Back to Journals » Cancer Management and Research » Volume 11
Nonspecific immunoglobulin G is effective in preventing and treating cancer in mice
Authors Xu Q, Zhang Z, Chen Z, Zhang B, Zhao C, Zhang Y, Zhao C, Deng X, Zhou Y, Wu Y, Gu J
Received 20 September 2018
Accepted for publication 24 January 2019
Published 7 March 2019 Volume 2019:11 Pages 2073—2085
DOI https://doi.org/10.2147/CMAR.S188172
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Chien-Feng Li
Qian Xu,1,* Zaiping Zhang,1,* Zhiming Chen,1,* Biying Zhang,1 Chanyuan Zhao,1 Yimin Zhang,1 Conghui Zhao,2 Xiaodong Deng,1 Yao Zhou,1 Yanyun Wu,1 Jiang Gu1–3
1Provincial Key Laboratory of Molecular Pathology and Personalized Medicine, Shantou University Medical College, Shantou 515041, Guangdong, China; 2Department of Pathology, Beijing University Health Science Center, Beijing 100083, China; 3Jinxin Research Institute for Reproductive Medicine and Genetics, Chengdu Jinjiang Hospital for Maternal and Child Health Care, Chengdu 610066, Sichuan, China
*These authors contributed equally to this work
Background: Previous accidental findings showed that administration of immunoglobulin G (IgG) in treating autoimmune diseases was able to inhibit cancers that happened to grow in these patients. However, such treatment has not been used to treat cancer patients clinically. The mechanism and optimal dosages of this treatment have not been established. Subsequent animal experiments confirmed this effect, but all previous studies in animal models used human IgG which was heterogeneous to the animal hosts and therefore could adversely interfere with the results.
Materials and methods: We tested different dosages of mouse IgG in treating and preventing three syngeneic cancer types (melanoma, colon cancer, and breast cancer) in three immune potent mouse models. The expression of Ki67, CD34, VEGF, MMPs, and cytokines in tumor tissues were examined with immunohistochemistry or quantitative real-time PCR to evaluate tumor proliferation, vascularization, metastasis, and proinflammatory response in the tumor microenvironment.
Results: We found that low-dose IgG could effectively inhibit cancer progression, regulate tumor vessel normalization, and prolong survival. Administration of IgG before cancer cell inoculation could also prevent the development of cancer. In addition, IgG caused changes in a number of cytokines and skewed macrophage polarization toward M1-like phenotype, characterized by proinflammatory activity and inhibition of proliferation of cancer cells.
Conclusion: Our findings suggest that nonspecific IgG at low dosages could be a promising candidate for cancer prevention and treatment.
Keywords: IVIg, cancer therapy, macrophages, mouse model, immunotherapy
Introduction
Immunotherapy is the method of choice for many cancer types, and antibodies have been the main regiment for such treatment.1,2 Most of the recent advances in immunotherapy have been employing specific antibodies against particular molecules in effector immune cells or cancer cells facilitating enhanced cancer cell recognition and destruction.3,4 It has been shown that non-cancer-specific immunoglobulin G (IgG) might also have anticancer effects, but information in this regards has been scarce.
Intravenous Ig (IVIg) is a pool of Ig collected from over 1,000 healthy adults and has been used widely as a treatment for immunodeficiency and autoimmunity diseases for over 40 years without much side effect.5,6 It has also been reported that IVIg can inhibit tumor growth and metastasis in patients and animal models.7–17 The initial discovery of this effect was accidental as some cancer patients also suffered from autoimmune disease. Physicians gave IVIg to these patients for autoimmune treatment but unexpectedly observed cancer regression.6,18–20 Then, scientists were inspired to test different doses of IVIg in tumor-bearing animals and found that high-dose IVIg (400 mg/kg or more) could effectively inhibit tumor growth. However, this promising observation was not followed up by clinical trials. Mouse studies were performed; however, IgG used in these studies were from human that is heterogenic to mice and might cause additional untoward reactions.21 Up-to-now the mechanism of non-cancer-specific IgG in inhibiting cancer growth has not been elucidated. The possibility of IVIg in preventing carcinogenesis has not been tested. Indeed, despite widespread and long-term clinical usage of IVIg in treating immune-related diseases, the exact mechanisms of IVIg in affecting the immune responses and achieving clinical benefits have been a matter of debate.22–24
In this study, we used mouse IgG (mIgG) to treat cancers in mouse models. A range of concentrations was tested. We also investigated if IgG could prevent cancer from developing if administrated prior to cancer cell inoculation. In addition, we examined the changes of cytokines in the animal models and the effects of IgG on macrophages in vitro. We found that IgG was effective in inhibiting cancer progression at a very low dosage. IgG could also prevent cancer from developing when given several weeks before cancer cell implantation as well as prolong the survival of cancer-bearing mice if administered after tumor cell inoculation. In addition, we found that IgG could polarize macrophages from M2 to M1 phenotype, providing insight into the mechanisms to explain the beneficial effects obtained with non-cancer-specific IgG in treating cancer.
Materials and methods
Animals and IgG preparation
Adult female BALB/c mice and C57BL/6 mice were obtained from Beijing Vital River Animal Technology Co. Ltd. (Beijing, China) and housed in the Animal Laboratory Center of Shantou University Medical College. Animal experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals of Shantou University Medical College. The protocol was approved by the Committee of the Ethics of Animal Experiments of the Shantou University Medical College. Mice aged between 6 and 8 weeks and weighing 22±2 g were used in all experiments. Human IgG preparation used in the study was purchased from Shanghai RAAS blood products co., Ltd. Mouse IgG preparation used in the study was purchased from Beijing Solarbio Science & Technology Co., Ltd. We prepared different concentrations of IgG in PBS buffer, quantified with BCA protein assay kit (Pierce™; Thermo Fisher Scientific, Waltham, MA, USA). filtered with 0.22 µm filter, and stored at –20°C.
Experimental design
To compared the effects of mouse IgG and human IgG, BALB/c mice were injected intravenously (tail vein) with 5×105 CT26 colon cancer cells on day 0, and were injected intravenously (tail vein) with human IgG or mIgG (50 mg/kg, n≥18) on days 5, 10, 15, and 20 with PBS as control. Mice were weighed on day 21.
The experiments were divided into three groups: preventive group in which IgG was given prior to inoculation of tumor cells, inhibitory group in which IgG was given at the same time as the tumor cells were inoculated, and therapeutic group in which IgG was given after the tumor cells were inoculated. Each group was inoculated with syngeneic mouse cancer types (mouse B16-F10 melanoma, mouse CT26 colon cancer, or mouse 4T1 breast cancer). More than two hundred mice were used in this study. The design of the experiment is shown in Figure S1.
For 4T1 breast cancer model, mice were injected subcutaneously (into mammary fat pad) with 5×105 4T1 breast cancer cells on day 0, and were injected intravenously (tail vein) with mIgG (25, 50, 100, and 400 mg/kg) or PBS (control) on days 5, 10, 15, 20, and 25. Mice were sacrificed on day 30 and tumors were weighed (n=10, Figure S2A).
For B16-F10 melanoma model, in the inhibitory experiment, mice were injected intravenously (tail vein) with 1.5×105 tumor cells on day 0 and given mIgG (100 mg/kg) or PBS (control) on days 1, 5, 9, 13, and 17. Mice were sacrificed on day 21 and lungs were weighed (n=20, Figure S2B). For survival analysis, mice were injected intravenously (tail vein) with 1.5×105 tumor cells on day 0 and given mIgG (100 mg/kg) or PBS (control) on days 1, 5, 9, 13, and 17 (n=20). Mice were monitored daily until death or the end of the study. In prevention experiment, mice were injected intravenously (tail vein) with mIgG (50, 100, 200, and 400 mg/kg) or PBS (control) on day –1, and then injected intravenously (tail vein) with 1.5×105 tumor cells on day 0 (n≥6, Figure S2D). Mice were sacrificed on day 21 and lungs were weighed. Then we tested different time schedule (n≥6, Figure S2D–G) of injection to achieve the best effect with the optimal dose (100 mg/kg). Mice were sacrificed on day 21 and lungs were weighed. For survival analysis, mice were injected intravenously (tail vein) with mIgG (100 mg/kg) or PBS (control) on days –15 and –1, and then injected intravenously (tail vein) with 1.5×105 tumor cells on day 0 (n=11). Mice were monitored daily until death or the end of the study.
For CT26 colon cancer model, in the inhibitory experiment, mice were injected intravenously (tail vein) with 3×105 tumor cells on day 0 and given mIgG (100 mg/kg) or PBS (control) on days 0, 4, 8, 12, and 16. Mice were sacrificed on day 21 and lungs were weighed (n=20, Figure S2C). For survival analysis, mice were injected intravenously (tail vein) with 2×105 tumor cells on day 0 and given mIgG (100 mg/kg) or PBS (control) on days 1, 5, 9, 13, 17, 21, and 25 (n=11). Mice were monitored daily until death or the end of the study. In prevention experiment, mice were injected intravenously (tail vein) with mIgG (100 mg/kg) or PBS (control) on days –8, –4, and –1, and then injected intravenously (tail vein) with 2×105 tumor cells on day 0. Mice were sacrificed on day 21 and tumors were weighed (n=22, Figure S2H).
Histopathology and immunohistochemistry
Mouse tumors and lungs were dissected and fixed in 4% paraformaldehyde and embedded in paraffin. Tissue sections at 4 µm were immunostained following standard procedures.25 We used Ki-67 (ab15580; Abcam, Cambridge, UK), CD34 (GTX61737; GeneTex, Irvine, CA, USA), MMP2 (GTX104577; GeneTex), MMP9 (GTX61537; GeneTex), CD206 (ab64693; Abcam), and inducible nitric oxide synthase (iNOS, ab15323; Abcam) as primary antibodies. Goat anti-rabbit IgG conjugated with horseradish peroxidase (Boster, Wuhan, China) was applied as secondary antibody. Positive reactions were colorized with 3-amino-9-ethylcarbazole (Zymed Laboratories, South San Francisco, CA, USA) and counterstained with hematoxylin. The slides were photographed with a Motic light microscope (Motic, Xiamen, China).
Quantitative real-time PCR (RT-qPCR)
Mice in each group were euthanized and tumors were photographed and weighed after removal. After snap-freezing in liquid nitrogen, total RNA was extracted from tumors with the RNAiso Plus kit (9109; Takara, Tokyo, Japan) following manufacturer’s guidelines. cDNA was synthesized using PrimeScript™ RT reagent Kit (RR037A; Takara). RT-q PCR was performed using SYBR® Premix Ex Taq™ II (RR820A; Takara) on 7500 Fast Real-Time PCR system (Thermo Fisher Scientific). Primers for real-time PCR are listed in Table 1. Results were expressed using the 2−ΔΔCT method for quantitation.
 | Table 1 Primers used in RT-qPCR assay Abbreviation: RT-qPCR, quantitative real-time PCR. |
Cell culture
Mouse breast cancer cell line 4T1 and mouse colon cancer cell line CT26 were obtained from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). Mouse melanoma cell line B16F10 was purchased from Wuhan Procell Life Science & Technology Co., Ltd, and mouse macrophage line RAW246.7 was purchased from Shanghai Gefan Biotechnology Co., Ltd. All cell lines used in this study were grown in RPMI-1640 medium (SH30809.01, Hyclone, Logan UT, USA) supplemented with 10% FBS (Thermo Fisher Scientific) and 100 IU/mL penicillin and 100 µg/mL streptomycin (Thermo Fisher Scientific) at 37°C in a 5% CO2 atmosphere.
Transwell co-culture assay
Transwell co-culture assay was performed as described previously.26 Briefly, mouse macrophages RAW246.7 cells seeded in 24-well plate were treated with 0.5 mg/mL mIgG or equal concentration of BSA as control for 24 hours. Medium was then replaced with fresh RPMI-1640 (supplemented with 8% FBS), and transwell inserts with 8 µm microporous membrane (cat no 3422; Corning Incorporated, Corning, NY, USA) were placed in the 24-well plate. Subsequently, 3×104 B16-F10 cells or CT26 cells in serum-free RPMI-1640 medium were seeded in the upper compartment of transwell inserts. After incubation at 37°C and 5% CO2 for 24 hours, unmigrated cells on the upper surface of microporous membrane were removed by cotton swabs. Migrated cells on the lower surface of microporous membrane were fixed with 4% paraformaldehyde for 20 minutes and subsequently stained with 0.1% crystal violet for 15 minutes. Transwell inserts were then rinsed with PBS thrice to remove excess stain and images of migrated cells were captured using Axiovert 40 CFL Microscope (Carl Zeiss AG, Jena, Germany) with CCD camera at 100× magnification. For the quantification of cell migration, migrated cells were solubilized with 33% acetic acid and absorbance was measured at OD595.
Cell proliferation assay
Mouse macrophages RAW246.7 seeded in six-well plates were treated with 0.5 mg/mL mIgG/BSA for 24 hours, and then the culture medium was renewed. After 24 hours of incubation, supernatants were collected and centrifuged to remove cells. 4T1 breast cancer cells seeded in 96-well plates were treated with 0.5 mg/mL mIgG/BSA or cultured with supernatants mentioned earlier. Cell proliferation was evaluated with a CCK8 kit (Dojindo, Kumamoto, Japan).
ELISA
Expressions of cytokines including IL-6, IL-10, and TNF-α in culture supernatants of macrophages mentioned earlier was measured with ELISA kits (4A Biotech Co. Ltd, Beijing, China) according to the manufacturer’s instructions.
Protein array
Culture supernatants of macrophages mentioned previously were measured for the presence of cytokines using Proteome ProfilerTM Array Mouse Cytokine Array Panel A (ARY006; R&D Systems, Inc., Minneapolis, MN, USA). Protein array was performed following protocol supplied by the manufacturers.
Statistical analyses
Statistical analyses were performed using GraphPad Prism 7.0. Statistical significance was calculated with Student’s t-test. Data represent mean ± standard error of mean of representative experiments unless otherwise stated, setting P<0.05 as statistically significant.
Results
Mouse IgG is more suitable than human IgG for use in mouse models
In a separate experiment, we compared the effects of injecting mice with mouse IgG with those injected with human IgG. We found that human IgG injection led to deteriorated health condition (Figure 1A) with significant loss of body weight (Figure 1B). As this would invariably affect the results of our subsequent experiments, we decided to use mouse IgG for our study.
 | Figure 1 Mouse IgG is more suitable than human IgG for use in mouse models. Notes: Compared with mice injected with mouse IgG, mice injected with human IgG displayed deteriorated health (A) with significant loss of body weight (B, n≥18 mice per group), which would affect the result interpretation. Therefore, we used mouse IgG in our study. ***P<0.001. Abbreviations: Ctrl, control; IgG, immunoglobulin G; ns, not significant. |
Mouse IgG inhibited tumor progression in vivo
According to our results, mIgG treatment significantly inhibited tumor progression and proliferation of blood vessels when compared with the controls. The tumors were reduced in weight and size for both the solid mass of the subcutaneous breast cancer (Figure 2A, B) and the lung metastases of melanoma (Figure 2C, D) and colon cancer (Figure 2E, F). Decreased expressions of Ki-67 (Figure 2G, H), MMP2, and MMP9 (Figure 2I–K) were observed in the mIgG-treated group, which revealed that tumor cell proliferation, metastasis, and invasion were effectively inhibited in the mIgG-treated group in comparison to the control group. Down-regulation of CD34 (Figure 2L, M) and VEGF (Figure 2N) was observed in the mIgG-treated group, which indicated that vascularization was also significantly reduced in areas of tumors in the mIgG-treated group in comparison to the control group. Altogether, data from the three mouse models revealed that mouse IgG could inhibit tumor progression in vivo.
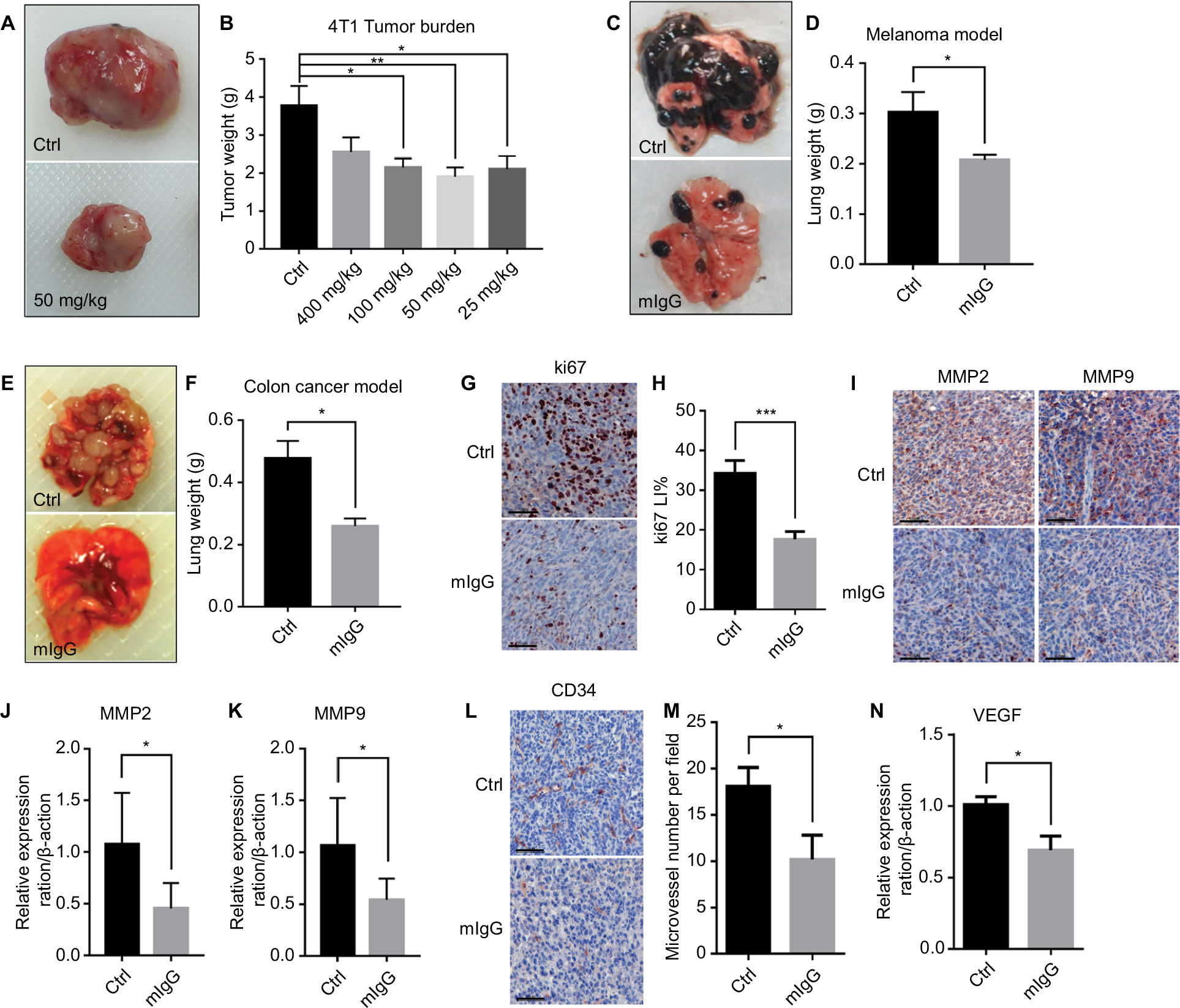 | Figure 2 Mouse IgG inhibits tumor progression and metastasis in vivo. Notes: Administration of mouse IgG inhibited tumor growth in 4T1 model (A, B, n=10, *P<0.05, **P<0.01), suppressed tumor invasion in B16F10 lung metastasis model (C, D, n=20, *P<0.05), and CT26 lung metastasis model (E, F, n=20, **P<0.01). Ki-67 immunostaining (G) revealing fewer proliferating tumor cells in mIgG-treated than control 4T1 tumors (bar: 60 µm). Ki67 LI = Ki67+/total cells (H, ***P<0.001). MMP2 and MMP9 immunostaining (I, bar: 60 µm) and the expressions of MMP2 and MMP9 in 4T1 tumor determined by quantitative real-time PCR (J, K, *P<0.05), revealing less metastasis in mIgG-treated than in control 4T1 tumors. CD34 immunostaining (L, bar: 60 µm, M, *P<0.05) and the expression of VEGF in 4T1 tumors determined by quantitative real-time PCR (N, *P<0.05), revealing less angiogenesis in mIgG-treated than in control 4T1 tumors. Abbreviations: Ctrl, control; IgG, immunoglobulin G; mIgG, mouse IgG. |
In the melanoma and colon cancer models, we tested the effect of systemic administration of IgG on animal survival. In the melanoma model, mice that received mIgG treatment achieved a median survival of 30 days vs 26.5 days in control group (Figure 3A, P<0.05). In the colon cancer model, mice in the IgG treatment group had a median survival of 70 days, while the control group had a median survival of 49 days (Figure 3B, P<0.01). These results demonstrated that administration of mIgG in cancer-bearing mice effectively prolonged their survival duration.
 | Figure 3 Mouse IgG prolonged the survival of cancer-bearing mice. Note: Administration of mouse IgG prolonged the survival durations in the B16-F10 melanoma model (A, n=20) and the CT26 colon cancer model (B, n=11). Abbreviations: Ctrl, control; IgG, immunoglobulin G; mIgG, mouse IgG. |
IgG prevented the development of tumor in vivo
In the melanoma and colon cancer animal models, we tested the possibility of using IgG to prevent the development of cancer. In melanoma model, we injected mIgG intravenously first and then inoculated low-dose tumor cells (1.5×105) to simulate early cancer development. We tested different dosages of IgG including 50, 100, 200, and 400 mg/kg and found that 100 mg/kg was the optimal dosage (Figure 4A, B). Then we tested different time schedule of injection to achieve the best effect (Figure 4C–F). As shown in Figure 4E, injection of 100 mg/kg mIgG twice with an interval of 14 days prior to tumor inoculation was found to be the best strategy to alleviate pulmonary metastasis. In the colon cancer model, injection of 100 mg/kg mIgG thrice on days –8, –4, and –1 prior to tumor inoculation gave satisfactory preventive effect (Figure 4H). In addition, preventive administration of mouse IgG could also effectively prolong the survival of tumor-bearing mice in the melanoma model (Figure 4G).
 | Figure 4 Mouse IgG prevented the occurrence of tumor in vivo. Notes: We tested different dosages of mIgG including 50 mg/kg, 100 mg/kg, 200 mg/kg, and 400 mg/kg (A, B, n≥6), and different routes of administration including injection once before tumor inoculation (C, n=6), injection twice with 2 days interval before tumor inoculation (D, n=7), injection twice with 2 weeks interval before tumor inoculation (E, n=7), injection twice with 3 weeks interval before tumor inoculation (F, n=7) in B16-F10 melanoma model (**P<0.01; ***P<0.001). It showed that injection of mIgG (100 mg/kg) twice with 2 weeks interval gave the optimal result in preventing the development of tumor (E) and prolonging the duration of survival (G, n=11). Injection of mIgG prior to tumor inoculation also worked in the CT26 colon cancer model (H, n=22; *P<0.05). Abbreviations: Ctrl, control; IgG, immunoglobulin G; mIgG, mouse IgG. |
Low dosage of IgG produced better effects than high dosages in inhibiting cancer progression
In the breast cancer mouse model, we tested different dosages on the effect of cancer inhibition. It was found that low dosage (25, 50, and 100 mg/kg) was consistently better than high dosages (400 mg/kg) in inhibiting cancer progression (Figure 2B). Similarly, in the melanoma model for preventive experiment, low dosage (100 mg/kg) was consistently better than high dosages (400 mg/kg) in preventing the development of tumor (Figure 4B). Therefore, in subsequent experiments, we used the low-dose mIgG to treat animals and analyzed the results.
IgG promoted proinflammatory response in the tumor microenvironment
To study the mechanisms of the inhibitory effect of IgG on cancer growth, we examined the effect of IgG on immune responses. A number of cytokines in the mouse tumor were examined with qPCR analysis. We found that mIgG injection led to increased expressions of TNF-α, INF-γ, and IL-1β which cause inflammatory reaction (Figure 5A–C). In addition, the expression of GM-CSF, which promotes differentiation and maturation of granulocyte and monocyte, was increased in the IgG-treated groups in comparison to the controls (Figure 5D). SDF-1 with strong chemotactic lymphocyte function showed no difference in expression (Figure 5E). These results indicated that mIgG treatment enhanced inflammatory responses in the tumor microenvironment.
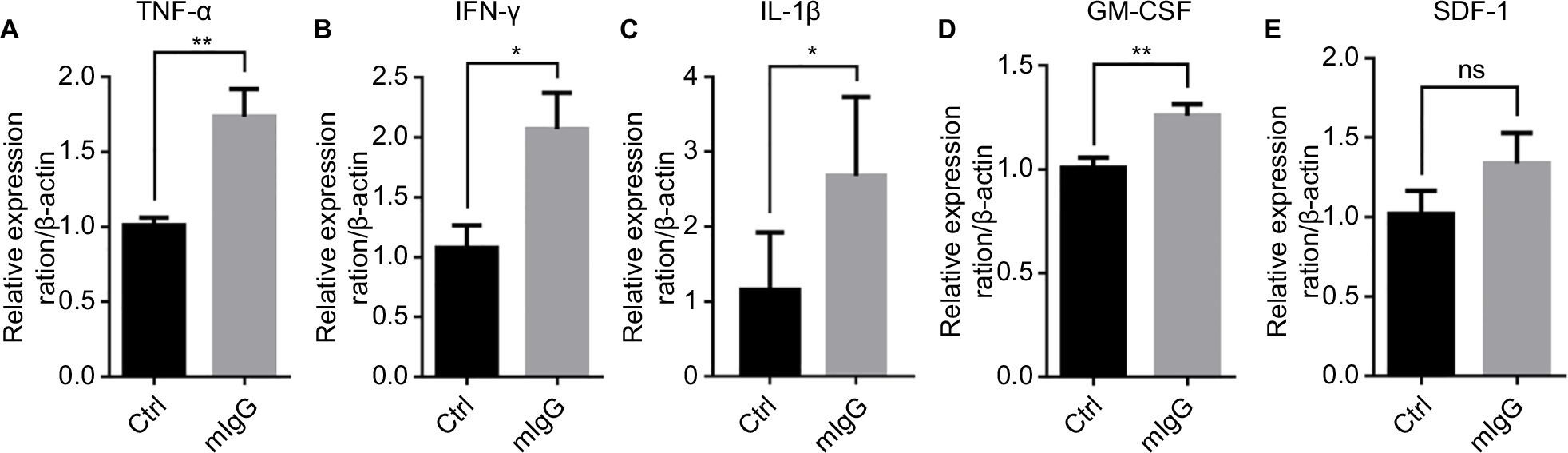 | Figure 5 Mouse IgG promotes proinflammatory response in the tumor microenvironment. Notes: The expressions of TNF-α (A), INF-γ (B), IL-1β (C), GM-CSF (D), and SDF-1 (E) in the 4T1 breast cancer model with or without mIgG treatment, as determined with quantitative real-time PCR. Relative expression ratio of cytokine/β-actin indicates the expression of each cytokine vs β-actin in the presence or absence of mIgG (*P<0.05, **P<0.01, NS P>0.05). Abbreviations: Ctrl, control; IgG, immunoglobulin G; INF-γ, interferon gamma; mIgG, mouse IgG; ns, not significant; TNF-α, tumor necrosis factor alpha. |
IgG polarized macrophages from M2 to M1 and inhibited cancer growth in vitro
To unravel the mechanisms for the effect of IgG on tumor inhibition, we first treated cultured 4T1 mouse breast cancer cells with mIgG in vitro. No inhibition of cancer cells was observed (Figure 6A). We then tested the effect of IgG on macrophages. We treated mouse macrophage RAW246.7 with mIgG (0.5 mg/mL, similar to the optimal dosage used in the in vivo experiment) for 24 hours, and then renewed the culture medium for another 24 hours. We collected the supernatant and found that the supernatant inhibited the proliferation of breast cancer cells significantly (Figure 6B). In addition, we co-cultured mIgG-treated macrophages with tumor cells (CT26 cells and B16-F10 cells) and found that the migration of tumor cells was effectively inhibited (Figure 6D–F). Then different cytokines in cell culture supernatants were measured by ELISA. We found that there was a significant increase of TNF-α and IL-6 secreted in cell culture supernatants and a decrease of IL-10 (Figure 6C). To verify the results obtained in ELISA, we performed protein array analysis for a wider range of proteins with same supernatants. Identical results in TNF-α, IL-6, and IL-10 were obtained. Several other proteins were also increased, including C5/C5a, MIP-2, TIMP-1, CCL5, and IL-1α. The expressions of some other proteins including CXCL12 and CCL1 were suppressed (Figure 6G). Meanwhile, we found that iNOS, a well recognized marker of M1 macrophages, was highly upregulated in tumors derived from mIgG-treated mice (Figure 6H, K). In contrast, decreased expression of Arg1 (Figure 6I), Ym1 (Figure 6J), and CD206 (Figure 6K), as M2 markers, were found in tumors derived from mIgG-treated mice. Therefore, it appeared that mIgG altered the polarization of macrophages in the tumor microenvironment and enriched proinflammatory cytokine expression to limit tumor growth.
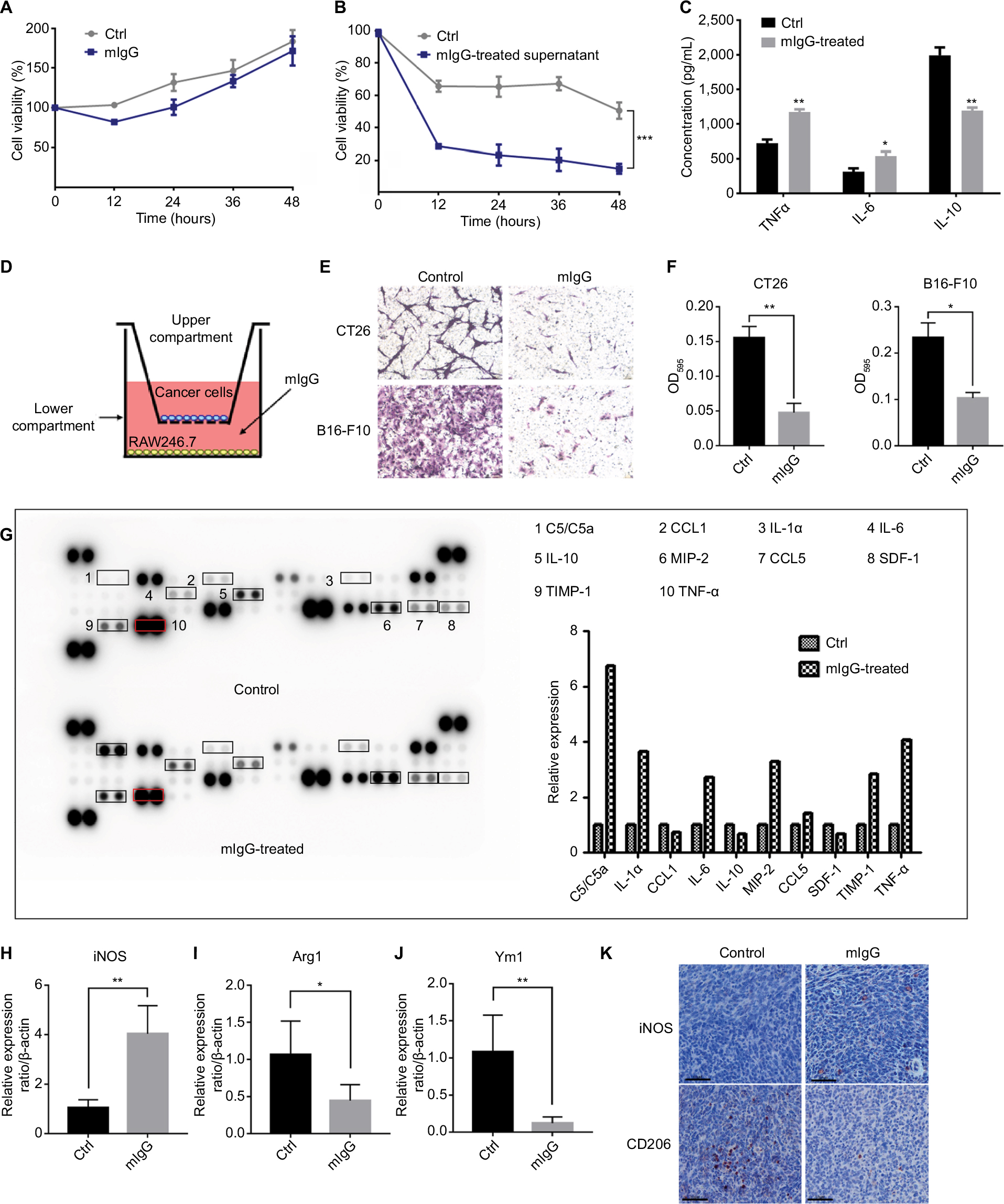 | Figure 6 Role of IgG in antitumor effect of macrophages. Notes: Mouse IgG had no effect when tumor cells were treated directly (A). But 4T1 breast cancer cells cultured with the supernatants of mouse macrophages treated with mIgG (0.5 mg/mL, 0.5 mg/mL, BSA as control) showed that mIgG suppressed tumor cell proliferation via macrophages (B, ***P<0.001). The concentration of IL-6, IL-10, and TNF-α in culture supernatants of mouse macrophages treated with mIgG or BSA (control) (C, *P<0.05, **P<0.01). Mouse macrophages RAW246.7 cells were treated with/without mIgG for 24 hours and 3×104 CT26 or B16F10 cells were seeded in the upper compartment of transwell inserts (D). After 24 hours of incubation, migrated cells were fixed and subsequently stained with crystal violet (E). Quantitative assay of migrated cells by solubilizing crystal violet with 33% acetic acid and absorbance (OD595) was measured by a microplate reader (F, *P<0.05, **P<0.01). Culture supernatants of mouse macrophages treated with mIgG or BSA (control) were measured by mouse cytokine array analysis (G). The graph shows the relative fold changes of protein concentration with significant difference upon mIgG treatment. Pretreatment control was normalized to 1. The expressions of iNOS (H), Arg1 (I), and Ym1 (J) in the 4T1 tumors with or without mIgG treatment, as determined with quantitative real-time PCR (*P<0.05, **P<0.01). iNOS and CD206 immunostaining (K) revealing fewer M2 macrophages and more M1 macrophages in mIgG-treated than control 4T1 tumors (bar: 60 µm). Abbreviations: Ctrl, control; IgG, immunoglobulin G; iNOS, inducible nitric oxide synthase; mIgG, mouse IgG; TNF-α, tumor necrosis factor alpha. |
Discussion
In this study, several original observations were made in addition to confirming that IgG could indeed inhibit cancer growth in mice. The findings of our study are significant for cancer therapy for a number of reasons. In contrast to previous reports, we used mouse IgG instead of human IgG and excluded the possible uncontrollable effect of allogenic reaction of the host to foreign antibodies in the animal experiments. In addition, we found that low dosages (50 and 100 mg/kg) of IgG is more effective than high dosages (400 mg/kg) in inhibiting cancer, and this would translate to substantial saving for patients when this treatment is used clinically. This would also make the treatment more feasible and practical as the previously recommended dosage of 1–2 g/kg body weight would consume a huge amount of human IgG that would make the treatment unattainable for many patients. We also found that regular IgG treatment alone could prolong the life of cancer-bearing mice. This would suggest that late-stage cancer patients might benefit from using IVIg in improving quality of life and prognosis. Most importantly, we found that low-dose IgG (100 mg/kg) treatment prior to cancer cell inoculation had the effect of preventing the development of cancers in mice, while without such treatment all other mice developed cancer following low-dose cancer cell inoculation. This indicates that prophylactic anticancer treatment could be tested in population with high cancer incidence, such as people with advanced age, smoking, occupational hazardous exposure, family history of cancer, etc. In general, those who were confirmed to have a high risk of cancer could be candidates for IgG treatment as a cancer preventive measure. The inhibitory effect was obtained in all the three cancer types tested, suggesting that the beneficial effect could be universal but not cancer type specific. The fact that IVIg treatment has been safely employed to treat many diseases clinically without many side effects makes IVIg a safe and reliable choice to treat cancer at all stages, as well as preventing cancer in the general population.
There are similarities and differences between our findings and the previous reports. Shoenfeld and Fishman first reported that administration of high-dose IVIg (25 mg/mouse) inhibited tumor metastasis in B16-F10 melanoma and MCA-105 sarcoma mouse models.12 Damianovich et al observed that mice exposed to low-dose IVIg (100, 10, and 2 mg/kg) had a significantly lower mean lung weight than the untreated ones.27 However, human IgG is a heterologous protein to mouse which would develop an immune response against human proteins.21,28,29 This would interfere with the results in animal experiments. In our study, we found that mouse IgG is more suitable than human IgG in treating mIgG, so we used different dosages of mouse IgG instead of human IgG. We found that certain low-dose IVIg treatment could achieve the best antitumor effect, while lower or higher dosages were less effective.
Despite the extensive usage of IVIg clinically to treat many diseases, the mechanisms of its action have not been fully understood, and the antitumor mechanism remains controversial. IVIg was reported to inhibit tumor cell proliferation in vitro and was thought to be due to modulation of programmed cell death.11,27,30 Expression of cytokines modulates immune cell activity, which might contribute to potential antitumoral actions of IVIg. Shoenfeld and Fishman speculated that the antitumor effect was mainly induced by upregulation of IL-12, a molecule with anti-angiogenic and NK cell-activating properties.12 Domínguez-Soto et al indicated that IVIg was capable of driving an M2 (protumoral)-to-M1 (antitumoral) switch in macrophages, which could explain the antitumoral activity of IVIg.31 Yasuma et al observed that IVIg, especially IgG1, could suppress angiogenesis via FcγRI in several animal tumor models.32 MMP-9 downregulation induced by IVIg was also reported to limit tumor spreading.33
Previous studies on IVIg suggested that its mechanism might be complicated. Our observation on cytokines and macrophages might not be inclusive but would provide some of the explanations to its actions. It appears that IgG might inhibit cancer growth by promoting inflammation, as we found that cytokines like TNF-α, IFN-γ, IL-1β, and GM-CSF were upregulated in the mIgG-treated tumor microenvironment. However, different from previous reports,10,11,27 we found that mouse IgG does not directly inhibit the growth of tumor cells in vitro. Therefore, the preventive effect of IgG was most likely an immunologic mechanism responsible for the decreased proliferation and metastasis of tumor cells in vivo. Based on our observation, IVIg exerts proinflammatory effects at least partially through macrophages. It is well known that macrophages can be polarized into inflammatory M1 from anti-inflammatory M2 phenotypes. Polarized macrophages are different in their functions, effector molecule expressions, and cytokine production.34 The classically activated M1 macrophages are potent effector cells that kill tumor cells and produce copious amounts of proinflammatory cytokines, including TNF-α, IL-1, IL-6, and IL-12. In contrast, M2 cells inhibit inflammatory responses; promote angiogenesis, tissue remodeling, and repair; and typically produce IL-10, etc.35–37 Tumor-associated macrophages are known as a polarized M2 macrophage population with poor antigen presenting capacity but can promote tumor-cell proliferation.38 In our experiments, macrophages exposed to IgG expressed more proinflammatory cytokines like IL-1, IL-6, and TNF-α and less anti-inflammatory factor like IL-10. In addition, administration of mIgG in mouse model significantly increased M1 hallmarks like iNOS; reduced the expression of M2 markers including Arg1, Ym1, and mannose receptor (CD206); inhibited the formation of blood vessels in tumor; and downregulated the secretion of MMPs. These results suggest that IgG may stimulate tumor-associated macrophages to polarize from a protumoral M2 state into an antitumoral M1 state, produce proinflammatory cytokines, and suppress angiogenesis to inhibit tumor progression.
Conclusion
Our study suggests that IVIg can inhibit cancer progression as well as prolong cancer host survival. In our experiments, low-dose mIgG was better than high-dose mIgG in achieving this effect. In addition, low-dose administration of IgG has the beneficial effect of preventing the development of cancers. Our findings also suggest that stimulation of macrophages to polarize from M2 to M1 might be one of the mechanisms with which IgG exerts its action. This and previous reports strongly suggest that well organized clinical trials of IVIg to treat cancer patients and to prevent cancer from developing are warranted to verify the benefits and possible limitation of this promising strategy to combat cancer.
Acknowledgments
This research was funded by the National Natural Science Foundation of China (81872334) and Li Ka Shing Foundation. The authors would like to thank the Laboratory Animal Center and the Center for Core Facilities of Shantou University Medical College for supporting and providing help to this research.
Disclosure
The authors report no conflicts of interest in this work.
References
Yang Y. Cancer immunotherapy: harnessing the immune system to battle cancer. J Clin Invest. 2015;125(9):3335–3337. | ||
Chen G, Bodogai M, Tamehiro N, Shen C, Dou J. Cancer immunotherapy: theory and application. J Immunol Res. 2018;2018:1–2. | ||
Lee YT, Tan YJ, Oon CE. Molecular targeted therapy: treating cancer with specificity. Eur J Pharmacol. 2018;834:188–196. | ||
Weiner GJ. Building better monoclonal antibody-based therapeutics. Nat Rev Canc. 2015;15(6):361–370. | ||
Kazatchkine MD, Kaveri SV. Immunomodulation of autoimmune and inflammatory diseases with intravenous immune globulin. N Engl J Med. 2001;345(10):747–755. | ||
Shoenfeld Y, Katz U. IVIg therapy in autoimmunity and related disorders: our experience with a large cohort of patients. Autoimmunity. 2005;38(2):123–137. | ||
Subhadra C, Dudek AZ, Rath PP, Lee MS. Improvement in visual fields in a patient with melanoma-associated retinopathy treated with intravenous immunoglobulin. J Neuroophthalmol. 2008;28(1):23–26. | ||
Chapel HM, Lee M. Immunoglobulin replacement in patients with chronic lymphocytic leukemia (CLL): kinetics of immunoglobulin metabolism. J Clin Immunol. 1992;12(1):17–20. | ||
Murie-Fernández M, Gurpide A, de la Cruz S, de Castro P. Total remission of thymus carcinoma after treatment with intravenous immunoglobulin. Clin Transl Oncol. 2006;8(9):697–699. | ||
Merimsky O, Meller I, Inbar M, Bar-Yehuda S, Shoenfeld Y, Fishman P. A possible role for IVIg in the treatment of soft tissue sarcoma: a clinical case and an experimental model. Int J Oncol. 2002;20(4):839–843. | ||
Fishman P, Bar-Yehuda S, Shoenfeld Y. IVIg to prevent tumor metastases (review). Int J Oncol. 2002;21(4):875–880. | ||
Shoenfeld Y, Fishman P. Gamma-globulin inhibits tumor spread in mice. Int Immunol. 1999;11(8):1247–1252. | ||
Sapir T, Shoenfeld Y. Uncovering the hidden potential of intravenous immunoglobulin as an anticancer therapy. Clin Rev Allergy Immunol. 2005;29(3):307–310. | ||
Schachter J, Katz U, Mahrer A, et al. Efficacy and safety of intravenous immunoglobulin in patients with metastatic melanoma. Ann N Y Acad Sci. 2007;1110(1):305–314. | ||
Carmeli Y, Mevorach D, Kaminski N, Raz E. Regression of Kaposi’s sarcoma after intravenous immunoglobulin treatment for polymyositis. Cancer. 1994;73(11):2859–2861. | ||
Shoenfeld Y, Levy Y, Fishman P. Shrinkage of melanoma metastases following high dose intravenous immunoglobulin treatment. Isr Med Assoc J. 2001;3(9):698–699. | ||
Ogihara Y, Ogata S, Nomoto K, et al. Transcriptional regulation by infliximab therapy in Kawasaki disease patients with immunoglobulin resistance. Pediatr Res. 2014;76(3):287–293. | ||
Krause I, Shoenfeld Y. Intravenous immunoglobulin treatment for fibrosis, atherosclerosis, and malignant conditions. Methods Mol Med. 2005;109:403–408. | ||
Sherer Y, Levy Y, Shoenfeld Y. IVIG in autoimmunity and cancer – efficacy versus safety. Expert Opin Drug Saf. 2002;1(2):153–158. | ||
Sobieszczańska M, Tubek S, Poplicha D, Grabelus A, Pawełczak J. Henoch-Schönlein purpura (Hsp) and high-dose immunoglobulin treatment in patient with familiar prostatic adenocarcinoma. Hum Vaccin Immunother. 2014;10(2):358–359. | ||
Hermeling S, Jiskoot W, Crommelin D, Bornaes C, Schellekens H. Development of a transgenic mouse model immune tolerant for human interferon beta. Pharm Res. 2005;22(6):847–851. | ||
Iqbal S, Lenz HJ. Integration of novel agents in the treatment of colorectal cancer. Cancer Chemother Pharmacol. 2004;54(Suppl 1): S32–39. | ||
Schwab I, Nimmerjahn F. Intravenous immunoglobulin therapy: how does IgG modulate the immune system? Nat Rev Immunol. 2013;13(3):176–189. | ||
Clynes R. IVIg therapy: Interfering with interferon-gamma. Immunity. 2007;26(1):4–6. | ||
Chen Z, Huang X, Ye J, et al. Immunoglobulin G is present in a wide variety of soft tissue tumors and correlates well with proliferation markers and tumor grades. Cancer. 2010;116(8):1953–1963. | ||
Tanoue T, Nishitani Y, Kanazawa K, Hashimoto T, Mizuno M. In vitro model to estimate gut inflammation using co-cultured Caco-2 and RAW264.7 cells. Biochem Biophys Res Commun. 2008;374(3):565–569. | ||
Damianovich M, Solomon AS, Blank M, Shoenfeld Y. Attenuation of colon carcinoma tumor spread by intravenous immunoglobulin. Ann N Y Acad Sci. 2007;1110(1):567–577. | ||
Ottesen JL, Nilsson P, Jami J, et al. The potential immunogenicity of human insulin and insulin analogues evaluated in a transgenic mouse model. Diabetologia. 1994;37(12):1178–1185. | ||
Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of therapeutic proteins: influence of aggregation. J Immunotoxicol. 2014;11(2):99–109. | ||
Prasad NK, Papoff G, Zeuner A, et al. Therapeutic preparations of normal polyspecific IgG (IVIg) induce apoptosis in human lymphocytes and monocytes: a novel mechanism of action of IVIg involving the Fas apoptotic pathway. J Immunol. 1998;161(7):3781–3790. | ||
Domínguez-Soto A, de las Casas-Engel M, Bragado R, et al. Intravenous immunoglobulin promotes antitumor responses by modulating macrophage polarization. J Immunol. 2014;193(10):5181–5189. | ||
Yasuma R, Cicatiello V, Mizutani T, et al. Intravenous immune globulin suppresses angiogenesis in mice and humans. Signal Transduction and Targeted Therapy. 2016;1:15002. | ||
Shapiro S, Shoenfeld Y, Gilburd B, Sobel E, Lahat N. Intravenous gamma globulin inhibits the production of matrix metalloproteinase-9 in macrophages. Cancer. 2002;95(9):2032–2037. | ||
Mantovani A, Sica A, Macrophages SA. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22(2):231–237. | ||
Eubank TD, Roberts RD, Khan M, et al. Granulocyte macrophage colony-stimulating factor inhibits breast cancer growth and metastasis by invoking an anti-angiogenic program in tumor-educated macrophages. Cancer Res. 2009;69(5):2133–2140. | ||
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–896. | ||
Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86(5):1065–1073. | ||
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23(11):549–555. |
Supplementary materials
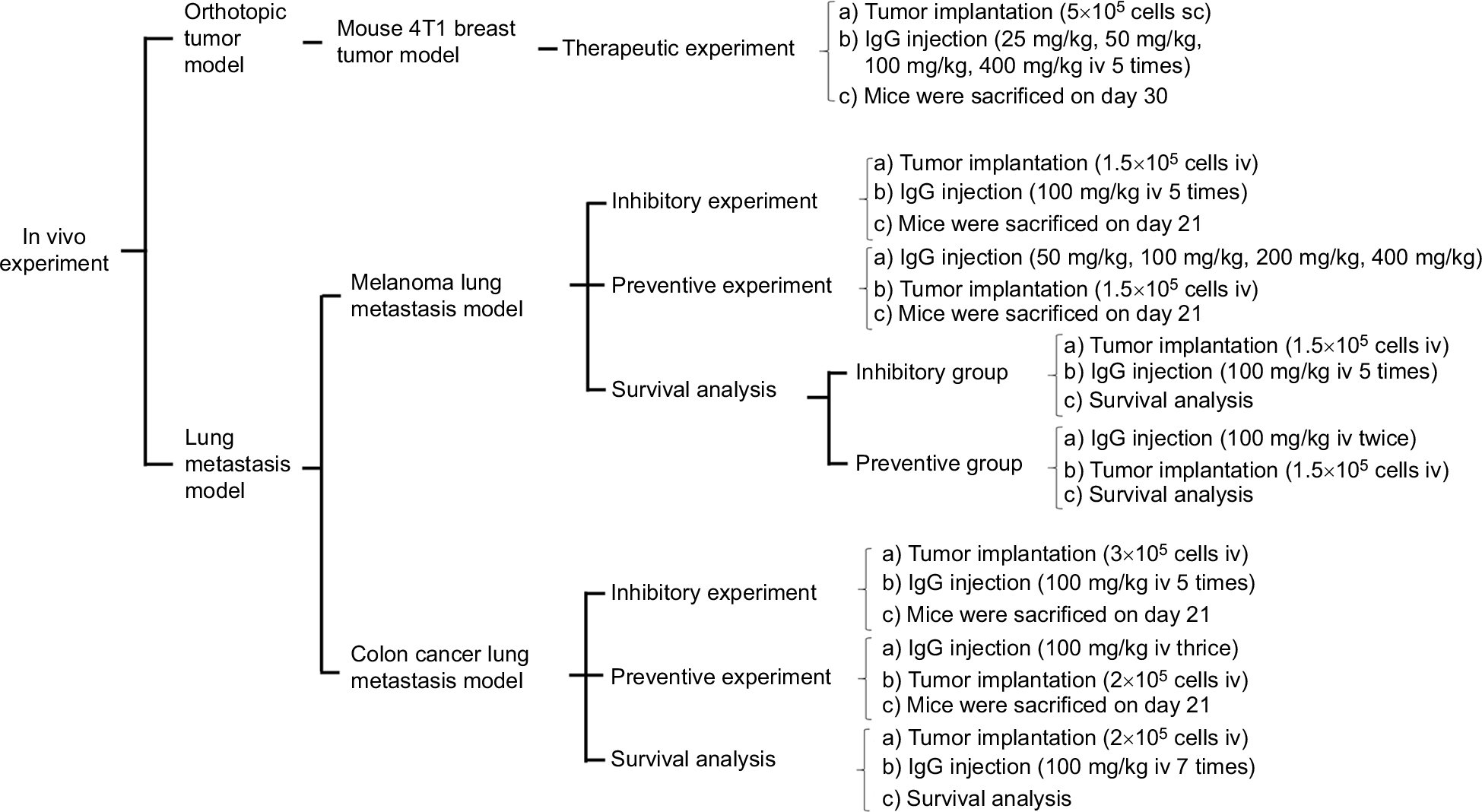 | Figure S1 The diagram shows the experimental design of various groups of mice treated with different protocols. Notes: We tested three tumor types, two ways of inoculation, and different dosages of IgG for treatment. We also tested different survival durations and cancer prevention strategies. Abbreviations: IgG, immunoglobulin G; sc, subcutaneous; iv, intravenous. |
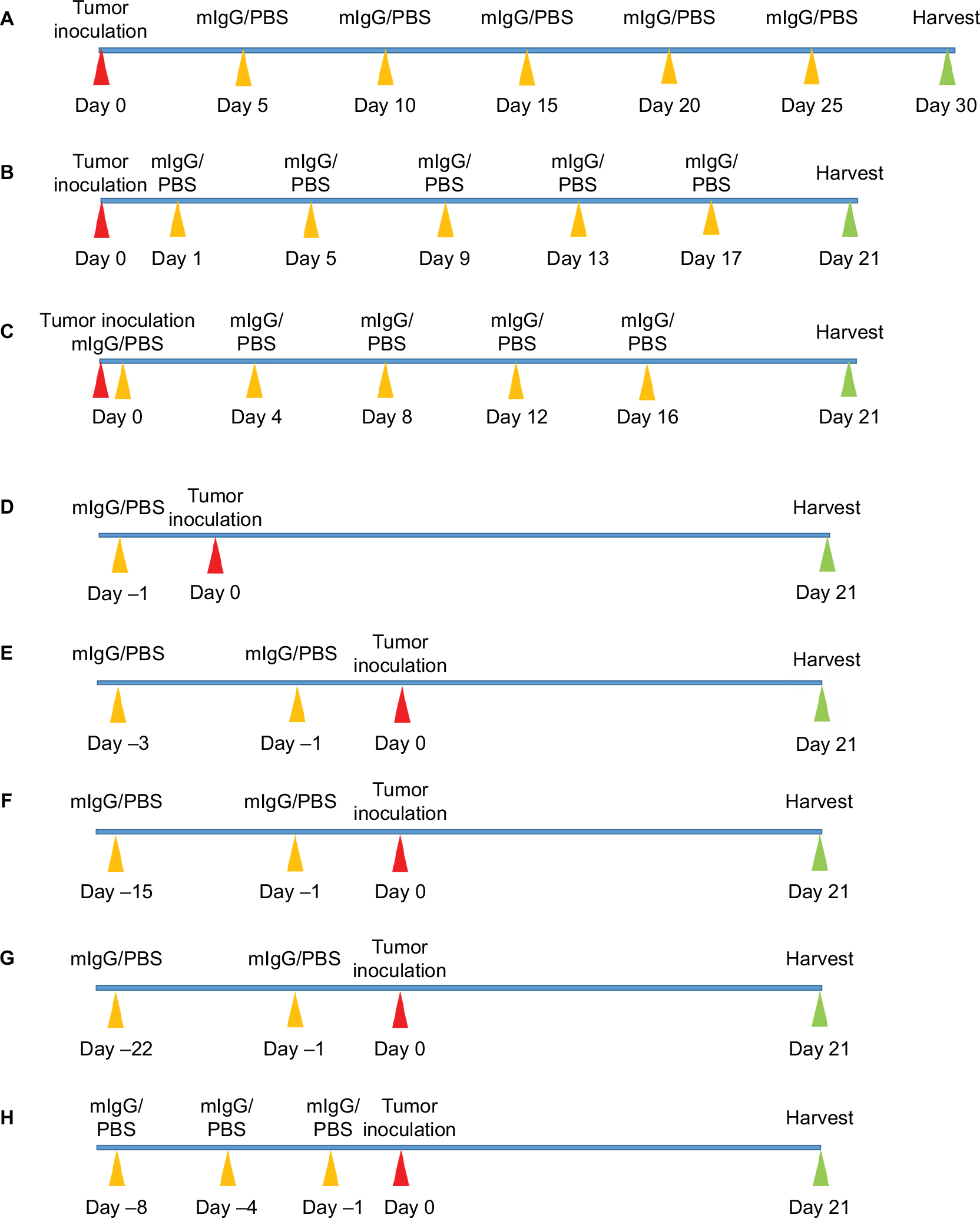 | Figure S2 Timelines of animal experiments. Notes: Red arrows indicate the time of tumor inoculation, yellow arrows indicate IgG administration, and green arrows indicate termination. The timelines of breast cancer mouse model (A), melanoma mouse model (B), and colon cancer mouse model (C) for therapeutic experiment or inhibitory experiment. The different routes of administration including injection once before tumor inoculation (D), injection twice with 2 days interval before tumor inoculation (E), injection twice with 2 weeks interval before tumor inoculation (F), and injection twice with 3 weeks interval before tumor inoculation (G) in melanoma mouse model for preventive experiment. The timelines of colon cancer mouse model for preventive experiment (H). Abbreviations: IgG, immunoglobulin G; mIgG, mouse IgG. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.