Back to Journals » International Journal of Nanomedicine » Volume 11
Novel antimicrobial peptide–modified azithromycin-loaded liposomes against methicillin-resistant Staphylococcus aureus
Authors Liu X, Li Z ![]() , Wang X, Chen Y, Wu F
, Wang X, Chen Y, Wu F ![]() , Men K
, Men K ![]() , Xu T, Luo Y, Yang L
, Xu T, Luo Y, Yang L
Received 24 February 2016
Accepted for publication 27 May 2016
Published 14 December 2016 Volume 2016:11 Pages 6781—6794
DOI https://doi.org/10.2147/IJN.S107107
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Lei Yang
Xiaowei Liu,1,* Zhan Li,1,* Xiaodong Wang,2,3 Yujuan Chen,2,3 Fengbo Wu,2,3 Ke Men,1 Ting Xu,2,3 Yan Luo,1 Li Yang1
1State Key Laboratory of Biotherapy/Collaborative Innovation Center for Biotherapy, 2Department of Thyroid and Breast Surgery, 3Department of Pharmacy, West China Hospital, Sichuan University, Chengdu, People’s Republic of China
*These authors contributed equally to this work
Abstract: Infections caused by multidrug-resistant bacteria, such as methicillin-resistant Staphylococcus aureus (MRSA), have become a public threat; therefore, development of new antimicrobial drugs or strategies is urgently required. In this study, a new antibacterial peptide DP7-C (Chol-suc-VQWRIRVAVIRK-NH2) and DP7-C-modified azithromycin (AZT)-loaded liposomes (LPs) are developed for the treatment of MRSA infection, and it was found that DP7-C inserted into the LP lipid bilayer not only functioned as a carrier to encapsulate the antibiotic AZT but also synergized the antibacterial effect of the encapsulated AZT. In vitro assays showed that DP7-C-modified LPs possessed sustained drug release profile and immune regulatory effect and did not show obvious cytotoxicity in mammal cells, but they did not possess direct antibacterial activity in vitro. In vivo studies revealed that DP7-C-modified LPs did not exhibit obvious side effects or toxicity in mice but were able to significantly reduce the bacterial counts in an MRSA-infectious mouse model and possessed high antibacterial activity. In particular, DP7-C-modified AZT-loaded LPs showed more positive therapeutic effects than either DP7-C-modified blank LPs or nonmodified AZT-loaded LPs treatment alone. Molecular mechanism studies demonstrated that DP7-C formulations effectively upregulated the production of anti-inflammatory cytokines and chemokines without inducing harmful immune response, suggesting that DP7-C was synergistic with AZT against the bacterial infection by activating the innate immune response. Most importantly, although DP7-C activated the innate immune response, it did not possess direct antibacterial activity in vitro, indicating that DP7-C did not possess the potential to induce bacteria resistance. The findings indicate that DP7-C-modified AZT-loaded LPs developed in this study have a great potential required for the clinical treatment of MRSA infections.
Keywords: DP7-C, azithromycin, antimicrobial resistance, MRSA infections, immune-regulation
Introduction
Antimicrobial resistance has reached alarming levels in many parts of the world, which has induced high morbidity and mortality.1–3 In particular, infections caused by methicillin-resistant Staphylococcus aureus (MRSA), vancomycin-resistant Enterococcus, and other resistant bacteria have posed significant clinical challenges to physicians.4 However, although there is a rapid increase in antimicrobial resistance, only a limited number of new antibiotic drugs are available; many new drugs having failed to be developed further into the market because of poor hydrophilicity, high toxicity, highly expensive production costs, and less financially lucrative.5–7 Several strategies, such as the combination of antibiotics with other antibiotics8–10 or compound drugs11,12 and synergistically applying new mechanisms of old drugs with antibiotics,13,14 have been reported to alleviate the clinical crisis. Another approach is nanotechnology or combination of nanotechnology with antibiotics,15–18 which provides promising weapons to fight against bacterial infection. Nevertheless, the antibacterial drugs through the aforementioned approaches may induce antimicrobial resistance and could be toxic when used at high doses6,7 as the antibacterial activities of these drugs primarily depend on their direct cytostatic/cytotoxic effects on bacteria. Hence, it is of great importance to develop novel strategies to effectively combat untreatable multidrug-resistant bacterial infection.
Recently, cationic host-defense peptides and their synthetic cationic antimicrobial peptide (AMP) derivatives have been investigated for their potential values in bacterial infection therapy and the prophylaxis of bacterial infections. AMPs are cationic amphipathic molecules with positive charges and a few hydrophobic residues17,19 and have broad-spectrum antimicrobial activities and less bacterial resistance.20–22 Unlike traditional antibiotic drugs that use direct killing mechanism, AMPs modulate the innate immune response as immunomodulators or interact with prokaryotic membranes to disrupt the membrane function, which leads to cell killing.23–26 Several of these peptides such as LL-37,27,28 VDR-1,23 and others,29,30 are currently undergoing clinical trials. Thus, AMPs are being considered as a new generation of antimicrobials, which have excellent therapeutic potential.
In our previous study, a new cationic AMP DP7 (VQWRIRVAVIRK) had been developed, which has a broad-spectrum antimicrobial activity and potent protective activity against bacterial infection by disrupting bacterial membrane.22 Unfortunately, DP7 has some side effects, such as high dosage and liver hemorrhagic and toxic deaths when administered by intravenous (IV) injection,22 which limit its clinical application. It is further discovered that the hemorrhaging is probably due to DP7’s disruption of the red blood cell membrane unselectively. As DP7 is a cationic AMP, it can be easily modified when conjugated with a hydrophobic fragment to generate an amphipathic compound. The amphipathic compounds can be used further to make nanomaterials with reduced toxicity.31 In this study, to improve antibacterial activity and to circumvent the systemic side effects of DP7, hydrophobic cholesterol was conjugated at the N-terminal of DP7 to produce an amphiphilic cholesteryl peptide DP7-C (Figure S1). Then, DP7-C was used to modify antibiotic-loaded liposomes (LPs) for achieving antibiotic and antibacterial peptide incorporated, better antibacterial efficacy, and sustained release of the antibacterial drugs. Among various antibiotics, azithromycin (AZT) has a broader Gram-negative antibacterial spectrum and a lower allergic rate. AZT works as a bacteriostatic, is able to bind the 50S ribosomal subunit of bacteria, and inhibits the bacterial protein synthesis.32–35 In addition, our recent study has demonstrated that DP7 synergizes with AZT in bactericidal and therapeutic activity against multidrug-resistant bacteria.
In this study, DP7-C was inserted into the lipid bilayer of LPs, with the additional incorporation of AZT to enhance its antibacterial activity. To our knowledge, this is the first report showing the incorporation of antibiotic and AMP into nanoformulations to prevent certain bacterial infections. Interestingly, the function of DP7-C in the LP was twofold: first, it served as a material to formulate cationic LPs to encapsulate drugs and caused the drugs to be released in a sustained manner; second, it activated the host immune responses and synergized with AZT against bacterial infections. The pharmaceutical properties of prepared LPs were characterized by size, morphology, zeta potential, encapsulation efficiency (EE), and in vitro cytotoxicity. In addition, the in vitro susceptibility to bacterial and in vivo antimicrobial of LPs in an MRSA-infectious murine model was assessed. Moreover, the molecular mechanisms of DP7-C formulation in human and mouse peripheral blood mononuclear cells (PBMCs) were investigated. Finally, the safety and toxicity evaluation was carried out by a histological examination of main organs and blood test for the animals. Our data demonstrated that the newly developed DP7-C modified AZT-loaded LPs (AZT-D-LPs) possessed excellent antibacterial efficacy and did not exhibit obvious side effects, suggesting that it is an excellent candidate that can be developed for the treatment of MRSA infections.
Materials and methods
Materials and animals
Soybean phosphatidylcholine (stored at −20°C) was purchased from Avanti Polar Lipids Inc. (Alabaster, AL, USA). AZT (purity >99%) was purchased from Hubei Pharmaceutical Co. Ltd. (Hubei, People’s Republic of China). Cholesterol, chloroform, acetonitrile, and potassium monobasic (KH2PO4) were purchased from KeLong Chemical (Chengdu, People’s Republic of China). All the reagents and chemicals used for the analysis were of high-performance liquid chromatography (HPLC) grade and passed through a 0.22-μm filter. Mueller–Hinton broth (MHB) and Mueller–Hinton agar (MHA) were purchased from Qingdao Hope Bio-Technology Co. Ltd. (Shandong, People’s Republic of China).
Methicillin-susceptible S. aureus (MSSA, American Type Culture Collection [ATCC] 25923), MRSA (ATCC 33591), and Escherichia coli (ATCC 25922) were obtained from the ATCC (Manassas, VA, USA); clinical isolates (Sau2, Sau5, Sau7, Sau9, S3487, and E. coli) were obtained from the Burn Unit, Southwest Hospital of China (Sichuan, People’s Republic of China). All bacteria were cultured in MHB (25 mg/L) or MHA (12.5 mg/L) in a humidified environment at 37°C.
Human PBMCs were isolated from healthy voluntaries, and mouse PBMCs were isolated from BALB/c mice. Briefly, venous blood samples from voluntaries or mice were collected into heparin-containing Vacutainer® tubes (Jiangsu Yuli Medical Instrument Co., Ltd, Jiangsu, People’s Republic of China). Each sample was then diluted with an equal volume of Roswell Park Memorial Institute 1640 medium (Thermo Fisher Scientific, Waltham, MA, USA) supplemented with 10% (v/v) heat-inactivated fetal bovine serum and 1% penicillin–streptomycin and separated by density-gradient centrifugation, to harvest PBMCs. Finally, PBMCs (1×106 cells/mL) were added in six-well tissue culture plates and treated with different LPs formulations.
Human embryo kidney cell line HEK293 and human hepatocyte cell line LO2 were obtained from the American Type Culture Collection. All cells were cultured in Dulbecco’s Modified Eagle’s Medium (Gibco®; Invitrogen Corp., Carlsbad, CA, USA) supplemented with 10% fetal bovine serum and maintained in a humidified atmosphere of 5% CO2 at 37°C.
Female BALB/c mice (specific pathogen-free conditions) were purchased from Chengdu Dossy Experimental Animals Co. Ltd. (Sichuan, People’s Republic of China). The mice were housed and maintained under specific pathogen-free conditions in facilities and treated humanely throughout the studies. All animal experiments were performed according to the protocols approved by the Ethics Review Committee of Animal Experimentation of Sichuan University. All our animal-handling procedures were performed according to the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and followed the guidelines of the Animal Welfare Act.
Peptide synthesis
DP7-C (Chol-suc-VQWRIRVAVIRK-NH2) was synthesized by standard solid-phase peptide synthesis method at Shanghai Science Peptide Biological Technology Co. Ltd. (Shanghai, People’s Republic of China).22 The hydrophobic monocholesterol ester of succinate was conjugated at the Rink-4-Methylbenzhydrylamine resin-linked DP7 peptide. Synthesized peptides were purified to 95% by HPLC, and their molecular weights were determined by mass spectrometry. The peptide was dissolved in water to a concentration of 5 mg/mL, stored at −20°C, and then diluted to the indicated concentrations with phosphate-buffered saline (PBS; pH 7.4) before use.
Preparation of LPs
The AZT-loaded LPs (AZT-LPs), DP7-C modified blank LPs (D-LPs), and AZT-D-LPs were prepared by a film dispersion method,31,36 and DP7-C was inserted into the lipid bilayers of LPs in the process of hydration (Figure 1). The resultant cholesterol peptide DP7-C was an amphiphilic peptide containing both hydrophobic and hydrophilic residues. The hydrophobicity of cholesterol transferred the cholesterol peptide DP7-C into the bilayers of LPs in the process of hydration. In brief, AZT, soybean phosphatidylcholine, and cholesterol in a mass ratio of 3:1 (lipid to cholesterol) were dissolved in chloroform, and the organic solvent was evaporated on a rotary evaporator to form a thin film; then, the thin film was hydrated in PBS for 40 minutes to prepare AZT-LPs or hydrated in DP7-C solution at a concentration of 125-500 μg/mL to prepare AZT-D-LPs. Then, the suspension of lipids was sonicated for 3 minutes (cycles of 3 seconds on and 3 seconds off) in an ultrasonic water bath with an amplitude of 85 Hz. Finally, LPs were stored at 4°C until use.
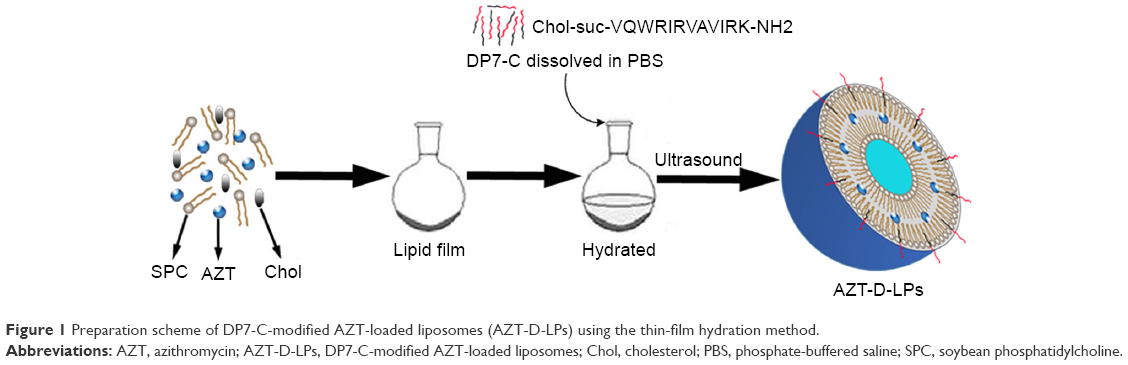 | Figure 1 Preparation scheme of DP7-C-modified AZT-loaded liposomes (AZT-D-LPs) using the thin-film hydration method. |
Characterization of LPs
The particle size and zeta potential of LPs were measured by a Zetasizer Nano ZS90 laser particle size analyzer (Malvern Instruments Ltd., Malvern, Worcestershire, UK). All data were the mean of three test runs and were expressed as mean ± standard deviation.
The morphological characteristics of D-LPs, AZT-LPs, and AZT-D-LPs were observed under a transmission electron microscope (TEM; H-6009IV, Hitachi, Japan) as described earlier.37 The drug content and the EE were determined by HPLC (Shimadzu, Kyoto, Japan), and a Shimadzu SPD-10A UV-VIS detector was used for detection. Briefly, 400 μL AZT-encapsulated LPs were dissolved in 1.6 mL CH3CN and vortexed. The solution was filtered through a 0.22-μm filter to remove polymer debris, and then, the AZT was separated chromatographically and quantified on a reversed-phase C18 column (4.6×150 mm, 5 μm, Sunfire Columns [Shimadzu, Kyoto, Japan]), with a mobile phase of 55:45 (v/v) CH3CN: 0.05 M KH2PO4 (adjusted to pH 7.2 with 20% H3PO4 before mixing with CH3CN). Sample was eluted at a flow rate of 1.0 mL/min with ultraviolet detection at 210 nm.
The EE of the AZT-encapsulated LPs was determined by HPLC as described earlier and calculated using the following equation:
|
|
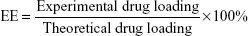
In vitro release of the AZT
The release profile of AZT-LPs and AZT-D-LPs was investigated by a dialysis method. Briefly, 1.5 mL of free AZT, AZT-LPs, or AZT-D-LPs was placed in a dialysis bag (molecular mass cutoff: 3.0 kDa). The dialysis bags were suspended in 10 mL of PBS (pH 7.4) and shaken at 80 rpm at 37°C. At designated time points (ie, 1, 2, 4, 12, 24, 48, 72, and 96 hours), 1.5 mL of the release medium was withdrawn and replaced with fresh PBS. The released amount of AZT was determined by HPLC as described earlier. The mean of three test runs was calculated and expressed as mean ± standard deviation.
Cytotoxicity study of mammalian cells
In vitro cytotoxicity of the LPs toward HEK293 cells and LO2 cells was investigated using an MTT assay. Briefly, cells were seeded in 96-well plates at a density of 5×103 cells/well in 100 μL of medium and incubated for 24 hours at 37°C. The next day, cells were treated with free AZT, different AZT, and/or DP7-C formulations or an equal volume of PBS as a control. After an incubation of 24 hours, the medium used was removed from each well, replaced with 200 μL of 1:10 MTT reagent/media mixture, and incubated for another 4 hours. Then, the medium was removed, and 150 μL of dimethyl sulfoxide was added. Finally, the absorbance at 570 nm was recorded using a microplate reader (Multiskan™ MK3; Thermo Fisher Scientific) to calculate the cell viability.
In vitro antibacterial study
The minimum inhibitory concentrations (MICs) of free AZT, AZT, and/or DP7-C LP formulations were measured using an agar dilution method in MHB medium from the National Committee for Clinical Laboratory Standards M7-A7, as described earlier.22 Briefly, the free AZT, AZT, and/or DP7-C LP formulations were serially diluted in MHB, and 100 μL of each concentration was added to 96-well plates. Bacteria were grown on an MHA plate overnight and were added to the 96-well plate at a number of 5×106 colony-forming units (CFUs)/well in 100 μL of the medium. Then, the concentrations of AZT and/or DP7-C were in the range of 0.25–256 μg/mL (final concentrations). Following incubation at 37°C for 20 hours, the absorbance at 595 nm was measured using a microplate reader (Multiskan MK3; Thermo Fisher Scientific) to calculate the MIC. The MIC of LPs was the concentration at which 80% growth inhibition relative to the growth control occurred.22
In vivo antibacterial effect study
MRSA-infectious murine model was established using BALB/c mice, which were widely used to assess antibiotic efficacy.22,23 Briefly, MRSA ATCC 33591 (~1×108 CFUs/0.5 mL PBS) was inoculated to each mouse by intraperitoneal injection. PBS, AZT, and/or DP7-C formulations were IV injected into these bacterial infection-bearing mice via the tail vein at 1 hour after an MRSA infection (DP7-C dosage of 0.1 or 2.5 mg/kg; AZT dosage of 0.625, 1.25, or 2.5 mg/kg). Mice were sacrificed 24 hours later, and the number of bacteria in the peritoneal lavage fluid was counted. Briefly, the peritoneal lavage fluid was serially diluted in normal saline and seeded on MHA plates. After incubation at 37°C overnight, the CFUs of the bacteria were counted. In vivo drug combination studies were analyzed by CalcuSyn 2.0 (BIOSOFT, Cambridge, UK), as previously described.38
Detection of cytokines and chemokines by quantitative real-time polymerase chain reaction
PBMCs were seeded in six-well tissue culture plates at 1×106 cells/mL in Roswell Park Memorial Institute 1640 medium and treated with D-LPs (DP7-C dosage of 120 μg/mL) in the presence or absence of lipopolysaccharide (LPS, 10 ng/mL) for 6 hours. Of note, for LPS experiments, PBMCs were pretreated for 1 hour with D-LPs before adding LPS. Following stimulation, the cells were centrifuged at 3,000 rpm for 5 minutes, and RNA was isolated from PBMCs for quantitative real-time polymerase chain reaction (qPCR) analysis. Briefly, RNA was isolated with RNA easy mini kit, treated with RNase-free DNase (AXYGEN, Union City, CA, USA), eluted in RNase-free water, and reverse-transcribed with the AccuRT Genomic DNA Removal Kit (ABM, Richmond, BC, Canada), according to the manufacturers’ instructions. Gene expression was quantified by qPCR according to the manufacturer’s instructions. Fold changes for each gene were calculated using the comparative Ct method and were normalized to endogenous β-actin or glyceraldehyde 3-phosphate dehydrogenase expression and relative to the gene expression in unstimulated cells (normalized to 1).23,39 The primers used for qPCR are listed in Tables S1 and S2.
Safety and toxicity evaluations of LPs in mice
To evaluate the potential toxicity and side effects of drug-loaded LPs, all mice were continuously observed for relevant indices such as appearance, independent activity, and mortality. After the mice were sacrificed, the main organs (ie, heart, lung, liver, spleen, and kidney) were harvested and fixed in 4% paraformaldehyde. These tissues were sectioned and stained with hematoxylin and eosin for histopathological examination.
A total of 20 healthy BALB/c mice were randomly divided into four groups (n=5) and were IV injected with PBS, AZT-LPs (AZT dosage of 1.25 mg/kg), D-LPs (DP7-C dosage of 2.5 mg/kg), AZT-D-LPs (dosage of 1.25 mg AZT/kg and 2.5 mg DP7-C/kg). They were sacrificed 24 hours after the injections, and the whole blood samples were obtained for hematological analysis by a Celltac α MEK-6318K fully automatic hematology analyzer (Nihon Kohden Corp., Shinjuku, Tokyo, Japan), and the serum was obtained for serological and biochemical analyses by an automatic analyzer (Hitachi High-Technologies Corp., Minato, Tokyo, Japan).
Statistical analysis
All the data were presented as mean ± standard deviation. Statistical comparisons were evaluated by one-way analysis of variance or paired t-tests. The bacterial colony counts from the animal studies were transformed in Log,10 and the significance between groups was determined by one-way analysis of variance. All the analyses were performed on Statistical Package for Software Analysis software, Version 19.0 (IBM, Armonk, NY, USA), and P-values <0.05 were statistically significant.
Results
Preparation and characterization of D-LPs
The biodegradable DP7-C was successfully synthesized by standard solid-phase peptide synthesis method. The N-terminus of DP7-C was conjugated with one molecule of cholesterol for anchoring in the lipid bilayer of the LPs, and the mass spectrum and the peptide sequence of DP7-C are shown in Figure S1.
A series of D-LPs were prepared by fabricating soybean phosphatidylcholine, cholesterol, DP7-C, and/or AZT. Figure 1 presents a schematic diagram of the preparation of AZT-D-LPs. It was found that DP7-C mass ratio in the feed affected the properties of the developed LPs; when the DP7-C mass ratio in the feed was ≥10%, the particle size of AZT-D-LPs would significantly increase and the dry lipid film would be difficult to be hydrated in PBS. In addition, when 5% DP7-C was used to modify different drug loading of AZT-D-LPs, there would be no obvious difference in particle size, polydispersity index, and EE compared with nonmodified AZT-LPs (Table S3), suggesting that membrane-incorporated DP7-C did not interfere with the formation of LPs or drug encapsulation. Thus, 5% DP7-C was chosen in the feed for future application.
The EE of D-LPs or nonmodified AZT-LPs was >97%, indicating that AZT was well encapsulated into the LPs and not affected by the presence of DP7-C. Based on the dynamic light scattering measurement, the particle size of AZT-D-LPs was ~100 nm, which resulted in a narrow particle size distribution (Figure 2A). As observed by TEM (Figure 2B), LPs developed in this study displayed uniform and near spherical shapes. The diameters of LPs observed by TEM were in good agreement with the results of dynamic light scattering measurement. The AZT-D-LPs had a zeta value of ~5 mV (Figure 2C), whereas the unmodified AZT-LPs were slightly negatively charged and nearly neutral. The zeta-potential values of DP7-C modified liposomes were slightly higher than that of nonmodified liposomes (Table S3). Because DP7-C is a cationic AMP with positive charges, when DP7-C was inserted into the LPs, the LPs became positively charged. The weak positive charge property could avoid the interaction of LPs with plasma proteins and hemolysis and would be beneficial to the behavior of LPs in vivo to some degree.40,41 These electrostatic interactions may contribute to the stability of AZT-D-LPs. These cationic AZT-D-LPs were more stable than the neutral AZT-LPs, AZT-LPs tended to aggregate and form precipitates in 7 days at 4°C, but DP7-C modified LPs remained unchanged and transparent (data not shown). This was consistent with a previous report that the peptide-modified LPs showed increased stability.41,42
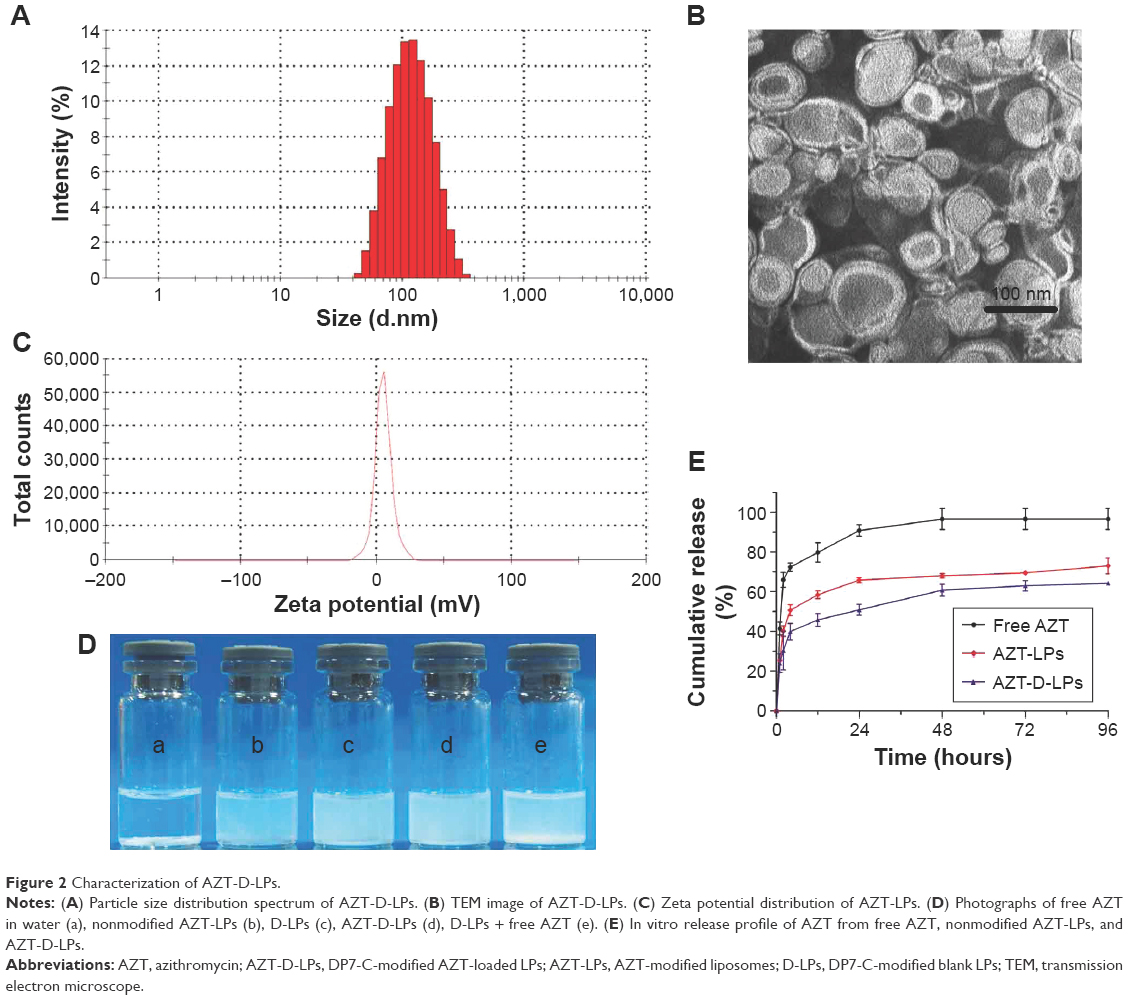 | Figure 2 Characterization of AZT-D-LPs. |
One of the major purposes of the encapsulation of AZT in LPs was to make AZT completely dispersible in aqueous media. It was also expected that DP7-C should have no influence on the property of LPs while inserted into the lipid bilayer of LPs. Figure 2D shows the appearance of the prepared LP aqueous solution. AZT itself could not be dissolved in water, as turbid white precipitates appeared (Figure 2D-a). In contrast, AZT-encapsulated LPs were fully dispersible in aqueous media. As shown in Figure 2D-b, AZT-LPs was opalescent and without precipitates. Like AZT-LPs, both D-LPs and AZT-D-LPs were also opalescent (Figure 2D-c and d), and there is no significant difference with AZT-LPs. Opalescence is one of the characteristics of LP and is mainly influenced by materials, concentration, and size of LPs, and D-LPs and nonmodified LPs developed in this study with similar opalescence and size (~100 nm) indicate that DP7-C had no influence on the appearance of LPs (Figure 2E-c and d). However, it was noticed that when AZT powder was mixed with D-LPs, turbid white precipitates were still observed (Figure 2D-e). Thus, the incorporation of AZT and DP7-C into the LPs (to form AZT-D-LPs) made AZT completely dispersible in aqueous media.
In vitro release profile of AZT
To confirm whether AZT-encapsulated LPs could be released, the drug release was examined by a dialysis method in vitro. AZT released from free AZT, AZT-LPs, and AZT-D-LPs was continuously monitored for up to 96 hours. As shown in Figure 2E, compared with the fast release behavior of free AZT, both AZT-LPs and AZT-D-LPs released AZT very slowly and did not have obvious burst release. For the first 24 hours, the cumulative release rate of AZT from free AZT was ~90.75%±2.89%, whereas the cumulative release rates of AZT-LPs and AZT-D-LPs are 65.76%±1.26% and 50.87%±2.72%, respectively. In addition, the release of AZT from AZT-D-LPs was slightly slower than that from AZT-LPs, indicating that DP7-C might slow down the drug release of LPs. Probably, the slightly positively charged AZT-D-LPs were more stable than the neutral AZT-LPs. Taken together, the sustained release profile of AZT-D-LPs may be exploited for the treatment of bacterial infections.
In vitro antibacterial effect of LPs
The antibacterial activity of free AZT, AZT-LPs, D-LPs, and AZT-D-LPs was investigated by bacteria inhibition experiments. Clinical isolates from both patients and laboratory reference strains were used in the current study. As shown in Table 1, the MIC values of AZT-LPs were slightly lower than that of free AZT. D-LPs had MIC values of >256 μg/mL for both Gram-positive and Gram-negative bacteria. AZT-D-LPs and AZT-LPs had similar MIC values, suggesting that the DP7-C might lack direct antibacterial activity in vitro.
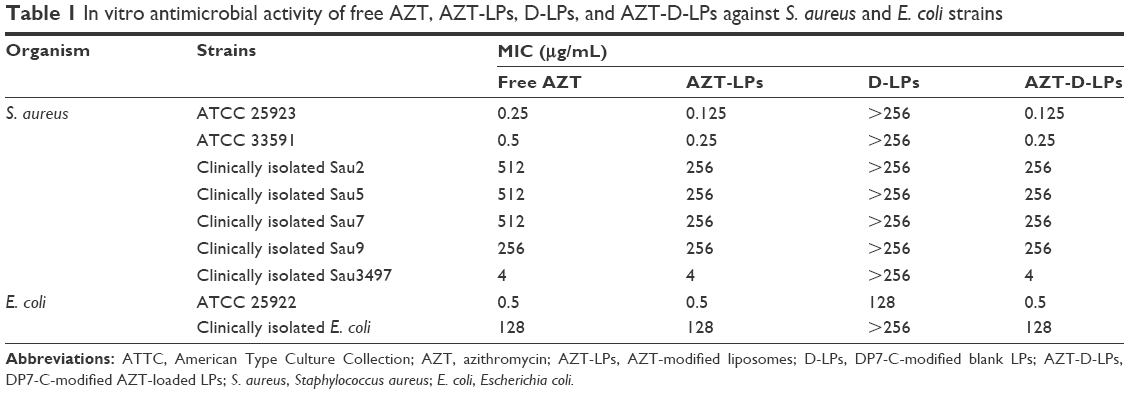 | Table 1 In vitro antimicrobial activity of free AZT, AZT-LPs, D-LPs, and AZT-D-LPs against S. aureus and E. coli strains |
In vivo cooperative antibacterial effect of DP7-C in combination with AZT
It was observed that DP7-C had no antibacterial activity in vitro, even in standard MHB media; however, whether DP7-C has antibacterial effect in vivo was not determined. In order to find this, a model using MRSA, a major cause of nosocomial infections, which is widely used to assess antibiotic efficacy, was established.23,43 PBS or D-LPs (DP7-C dosage of 0.1 or 2.5 mg/kg) was given by IV injection, 1 hour after MRSA challenge. Animals were sacrificed 20 hours later, and the number of the bacteria in the peritoneal lavage fluid was counted. It was observed that treatment with D-LPs at a dose of 2.5 mg/kg significantly (P<0.05) decreased CFUs of S. aureus (Figure 3A). However, no significant difference was observed when the D-LPs were given at a dose of 0.1 mg/kg. This indicated that DP7-C possessed high antimicrobial activity in the murine infection model.
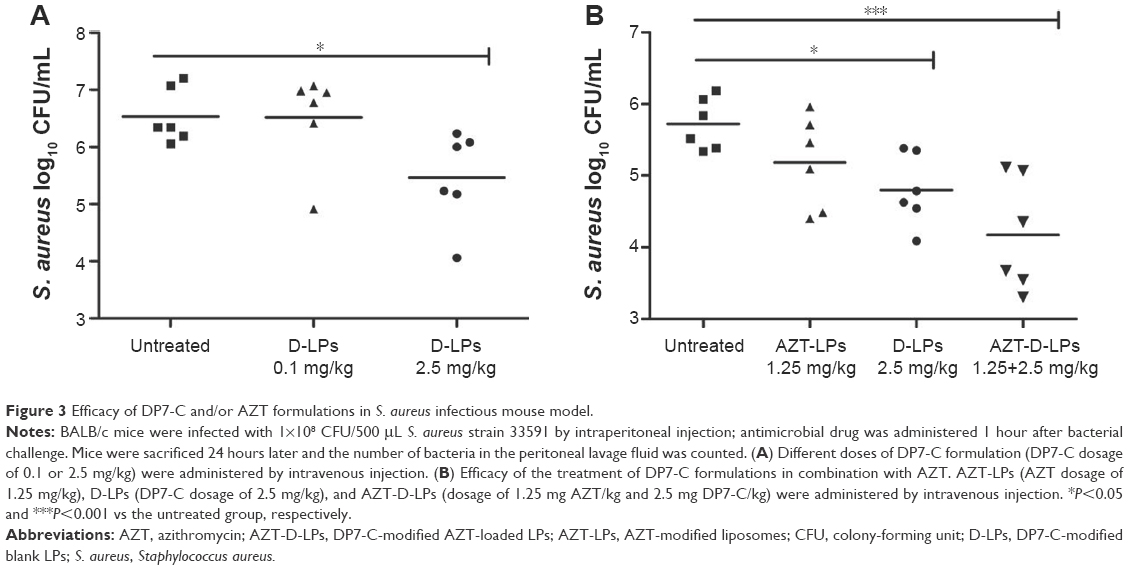 | Figure 3 Efficacy of DP7-C and/or AZT formulations in S. aureus infectious mouse model. |
In addition, the combinatory antibacterial effects of DP7-C and AZT were evaluated. To identify an effective dosage of AZT for the combination with DP7-C, DP7-C at a dose of 2.5 mg/kg and different doses of AZT (from 0.625 to 2.5 mg/kg) were incorporated into AZT-D-LPs. Equal doses of DP7-C or AZT were loaded in D-LPs or AZT-LPs as controls to treat the MRSA-infectious mice. As shown in Figure S2A, AZT-LPs inhibited the bacterial growth in a dose-dependent manner; AZT-D-LPs showed a more positive therapeutic effect than either AZT-LPs or D-LPs alone (Figure S2A and B). The formulation of AZT-D-LPs with 1.25 mg AZT/kg +2.5 mg DP7-C/kg showed a similar effect as that with 2.5 mg AZT/kg +2.5 mg DP7-C/kg. In particular, as shown in Figure 3B, treatment with a subeffective dose of AZT-LPs (AZT dosage of 1.25 mg/kg) did not significantly affect the bacterial burden (P>0.05), but treatment with AZT-D-LPs (dosage of 1.25 mg AZT/kg +2.5 mg DP7-C/kg) decreased the CFU of S. aureus very significantly (P<0.001). Also, treatment with AZT-D-LPs (dosage of 1.25 mg AZT/kg +2.5 mg DP7-C/kg) showed a more positive therapeutic effect than either AZT-LPs (AZT dosage of 1.25 or 2.5 mg/kg) or D-LPs (DP7-C dosage of 2.5 mg/kg) alone (Figure 3B). DP7-C-loaded LPs synergized the antibacterial effect of AZT against MRSA (confidence interval =0.57). In detail, treatment with the AZT-D-LPs resulted in ~96% reduction in CFU levels, compared with the control treatments (P<0.001 for AZT-D-LPs vs control; P=0.014 for AZT-D-LPs vs AZT-LPs). Combinatory treatment of subeffective doses of both AZT and DP7-C enhanced the protection compared to the treatment with either AZT-LPs or D-LPs alone, indicating the potential to use DP7-C formulations when the effect of antibiotic therapy alone is inadequate.
Mechanism of D-LPs
AMP displayed antimicrobial activities by disrupting the bacterial membrane functions or modulated the host immune response.44 According to Figure 3, DP7-C formulations displayed high antibacterial activities in vivo. However, the bacteria inhibition data (Table 1) indicated that DP7-C did not have a direct antibacterial effect. Thus, it was hypothesized that DP7-C may exert its potent antibacterial activity in vivo by activating the host immune function. To test this hypothesis, PBMCs, which are widely used to test the immune modulatory activities of host-defense peptides,23,44 were treated with or without D-LPs (DP7-C dosage of 120 μg/mL) for 6 hours. Gene expression was determined by qPCR. As shown in Figure 4A, mRNA levels of anti-inflammatory cytokine interleukin (IL)-10 were highly upregulated (135-fold) by the treatment with D-LPs, whereas the proinflammatory cytokines IL-6, IL-8, and tumor necrosis factor-alpha (TNF-α) were downregulated significantly (P<0.001), consistent with its ability to induce the anti-inflammatory cytokine IL-10. The chemokine cytokine monocyte chemoattractant protein-1 was upregulated approximately twofold. In addition, treatment with D-LPs also dramatically upregulated the expression of the IL-1β (908-fold), granulocyte colony-stimulating factor (196-fold) and interferon-gamma (IFN-γ; 517-fold). Collectively, the results imply that DP7-C has the ability to modulate the immune function.
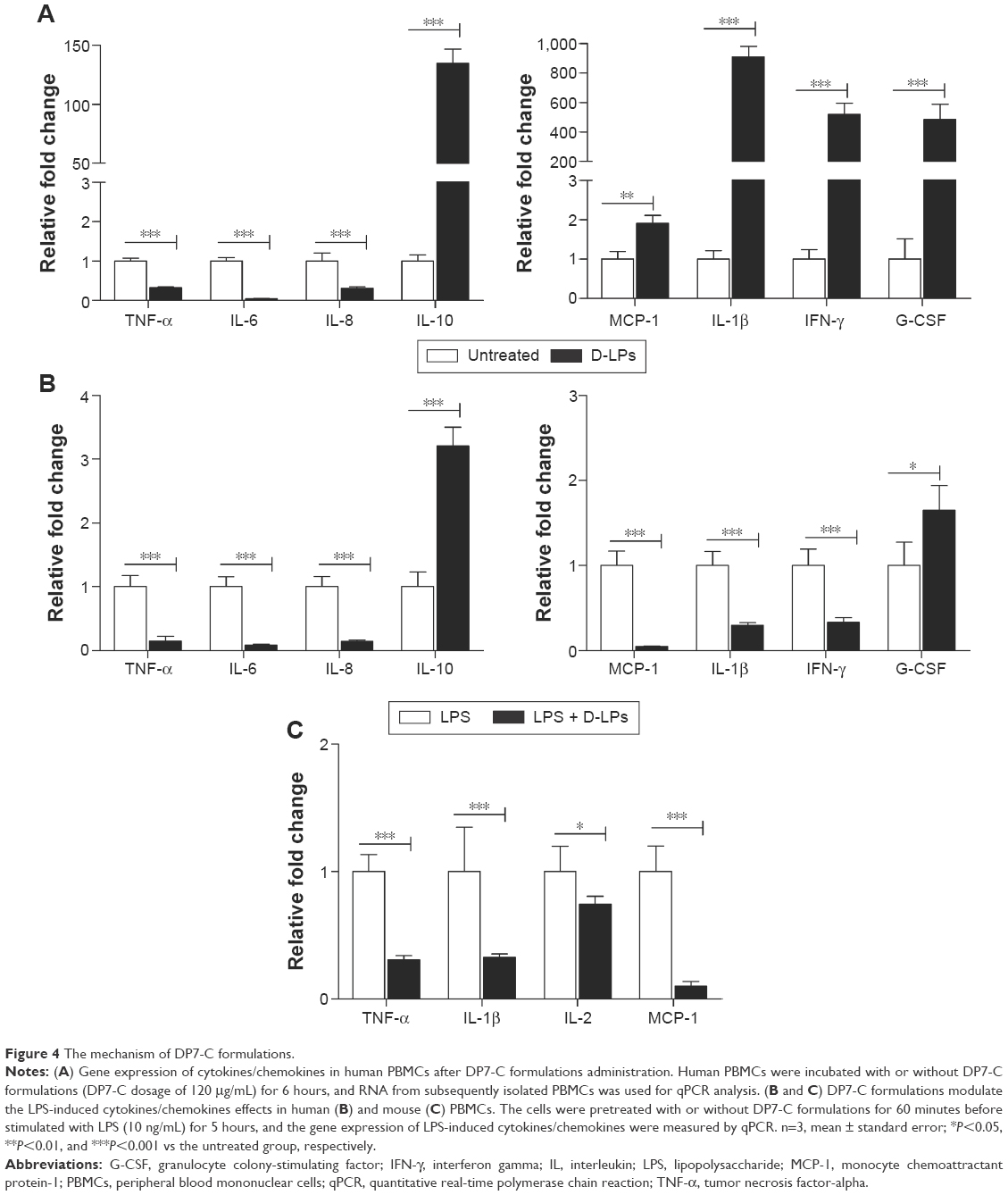 | Figure 4 The mechanism of DP7-C formulations. |
LPS is a Gram-negative bacteria signature molecule, which can imitate the bacterial infection.44 To substantiate the aforementioned finding, whether DP7-C has a potential to modify innate immunity and cause harmful inflammation by analyzing its impact on LPS-induced chemokines and inflammatory cytokine production in PBMCs was determined. As shown in Figure 4B, LPS-induced gene expression of anti-inflammatory cytokine IL-10 was upregulated by D-LPs approximately threefold in human PBMCs. In contrast, LPS-induced gene expression of TNF-α, IL-6, IL-8, IL-1β, MCP-1, and IFN-γ was significantly downregulated (P<0.001) upon D-LPs treatment. The results indicated that DP7-C could balance LPS-induced inflammation, as the gene expression of IL-1β, MCP-1, and IFN-γ was highly upregulated by DP7-C in the absence of LPS (Figure 4A). LPS-induced gene expression of granulocyte colony-stimulating factor was also increased by D-LP treatment (Figure 4B). In addition, DP7-C reduced (P<0.001) LPS-induced gene expression of TNF-α, IL-1β, IL-2, and MCP-1 in mouse PBMCs (Figure 4C), in line with its ability to reduce the expression of proinflammatory cytokines in human cells.
Toxicity evaluations of LPs
The cytotoxicity of LPs in mammalian cells was assessed using two normal human cell lines (HEK293 and LO2). Cells were treated with different AZT-LPs and/or DP7-C-loaded LPs, and cell viability was measured by an MTT assay. As shown in Figure S3, all the LPs exhibited negligible cytotoxicity, even at a high concentration of 250 μg/mL D-LPs or AZT-LPs. Of interest, the cytotoxicity of AZT-LPs, D-LPs, or AZT-D-LPs was slightly lower than that of free AZT.
The systemic toxicity of AZT and/or DP7-C formulations was further investigated in mice. During the experiment, no death, no significant appearance change, and no gross side effects were observed in any group after IV administration of the LPs. Histological examination did not find obvious pathological lesions or necrosis in the heart, liver, spleen, lung, and kidney of the mice treated with AZT-LPs, D-LPs, AZT-D-LPs, or PBS, as well as in the untreated animals (Figures 5 and S4). To further study the side effects of LPs on the physiological function of mice, blood test and serum biochemical profile test were carried out. As shown in Table S4, white blood cells, red blood cells, hemoglobin, platelet, and mean corpuscular hemoglobin counts were all in normal ranges after treatment with the LPs, although the levels of white blood cells and platelet increased slightly, compared with control (PBS). For the biomarkers (Table S5), aspartate transaminase, alanine aminotransaminase, and lactate dehydrogenase were used as an indicator for the liver functions;45 creatine kinase was used for the diagnosis of cardiac diseases;46 the change in creatinine and blood urea nitrogen indicated kidney injury;47 and glucose, total cholesterol, triglyceride, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol were used for the evaluation of glucose and lipid metabolism. As shown in Table S5, all the biochemical indices were found at the normal levels upon treatment with the LPs, and there were no differences compared with control (PBS). The data indicated that the hepatic function, renal function, and blood system were not affected by the treatments with AZT and/or DP7-C formulations. Thus, AZT-LPs, D-LPs, and AZT-D-LPs were all safe formulations by IV administration.
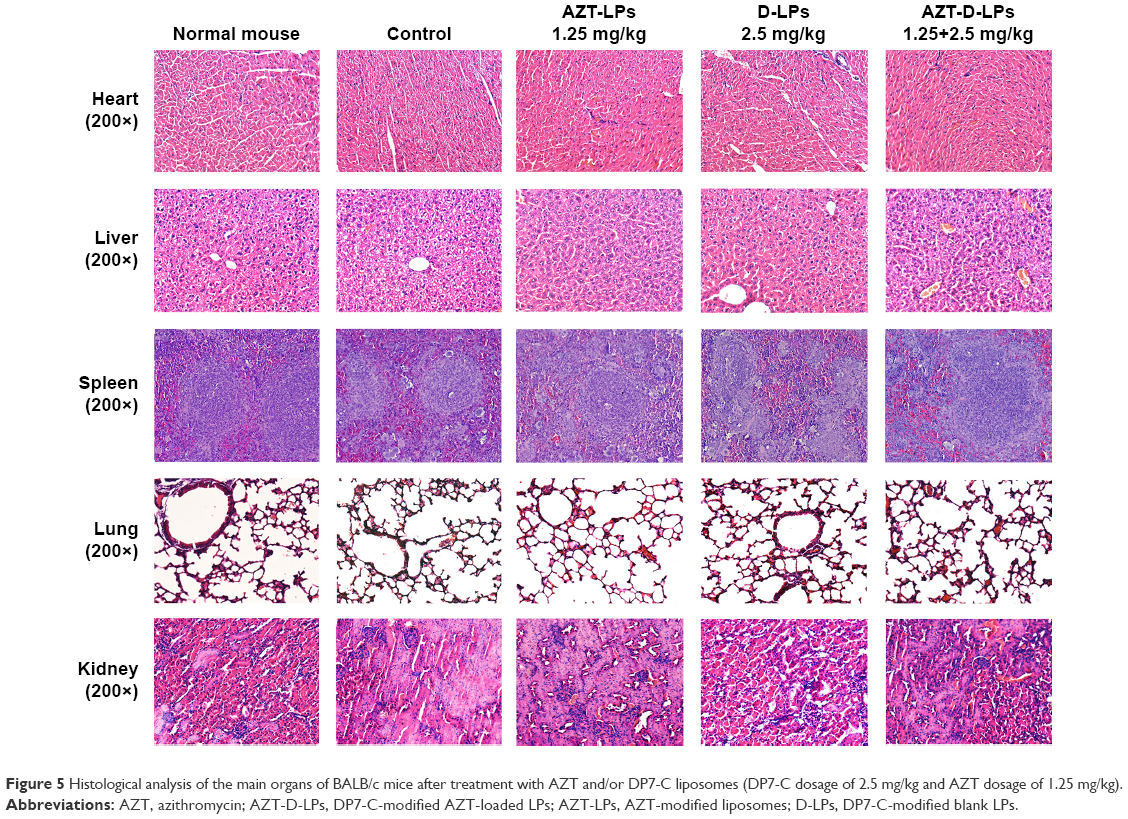 | Figure 5 Histological analysis of the main organs of BALB/c mice after treatment with AZT and/or DP7-C liposomes (DP7-C dosage of 2.5 mg/kg and AZT dosage of 1.25 mg/kg). |
Discussion
Developing antibacterial drugs with high antibacterial effect and low toxicity is critical for clinical applications. In order to achieve this, in this study, the cationic antibacterial peptide DP7-C was designed and synthesized, which was then inserted into LP lipid bilayer as a carrier to encapsulate the antibiotic AZT, and AZT-D-LPs were successfully developed. The pharmaceutical properties including size, surface charge, and EE are important factors that determine the pharmacokinetics of nanoparticles.48 The dynamic light scattering and TEM analyses indicated that LPs developed in this study were monodisperse and spherical in shape (Figure 2B). All the D-LPs were slightly positively charged (Table S3), probably because DP7-C is a cationic AMP (positively charged). The positive charge characteristic would help stabilize the LPs, as the electrostatic interaction or the stereo-specific blockade can make positively charged LPs more stable.49,50 Using HPLC, it was found that AZT was well encapsulated into AZT-D-LPs, with EE of >97% (Table S3), and the membrane-incorporated DP7-C did not interfere with the formation of LPs or drug encapsulation (Table S3). Confirmed by the appearance of AZT-LP aqueous solution, encapsulation of AZT in both D-LPs and non-modified LPs made AZT completely dispersible in water (Figure 2D). The release of AZT from AZT-D-LPs was slow but sustained (Figure 2E). Unlike free AZT, AZT-D-LPs had a much lower initial burst release (Figure 2E). However, like the well-studied human LL-3719,51 and IDR-1,23 DP7-C did not have direct antimicrobial activity, even in standard MHB media (Table 1).
S. aureus is a major cause of nosocomial infections, which is widely used to assess the antibiotic efficacy.23,43 In vivo antibacterial experiments showed that D-LPs had potent antibacterial effect and showed significant synergistic effects with AZT, which markedly potentiate the antibacterial activity of AZT. Treatment with AZT-D-LPs showed a more positive therapeutic effect in mice than treatment with AZT-LPs or D-LPs alone (Figure 3). Previous studies have shown that AZT can synergize with cationic AMPs to enhance bactericidal and therapeutic activities.52–55 Most importantly, the synergistic action of DP7-C with AZT can reduce the need for high dosages of AZT and minimize adverse drug effects, both of which are beneficial attributes for therapeutic strategies to fight against multidrug-resistant bacteria.23,53 Moreover, our in vitro and in vivo studies also showed that AZT-D-LPs exhibited negligible cytotoxicity in mammal cells and no side effects on the blood system and metabolic enzymes and did not exhibit any obvious pathological effect on the main organs of mice. These results suggest the AZT-D-LPs are a safe formulation and have a great potential for the clinical treatment of MRSA infections.
It is interesting that D-LPs did not show any in vitro antimicrobial activity, but they displayed a potent therapeutic effect in the systemic infectious mouse models. The molecular mechanism studies indicated that D-LPs possessed the ability to neutralize the harmful inflammation caused by LPS-induced gene expression of TNF-α, IL-6, IL-8, IL-1β, MCP-1, and IFN-γ, consistent with the ability to upregulate the expression of anti-inflammatory IL-10 and granulocyte colony-stimulating factor (Figure 4B). The basis of this anti-inflammatory effect may be due to the anti-inflammatory mechanisms of IL-10. In addition, the gene expression of IL-1β, MCP-1, and IFN-γ was highly upregulated by D-LPs in the absence of LPS, suggesting that DP7-C could balance the immune response, rather than merely stimulating or suppressing it. The antibacterial potential of AZT-D-LPs was achieved by the immunomodulatory activities of DP7-C formulation and collaboration with the antimicrobial effect of AZT; DP7-C selectively modulated the innate immune response and collaborated with AZT, which resulted in more effective clearance of the bacterial debris (Figure 6). In detail, after IV injection, both AZT and DP7-C were released from AZT-D-LPs. The released AZT could bind the 50S ribosomal subunit of bacteria and inhibit the bacterial protein synthesis,32–35 causing bacterial death. The DP7-C effectively modulated the innate immune response and neutralized the harmful inflammation, which may transmit effector cells and proteins to the infection sites. AZT-D-LPs, which contained both the antibiotic AZT and antibacterial peptide DP7-C, combined the immunomodulatory activities of DP7-C and antimicrobial effect of AZT, thereby providing prophylaxis and efficient treatment for bacterial infections.
 | Figure 6 Proposed mechanism of action of AZT-D-LPs. |
In clinical applications, AZT-D-LPs may have several advantages because of its higher antibacterial effect and a sustained drug release. First, DP7-C developed in this study was inserted into the lipid bilayer of LPs as a carrier of the antibiotic drug and also modulated the innate immunity to prevent the bacterial infection in vivo, which may overcome the resistance of conventional antibiotic, as the peptides lacked direct antimicrobial activity.23,51 Second, AZT encapsulated in D-LPs was released in a sustained manner and synergized with DP7-C, which could markedly potentiate the bactericidal activity of AZT in vivo. Third, in vitro cytotoxicity in normal human cells and in vivo blood test, serological and biochemical analyses, as well as histological examinations showed that AZT-D-LPs were a safe formulation. Finally, our method to make AZT-D-LPs was simple and inexpensive. All of these indicate that the novel AZT-D-LPs could serve as a potential candidate for the clinical treatment of MRSA infection.
Conclusion
In this study, for the first time, novel antimicrobial AZT-D-LPs were successfully developed, by incorporating a newly cationic antibacterial peptide DP7-C into AZT-LPs. Results of pharmaceutical properties, in vitro release profile, and cytotoxicity studies demonstrated that AZT-D-LP had a small-size, controlled release profile and satisfied safety. Moreover, our results showed that AZT-D-LPs possessed a high antibacterial activity against MRSA infection, and DP7-C formulations showed high antimicrobial activities and significant synergistic effects with AZT in vivo. Molecular mechanisms of DP7-C formulations were achieved by the promotion of chemokine and anti-inflammatory cytokine induction and suppression of inflammatory cytokine production, suggesting that DP7-C could balance the immune response. The D-LPs not only acted as a carrier of AZT but also was an immunomodulator of innate immune response. Most importantly, DP7-C formulation lacked direct antimicrobial activity; therefore, the bacterial resistance may be avoided. In summary, antibacterial drug AZT-D-LPs developed in this study showed high antibacterial activity, immunoregulation effect, and satisfied safety. Therefore, AZT-D-LP is an ideal candidate for the treatment of MRSA infections, which may alleviate bacterial resistance crisis. The strategy of incorporating antibacterial peptide and antibiotic into LP provides a new platform for the development of new interventions against bacterial infections. In addition, D-LPs can serve as promising carriers of antibiotics or anticancer drugs and may benefit patients after tumor surgery.
Acknowledgments
The study was supported mainly by the National Natural Science Foundation of China (31570927), National Major Scientific and Technological Special Project for “Significant New Drugs Development” (2013ZX09102030). Yinbo Zhao, Qian Lei, and Xiuran Zhen (State Key Laboratory of Biotherapy/Collaborative Innovation Center for Biotherapy, West China Hospital, Sichuan University) are acknowledged for their help with the experiments, useful suggestions, and valuable discussions.
Author contributions
All authors contributed toward data analysis, drafting and revising the paper and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
World Health Organization. Antimicrobial Resistance: Global Report on Surveillance. World Health Organization; 2014. Available from: http://www.who.int/drugresistance/documents/surveillancereport/en/. Accessed August 31, 2016. | ||
Buick S, Joffe AM, Taylor G, Conly J. A consensus development conference model for establishing health policy for surveillance and screening of antimicrobial-resistant organisms. Clin Infect Dis. 2015;60(7):1095–1101. | ||
Lai C-C, Lee K, Xiao Y, et al. High burden of antimicrobial drug resistance in Asia. J Glob Antimicrob Resist. 2014;2(3):141–147. | ||
Chang H-H, Cohen T, Grad YH, Hanage WP, O’Brien TF, Lipsitch M. Origin and proliferation of multiple-drug resistance in bacterial pathogens. Microbiol Mol Biol Rev. 2015;79(1):101–116. | ||
Boucher HW, Talbot GH, Benjamin DK, et al. 10×20 Progress – development of new drugs active against gram-negative bacilli: an update from the Infectious Diseases Society of America. Clin Infect Dis. 2013;56(12):1685–1694. | ||
Roemer T, Boone C. Systems-level antimicrobial drug and drug synergy discovery. Nat Chem Biol. 2013;9(4):222–231. | ||
Spellberg B, Powers JH, Brass EP, Miller LG, Edwards JE. Trends in antimicrobial drug development: implications for the future. Clin Infect Dis. 2004;38(9):1279–1286. | ||
Akova M, Daikos G, Tzouvelekis L, Carmeli Y. Interventional strategies and current clinical experience with carbapenemase-producing Gram-negative bacteria. Clin Microbiol Infect. 2012;18(5):439–448. | ||
Giamarellou H, Galani L, Baziaka F, Karaiskos I. Effectiveness of a double-carbapenem regimen for infections in humans due to carbapenemase-producing pandrug-resistant Klebsiella pneumoniae. Antimicrob agents chemother. 2013;57(5):2388–2390. | ||
Pieren M, Tigges M. Adjuvant strategies for potentiation of antibiotics to overcome antimicrobial resistance. Curr Opin Pharmacol. 2012;12(5):551–555. | ||
Ejim L, Farha MA, Falconer SB, et al. Combinations of antibiotics and nonantibiotic drugs enhance antimicrobial efficacy. Nat Chem Biol. 2011;7(6):348–350. | ||
Farha MA, Brown ED. Discovery of antibiotic adjuvants. Nat Biotechnol. 2013;31(2):120–122. | ||
Bush K, Courvalin P, Dantas G, et al. Tackling antibiotic resistance. Nat Rev Microbiol. 2011;9(12):894–896. | ||
Imperi F, Massai F, Pillai CR, et al. New life for an old drug: the anthelmintic drug niclosamide inhibits Pseudomonas aeruginosa quorum sensing. Antimicrob Agents Chemother. 2013;57(2):996–1005. | ||
Wu F, Meng G, He J, Wu Y, Wu F, Gu Z. Antibiotic-loaded chitosan hydrogel with superior dual functions: antibacterial efficacy and osteoblastic cell responses. ACS Appl Mater Interfaces. 2014;6(13):10005–10013. | ||
Zhao Y, Jiang X. Multiple strategies to activate gold nanoparticles as antibiotics. Nanoscale. 2013;5(18):8340–8350. | ||
Zhou C, Wang M, Zou K, Chen J, Zhu Y, Du J. Antibacterial polypeptide-grafted chitosan-based nanocapsules as an “armed” carrier of anticancer and antiepileptic drugs. ACS Macro Letters. 2013;2(11):1021–1025. | ||
Li C, Zhang X, Huang X, Wang X, Liao G, Chen Z. Preparation and characterization of flexible nanoliposomes loaded with daptomycin, a novel antibiotic, for topical skin therapy. Int J Nanomedicine. 2013;8:1285–1292. | ||
Hancock RE, Sahl H-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24(12):1551–1557. | ||
Hancock RE, Scott MG. The role of antimicrobial peptides in animal defenses. Proc Natl Acad Sci U S A. 2000;97(16):8856–8861. | ||
Morris CJ, Beck K, Fox MA, Ulaeto D, Clark GC, Gumbleton M. Pegylation of antimicrobial peptides maintains the active peptide conformation, model membrane interactions, and antimicrobial activity while improving lung tissue biocompatibility following airway delivery. Antimicrob Agents Chemother. 2012;56(6):3298–3308. | ||
Wu X, Wang Z, Li X, et al. In vitro and in vivo activities of antimicrobial peptides developed using an amino acid-based activity prediction method. Antimicrob Agents Chemother. 2014;58(9):5342–5349. | ||
Scott MG, Dullaghan E, Mookherjee N, et al. An anti-infective peptide that selectively modulates the innate immune response. Nat Biotechnol. 2007;25(4):465–472. | ||
Braff MH, Hawkins MA, Di Nardo A, et al. Structure-function relationships among human cathelicidin peptides: dissociation of antimicrobial properties from host immunostimulatory activities. J Immunol. 2005;174(7):4271–4278. | ||
Karlsson AJ, Pomerantz WC, Weisblum B, Gellman SH, Palecek SP. Antifungal activity from 14-helical β-peptides. J Am Chem Soc. 2006;128(39):12630–12631. | ||
Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415(6870):389–395. | ||
Nagaoka I, Hirota S, Niyonsaba F, et al. Augmentation of the lipopolysaccharide-neutralizing activities of human cathelicidin CAP18/LL-37-derived antimicrobial peptides by replacement with hydrophobic and cationic amino acid residues. Clin Diagn Lab Immunol. 2002;9(5):972–982. | ||
Vignoni M, de Alwis Weerasekera H, Simpson MJ, et al. LL37 peptide@silver nanoparticles: combining the best of the two worlds for skin infection control. Nanoscale. 2014;6(11):5725–5728. | ||
Hancock RE, Lehrer R. Cationic peptides: a new source of antibiotics. Trends Biotechnol. 1998;16(2):82–88. | ||
Travis S, Yap LM, Hawkey C, et al. RDP58 is a novel and potentially effective oral therapy for ulcerative colitis. Inflamm Bowel Dis. 2005;11(8):713–719. | ||
Chen WL, Yuan ZQ, Liu Y, et al. Liposomes coated with N-trimethyl chitosan to improve the absorption of harmine in vivo and in vitro. Int J Nanomedicine. 2016;11:325. | ||
Tateda K, Ishii Y, Matsumoto T, et al. Direct evidence for antipseudomonal activity of macrolides: exposure-dependent bactericidal activity and inhibition of protein synthesis by erythromycin, clarithromycin, and azithromycin. Antimicrob Agents Chemother. 1996;40(10):2271–2275. | ||
Azhdarzadeh M, Lotfipour F, Zakeri-Milani P, Mohammadi G, Valizadeh H. Anti-bacterial performance of azithromycin nanoparticles as colloidal drug delivery system against different gram-negative and gram-positive bacteria. Adv Pharmal Bull. 2012;2(1):17. | ||
Zakeri-Milani P, Ghanbarzadeh S, Lotfi poor F, Milani M, Valizadeh H. Pharmacokinetic study of two macrolide antibiotic oral suspensions using an optimized bioassay procedure. J Bioequiv Bioavailab. 2010;2:111–115. | ||
Sugie M, Asakura E, Zhao YL, et al. Possible involvement of the drug transporters P glycoprotein and multidrug resistance-associated protein Mrp2 in disposition of azithromycin. Antimicrob Agents Chemother. 2004;48(3):809–814. | ||
He Z, Yu Y, Zhang Y, et al. Gene delivery with active targeting to ovarian cancer cells mediated by folate receptor α. J Biomed Nanotechnol. 2013;9(5):833–844. | ||
Gou M, Men K, Shi H, et al. Curcumin-loaded biodegradable polymeric micelles for colon cancer therapy in vitro and in vivo. Nanoscale. 2011;3(4):1558–1567. | ||
Bruzzese F, Di Gennaro E, Avallone A, et al. Synergistic antitumor activity of epidermal growth factor receptor tyrosine kinase inhibitor gefitinib and IFN-α in head and neck cancer cells in vitro and in vivo. Clin Cancer Res. 2006;12(2):617–625. | ||
Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29(9):e45–e45. | ||
Gao H, He Q. The interaction of nanoparticles with plasma proteins and the consequent influence on nanoparticles behavior. Expert Opin Drug Deliv. 2014;11(3):409–420. | ||
Liu Y, Mei L, Yu Q, et al. Multifunctional tandem peptide modified paclitaxel-loaded liposomes for the treatment of vasculogenic mimicry and cancer stem cells in malignant glioma. ACS Appl Mater Interfaces. 2015;7(30):16792–16801. | ||
Zhang Q, Zhang X, Chen T, et al. A safe and efficient hepatocyte-selective carrier system based on myristoylated preS1/21-47 domain of hepatitis B virus. Nanoscale. 2015;7(20):9298–9310. | ||
Tsuji M, Takema M, Miwa H, Shimada J, Kuwahara S. In vivo antibacterial activity of S-3578, a new broad-spectrum cephalosporin: methicillin-resistant Staphylococcus aureus and Pseudomonas aeruginosa experimental infection models. Antimicrob Agents Chemother. 2003;47(8):2507–2512. | ||
Wieczorek M, Jenssen H, Kindrachuk J, et al. Structural studies of a peptide with immune modulating and direct antimicrobial activity. Chem Biol. 2010;17(9):970–980. | ||
Goldberg D, Winfield D. Diagnostic accuracy of serum enzyme assays for myocardial infarction in a general hospital population. Br Heart J. 1972;34(6):597. | ||
Ozer JS, Chetty R, Kenna G, et al. Enhancing the utility of alanine aminotransferase as a reference standard biomarker for drug-induced liver injury. Regul Toxicol Pharmacol. 2010;56(3):237–246. | ||
Xin H, Sha X, Jiang X, Zhang W, Chen L, Fang X. Anti-glioblastoma efficacy and safety of paclitaxel-loading angiopep-conjugated dual targeting PEG-PCL nanoparticles. Biomaterials. 2012;33(32):8167–8176. | ||
Li S-D, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5(4):496–504. | ||
Kim BY, Rutka JT, Chan WC. Nanomedicine. N Engl J Med. 2010;363(25):2434–2443. | ||
Gong C, Yang T, Yang X, et al. Carrier-free nanoassemblies of a novel oxazolidinone compound FYL-67 display antimicrobial activity on methicillin-resistant Staphylococcus aureus. Nanoscale. 2013;5(1):275–283. | ||
Bowdish DM, Davidson DJ, Lau YE, Lee K, Scott MG, Hancock RE. Impact of LL-37 on anti-infective immunity. J Leuko Biol. 2005;77(4):451–459. | ||
Cederlund A, Gudmundsson GH, Agerberth B. Antimicrobial peptides important in innate immunity. FEBS J. 2011;278(20):3942–3951. | ||
Lin L, Nonejuie P, Munguia J, et al. Azithromycin synergizes with cationic antimicrobial peptides to exert bactericidal and therapeutic activity against highly multidrug-resistant Gram-negative bacterial pathogens. EBioMedicine. 2015;2(7):688–696. | ||
Matijašić M, Kos VM, Nujić K, et al. Fluorescently labeled macrolides as a tool for monitoring cellular and tissue distribution of azithromycin. Pharmacol Res. 2012;66(4):332–342. | ||
Sochacki KA, Barns KJ, Bucki R, Weisshaar JC. Real-time attack on single Escherichia coli cells by the human antimicrobial peptide LL-37. Proc Natl Acad Sci U S A. 2011;108(16):E77–E81. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.