. | |
Nakagawa T, Li JH, Garcia G, et al. TGF-beta induces proangiogenic and antiangiogenic factors via parallel but distinct Smad pathways. Kidney Int. 2004;66:605–613. | |
Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3:401–410. | |
Kieran MW, Kalluri R, Cho YJ. The VEGF pathway in cancer and disease: responses, resistance, and the path forward. Cold Spring Harb Perspect Med. 2012;2:a006593. | |
Hicklin DJ, Ellis LM. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J Clin Oncol. 2005;23:1011–1027. | |
Ugurel S, Rappl G, Tilgen W, Reinhold U. Increased serum concentration of angiogenic factors in malignant melanoma patients correlates with tumor progression and survival. J Clin Oncol. 2001;19:577–583. | |
Rajabi P, Neshat A, Mokhtari M, Rajabi MA, Eftekhari M, Tavakoli P. The role of VEGF in melanoma progression. J Res Med Sci. 2012;17:534–539. | |
Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–4380. | |
Willett CG, Boucher Y, di Tomaso E, et al. Direct evidence that the VEGF-specific antibody bevacizumab has antivascular effects in human rectal cancer. Nat Med. 2004;10:145–147. | |
Chekhonin VP, Shein SA, Korchagina AA, Gurina OI. VEGF in tumor progression and targeted therapy. Curr Cancer Drug Targets. 2013;13:423–443. | |
Planchard D. Bevacizumab in non-small-cell lung cancer: a review. Expert Rev Anticancer Ther. 2011;11:1163–1179. | |
Shih T, Lindley C. Bevacizumab: an angiogenesis inhibitor for the treatment of solid malignancies. Clin Ther. 2006;28:1779–1802. | |
Ferrara N, Hillan KJ, Gerber HP, Novotny W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov. 2004;3:391–400. | |
Adams JC, Lawler J. The thrombospondins. Cold Spring Harb Perspect Biol. 2011;3:a009712. | |
Jin RJ, Kwak C, Lee SG, et al. The application of an anti-angiogenic gene (thrombospondin-1) in the treatment of human prostate cancer xenografts. Cancer Gene Ther. 2000;7:1537–1542. | |
Lee YJ, Koch M, Karl D, et al. Variable inhibition of thrombospondin 1 against liver and lung metastases through differential activation of metalloproteinase ADAMTS1. Cancer Res. 2010;70:948–956. | |
Bornstein P. Thrombospondins as matricellular modulators of cell function. J Clin Invest. 2001;107:929–934. | |
Rofstad EK, Henriksen K, Galappathi K, Mathiesen B. Antiangiogenic treatment with thrombospondin-1 enhances primary tumor radiation response and prevents growth of dormant pulmonary micrometastases after curative radiation therapy in human melanoma xenografts. Cancer Res. 2003;63:4055–4061. | |
Horiguchi H, Yamagata S, Rong Qian Z, Kagawa S, Sakashita N. Thrombospondin-1 is highly expressed in desmoplastic components of invasive ductal carcinoma of the breast and associated with lymph node metastasis. J Med Invest. 2013;60:91–96. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.