Back to Journals » OncoTargets and Therapy » Volume 12
Nonhomologous end joining key factor XLF enhances both 5-florouracil and oxaliplatin resistance in colorectal cancer
Authors Liu Z, Yu M, Fei B, Sun J, Wang D
Received 31 October 2018
Accepted for publication 16 January 2019
Published 20 March 2019 Volume 2019:12 Pages 2095—2104
DOI https://doi.org/10.2147/OTT.S192923
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr William C. Cho
Zhuo Liu,1 Miao Yu,1 Bingyuan Fei,1 Jing Sun,2 Dongxin Wang3
1Department of Gastrointestinal Colorectal and Anal Surgery, China–Japan Union Hospital of Jilin University, Changchun, Jilin 130021, China; 2Department of Biochemistry and Molecular Medicine, The George Washington University, Washington, DC 20052, USA; 3Department of Anesthesiology, Jilin Cancer Hospital, Changchun, Jilin 130021, China
Background: Colorectal cancer (CRC) is the third commonly diagnosed cancer with a high risk of death. After curative surgery, 40% of patients will have metastases or develop recurrence. Therefore, chemotherapy is significantly responsible as the major therapy method. However, chemoresistance is found in almost all metastatic patients and remains a critical obstacle to curing CRC.
Materials and methods: Cell viability is analyzed by sulforhodamine B staining assay. The nonhomologous end joining (NHEJ) repair ability of each cell line was determined by NHEJ reporter assay. mRNA expression levels of NHEJ factors are detected by real-time quantitative polymerase chain reaction. The protein expression levels were observed by western blot assay.
Results: Our study found that 5-florouracil (5-Fu) and oxaliplatin (OXA)-resistant HCT116 and LS174T cells showed upregulated efficiency of DNA double-strand repair pathway NHEJ. We then identified that the NHEJ key factor XLF is responsible for the chemoresistance and XLF deficiency sensitizes CRC cells to 5-Fu and OXA significantly.
Conclusion: Our research first demonstrates that the NHEJ pathway, especially its key factor XLF, significantly contributes to chemoresistance in CRC.
Keywords: XLF, nonhomologous end joining, 5-florouracil, oxaliplatin, chemoresistance, colorectal cancer
Introduction
Colorectal cancer (CRC) is one of the major causes of death worldwide.1,2 It is the third most common cancer in men and women.1 Because of changes in risk factors, such as reduced smoking and a healthier diet, the death rate from CRC has decreased for decades. However, the incidence of CRC is still growing, and tumors recur after surgery while chemotherapy still needs to overcome chemoresistance.3 Unfortunately, the majority of patients diagnosed with CRC are at advanced stages that require chemotherapy.4 5-Fluorouracil (5-Fu) has been used in first-line chemotherapy, against a variety of cancers, for more than five decades by inhibiting thymidylate synthase that causes thymineless death in cancer cells.5 Oxaliplatin (OXA) is another first-line chemotherapy drug used in CRC, most commonly in combination with anticancer drugs such as 5-Fu.6 Sadly, multidrug resistance has become a critical obstacle in this chemotherapy strategy.7 Therefore, identification of new targets to generate synergistic inhibition for cell survival is desperately needed to improve chemotherapy results in CRC.
Both 5-Fu and OXA generate cell death through replication stress, instability of genomes, and DNA damage, eventually.8 Therefore, a DNA repair mechanism could play an important role in chemoresistance. The DNA double-strand break (DSB) is the most toxic DNA lesion that can cause cell death.9 Thus, we are going to investigate the role of double-strand break repair (DSBR) pathways in CRC. Human cells possess two major DSBR pathways: homologous recombination (HR) and nonhomologous end joining (NHEJ).10,11 HR precisely repairs DSBs using a sister chromatid to serve as a template.12 Therefore, HR is limited to the late S–G2 phase of the cell cycle while NHEJ, which is template independent, is performed to repair DSBs through most of the cell cycle.13 HR is initiated via single DNA strand invasion directed by Rad51 to generate a long 3′ overhang.14 The first several steps of strand invasion also require Exo1, Dna2, and RPA, followed by DNA synthesis by DNA polymerases.15,16 After end resection and DNA synthesis, HR will use distinct subpathways – the DSBR pathway or the synthesis-dependent strand annealing (SDSA) pathway – to finish final ligation.14 NHEJ is initiated by Ku70/80 heterodimers (Ku) interacting with both ends of DSB strands.17 Ku recruits other NHEJ key factors including DNA-dependent protein kinase, catalytic subunits (DNAPKcs), XLF, XRCC4, and ligase IV.18–22 Between DNA tethering and final ligation, DSBs usually go through DNA end processing to generate ligatable DNA ends that require different DNA end processors upon distinct DNA damage types.23–27
XLF is a 33-kDa protein located in chromosome 2 and exists in the nucleus as a dimer via its coil–coil domain of the monomers.28 It has been known as a scaffold protein participating in NHEJ since 2006.29,30 XLF enzymatic activity has not yet been found; the most significant contribution of XLF to NHEJ is stimulating the final ligation step by interacting with ligase IV.30,31 Interestingly, XLF is believed to form a long filament with XRCC4 at DNA breaks to help DNA gap synapsis.32 Although XLF deficiency in mice does not affect V(D)J recombination like all other NHEJ key factors, XLF deficiency in human still prevents NHEJ and results in immunodeficiency.33 The role of XLF in NHEJ is essential but is still not thoroughly clear. Our research identified that XLF is overexpressed in chemoresistant CRC cells and significantly contributes to 5-Fu and OXA resistance.
Materials and methods
Cell lines and cell cultures
HCT116 cells (ATCC CCL-247; American Type Culture Collection, Manassas, VA, USA) and LS174T cells (ATCC CL-188) were cultured at 37°C in 5% CO2 atmosphere in McCoy’s 5a Medium (ATCC 30-2007) and Eagle’s Minimum Essential Medium (ATCC 30-2003), respectively with 10% FBS for less than 6 months. HCT116 and LS174T 5-Fu-resistant cells were generated by incubating with 5 μM and 10 μM 5-Fu, respectively, for 3 months. HCT116 and LS174T OXA-resistant cells were generated by incubating with 7 μM and 15 μM OXA, respectively for 3 months.
Cell viability assay
Cells were seed at 4×103 cells/well and cultured overnight to allow adherence. Cells were incubated with drug for 72 h and the cell viability was detected using the sulforhodamine B (SRB) assay. Cells were fixed using 100 μL/well of 10% trichloroacetic acid at 4°C for 1 h. The plate was washed four times with slow-running tap water and air-dried for 1 h at room temperature (RT) or for 20 min in a fume hood. Cells were stained by 100 μL/well of 0.02% SRB in 1% acetate acid for 1 h at RT. Plates were washed three times with 200 μL/well of 1% acetate acid and air-dried. Then, 200 μL/well of 10 mM Tris HCl, pH 10.5, was added to each well to extract SRB with 1 h of shaking on an orbital shaker. The absorbance was measured at 510 nm by a microplate reader (Synergy 2; BioTek Instruments Inc., Winooski, VT, USA).
Generation of XLF-deficient HCT116 cell line using CRISPR/Cas9
Cas9 along with XLF guide RNA plasmid was constructed by ligating oligonucleotide duplexes, which target exon1 of XLF, into BbsI cut pX330-U6-Chimeric_BB-CBh-hSpCas9 (Addgene #42230; Addgene, Watertown, MA, USA). The plasmid was transfected into the HCT116 cell line along with pcDNA3.1.puro using Lipofectamine 2000 and incubated for 72 h. Successfully transfected cells were selected by media containing 100 μg/mL puromycin for 48 h. Cells were harvested and seeded in a 96-well plate at concentrations of 100 cells/mL, 333 cells/mL, or 1,000 cells/mL, and incubated for 2–3 weeks. Individual clones were passaged, expanded, and screened for XLF expression.
HR and NHEJ reporter assay
HR reporter has been described previously.34 NHEJ reporter was obtained from Addgene (Addgene 44026). Briefly, 10 μg of NHEJ reporter plasmid was linearized with 50 U of NheI in 50 μL for 6 h at 37°C. Linearized DNA was purified using a QIAquick Gel Extraction Kit (ID:28704; QIAGEN, Venlo, the Netherlands) gel extraction kit and 1 μg was transfected into HCT116 and LS174T cells using Lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA, USA) following the manufacturer’s recommendations. Cells with chromosomally integrated reporter constructs were selected by incubating in media with 1 mg/mL geneticin 24 h after transfection for 2 weeks. Plasmid integrated cells were seeded at 3×105 cells/mL in a 6-well plate and cultured for 24 h to allow adherence. Then, 2 μg/well of I-SceI was transfected into the cell using Lipofectamine 3000 and incubated for 48 h. Cells were harvested using trypsin and resuspended in PBS by pipetting 10 times. Green fluorescent protein (GFP)-positive cells were counted by flow cytometry (Beckman Coulter Inc., Brea, CA, USA).
Western blot assay
This method has been described in a previous work.35 Briefly, protein samples were denatured using SDS-PAGE sample buffer, and boiled for 5 min. The samples were then loaded and separated on a 7% polyacrylamide gel (29:1) (1610156; Bio-Rad Laboratories Inc., Hercules, CA, USA) at 120 V for 1.5 h in an electrophoresis apparatus (Bio-Rad). Separated samples were transferred to nitrocellulose membrane at 100 V at 4°C for 1 h. The membrane was blocked using 3% nonfat milk solution diluted in PBS with 0.1% Tween20 and probed by the relevant antibody followed by horseradish peroxidase-conjugated rabbit secondary antibody. The protein signal was developed by SuperSignal™ West Pico PLUS Chemiluminescent Substrate (Thermo Fisher Scientific #34580) and detected by ChemiDoc™ (Bio-Rad).
Results
Generation of chemoresistant CRC cell lines
To identify a pathway or target to overcome chemoresistance, we first generated 5-FU and OXA-resistant cell lines using epithelial CRC cell lines HCT116 (HCT116 Fu-R and HCT116 OXA-R) and LS174T (LS174T Fu-R and LS174T OXA-R). To evaluate resistance, we used the SRB cell survival assay to determine the half-maximal inhibitory concentration (IC50) of cells to Fu and OXA. As compared to the wild-type cell line (HCT116 WT), HCT116 Fu-R showed 3.5-fold resistance to 5-Fu (Figure 1A). Similarly, HCT116 OXA-R cells showed 3.9-fold resistance to OXA (Figure 1B). We also successfully generated 5-Fu and OXA-resistant LS174T cell lines with 3.7-fold and 4-fold increased IC50, respectively (Figure 1C and D).
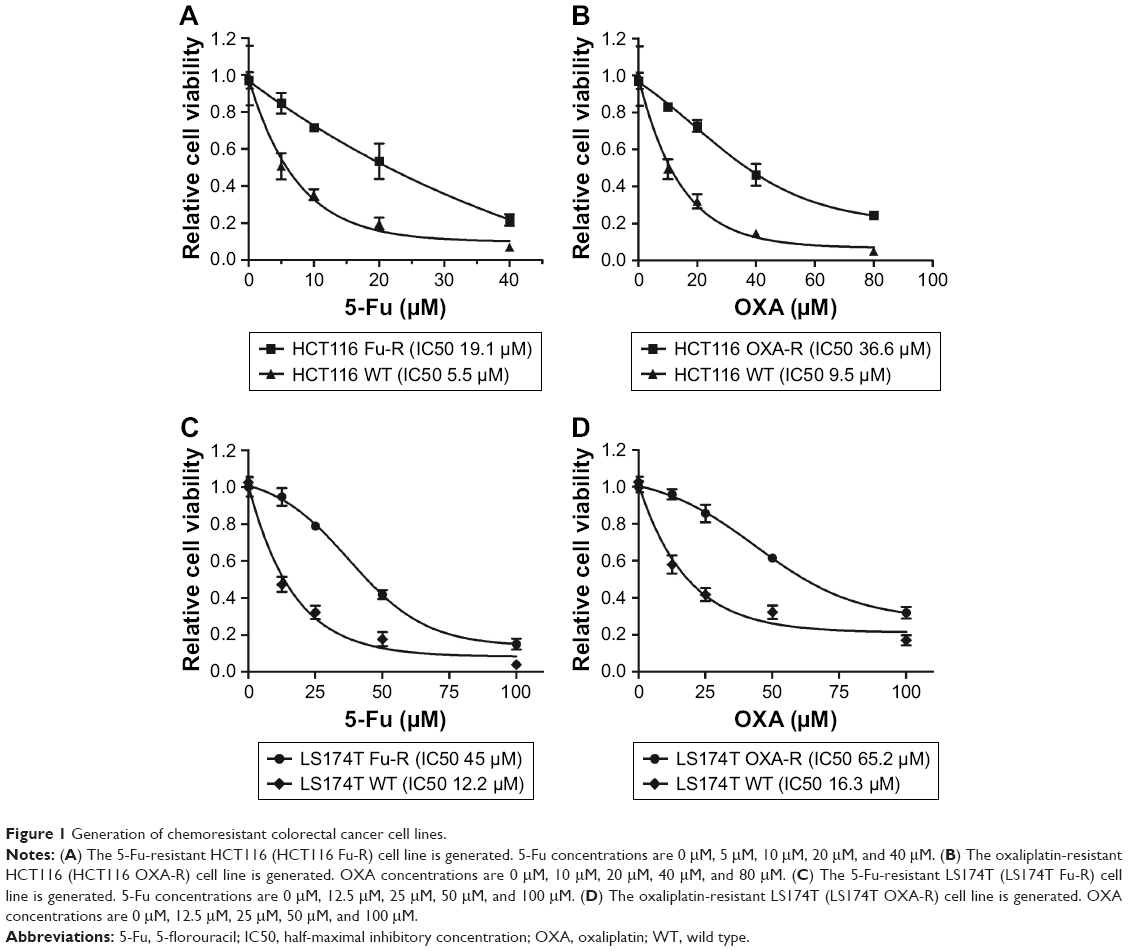 | Figure 1 Generation of chemoresistant colorectal cancer cell lines. |
We further used these cell lines to measure DNA repair efficiency and screen for DNA repair factors that are responsible for chemoresistance.
NHEJ pathway efficiency is significantly increased in chemoresistant CRC cell lines
To evaluate whether DNA repair pathways contribute to chemoresistance of CRC cells, we used plasmid-based reporter assays to measure the efficiency of the two DNA DBSR pathways: HR and NHEJ. We first linearized the plasmid and then transfected the linearized plasmid into the HCT116 WT, HCT116 Fu-R, HCT116 OXA-R, LS174T WT, LS174T Fu-R, and LS174T OXA-R cell lines. The plasmid could have integrated into chromosomes, and cells with successful integration were selected using hygromycin. These stably transfected cell lines were treated with I-SceI restriction enzyme to introduce DNA DSBs. After DNA repair, rejoined DNA will result in restored GFP expression. We used flow cytometry to count the GFP-positive cells out of 80,000 cells. We observed that chemoresistant cells do not show significant change of HR efficiency (data not shown). However, Fu and OXA-resistant cell lines showed significant increase of NHEJ efficiency. Specifically, Fu and OXA-resistant HCT116 cells showed 3-fold and 2.6-fold increased NHEJ efficiency as compared to the wild-type cell line (Figure 2A). In addition, compared to LS174T WT cells, Fu-resistant cells have 2.2-fold more efficient NHEJ whereas OXA-resistant cells have 3.2-fold more efficient NHEJ (Figure 2B).
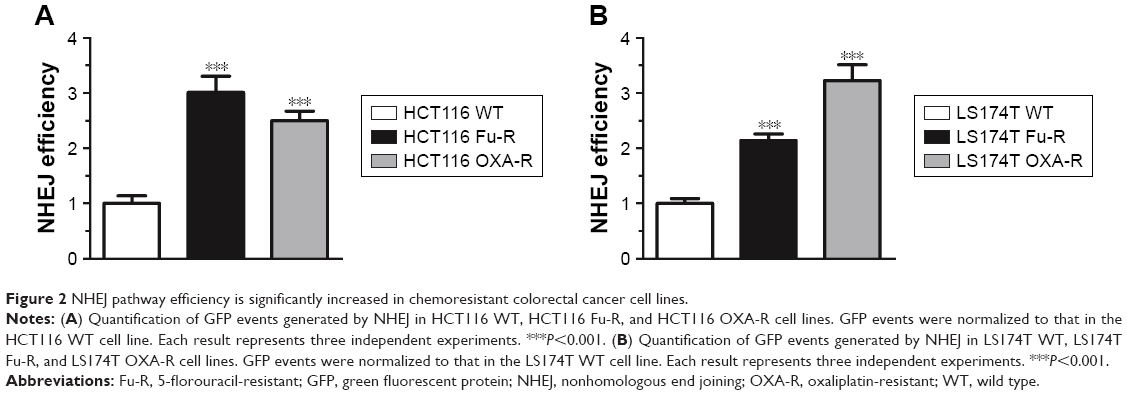 | Figure 2 NHEJ pathway efficiency is significantly increased in chemoresistant colorectal cancer cell lines. |
Our results suggest that, other than base excision repair, the DNA DSBR pathway also contributes to chemoresistance.
XLF expression is upregulated in chemoresistant CRC cells
Since NHEJ efficiency is enhanced in chemoresistant CRC cells, we then wanted to find out whether expression of NHEJ factors is upregulated. We also wanted to identify which factor of NHEJ has the potential to be responsible for chemoresistance. We performed a quantitative polymerase chain reaction (qPCR) to screen the mRNA level of NHEJ factors, including Ku80, DNAPKcs, XLF, XRCC4, and ligase IV. We found that, compared to HCT116 WT cells, the mRNA level of XLF showed a significant increase in both Fu-R and OXA-R cells (Figure 3A). Interestingly, the mRNA level of XLF was also upregulated in Fu- and OXA-resistant LS174T cells as compared to LS174T WT cells (Figure 3B).
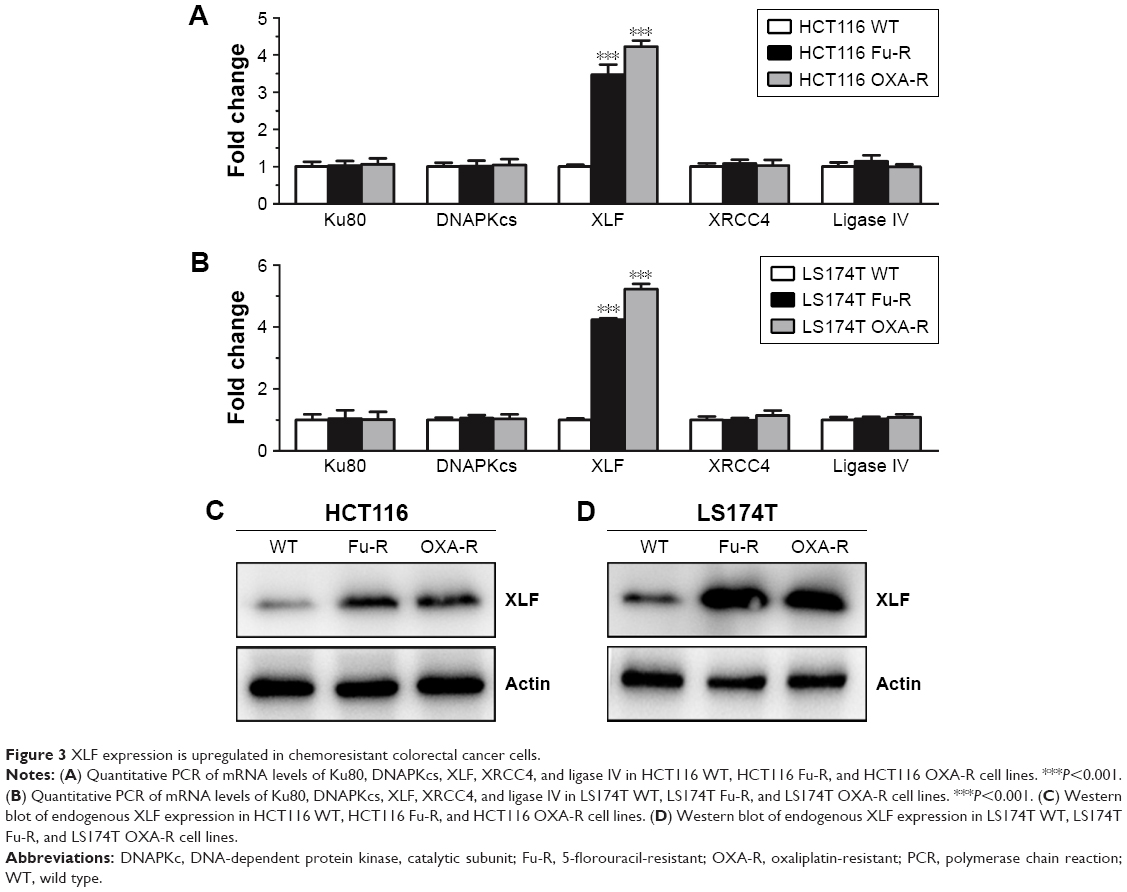 | Figure 3 XLF expression is upregulated in chemoresistant colorectal cancer cells. |
To further evaluate protein expression of XLF, we used western blot assay with antibody against XLF to compare the XLF expression in the HCT116 WT, HCT116 Fu-R, and HCT116 OXA-R cell lines. As we expected, XLF protein expression is upregulated in the HCT116 Fu-R and HCT116 OXA-R cell lines (Figure 3C). Similarly, XLF protein expression is increased in Fu- and OXA-resistant LS174T cells compared to LS174T WT cells (Figure 3D). To examine whether XLF is correlated with chemoresistance in CRC patients, we analyzed XLF mRNA expression levels from 5-Fu-sensitive (n=30) and 5-Fu-resistant (n=45) patient samples. As shown in Figure S1, chemoresistant patients have a significant increase of XLF mRNA levels.
Our result suggests that XLF expression is correlated with 5-Fu and OXA resistance in CRC cells.
XLF deficiency generates a synergistic effect with 5-Fu and OXA
To determine whether XLF is responsible for chemoresistance in CRC cells, we used the CRISPR/Cas9 technique to knockout XLF in the HCT116 Fu-R and HCT116 OXA-R cell lines. We identified 10 XLF knockout clones in each cell line. We randomly picked two clones in each cell line and evaluated XLF expression using western blot assay. Both clones lack XLF compared to the HCT116 Fu-R and HCT116 OXA-R cell lines (Figure 4A and B).
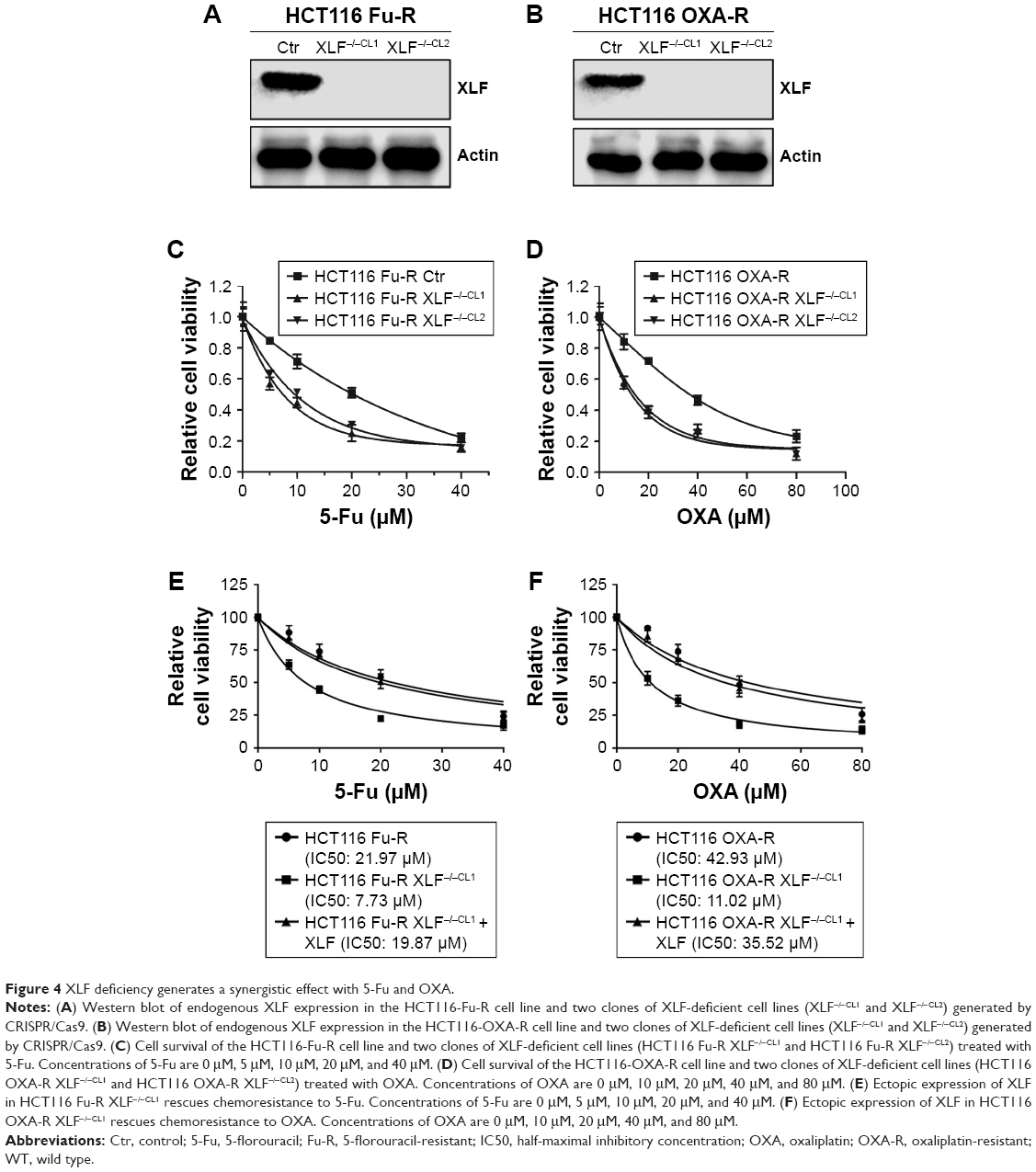 | Figure 4 XLF deficiency generates a synergistic effect with 5-Fu and OXA. |
Since we found out that XLF expression is correlated with chemoresistance, we hypothesize that XLF knockout cells will be more sensitive to 5-Fu and OXA. Thus, we performed the SRB cell survival assay to evaluate drug potency. We observed that XLF-deficient HCT116 Fu-R cell lines (HCT116 Fu-R XLF−/−CL1 and HCT116 Fu-R XLF−/−CL2) are hypersensitive to 5-Fu (Figure 4C). Similarly, XLF deficiency also sensitized OXA-resistant cells to OXA by 5-fold (Figure 4D). To minimize the off-target effect of XLF knockout, we ectopically expressed full-length XLF in HCT116 Fu-R XLF−/−CL1 and HCT116 OXA-R XLF−/−CL1 cells and examined the cell viability to 5-Fu and OXA, respectively. We found that complementation of XLF in XLF-KO cells rescued chemoresistance to 5-Fu and OXA (Figure 4E and F), suggesting XLF indeed contributes to chemoresistance in HCT116 cells.
To determine whether XLF depletion can sensitize wild-type cells to chemotherapies, we knocked down XLF in HCT116 WT cells and examined cell survival in response to 5-Fu and OXA. We observed that XLF depletion indeed sensitized wild-type cells to 5-Fu and OXA by 2.69-fold and 2.60-fold, respectively (Figure S2A and B). Similarly, we observed that XLF knockdown sensitized HT29 cells to 5-Fu (Figure S2C), suggesting the role of XLF in chemoresistance in CRC is relatively universal.
Our data indicate that XLF is one of the DNA repair factors that significantly contributes to chemoresistance in CRC.
Discussion
Chemoresistance, especially cross-resistance, is the major cause of death in metastatic CRC and patients with recurrent CRC.36 It is urgent to find new targeting strategies to overcome chemoresistance. We hypothesized that DNA repair pathways play important roles in chemoresistance since they facilitate DNA recovery after anticancer drug treatments, which target DNA in cancer cells. Since DSB is the most severe DNA damage in cells, DSBR pathways are responsibile for survival of cancer cells. We found that the NHEJ factor, XLF is positively associated with 5-Fu and OXA drug resistance in HCT116 and LS174T cells. However, the mechanism of XLF upregulation in chemoresistant cells remains unclear. The research of XLF regulation is limited and the results vary between cancer types. Zhang et al37 showed that positive cofactor 4 decreases XLF expression in esophageal squamous cell carcinoma. In addition, Liu et al38 demonstrated that depletion of WRN in HeLa cells led to a decrease of XLF mRNA and its promoter activity. Sulkowski et al39 found that PTEN induces expression of XLF at both mRNA and protein levels. We can evaluate whether these regulations are consistent in colon cancer cells. If these regulations cannot be observed in colon cancer cells, we could use an RNA-Seq assay to identify genes that are upregulated in chemoresistant colon cancer. Also, we could overlap possible transcription factors of XLF with the RNA-Seq results to elucidate the transcription factor that is responsible for XLF upregulation in chemoresistant colon cancer cells.
It has been well established that impaired mismatch repair, especially involving SMUG1 and MBD4, is correlated with chemoresistance in CRC.40–42 However, DSBR pathways, which are extremely critical for cell survival, are underexamined for chemoresistance in CRC. Chen et al43 found that Dicer, which regulates NHEJ, is positively correlated to chemoresistance of colon cancer cells. The potential roles of NHEJ key factors in chemoresistance in CRC are relatively unexplored. Our study emphasized the contribution of DSBR to chemoresistance in CRC and indicated that inhibition of NHEJ, specifically XLF, could generate significant synergistic effects with first-line chemotherapy drugs, such as 5-Fu and OXA. Combinations of 5-Fu and OXA are commonly used as the first-line chemotherapeutic method.44 Interestingly, XLF-deficient cells are sensitive to 5-Fu and OXA; therefore, XLF inhibition or deficiency could be used as another combination choice with either 5-Fu or OXA instead of serving as the third component in a 5-Fu and OXA combination regimen.
Conclusion
Our study demonstrated that NHEJ participates in the chemoresistance of CRC. We generated 5-Fu- and OXA-resistant CRC cell lines and observed that the NHEJ key factor XLF is elevated at both mRNA and protein levels in these drug-resistant cell lines. Furthermore, XLF deficiency has a significant synergy with 5-Fu or OXA. Our research identified a new target that could contribute to overcome 5-Fu and OXA chemoresistance in CRC.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Acknowledgment
This work was supported by funding from the Jilin Province Science and Technology Institute (20160101034JC and 20170204050SF to Zhuo Liu and Dongxin Wang, respectively).
Author contributions
ZL designed experiments and interpreted results. ZL, MY, BF, and JS performed experiments. ZL, MY, BF, and JS wrote and edited the manuscript. ZL and DW supervised the project and acquired funding. All authors contributed to data analysis, drafting and revising the article, gave final approval of the version to be published, and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67(3):177–193. | ||
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67(1):7–30. | ||
Sargent D, Sobrero A, Grothey A, et al. Evidence for cure by adjuvant therapy in colon cancer: Observations based on individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol. 2009;27(6):872–877. | ||
Giacchetti S, Perpoint B, Zidani R, et al. Phase III multicenter randomized trial of oxaliplatin added to chronomodulated fluorouracil–leucovorin as first-line treatment of metastatic colorectal cancer. J Clin Oncol. 2000;18(1):136–147. | ||
Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3(5):330–338. | ||
Peinert S, Grothe W, Stein A, et al. Safety and efficacy of weekly 5-fluorouracil/folinic acid/oxaliplatin/irinotecan in the first-line treatment of gastrointestinal cancer. Ther Adv Med Oncol. 2010;2(3):161–174. | ||
Umsumarng S, Pintha K, Pitchakarn P, et al. Inhibition of P-glycoprotein mediated multidrug resistance by stemofoline derivatives. Chem Pharm Bull. 2013;61(4):399–404. | ||
Hills SA, Diffley JF. DNA replication and oncogene-induced replicative stress. Curr Biol. 2014;24(10):R435–R444. | ||
Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12(9):440–450. | ||
Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem. 2010;79(1):181–211. | ||
Moynahan ME, Jasin M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat Rev Mol Cell Biol. 2010;11(3):196–207. | ||
West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol. 2003;4(6):435–445. | ||
Mathiasen DP, Lisby M. Cell cycle regulation of homologous recombination in Saccharomyces cerevisiae. FEMS Microbiology Reviews. 2014;38(2):172–184. | ||
Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat Rev Mol Cell Biol. 2006;7(10):739–750. | ||
Wold MS. Replication protein A: a heterotrimeric, single-stranded DNA-binding protein required for eukaryotic DNA metabolism. Annu Rev Biochem. 1997;66(1):61–92. | ||
Mimitou EP, Symington LS. Nucleases and helicases take center stage in homologous recombination. Trends Biochem Sci. 2009;34(5):264–272. | ||
Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412(6847):607–614. | ||
Palmbos PL, Wu D, Daley JM, Wilson TE. Recruitment of Saccharomyces cerevisiae Dnl4-Lif1 complex to a double-strand break requires interactions with Yku80 and the Xrs2 FHA domain. Genetics. 2008;180(4):1809–1819. | ||
Yano K, Morotomi-Yano K, Wang SY, et al. Ku recruits XLF to DNA double-strand breaks. EMBO Rep. 2008;9(1):91–96. | ||
Zha S, Boboila C, Alt FW. Mre11: roles in DNA repair beyond homologous recombination. Nat Struct Mol Biol. 2009;16(8):798–800. | ||
Weterings E, Verkaik NS, Brüggenwirth HT, Hoeijmakers JH, van Gent DC. The role of DNA dependent protein kinase in synapsis of DNA ends. Nucleic Acids Res. 2003;31(24):7238–7246. | ||
Defazio LG, Stansel RM, Griffith JD, Chu G. Synapsis of DNA ends by DNA-dependent protein kinase. Embo J. 2002;21(12):3192–3200. | ||
Nick McElhinny SA, Ramsden DA. Sibling rivalry: competition between Pol X family members in V(D)J recombination and general double strand break repair. Immunol Rev. 2004;200(1):156–164. | ||
Daley JM, Laan RL, Suresh A, Wilson TE. DNA joint dependence of Pol X family polymerase action in nonhomologous end joining. J Biol Chem. 2005;280(32):29030–29037. | ||
Bahmed K, Seth A, Nitiss KC, Nitiss JL. End-processing during non-homologous end-joining: a role for exonuclease 1. Nucleic Acids Res. 2011;39(3):970–978. | ||
Li J, Summerlin M, Nitiss KC, Nitiss JL, Hanakahi LA. Tdp1 is required for efficient non-homologous end joining in human cells. DNA Repair. 2017;60:40–49. | ||
Heo J, Li J, Summerlin M, et al. Tdp1 promotes assembly of non-homologous end joining protein complexes on DNA. DNA Repair. 2015;30:28–37. | ||
Li Y, Chirgadze DY, Bolanos-Garcia VM, et al. Crystal structure of human XLF/Cernunnos reveals unexpected differences from XRCC4 with implications for NHEJ. Embo J. 2008;27(1):290–300. | ||
Revy P, Malivert L, de Villartay JP. Cernunnos-XLF, a recently identified non-homologous end-joining factor required for the development of the immune system. Curr Opin Allergy Clin Immunol. 2006;6(6):416–420. | ||
Ahnesorg P, Smith P, Jackson SP. XLF interacts with the XRCC4-DNA ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124(2):301–313. | ||
Riballo E, Woodbine L, Stiff T, Walker SA, Goodarzi AA, Jeggo PA. XLF-Cernunnos promotes DNA ligase IV-XRCC4 re-adenylation following ligation. Nucleic Acids Res. 2009;37(2):482–492. | ||
Hammel M, Yu Y, Fang S, Lees-Miller SP, Tainer JA. XLF regulates filament architecture of the XRCC4·ligase IV complex. Structure. 2010;18(11):1431–1442. | ||
Oksenych V, Alt FW, Kumar V, et al. Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proc Natl Acad Sci U S A. 2012;109(7):2455–2460. | ||
Seluanov A, Mao Z, Gorbunova V. Analysis of DNA double-strand break (DSB) repair in mammalian cells. J Vis Exp. 2010(43). | ||
Sun J, Fu X, Wang Y, et al. Erianin inhibits the proliferation of T47D cells by inhibiting cell cycles, inducing apoptosis and suppressing migration. Am J Transl Res. 2016;8(7):3077–3086. | ||
Hsu HH, Chen MC, Baskaran R, et al. Oxaliplatin resistance in colorectal cancer cells is mediated via activation of ABCG2 to alleviate ER stress induced apoptosis. J Cell Physiol. 2018;233(7):5458–5467. | ||
Zhang T, Liu X, Chen X, et al. Inhibition of PC4 radiosensitizes non-small cell lung cancer by transcriptionally suppressing XLF. Cancer Med. 2018;7(4):1326–1337. | ||
Liu D, Deng X, Yuan C, Chen L, Cong Y, Xu X. Werner syndrome protein positively regulates XRCC4-like factor transcription. Mol Med Rep. 2014;9(5):1648–1652. | ||
Sulkowski PL, Scanlon SE, Oeck S, Glazer PM. PTEN regulates nonhomologous end joining by epigenetic induction of NHEJ1/XLF. Mol Cancer Res. 2018;16(8):1241–1254. | ||
Kawakami H, Zaanan A, Sinicrope FA. Implications of mismatch repair-deficient status on management of early stage colorectal cancer. J Gastrointest Oncol. 2015;6(6):676–684. | ||
An Q, Robins P, Lindahl T, Barnes DE. 5-fluorouracil incorporated into DNA is excised by the SMUG1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res. 2007;67(3):940–945. | ||
Cortellino S, Turner D, Masciullo V, et al. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc Natl Acad Sci U S A. 2003;100(25):15071–15076. | ||
Chen X, Li WF, Wu X, et al. Dicer regulates non-homologous end joining and is associated with chemosensitivity in colon cancer patients. Carcinogenesis. 2017;38(9):873–882. | ||
Cavanna L, Artioli F, Codignola C, et al. Oxaliplatin in combination with 5-fluorouracil (5-FU) and leucovorin (LV) in patients with metastatic gastric cancer (mgc). Am J Clin Oncol. 2006;29(4):371–375. |
Supplementary materials
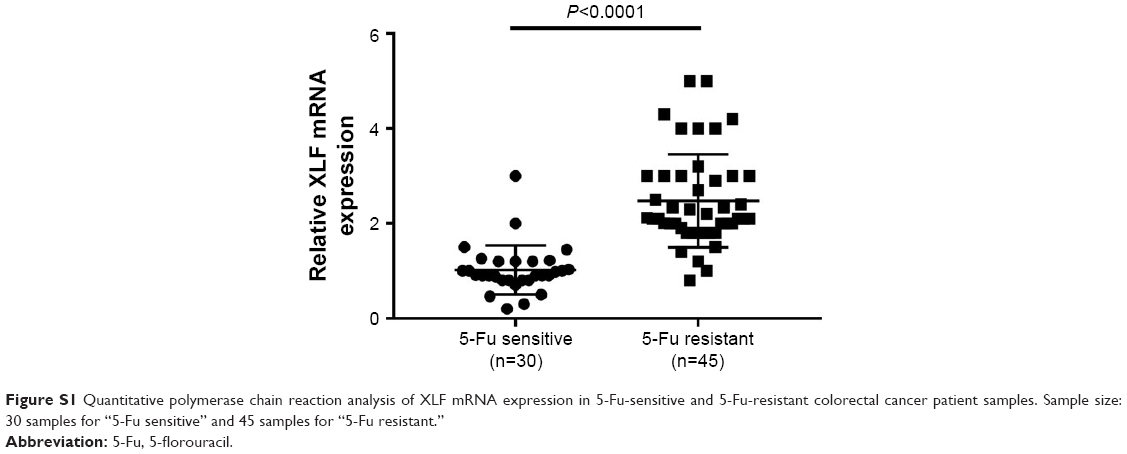 | Figure S1 Quantitative polymerase chain reaction analysis of XLF mRNA expression in 5-Fu-sensitive and 5-Fu-resistant colorectal cancer patient samples. Sample size: 30 samples for “5-Fu sensitive” and 45 samples for “5-Fu resistant.” |
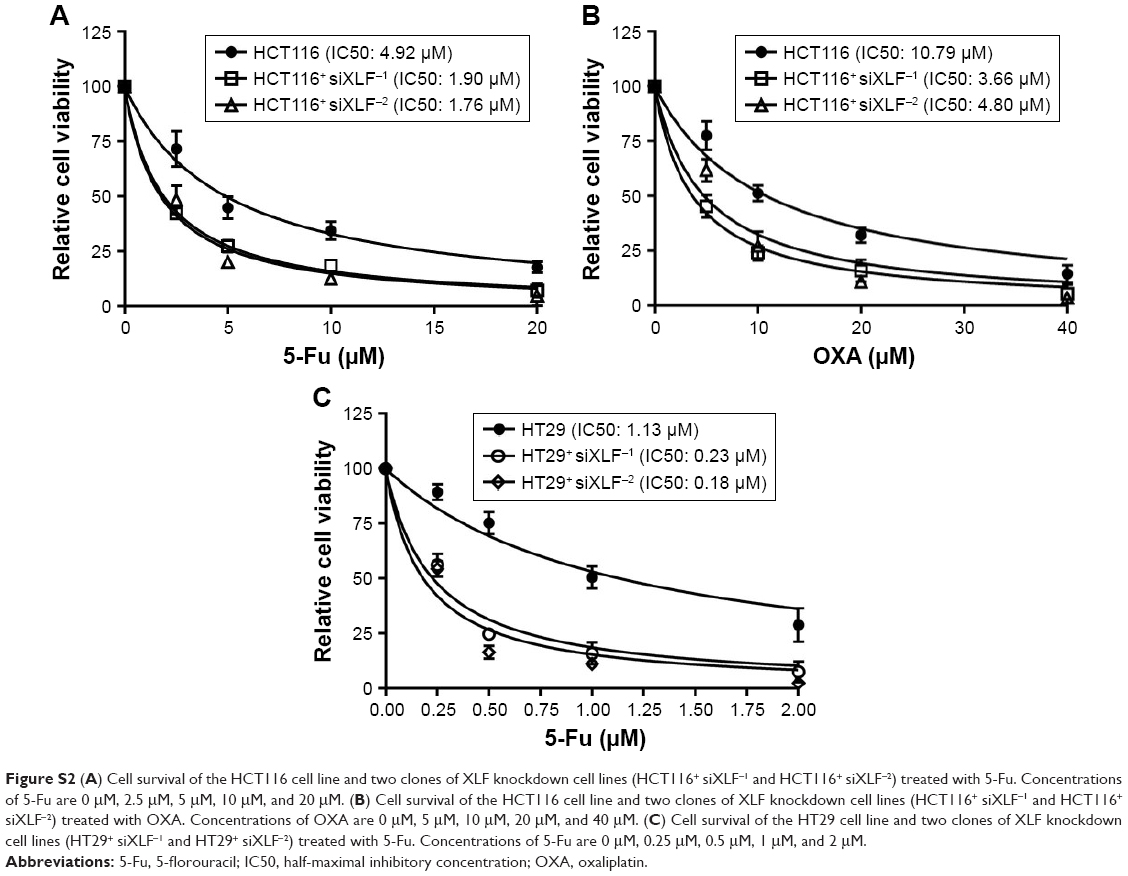 | Figure S2 (A) Cell survival of the HCT116 cell line and two clones of XLF knockdown cell lines (HCT116+ siXLF−1 and HCT116+ siXLF−2) treated with 5-Fu. Concentrations of 5-Fu are 0 μM, 2.5 μM, 5 μM, 10 μM, and 20 μM. (B) Cell survival of the HCT116 cell line and two clones of XLF knockdown cell lines (HCT116+ siXLF−1 and HCT116+ siXLF−2) treated with OXA. Concentrations of OXA are 0 μM, 5 μM, 10 μM, 20 μM, and 40 μM. (C) Cell survival of the HT29 cell line and two clones of XLF knockdown cell lines (HT29+ siXLF−1 and HT29+ siXLF−2) treated with 5-Fu. Concentrations of 5-Fu are 0 μM, 0.25 μM, 0.5 μM, 1 μM, and 2 μM. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.