Back to Journals » The Application of Clinical Genetics » Volume 11
Novel ATP7A gene mutation in a patient with Menkes disease
Authors Caicedo-Herrera G, Candelo E ![]() , Pinilla J, Vidal A, Cruz S, Pachajoa HM
, Pinilla J, Vidal A, Cruz S, Pachajoa HM ![]()
Received 13 July 2018
Accepted for publication 10 October 2018
Published 22 November 2018 Volume 2018:11 Pages 151—155
DOI https://doi.org/10.2147/TACG.S180087
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Martin Maurer
Gabriela Caicedo-Herrera,1 Estephania Candelo,1 Juan Pinilla,2 Andrés Vidal,2 Santiago Cruz,3 Harry Mauricio Pachajoa1–4
1Health Sciences Faculty, Universidad Icesi, Cali, Colombia; 2Dermatology, Fundación Valle Del Lili, Cali, Colombia; 3Paediatric Neurology, Fundación Valle Del Lili, Cali, Colombia; 4Department of Genetics, Fundación Valle Del Lili, Cali, Colombia
Background: Menkes disease is a congenital neurodegenerative disorder caused by ATP7A gene mutations. Clinical features include epilepsy, growth delay, reduced muscle strength, skin laxity, abnormal hair, and urologic abnormalities.
Case presentation: We describe an infant with developmental delay, neurologic degeneration, and kinky hair. Molecular test revealed a novel heterozygous mutation in exon 21 of the ATP7A gene. The genotype and phenotype of the patient were compared with those of the patients reported in the literature.
Conclusion: We propose that this mutation caused a dysfunctional protein resulting in classical Menkes disease. This case adds to the spectrum of pathogenic variants of the ATP7A gene known to cause disease.
Keywords: Menkes disease, ATP7A gene, copper-transporting ATPasa, genodermatosis
Background
Menkes disease ([MD] MIM 309400) is an X-linked recessive neurodegenerative disorder with onset in infancy caused by pathogenic variants in the ATP7A gene. The gene codes for a transmembrane (TM) copper-transporting ATPase that cycles between the transportation Golgi network and plasma membrane. MD involves deficient cuproenzyme activity.1,2 At least three different phenotypes have been described: classical MD with death in early childhood, mild MD with longer survival, and occipital horn syndrome (OHS). Classical MD is characterized by cerebral and cerebellar neurodegeneration, fair skin with kinky hair, and connective tissue abnormalities.2
The overall incidence of MD is reported to be one in 100,000–250,000 births. The true incidence of OHS is unknown, but it is believed to be lower than that of MD.1–3 Approximately 35–40 affected individuals are reported in the literature, so it is possible that OHS may be underdiagnosed. The vast majority of patients with MD are boys whose biological mothers are heterozygous carriers.3 We report the first case of MD in Colombia, an infant with findings of classical MD caused by a missense mutation not previously reported in the literature causing this form of the disease.
Case presentation
The patient is the second child of healthy, non-consanguineous Colombian parents. He was born at 40 weeks gestation. Pregnancy and cesarean delivery were uncomplicated, and the neonate had no obvious abnormalities. At the age of 3 months, he had difficulty holding up his head, and generalized hypotonia was noted. One month later, he presented with focal seizures requiring anticonvulsant medication. He was referred to our medical center for a neuropediatric evaluation. Physical examination showed generalized hypotonia, lack of eye contact, dry skin, kinky hair, and minor malformations including dolichocephaly, micrognathia, and pectus excavatum (Figure 1). Cognitive examination revealed decreased interaction with the family, inconstant smiling, no interest in toys or faces, and irritability.
 | Figure 1 Clinical findings. Notes: (A) Dolichocephaly, alopecia, and hypopigmented skin; (B) kinky, hypopigmented hair; (C) pili torti observed during dermatoscopy. |
Light microscopy of scalp hair at 5 months of age revealed abnormally silver-colored hairs, pili torti (hair shafts twisted 180°), nodosa trichorrhexis (transverse fractures of the hair shaft), and monilethrix (Figure 2). Blood tests indicated no significant acidosis or hyperammonemia. However, serum levels of both copper (2.94 μmol/L; normal range 4.35–9.18 μmol/L) and ceruloplasmin (5.9 ng/dL; normal range 20–60 ng/dL) were low. Levels of amino acids and organic acids were within the normal range. Brain magnetic resonance images showed delayed myelination and diffuse brain atrophy. The cerebral blood vessels were tortuous on magnetic resonance angiography (Figure 3). Electroencephalography (EEG) showed cortical dysfunction with epileptogenic activity in the right frontotemporal region. Abdominal ultrasonography revealed mild left hydronephrosis.
 | Figure 2 Microscopic hair analysis. Notes: (A) Trichorrhexis nodosa; (B) monilethrix. |
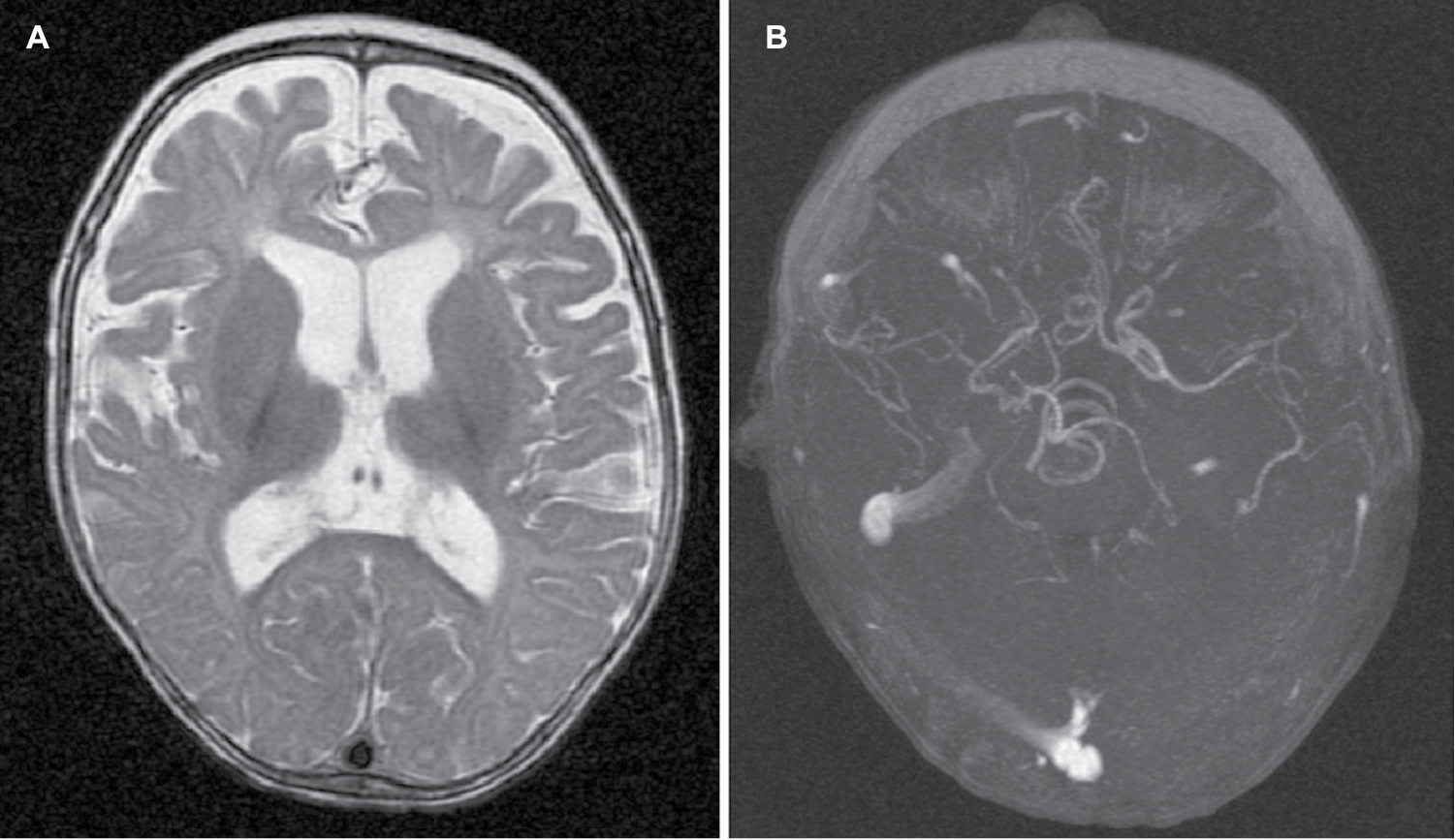 | Figure 3 Radiologic findings. Notes: (A) Diffuse cerebral atrophy; (B) tortuosity of cerebral blood vessels. |
On the basis of clinical, radiologic, and biochemical findings, the child was diagnosed at 6 months of age with MD. Treatment with 100 µg/kg/day of copper-histidinate was initiated, although treatment was sometimes interrupted because of difficulty obtaining the drug in Colombia. However, after 10 months of continuous treatment, the copper and ceruloplasmin levels normalized, and his hair characteristics and muscle tone were mildly improved. A video-EEG suggested West syndrome. At 2 years of age, he had recurrent respiratory infections attributed to a swallowing disorder and gastroesophageal reflux, requiring tracheostomy. The frequency of seizures decreased, but brain magnetic resonance images showed disease progression with increased brain atrophy and a middle cerebral artery aneurysm.
Molecular studies identified a novel missense mutation c.4067G>C (pArg1356Pro) in the ATP7A gene. The same mutation was found in his mother, who was asymptomatic and had normal copper and ceruloplasmin serum levels. The patient’s sister had urologic abnormalities (duplicated collecting system) but no other findings of MD. Genetic analysis for the sister was pending at the time of writing.
Discussion
Pathogenic mutations in ATP7A result in a variety of clinically recognizable phenotypes associated with copper transport disorders. The degree to which residual ATP7A function is maintained determines the severity of the phenotypes.3 To date, more than 311 different mutations have been reported in patients with MD.4 Approximately 22% of the ATP7A mutations are gross deletions. The rest are intragenic mutations: deletions or insertions (22%), duplications, missense (17%), nonsense (18%), and splice junction mutations (18%).3
Truncating variants, such as exon deletions or nonsense variants, result in severe classical MD with death in early childhood.5,6 Missense and aberrant splicing mutations produce a hypofunctional protein because of incomplete truncation, resulting in milder phenotypes.7,8 In our patient, a novel heterozygous missense mutation in the ATP7A gene was identified: c.4067G>C (pArg1356Pro). This variant has not been previously reported in the literature, and in this case, it was associated with findings of classical MD.
ATP7A gene mutations reduce copper absorption, leading to copper deficiency. This results in reduced activity of copper-dependent enzymes, leading to connective tissue abnormalities, tortuosity of blood vessels, and peculiar hair.3–9 These clinical features were present in our patient and support the diagnosis of classical MD (Table 1). The natural history of epilepsy in MD, a major clinical feature of the disease, has three stages: 1) an early stage at the age of 3 months with focal seizures progressing to epileptic spasms; 2) an intermediate stage (6–11 months) with epileptic spasms including West syndrome; and, finally, 3) a late stage characterized by multifocal seizures, tonic spasms, and myoclonus (at a mean age of 20–25 months).10 Our patient had epileptic spasms with EEG evidence of hypsarrhythmia and required three different anticonvulsants to control the seizures. Respiratory infections, including pneumonia, are reportedly one of the causes of severe morbidity or mortality in MD.3 At 2 years of age, our patient had suffered recurrent respiratory infections, with severe respiratory failure on one occasion requiring tracheostomy and supplemental oxygen.
 | Table 1 Copper enzymes, their role in MD, and the features in our patient Abbreviations: CNS, central nervous system; MD, Menkes disease. |
The ATP7A gene, located on locus Xq21.1 of the human chromosome, contains 23 exons distributed over approximately 140 kb of genomic DNA. It encodes a TM copper-transporting P-type ATPase of 1,500 amino acids. ATP7A contains six copper-binding sites in the cytoplasmic N-terminal. Similar to other members of the P-type family of ATPases, the ATP7A protein contains three additional cytoplasmic domains: the activation (A), phosphorylation (P), and nucleotide-binding (N) domains. The protein contains eight TM domains, forming a copper-transporting channel.3 The catalytic activity of ATP7A is likely to be mediated through coordinated action of the N, P, and A domains. The N and P domains reside between TM6 and TM7, and the A domain is located between TM4 and TM5.6 The mutation identified in our patient was located in exon 21, causing an arginine-to-proline substitution in the highly conserved TM7 domain at the C-terminal end of the protein. This mutation was not found in the 1,000 Genomes Project, ExAC, and EVSor in-house database and is therefore considered to be previously unreported. On the basis of the American College of Medical Genetics and Genomics variant interpretation guideline,11 it is classified as a variant of uncertain significance. However, it is predicted to be probably pathogenic, with a DANN score of 0.996 and validated by Mutation Taster, SIFT, and FATHMM. This variant occurs in a genomic position that is highly conserved in vertebrates and is predicted to have deleterious effects.
Similar mutations have been reported in exon 21 in association with OHS and the milder form of MD.12 These mutations are thought to affect conserved residues in the TMD7 domain and to have a number of effects. They might affect copper transport, have a decrease in normal activity, and affect translocation of the protein and fail to redistribute toward the cell periphery in response to copper, remained in transportation Golgi network.13 A similar ATP7A mutation (c.4085C>T), causing an alanine-to-valine substitution, has been reported.14,15 The phenotype in that case included cerebellar ataxia, pili torti, and vascular tortuosity. On follow-up, the patient had a predominant motor delay with extreme dysarthria requiring a communication board until age 7. He had commenced copper treatment at age 21 months and, from 3 years on, was on copper-histidinate injections. At 34 years of age, the patient had mild intellectual impairment. He has tolerated copper-histidinate injections for more than 30 years and demonstrated a slow improvement in his dysarthria and ataxia. His cerebral vascular tortuosity also lessened over time.15 Although his mutation was in the same exon as in our proband, the disease severity is different, with our patient having features of classical MD rather than the milder form in the other case.
In summary, we report a Colombian patient with developmental delay, neurologic degeneration, and hair abnormalities who had a novel missense mutation in the ATP7A gene. This finding adds to the spectrum of pathogenic ATP7A variants causing classical MD, in contrast to other reports suggesting that mutations in the TMD7 domain of the ATP7A gene are linked to OHS or mild MD.12 According to clinical findings in our patient, we propose that a variant in this domain can generate a seriously dysfunctional protein, resulting in classical MD. Additional reports are needed to confirm our observation.
Informed consent
Written informed consent was obtained from the patient’s parents for publication of his images and clinical data for scientific purposes. Data were collected in the context of studies performed in accordance with the Declaration of Helsinki Good Clinical Guidelines and protocol no 509 “registry of surveillance and survival of congenital defects of the Colombian South-West” approved by the Ethics Committee of Universidad Icesi (Act 192/2011).
Acknowledgments
We would like to thank the patient and his family for their participation in this study.
Disclosure
The authors report no conflicts of interest in this work.
References
Kaler SG, Holmes CS, Goldstein DS, et al. Neonatal diagnosis and treatment of Menkes disease. N Engl J Med. 2008;358(6):605–614. | ||
Tümer Z, Møller LB. Menkes disease. Eur J Hum Genet. 2010;18(5):511–518. | ||
Ojha R, Prasad AN. Menkes disease: what a multidisciplinary approach can do. J Multidiscip Healthc. 2016;9:371–385. | ||
Stenson PD, Mort M, Ball EV, et al. The human gene mutation database: 2008 update. Genome Med. 2009;1(1):13. | ||
Skjørringe T, Tümer Z, Møller LB. Splice site mutations in the ATP7A gene. PLoS One. 2011;6(4):Article e18599. | ||
Tümer Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum Mutat. 2013;34(3):417–429. | ||
Kaler SG. Inborn errors of copper metabolism. Handb Clin Neurol. 2013;113:1745–1754. | ||
Møller LB. Small amounts of functional ATP7A protein permit mild phenotype. J Trace Elem Med Biol. 2015;31:173–177. | ||
Kodama H, Fujisawa C, Bhadhprasit W. Inherited copper transport disorders: biochemical mechanisms, diagnosis, and treatment. Curr Drug Metab. 2012;13(3):237–250. | ||
Prasad AN, Levin S, Rupar CA, Prasad C. Menkes disease and infantile epilepsy. Brain Dev. 2011;33(10):866–876. | ||
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–423. | ||
Møller LB, Mogensen M, Horn N. Molecular diagnosis of Menkes disease: genotype-phenotype correlation. Biochimie. 2009;91(10):1273–1277. | ||
Møller LB, Bukrinsky JT, Mølgaard A, et al. Identification and analysis of 21 novel disease-causing amino acid substitutions in the conserved part of ATP7A. Hum Mutat. 2005;26(2):84–93. | ||
Procopis P, Camakaris J, Danks DM. A mild form of Menkes steely hair syndrome. J Pediatr. 1981;98(1):97–99. | ||
Tchan MC, Wilcken B, Christodoulou J. The mild form of Menkes disease: a 34 year progress report on the original case. JIMD Rep. 2013;9:81–84. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.