Back to Journals » Drug Design, Development and Therapy » Volume 12
Notch inhibitor can attenuate apparent diffusion coefficient and improve neurological function through downregulating NOX2-ROS in severe traumatic brain injury
Authors Zhang HM, Chen W, Liu RN, Zhao Y
Received 13 May 2018
Accepted for publication 10 August 2018
Published 8 November 2018 Volume 2018:12 Pages 3847—3854
DOI https://doi.org/10.2147/DDDT.S174037
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Anastasios Lymperopoulos
Hong-Mei Zhang,1,2 Wei Chen,3 Rui-Ning Liu,1 Yan Zhao1
1Emergency Center, Zhongnan Hospital of Wuhan University, Wuhan 430071, Hubei, People’s Republic of China; 2Emergency Department, The Second Medical College of Jinan University, Shenzhen People’s Hospital, Shenzhen 518020, Guangdong, People’s Republic of China; 3Department of Intensive Care Unit, Taihe Hospital, Hubei University of Medicine, Shiyan 442000, Hubei, People’s Republic of China
Introduction: Secondary brain injury is a major factor that affects the prognosis and outcome of traumatic brain injury (TBI) patients. Secondary brain edema is considered to be an initiating factor in secondary brain injury after TBI. A previous study has indicated that Notch signaling activation contributes to neuron death in mice affected by stroke; however, its role in neuronal oxidation stress for brain edema after TBI is not well established. Apparent diffusion coefficient (ADC) values can represent the brain edema after TBI.
Methods: We established a rat model of acute craniocerebral injury, using functional MRI to evaluate the ADC and cerebral blood flow values. The present study was designed to determine the effect of Notch inhibitor DAPT upon oxidation stress for brain edema after TBI. Rats were randomly distributed into five groups, control group, severe TBI group, severe TBI + vehicle group, severe TBI + DAPT group, and severe TBI + DPI group. All rats were sacrificed at 24 hours after TBI.
Results: Our data indicated that Notch signaling inhibitor DAPT significantly reduced the ADC values and improved the neurological function after TBI. In addition, DAPT decreased NOX2 levels and the ROS levels. Furthermore, DPI can decrease NOX2 levels and ROS levels.
Conclusion: This study indicated that DAPT Notch signal inhibitors can inhibit NOX2-ROS generation, reduce the ADC values, relieve cerebral edema, and improve nerve function.
Keywords: traumatic brain injury, secondary brain injury, apparent diffusion coefficient, Notch, cerebral blood flow
Introduction
Traumatic brain injury (TBI) is a serious public health problem, which has high mortality and disability rate. TBI can bring serious health and economic burden to society and individuals.1 After primary brain injury occurs, it is followed by a secondary brain injury cascade.2 Research has shown that secondary brain injury has become a major factor that affects the prognosis and outcome of the TBI patients.3 Secondary brain edema is considered to be an initiating factor in secondary brain injury.4 Therefore, early diagnosis and finding out of the mechanism of cerebral edema after TBI can provide new theoretical basis for treatment of TBI. Recently, with the rapid development of imaging techniques, in comparison with conventional MRI, functional MRI (fMRI) has been shown to provide, in addition to the morphology information, metabolism and physiological functional information in TBI.5 Diffuse-weighted imaging (DWI) can show water molecule movement noninvasively at cell level. Apparent diffusion coefficient (ADC) is used to represent the range and velocity of the diffusion movement of water molecules in different directions in DWI. Studies have showed that ADC can represent brain edema after TBI.6
Notch signaling pathway is involved in many physiological and pathological processes. Notch signaling pathway is a conservative pathway which participates in the development of multiple organ systems, including angiogenesis, nerve regeneration, and hematopoietic cascade.7 Research has shown that γ-secretase complex and Notch1 are involved in some neurodegenerative diseases, such as Alzheimer’s disease and ischemic stroke.8 Notch signal pathway inhibitor can inhibit apoptosis and inflammation-mediated ischemic brain injury.9 In our previous studies, we found that Notch inhibitor DAPT can improve the neural function of severe TBI (unpublished). Here, Notch signal with brain edema and the mechanism after TBI is discussed.
In this study, we used fMRI test the cerebral blood flow (CBF) and ADC values in the areas surrounding severe TBI, through the ADC values to judge the brain edema after TBI, to explore the possible mechanism of the Notch signaling pathway in TBI, and thus provide a new theoretical basis for the treatment of cerebral edema after TBI.
Materials and methods
Animals
The animals were 250–300 g SPF-grade Sprague Dawley rats purchased from Animal Center of Wuhan University (Wuhan, People’s Republic of China). The animal experiment was approved by the Animal Experiment Center and ethics committee of Zhongnan Hospital of Wuhan University and followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals. DAPT (MCE, Bridgewater, NJ, USA) solution (1 μg/μL) was prepared by dissolving DAPT powder in 0.01 M PBS containing 5% dimethyl sulfoxide. The solution was filtered and stereotactically injected into the right cerebral ventricle using the following coordinates: −0.8 mm anteroposterior, ±1.6 mm mediolateral, and −4.0 mm dorsoventral from the bregma. Control rats received the injection of PBS instead of DAPT in the same way.
Animal model
Animal models with different degrees of TBI were prepared as described previously.10 Briefly, the rats received intraperitoneal injection of 1% pentobarbital at a dose of 30 mg/kg. After the success of the anesthesia, rats were fixed on the brain stereotaxic device to cut the top of the skull skin to determine the bregma. Then a 5 mm diameter bone window was drilled, and dura mater was exposed. A 40 g hammer fell at 25 cm vertically along the outer tube leading to moderate or severe TBI. The control group received no brain damage. Half an hour after injury, DAPT (0.03 mg/kg BW), DPI (0.25 mg/kg), or PBS was injected into lateral ventricle, followed by the closing of bone window. Experimental grouping was as follows: control group (n=10), severe TBI (n=10), severe TBI + PBS (n=10), severe TBI + DAPT (n=10), and severe TBI + DPI (n=10).
Neurological function
The neurological function of the rats was evaluated using modified Neurological Severity Scores (NSS) as described previously.11
MRI methods
MRI studies were performed on a 7 .0 T magnet (Bruker, PharmaScan, Karlsruhe, Germany), equipped with an actively shielded gradient system (14 cm inner diameter). A surface coil (2.3×1.5 cm) was used for brain imaging. Rats were anesthetized using facemask inhalation of 1.8% isoflurane by isoflurane anesthesia system (JD Medical Dist. Co. Inc., Phoenix, AZ, USA). CBF images were obtained from continuous arterial spin labeling recovery (echo-planar fluid-attenuated inversion recovery) sequences. CBF images were reconstructed with Para Vision version 5.1 software (Bruker, PharmaScan), acquisition parameters were FV = 40×40 mm, TR =18,000 ms, TE =26 ms, matrix size =128×128, and the number of excitations =1.
Multislice coronal spin-echo DWI were acquired with an echo planar diffusion-trace MRI sequence (matrix size =128×128, TR =7,100 ms, TE =22 ms, FOV =33×33 mm2). DWI images were reconstructed with Para Vision version 5.1 software (Bruker, PharmaScan).
Western blot analysis
Total protein was isolated from brain tissues and quantitated by BSA method. Thirty microgram protein was separated on 10% SDS-PAGE and transferred to PVDF membranes (Millipore, Billerica, MA, USA). Next, the membranes were incubated at 4°C overnight with the following primary antibodies: GAPDH (1: 1,000 dilution), Notch1 (1: 1,000 dilution), Hes1 (1:800 dilution), Hes5 (1:500 dilution), and NOX2 (1: 1,000 dilution) (all from Proteintech Inc., Rosemont, IL, USA). The membranes were washed with TBST three times, then incubated with HRP-labeled goat anti-rabbit secondary antibody (Boster Biotech., Wuhan, People’s Republic of China) for 1 hour at room temperature. The membranes were developed using ECL kit (Pierce, Rockford, IL, USA) and exposed to X-ray film.
ROS detection
ROS Assay Kit is the most commonly used method for quantitative detection of reactive oxygen ROS levels in cells based on fluorescence dye dichlorodihydrofluorescein diacetate (DCFH-DA). DCFH-DA itself has no fluorescence and can pass freely across the cell membrane. After entering the cell, dichlorofluorescin (DCFH) can be hydrolyzed by esterase in the cell. DCFH does not penetrate cell membranes, so probes can easily accumulate in cells. The reactive oxygen in the cell can oxidize fluorescence-free DCFH to produce fluorescent DCF. The intensity of green fluorescence is proportional to the level of ROS. At maximum excitation wavelength of 480 nm, the maximum emission wavelength of 525 nm, using a fluorescence microscope, flow cytometry and laser confocal microscope detection such as fluorescent signal, according to the positive control Rosup fluorescent signals to analyze the real level of ROS.
Measurement of total ROS, including hydrogen peroxide, nitric oxide, peroxyl radical, and peroxynitrite anion, were assessed. Total ROS were detected using a commercial ROS Assay Kit (E003, Nanjing Jiancheng Bioengineering Institute, Nanjing, People’s Republic of China). Assay was performed in accordance with the manufacturer’s instructions.
Statistical analysis
Data were expressed as number, percentage or mean ± SD. Statistical analysis was performed and graphs were created using GraphPad Prism 7 (Graph Pad Software, La Jolla, CA, USA). Statistical differences between groups were determined using one-way ANOVA followed by Student’s t-test. A two-tailed P-value <0.05 was considered statistically significant.
Results
Evaluating the effects of TBI on ADC and CBF
To evaluate the effects of TBI on CBF, animals were prepared for fMRI 24 hours after injury. We found that the DWI signal (Figure 1A), the values of ADC (Figure 1B and D, P<0.01), and the values of CBF (Figure 1C and E, P<0.01) were increased in severe TBI animals compared to controls. Importantly, a “luxury perfusion” phenomena was observed in this study. Treatments with DAPT for severe TBI animals resulted in decreased ADC (Figure 1D, P<0.05), but no effect on CBF.
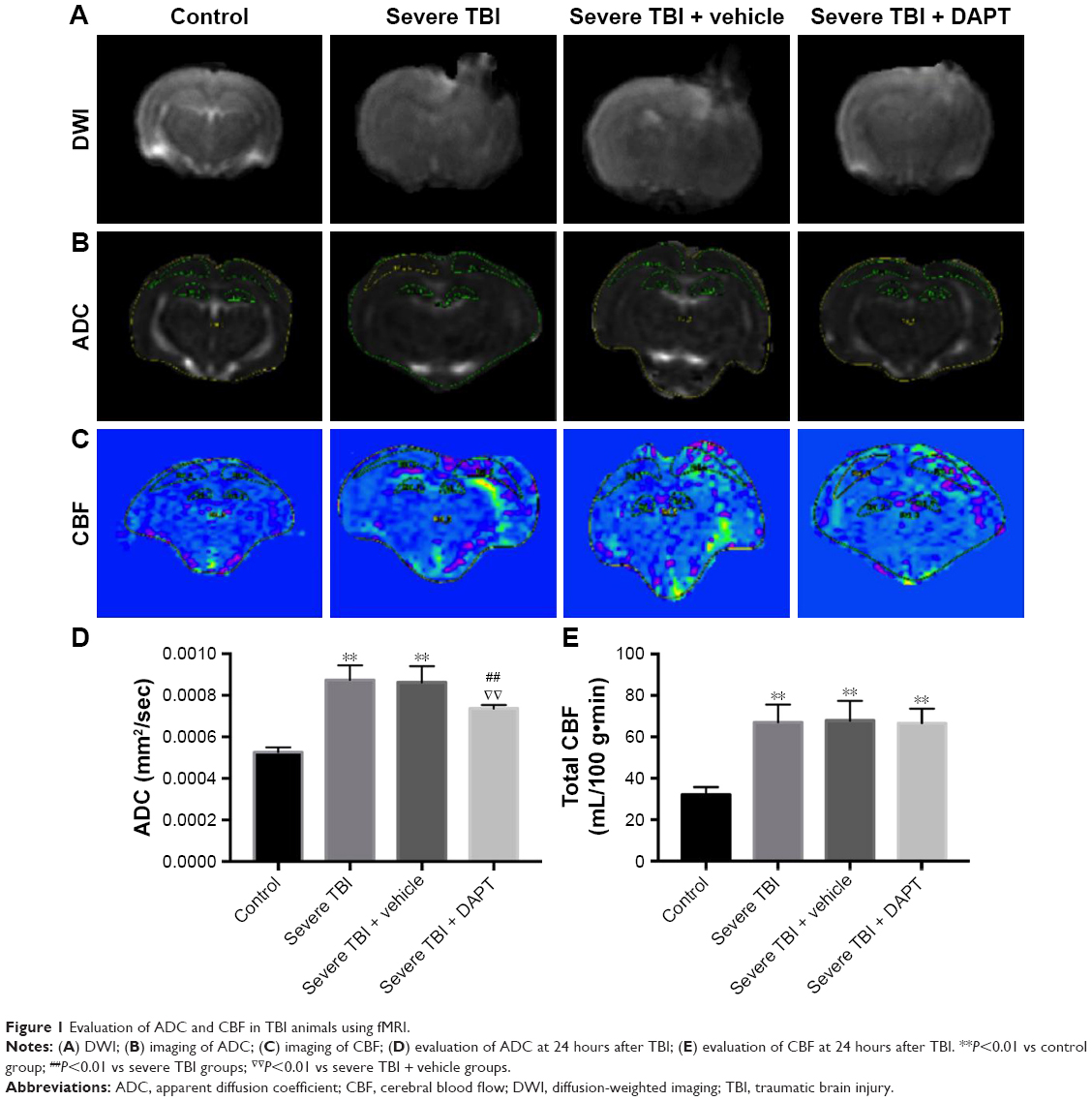 | Figure 1 Evaluation of ADC and CBF in TBI animals using fMRI. |
Acute TBI decreased neurological function; DAPT and DPI attenuated neurological function
The NSS of each animal was calculated. Our results showed that the values of NSS were significantly higher in severe TBI animals than normal controls (Figure 2, P<0.01). The NSS in severe TBI and severe TBI + vehicle groups were significantly higher than that in the DPI and DAPT groups (Figure 2, P<0.05, respectively), which indicated that both DPI and DAPT attenuated neurological function in severe TBI animals.
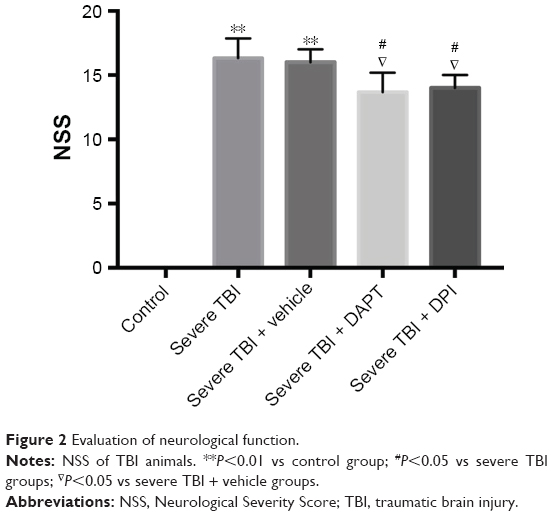 | Figure 2 Evaluation of neurological function. |
Notch signaling and NOX2 were upregulated in severe TBI; DAPT downregulated Notch signaling and NOX2; DPI downregulated NOX2
Western blot assay showed that severe TBI increased NICD (Figure 3, P<0.01), Hes1 (Figure 3, P<0.01), Hes5 (Figure 3, P<0.01), and NOX2 (Figure 3, P<0.01) expression at the protein level. DAPT treatment resulted in significant inhibition on NICD (Figure 3, P<0.01), Hes1 (Figure 3, P<0.05), Hes5 (Figure 3, P<0.05), and NOX2 (Figure 3, P<0.01) expression. Also, DPI treatment decreased NOX2 expression Figure 3 (P<0.01).
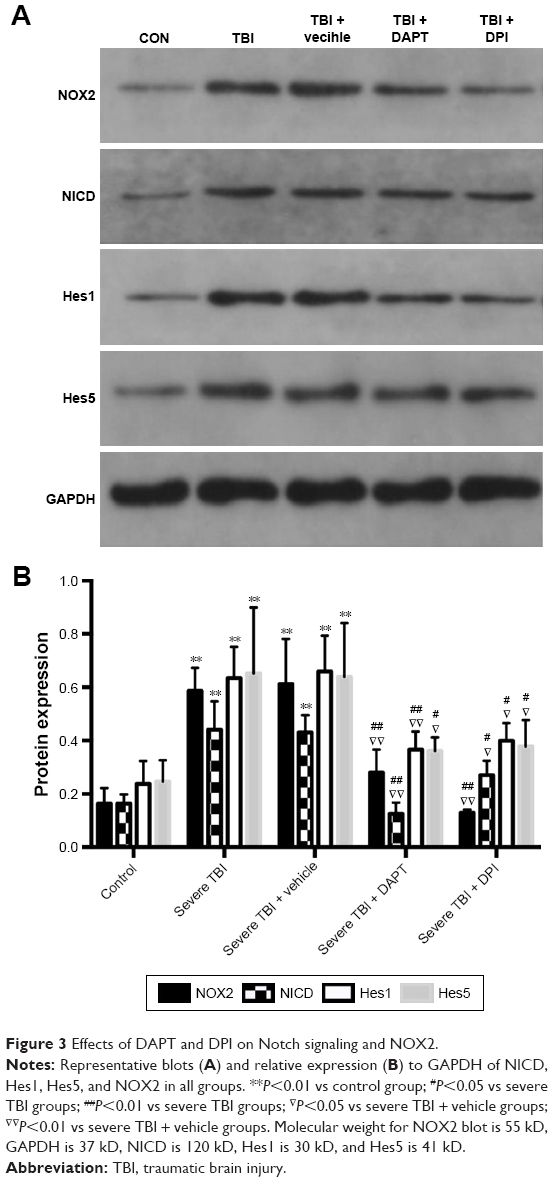 | Figure 3 Effects of DAPT and DPI on Notch signaling and NOX2. |
Severe TBI induced oxidative stress in brain
We measured total ROS in brain tissue. Our results showed that ROS levels in the severe TBI group were significantly higher than that in the control group (Figure 4, P<0.001). After administration of DPI, ROS levels were significantly lower than in the severe TBI group (Figure 4, P<0.001) and the severe TBI + vehicle group (Figure 4, P<0.001).
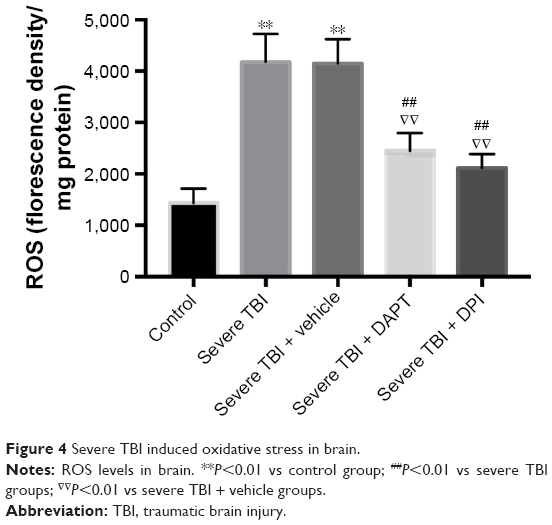 | Figure 4 Severe TBI induced oxidative stress in brain. |
Discussion
In our paper, we used fMRI to detect the whole-brain CBF and ADC values and the DWI signal (Figure 1A) after severe TBI and found that after 24 hours of TBI, whole-brain CBF was significantly higher, presented with the “luxury perfusion” status (Figure 1C and E), had higher ADC values (Figure 1B and D), and higher DWI signal (Figure 1A). Luxury perfusion is considered to be one of the most important pathologic mechanisms of acute brain injury, especially in stroke and acute surgery, and also in TBI. In both human and animal studies, luxury perfusion was observed.12 In the study by Mario et al,13 patients with severe craniocerebral trauma were shown to have CBF reaching its peak at 24 hours after the onset of brain injury. This is consistent with our research on animal experiments. Langfitt et al14 considered luxury perfusion as brain blood vessel movement paralysis, caused by cerebral vascular expansion and increased blood flow to the brain, and recent research has shown that this is due to acute cerebral vascular expansion after brain damage.15 Lassen16 considered luxury perfusion to be associated with acute metabolic acidosis in brain. In our study, Notch signaling inhibitor DAPT could not change the luxury perfusion status; the reason for this may be that DAPT cannot attenuate the brain metabolic acidosis after TBI. The cerebral edema after TBI is the most difficult problem in clinical treatment, including vasogenic brain edema and cytotoxic cerebral edema, both of which generally coexist, in different periods of pathology and one of the edema types predominated.17 Vasogenic cerebral edema refers to the change in the blood–brain barrier that leads to transient increase in water movement from intravascular into the extracellular space; cytotoxic cerebral edema refers to changes in the osmotic pressure of cells inside and outside, thus making water from outside the cell move into the cell.18 In cytotoxic cerebral edema, the total water content of the tissue is not increased, the dispersion of water molecules is limited, and the DWI signal is increased. Vascular cerebral edema is further aggravated by cytotoxic edema, further restricting water molecule dispersion, and leading to more obvious increase in DWI signal. In this study, the DWI signal of severe TBI increased obviously (Figure 1A), indicating severe cerebral edema had occurred.
ADC values can distinguish between vasogenic cerebral edema and cytotoxic cerebral edema. A high ADC value was found in vascular cerebral edema, and cytotoxic cerebral edema indicated ADC drop.19 In this study, a high ADC was also found 24 hours after the brain injury (Figure 1B and D), indicating that the vasogenic cerebral edema was the main cause. Vasogenic cerebral edema is associated with the loss of the function cerebral autoregulation and blood–brain barrier damage,20 while cytotoxic brain edema is associated with brain tissue cell energy ion pump failure, leading to brain swelling and rupture, thus leading to the decrease of cerebral blood volume.21 This study found that high ADC values at 24 hours after brain injury, suggestive of vasogenic cerebral edema, and cerebral autoregulation function was lost, accompanied by high cerebral brain flow, which may explain the luxury perfusion status observed in this study. In this study, the advantages of fMRI were used to determine the type of cerebral edema in combination with ADC and CBF, so as to provide an irreplaceable role for further study of cerebral edema mechanism and treatment.
After TBI, free radical production increases and oxidative stress level increases. The free radical damage caused by oxidative stress is one of the main pathologic mechanisms.22 The generation and injury caused by free radicals, the stress effect, is involved in the occurrence of secondary cerebral edema after TBI.23 Studies have shown that oxygen free radicals caused brain tissue slice moisture content to increase.24 Therefore, after TBI, inhibiting the free radical generation caused by oxidative stress can improve the occurrence of secondary cerebral edema. Oxidative stress refers to the loss of balance between the formation of ROS and the antioxidant capacity to prevent reactive oxygen injury. Brain tissue is sensitive to oxidative stress due to its high oxidative metabolic rate and high production of ROS. NADPH oxidase is one of the main sources of ROS production. Studies have shown that NADPH oxidase and its derived ROS are present in many organs and tissues. NADPH exists in many subtypes, and NOX2 exists in a variety of cell types, including endothelial cells, fibroblasts, immune cells, and nerve cells. Studies have shown that NOX2 plays a major role in cerebral ischemia reperfusion, which is the main source of ROS production.25 NOX2 participated in the early brain injury caused after subarachnoid; in TBI, NOX2 participated in the activation of the inflammasome and participated in the pathological process of brain trauma.26 In this study, we found that NOX2 was activated in 24 hours after severe brain injury (Figure 3), and ROS levels increased (Figure 4); inhibition of NOX2 led to ROS decline (Figure 4), suggesting that in 24 hours after severe brain injury, NOX2 is the main source of ROS in central nervous system.
Notch signaling pathway is highly conserved during evolution and plays an important role in cell differentiation, proliferation, apoptosis, synaptic plasticity, and cell survival.27 Notch receptors and Notch ligand binding trigger signal transduction. The Notch receptor and the ligand interactions lead to γ-secretase complex catalyzing and cracking Notch receptor protein in the transmembrane cell region, making NICD to be released from the inside of the cell membrane. NICD, released directly into the nucleus, interacts with transcription factors RBP-J, making it a transcription activation factor, inducing the expression of downstream target genes such as Hes.28 It can be seen that γ-secretase is a key enzyme in the Notch pathway, which directly affects the activation of the pathway. Moreover, DAPT, a suppressor of γ-secretase, specifically inhibits the activation of Notch signaling pathway and downstream target genes. Notch receptor-mediated signaling pathway plays an important role in nervous system development, and previous studies have found that Notch signaling is involved in the occurrence of some neurodegenerative diseases, such as Alzheimer’s disease and ischemic stroke.29 Our preliminary results suggest that Notch signal inhibitor DAPT can improve the behavioral function of TBI.30 In this study, Notch inhibitors were shown to decrease ADC value in rats with severe brain injury, indicating that Notch inhibitors can attenuate the vasogenic cerebral edema of severe TBI. Studies have shown that Notch signaling pathway may be involved in the functional maintenance of vascular endothelial cells and can mediate vascular regeneration and vascular remodeling in the state of disease.31 In this study, the Notch signal after acute severe brain injury was activated, and Notch inhibitors can decrease the ADC value and attenuate the vasogenic cerebral edema in rats with severe brain injury. This led to the hypothesis that Notch signal may be involved in the dysfunction of cerebral vascular endothelial cells in acute severe brain trauma, thus inducing the occurrence of cerebral edema. Further to the related mechanism, DAPT inhibits NOX2 expression and the level of ROS, giving NOX2 inhibitor treatment, decreased the ROS, ADC values also dropped (Figures 3 and 4), and NSS were attenuated in severe TBI animals (Figure 4) suggests that NOX2-ROS production is the main factor lead to increased ADC after severe brain trauma, NOX2-ROS can also be considered as one of the main factors that leading to secondary brain edema of severe brain trauma. Research has shown that increased ROS generation is one of the important mechanisms of secondary damage injury of TBI; NOX2 is the main source of ROS produced in the brain,32 and therefore this paper assumes that NOX2 after TBI is one of the main sources of ROS. DPI can inhibit the activity of NADPH enzyme, nitric oxide synthase, xanthine oxidase, and NADPH CYP/CYP450 oxidoreductase. DPI is often used to inhibit the production of ROS mediated by various flavoenzyme. Studies have shown that DPI is a classic effective inhibitor of NOX2. In many studies, DPI is used as the inhibitor of NOX2.33 Therefore, DPI was also selected as the inhibitor of NOX2 in this study. Notch signal inhibitor DAPT can inhibit the NOX2 protein expression in rats with severe brain trauma, reduce NOX2 producing ROS, and thus reduce the ADC values of rats, thus attenuating vasogenic cerebral edema after traumatic brain edema, which can improve the outcome after secondary brain injury. The Notch signaling pathway is closely associated with various subtypes of NADPH and ROS. Some studies have shown that the Notch signaling pathway is involved in oxidative stress injury of renal ischemia reperfusion and oxidative stress in vascular endothelium.34 In the hepatic ischemia reperfusion injury, the ROS level is controlled by Notch signals. In human umbilical vein endothelial cells, Notch signaling pathway regulates NOX4 level.35 In this study, we found that Notch signal and NOX2-ROS expression were higher after TBI, inhibiting the Notch signaling pathway could induce NOX2-ROS expression to decrease obviously, and shows that in acute phase of TBI NOX2-ROS is controlled by the Notch signal. The inhibition of Notch signaling pathway can inhibit the production of NOX2-ROS, thus reducing level of the ADC value, and attenuate the vascular cerebral edema after trauma, thus alleviating secondary injury after TBI.
Conclusion
fMRI can judge the early edema state of severe TBI through the ADC values. DAPT Notch signal inhibitors can inhibit NOX2-ROS generation, reduce the ADC values, relieve cerebral edema, and improve nerve function.
Acknowledgments
This study was financially supported by the Emergency Diagnostic and Therapeutic Center of Central China. The authors would like to thank the Core Facilities for Medical Imaging of Capital Medical University (Beijing, People’s Republic of China) for the technical assistance in fMRI imaging.
Disclosure
The authors report no conflicts of interest in this work.
References
Lasry O, Liu EY, Powell GA, Ruel-Laliberté J, Marcoux J, Buckeridge DL. Epidemiology of recurrent traumatic brain injury in the general population: A systematic review. Neurology. 2017;89(21):2198–2209. | ||
Vasterling JJ, Jacob SN, Rasmusson A. Traumatic brain injury and posttraumatic stress disorder: conceptual, diagnostic, and therapeutic considerations in the context of co-occurrence. J Neuropsychiatry Clin Neurosci. 2018;30(2):91–100. | ||
Martin M, Cook F, Lobo D, et al. Secondary insults and adverse events during intrahospital transport of severe traumatic brain-injured patients. Neurocrit Care. 2017;26(1):87–95. | ||
Chen JW, Paff MR, Abrams-Alexandru D, Kaloostian SW. Decreasing the cerebral edema associated with traumatic intracerebral hemorrhages: use of a minimally invasive technique. Acta Neurochir Suppl. 2016;121:279–284. | ||
Stovell MG, Yan JL, Sleigh A, et al. Assessing metabolism and injury in acute human traumatic brain injury with magnetic resonance spectroscopy: current and future applications. Front Neurol. 2017;8:426. | ||
Wei XE, Li YH, Zhao H, Li MH, Fu M, Li WB. Quantitative evaluation of hyperbaric oxygen efficacy in experimental traumatic brain injury: an MRI study. Neurol Sci. 2014;35(2):295–302. | ||
Fazio C, Ricciardiello L. Inflammation and Notch signaling: a crosstalk with opposite effects on tumorigenesis. Cell Death Dis. 2016;7(12):e2515. | ||
Murakami K, Watanabe T, Koike T, Kamata M, Igari T, Kondo S. Pharmacological properties of a novel and potent γ-secretase modulator as a therapeutic option for the treatment of Alzheimer’s disease. Brain Res. 2016;1633:73–86. | ||
Mathieu P, Adami PV, Morelli L. Notch signaling in the pathologic adult brain. Biomol Concepts. 2013;4(5):465–476. | ||
Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res. 1981;211(1):67–77. | ||
Chen J, Li Y, Wang L, et al. Therapeutic benefit of intravenous administration of bone marrow stromal cells after cerebral ischemia in rats. Stroke. 2001;32(4):1005–1011. | ||
Lassen NA. The luxury-perfusion syndrome and its possible relation to acute metabolic acidosis localised within the brain. Lancet. 1966;288(7473):1113–1115. | ||
Marion DW, Darby J, Yonas H. Acute regional cerebral blood flow changes caused by severe head injuries. J Neurosurg. 1991;74(3):407–414. | ||
Langfitt TW, Gennarelli TA, Obrist WD, Bruce DA, Zimmerman RA. Prospects for the future in the diagnosis and management of head injury: pathophysiology, brain imaging, and population-based studies. Clin Neurosurg. 1982;29:353–376. | ||
Sharma VK, Teoh HL, Paliwal PR, Chong VF, Chan BP, Sinha AK. Reversed Robin Hood syndrome in a patient with luxury perfusion after acute ischemic stroke. Circulation. 2011;123(7):e243–e244. | ||
Lassen NA. The luxury-perfusion syndrome and its possible relation to acute metabolic acidosis localised within the brain. Lancet. 1966;2(7473):1113–1115. | ||
Finnie JW, Manavis J, Casson RJ, Chidlow G. Retinal microvascular damage and vasogenic edema produced by Clostridium perfringens type D epsilon toxin in rats. J Vet Diagn Invest. 2014;26(3):470–472. | ||
Kimberly WT, Battey TW, Pham L, et al. Glyburide is associated with attenuated vasogenic edema in stroke patients. Neurocrit Care. 2014;20(2):193–201. | ||
Weimer JM, Jones SE, Frontera JA. Acute cytotoxic and vasogenic edema after subarachnoid hemorrhage: a quantitative MRI study. AJNR Am J Neuroradiol. 2017;38(5):928–934. | ||
Hudak AM, Peng L, Marquez de La Plata C, et al. Cytotoxic and vasogenic cerebral oedema in traumatic brain injury: assessment with FLAIR and DWI imaging. Brain Inj. 2014;28(12):1602–1609. | ||
Kurita D, Haida M, Shinohara Y. Energy metabolism and cerebral blood flow during cytotoxic brain edema induced by 6-aminonicotinamide. Acta Neurochir Suppl. 2003;86:41–44. | ||
Yonutas HM, Vekaria HJ, Sullivan PG. Mitochondrial specific therapeutic targets following brain injury. Brain Res. 2016;1640(Pt A):77–93. | ||
Eve DJ, Steele MR, Sanberg PR, Borlongan CV. Hyperbaric oxygen therapy as a potential treatment for post-traumatic stress disorder associated with traumatic brain injury. Neuropsychiatr Dis Treat. 2016;12:2689–2705. | ||
Cruz-Haces M, Tang J, Acosta G, Fernandez J, Shi R. Pathological correlations between traumatic brain injury and chronic neurodegenerative diseases. Transl Neurodegener. 2017;6:20. | ||
Wang J, Ma MW, Dhandapani KM, Brann DW. Regulatory role of NADPH oxidase 2 in the polarization dynamics and neurotoxicity of microglia/macrophages after traumatic brain injury. Free Radic Biol Med. 2017;113:119–131. | ||
Zhang L, Li Z, Feng D, et al. Involvement of Nox2 and Nox4 NADPH oxidases in early brain injury after subarachnoid hemorrhage. Free Radic Res. 2017;51(3):316–328. | ||
Guruharsha KG, Kankel MW, Artavanis-Tsakonas S. The Notch signalling system: recent insights into the complexity of a conserved pathway. Nat Rev Genet. 2012;13(9):654–666. | ||
Borggrefe T, Lauth M, Zwijsen A, et al. The Notch intracellular domain integrates signals from Wnt, Hedgehog, TGFbeta/BMP and hypoxia pathways[J]. Biochim Biophys Acta. 1863;2016(2):303–313. | ||
Barrett JP, Henry RJ, Villapol S, et al. NOX2 deficiency alters macrophage phenotype through an IL-10/STAT3 dependent mechanism: implications for traumatic brain injury. J Neuroinflammation. 2017;14(1):65. | ||
Zhang HM, Liu P, Jiang C, et al. Notch signaling inhibitor DAPT provides protection against acute craniocerebral injury. PLoS One. 2018;13(2):e193037. | ||
Polacheck WJ, Kutys ML, Yang J, et al. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature. 2017;552(7684):258–262. | ||
Chandran R, Kim T, Mehta SL, et al. A combination antioxidant therapy to inhibit NOX2 and activate Nrf2 decreases secondary brain damage and improves functional recovery after traumatic brain injury[J]. J Cereb Blood Flow Metab. 2017:271678X–17738701X. | ||
Zhu Y, Fan S, Wang N, et al. NADPH oxidase 2 inhibitor diphenyleneiodonium enhances ROS-independent bacterial phagocytosis in murine macrophages via activation of the calcium-mediated p38 MAPK signaling pathway. Am J Transl Res. 2017;9(7):3422–3432. | ||
Vieceli Dalla Sega F, Aquila G, Fortini F, et al. Context-dependent function of ROS in the vascular endothelium: The role of the Notch pathway and shear stress. Biofactors. 2017;43(4):475–485. | ||
Cai WX, Liang L, Wang L, et al. Inhibition of Notch signaling leads to increased intracellular ROS by up-regulating Nox4 expression in primary HUVECs. Cell Immunol. 2014;287(2):129–135. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.