")
Back to Journals » International Journal of Nanomedicine » Volume 12
Noncovalent interaction-assisted drug delivery system with highly efficient uptake and release of paclitaxel for anticancer therapy
Authors Wei YP, Ma L, Zhang L, Xu X
Received 19 June 2017
Accepted for publication 3 August 2017
Published 25 September 2017 Volume 2017:12 Pages 7039—7051
DOI https://doi.org/10.2147/IJN.S144322
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Dongwoo Khang
Yuping Wei,1 Liang Ma,2 Liang Zhang,1,2 Xia Xu2
1State Key Laboratory of Biochemical Engineering, Institute of Process Engineering, Chinese Academy of Sciences, Beijing, 2School of Chemistry and Chemical Engineering, Anhui University of Technology, Maanshan, Anhui, People’s Republic of China
Abstract: An effective drug delivery system requires efficient drug uptake and release inside cancer cells. Here, we report a novel drug delivery system, in which paclitaxel (PTX) interacts with a novel cell penetrating peptide (CPP) through noncovalent interaction designed based on molecular simulations. This CPP/PTX complex confers high efficiency in delivering PTX into cancer cells not by endocytosis but by an energy-independent pathway. Once inside cells, the noncovalent interaction between PTX and the CPP may allow fast release of PTX within cells due to the direct translocation of CPP/PTX. This drug delivery system exhibits strong capacity for inhibition of tumor growth and offers a new avenue for the development of advanced drug delivery systems for anticancer therapy.
Keywords: paclitaxel, drug delivery, cell penetrating peptide, anticancer therapy, molecular simulations
Introduction
Due to its capacity of stabilizing the microtubule polymer and protecting it from disassembly,1 paclitaxel (PTX), an effective chemotherapeutic drug, has been widely used in treating various solid tumors, such as ovarian and breast cancer, small and non-small cell lung cancer, melanoma, head and neck cancer, and bladder cancer.2 Despite its clinical efficacy, clinical applications of PTX are hampered by the extremely poor water solubility (<0.01 mg/mL) and low permeability due to the presence of the P-gp efflux transport system in cellular barriers.3,4 Currently, to enhance its water solubility, PTX is formulated in a clinically approved formulation, in which PTX is solubilized in a 1:1 solution of Cremophor EL and dehydrated ethanol to arrive at solvent-based PTX (Taxol). However, the high amount of Cremophor EL for a single intravenous administration can cause many significant side effects, including hypersensitivity, nephrotoxicity, cardiotoxicity, and neurotoxicity.5 Therefore, it is necessary to develop a Cremophor®-free formulation of PTX with less toxicity and high efficiency.
In recent years, numerous attempts have been made to develop drug delivery systems for achieving efficient PTX delivery. Various formulations have been under development, such as liposome,6 micelle,7,8 biodegradable polymer, and hydrogel,9,10 which are free drug-loaded nano-carriers formulated without chemical linkage. Compared with free PTX,11,12 PTX-loaded nano-carriers have high aqueous solubility,13 cytotoxicity on tumor cells, and PTX tolerance dosage.14 Translocation of PTX-loaded nano-carriers into cells through endocytosis pathways,15 which is often dependent on the size, shape, surface chemistry, and even stiffness of nano-carriers,16 may cause difficulty in drug release inside cells. Additionally, due to endocytosis saturation, generation of nano-carriers with high-loading capability is usually required.12 Hence, the process for preparing nano-carriers is much more difficult for quality control.
Prodrug, which is formulated by chemical linkage, is a promising strategy for improving solubility and permeability and reducing toxicity of parent drugs.17 In comparison with nano-carrier drug delivery systems, prodrug formulation is much easier to scale up and more reproducible. Prodrugs, such as macromolecule-based prodrugs, which link PTX with a macromolecule through conjugation, can elevate tumor uptake and pharmacokinetics by enhanced permeability and the retention effect of macromolecules.18,19 However, this strategy may also suffer from a poor release of the covalently linked PTX. Hence, it is necessary to develop a novel drug delivery system with fast drug release inside cells.
Noncovalent drug delivery systems with the advantage of simple preparation procedures have drawn more attention in recent years. Noncovalent interactions, such as strong hydrophobic, electrostatic, and hydrogen bonding, play important roles in the construction of drug delivery systems. However, currently most researches focus on introducing noncovalent interactions into polymeric micelles to enhance their stability and bioactivity.20 Due to PTX-loaded micelles translocation through endocytosis pathway, the challenge of PTX release inside cells still remains.
Cell penetrating peptides (CPPs) are peptides usually containing 7–50 residues which can facilitate cellular uptake. Generally, they can be categorized into polycationic, amphipathic, and hydrophobic peptides. It has been demonstrated that the penetrating capacity of polycationic and amphipathic CPPs is particularly associated with the interaction between positively charged residues, especially arginine (Arg, R), and negatively charged membrane.4 Currently, CPPs-assisted strategy is promising for effective drug delivery. Generally, CPP-aided delivery system is constructed in a covalent pattern, in which CPP and its cargo are linked via covalent bond, which may lead to poor release of drugs. To avoid the poor release rate of PTX, noncovalent interactions between PTX and CPP are required.20 Furthermore, to overcome the slow release rate of PTX resulting from endocytosis, CPP which can deliver PTX into cells through direct translocation instead of endocytosis is desired.
Hence, in this study, a novel CPP interacting with PTX via noncovalent bond was designed based on molecular simulations. The translocation efficiency and cytotoxicity of CPP were determined. The intracellular PTX concentrations at different conditions were also measured using high performance liquid chromatography (HPLC). The cytotoxicity on tumor cells in vitro and in vivo was further evaluated. Additionally, the mechanism of CPP translocation into cells was inspected.
Materials and methods
Materials and reagents
Human cervical cancer cell line (HeLa) was obtained from Shanghai Institute of Biochemistry and Cell Biology, Chinese Academy of Sciences. Dulbecco’s Modified Eagle’s Medium (DMEM), fetal bovine serum (FBS), trypsin, and phosphate-buffered solution (PBS, pH 7.4) were all purchased from Hyclone (San Angelo, TX, USA). Taxol and PTX were donated by Beijing ShuangLu Pharmaceutical Co, Ltd. Peptides and fluorescein isothiocyanate (FITC)-labeled peptides were synthesized by GL Biochem (Shanghai, People’s Republic of China). Chlorpromazine (CPZ) was purchased from Heowns Biochemical Technology (Tianjin, People’s Republic of China), nystatin (Nys) from Topscience (Shanghai, People’s Republic of China), 2-deoxy-D-glucose (2-D-G) and dimethyl sulfoxide (DMSO) from Aladdin (Shanghai, People’s Republic of China), sodium azide (NaN3) from Micxy Chemical Co (Cheng Du, People’s Republic of China), paraformaldehyde (PFA), 4′,6-diamidino-2-phenylindole (DAPI), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT), and heparin from Solarbio (Beijing, People’s Republic of China). Mouse anti-β-tubulin primary antibody and FITC-labeled goat anti-mouse secondary antibody were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Female BALB/c mice were purchased from Beijing HFK Bioscience Co, Ltd (Beijing, People’s Republic of China).
Molecular simulations
Coordinates of PTX (TA1) and heparin (1HPN) were obtained from the Research Collaboratory for Structural Bioinformatics protein data bank. Topology files of PTX and heparin were generated using the PRODRG2 server.21 The structure of polypeptides was sketched using the Discovery Studio Visualizer 3.0 (DSV; Accelrys, San Diego, CA, USA). Molecular docking was carried out to investigate the interactions between the designed functional group and PTX by using AutoDock Vina.22 The number of docking modes was set to 20 within a 40×40×40 grid box (grid spacing 1.0 Å) and other settings were set as the defaults.
Molecular dynamics (MD) simulations were performed using the Gromacs 4.5.4 simulation package.23 The simulations for the systems of peptide-PTX and peptide-PTX-heparin were performed for 20 nanoseconds using the standard Gromos96 G43a1 force field in combination with the simple point charge water model. The peptide was first immersed in a box and followed by the addition of PTX and heparin. System stability was verified by root mean square deviation (RMSD) comparisons of the structures in the trajectory to the starting conformation. The Particle Mesh Ewald method was used to calculate long-range electrostatic interactions. Energy changes during the simulations were analyzed by the auxiliary program of Gromacs. The simulation results were visualized using DSV.
Cell culture
HeLa cells were cultured in the medium containing DMEM supplemented with 10% FBS at 37°C with 5% CO2. The culture medium was replaced every 3 days. Once the cells reached confluence, they were digested using trypsin and passaged at an appropriate split ratio.
Cytotoxicity assay
To determine the cytotoxicity of CPP on HeLa cells, the cells were seeded into 96-well plates at a density of 5×103 cells/well and cultured for 24 hours. Then, the medium was replaced with the fresh medium containing various concentrations of CPP from 50 to 1,000 μM. After 3 hours of incubation, the cells were washed twice using PBS and cultured in the fresh culture medium for 24 hours. To further test the effect of treatment time on cell viability, the cells were incubated with 100 and 1,000 μM CPP for 24 hours.
To evaluate the effect of incubation time with CPP loading PTX on HeLa cells, culture medium containing 100 μM CPP and 1.47±0.28 μg/mL (about 1.7±0.38 μM, determined by HPLC) free PTX was prepared.24 The cells were incubated with the culture medium for 30 seconds, 1 minute, 5 minutes, 30 minutes, and 24 hours, washed twice with PBS, and incubated with fresh culture medium for another 24 hours.
To investigate how the concentrations of CPP and PTX affect cell viability, HeLa cells were cultured in the medium containing 1.47 μg/mL (1,470 ng/mL) free PTX with CPP concentrations ranging from 5 to 1,000 μM, and in the medium composed of 100 μM CPP with different concentrations of PTX from 1.47 to 1,470 ng/mL. After 30 minutes of treatment, the cells were washed twice with PBS and incubated with fresh culture medium for another 24 hours.
The MTT method was used to assess cytotoxicity. The cells were washed twice with PBS and incubated for 3 hours with fresh culture medium containing MTT as previously described by Yan et al.25 After removal of culture medium, 100 μL of DMSO was introduced. The absorbance was measured at 492 nm (OD492) using a microplate reader (Tecan, Männedorf, Switzerland). Cells without treatment were used as the negative control. Cytotoxicity is presented as the percentage normalized to the negative control.
Cellular uptake of CPP
To visualize the cellular uptake, HeLa cells (4×104 cells) were seeded onto coverslips in 24-well plates. After 24 hours of incubation, the cells were exposed to serum-free culture medium containing FITC-labeled CPP or FITC only at concentrations of 10 and 100 μM at 37°C for 30 minutes. The cells were washed twice with PBS containing heparin to remove surface bound CPPs,26 fixed with 4% PFA, and then stained with DAPI labeling the nucleus in blue color. Cellular uptake was visualized using fluorescence microscopy (Nikon, Japan).
Uptake efficiency analysis
To evaluate uptake efficiency, the cells were incubated in culture medium with 100 μM FITC-labeled CPP or FITC only under 37°C for 30 minutes, then washed using PBS with heparin twice, trypsinized, and finally resuspended in PBS. The ratio of the cells in green color to the cells without green color was considered as the uptake efficiency, which was determined using a flow cytometer (Cyflow Cube 6; Partec, Munich, Germany). The untreated cells were used as the negative control.
Distribution of CPP in HeLa cells
To monitor the distribution of FITC-labeled CPP inside cells, HeLa cells were incubated with the culture medium containing 10 and 100 μM FITC-labeled CPP for 30 minutes. After washing with PBS and heparin, the cells were fixed with 4% PFA and immediately examined using a confocal microscope (Carl Zeiss Meditech AG, Jena, Germany).
Microtube analysis
After seeding (4×104 cells/well), the HeLa cells were cultured for 24 hours, treated with 10 μM CPP and 1.47 μg/mL PTX at 37°C for 30 minutes, washed with PBS twice, and then cultured for another 24 hours in fresh culture medium. Next, the cells were fixed with 4% PFA, followed by permeabilization with PBS containing 0.1% Triton X-100, and then blocked using PBS with 1% goat serum and 0.5% Tween 80 for 30 minutes at room temperature. Subsequently, the cells were stained with mouse anti-β-tubulin primary antibody for 1 hour, then FITC-labeled goat anti-mouse secondary antibody for 1 hour, and DAPI for 30 minutes at room temperature. The cells were imaged immediately using a confocal microscope (Carl Zeiss Meditech AG). For fluorescence activated cell sorting (FACS) analysis, the cells were first trypsinized, then fixed with 4% PFA, followed by immunofluorescence staining as previous described, and finally resuspended in PBS. FACS analysis was carried out using a flow cytometer (Cyflow Cube 6). The untreated cells were used as the negative control.
Cellular uptake pathway
To investigate the effect of low temperature on CPP/PTX uptake, after preincubation at 4°C for 30 minutes, the cells were cultured at 4°C for 30 minutes in serum-free culture medium containing 1.47 μg/mL PTX with 10 or 100 μM CPP. After incubation, the cells were washed twice using PBS and cultured in fresh culture medium for 24 hours at 37°C. The cells cultured at 37°C were used for comparison.
To deplete intracellular ATP pools, the cells were treated with metabolic inhibitors (10 mM NaN3 and 6 mM 2-D-G) in glucose- and serum-free culture medium for 30 minutes. After washing with PBS twice, the cells were incubated in the same culture medium but containing the metabolic inhibitor, 10 or 100 μM CPP with PTX (1.47 μg/mL) at 37°C for 30 minutes. To inhibit the endocytosis pathway, the cells were preincubated in serum-free culture medium containing endocytosis inhibitors (5 μM CPZ or 10 μM Nys) for 30 minutes. After washing twice with PBS, the cells were incubated in serum-free culture medium containing the endocytosis inhibitor, 10 and 100 μM CPP with PTX (1.47 μg/mL). After incubation, the cells were washed twice using PBS and cultured in fresh culture medium for 24 hours. Cells treated in the same culture medium containing 10 or 100 μM CPP with PTX (1.47 μg/mL) without any inhibitor at 37°C following the preincubation were used for comparison. The results are presented as percentage normalized to the control value.
Intracellular PTX assay
To determine the intracellular concentration of PTX, HeLa cells (2×105 cells) were seeded into 24-well plates. After 30 minutes incubation with Taxol (1.47 μg/mL), free PTX (1.47 μg/mL), and CPP (100 μM)/PTX (1.47 μg/mL), the cells were washed by PBS twice and intracellular PTX was extracted from HeLa cells by DMSO. PTX concentration was analyzed at a wavelength of 227 nm using a LC-6AD HPLC system (Shimadzu, Japan) with a C18 column (250×4.6 mm; Agilent Technologies, Santa Clara, CA, USA) with a linear gradient of methanol from 50% to 100%.
In vivo animal experiments
All animal experiments were approved by the Institute of Process Engineering, Chinese Academy of Sciences (IPE-CAS) Biotech Center Ethics Committee. The protocols for the in vivo animal experiments conformed to the guidelines provided in the Guide for the Care and Use of Laboratory Animals. All procedures involving animal studies were carried out in accordance with protocols approved by the Committee for Animal Research of Peking University, People’s Republic of China. HeLa cells (5×106) were inoculated subcutaneously in female BALB/c mice (4–6 weeks old and weighing 18–22 g). When the tumor volume reached around 50 mm3, the mice were randomly divided into four groups: group 1 was administered PBS solution, group 2 free PTX, group 3 Taxol (10 mg/kg), and group 4 CPP/PTX complex. All the formulations were filtered by 0.22 μM sterilized filter and 100 μL was administered everyday via a tail vein. During 2 weeks of treatment, the mice were weighed and the size of tumors in two dimensions was measured using a caliper every other day. Tumor volumes were calculated using the following formula: volume (mm3) = length × width × width/2.
In vivo bio-distribution assay
For the free PTX group and CPP/PTX group, the mice were sacrificed after 2 weeks of treatment. Blood, lungs, liver, kidney, heart, and spleen of the mice were collected. The collected tissues were washed three times in ice-cold saline, weighed, and stored in −20°C until further analysis. Plasma and tissue homogenate samples were precipitated using methanol. The extracted samples were centrifuged at 12,000 rpm for 10 minutes. PTX in the supernatant collected after centrifugation was analyzed at a wavelength of 227 nm using a LC-6AD HPLC system. The mobile phase consisted of methanol and water at 50:50 (v/v). Separation was performed using a C18 column (250×4.6 mm; Agilent Technologies) with a linear gradient of methanol from 50% to 100%.
Histology assay
After 2 weeks of treatment, lung, liver, kidney, heart, and spleen were collected from the sacrificed mice and washed three times using ice-cold saline. Then, the organs were fixed by 4% PFA. Before sectioning, water was removed from the sample in successive stages using increasing concentrations of alcohol and xylene in the last dehydration stage, and then embedded in paraffin. Sections were mounted on glass slides and then stained with hematoxylin/eosin. The stained samples were examined by a microscope (Nikon).
Statistical analysis
One-way analysis of variance was used to assess statistical significance. A probability of p<0.05 (*) was considered to be significant, while p<0.01 (**) was considered to be very significant.
Results and discussion
CPP design
Dual-functional CPP design
In this paper, a dual-functional CPP consisting of the cell penetrating sequence and the affinity sequence was designed to provide efficient uptake and release of PTX. Arg, R is an essential amino acid for CPP to cross the cell membrane.26–28 Furuhata et al revealed that R4 has the greatest penetration efficiency when CPPs consist of Arg with the number of 4, 6, 8, 10.29 The lowest binding free energy of R4 among (Arg)n (n=2–5) binding to membrane receptor also indicates that R4 has the greatest capacity for crossing the cell membrane.30 Due to the difficulty in peptide synthesis and toxicity caused by the increase in Arg residues,26 R4 was chosen as the cell penetrating sequence in this study.
CPP-assisted strategy has been used for intracellular delivery of various cargos such as small molecules, protein, and nucleic acids. To avoid interference from the covalent binding between CPPs and cargo to the biological activity of cargos,26,30 affinity sequence coupling with PTX via noncovalent interaction was developed based on molecular docking. A pentapeptide library was established. The binding free energies (Ebinding) between pentapeptide and PTX, required to be in the range of −6.9 to −9.5 kcal/mol for affinity ligand design, were used as a criterion for designing new affinity sequence.31,32 Seven candidates of RRRRQWWPY, RRRRRWWWW, RRRRRWWQY, RRRRQWWQW, RRRRQWWYW, RRRRRQWWW, and RRRRQWWWP were obtained for further simulations.
MD simulation
Heparin as a membrane receptor plays an important role in CPPs translocation as co-receptor.33 Thus, here, heparin was chosen as a membrane receptor for CPP translocation and PTX was chosen as the target drug. According to the RMSD criteria and geometry criteria described in a previous study,34 only the peptides with stable conformation and having the desired binding sites with PTX and heparin were selected for further cytotoxicity, penetrating capacity, and drug delivery experiments. The simulation results revealed that only RRRRQWWQW (Rq) was able to interact with both PTX and heparin (Figures 1 and S1). Based on the fluctuation of RMSD, Rq formed stable interactions with PTX at Trp6, Trp7, Qln8, and Trp9 after 17 nanoseconds (Figure 1A). Once heparin was introduced, the PTX-Rq-heparin system obtained equilibrium at 5 nanoseconds (Figure 1B). Conformation analysis revealed that Rq interacted with PTX at Qln5, Trp6, Trp7, Qln8, and Trp9, and interacted with heparin at Arg1 and Arg2. The total energy changes of PTX-Rq and PTX-Rq-heparin systems were −85.6003 and −89.8901 KJ/mol, respectively, indicating that Rq had great potential for interacting with PTX and further with heparin. Hence, Rq was selected as the CPP candidate for the intracellular delivery of PTX.
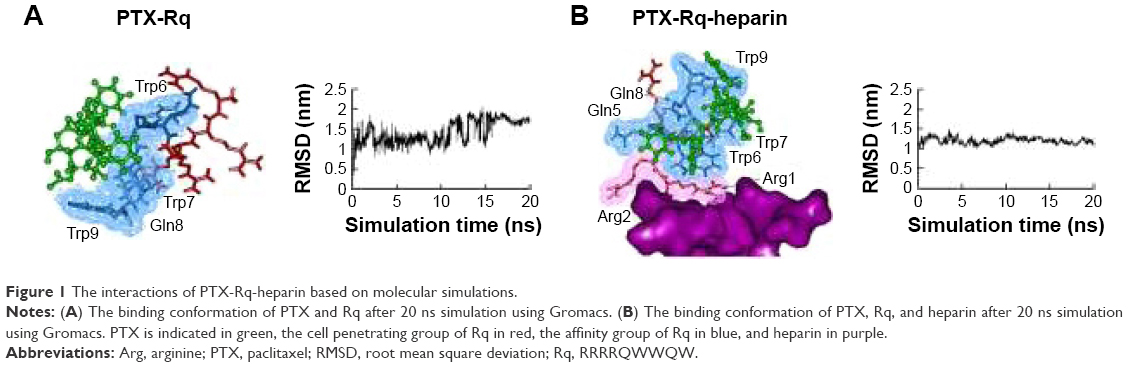 | Figure 1 The interactions of PTX-Rq-heparin based on molecular simulations. |
In vitro cytotoxicity of Rq
The cytotoxicity of CPPs largely depends on the type, quantity, and arrangement of sequence.27 Normally, CPPs which can cause cell damage even at micrometer concentration levels are not suitable for use as carriers in drug delivery systems.35 Hence, before determining the cellular uptake of Rq, cytotoxicity was evaluated using the MTT method. Nearly all the cells were viable after 3 and 24 hours incubation even at 1,000 μM (Figure 2). We further estimated the cytotoxicity of Rq to other cell types. There was no significant decrease in the cell viability after 24 hours incubation with 1,000 μM Rq (Figure S2), which was much less than that after 24 hours of incubation with IMT-P8 (Arg-rich CPP) at 20 μM,36 indicating that Rq was not toxic at physiological pH even at a high concentration of 1 mM. This negligible cytotoxicity of Rq may be caused by the presence of Qln in the affinity sequence of Rq as demonstrated in a previous study, in which the introduction of glutamine (Qln, Q) to amphipathic α-helical model CPPs (LK peptide) results in a reduction in hemolytic toxicities.37 Furthermore, the short length of Rq may be another reason for the low cytotoxicity.38
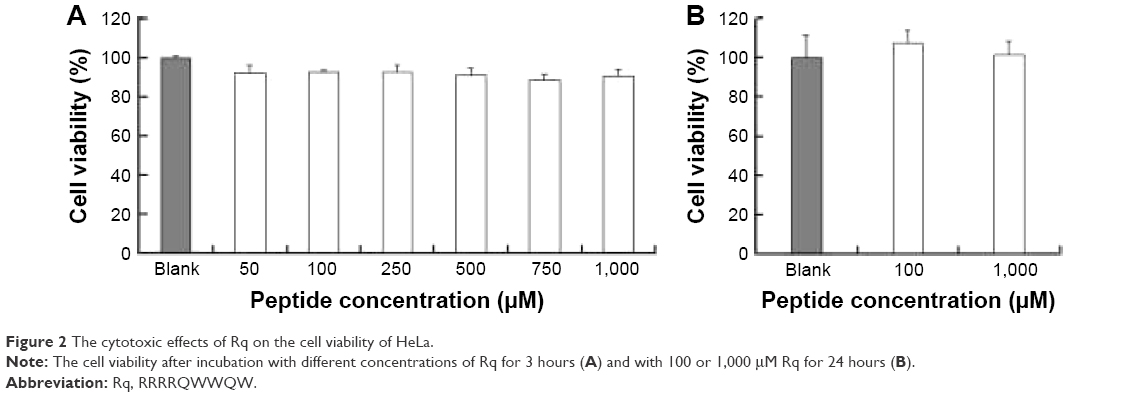 | Figure 2 The cytotoxic effects of Rq on the cell viability of HeLa. |
Cellular uptake of Rq
To validate the Rq translocation, Rq was linked with FITC at the C-terminal to prevent potential toxicity.35,39 As shown in Figure 3A, obvious green fluorescence was observed in the presence of both 10 and 100 μM Rq-FITC while no fluorescence was observed in the untreated cells and cells treated with FITC alone, indicating that Rq could efficiently penetrate into cells. Figure 3B and C show a significant difference in the percentage of green cells between the Rq-FITC group and FITC group. Around 62.65%±3.45% (p=0.001) and 73.35%±5.67% (p=0.001) appeared in green for 10 and 100 μM Rq-FITC, respectively, indicating that the uptake efficiency was related to the peptide concentration. Intracellular localization of Rq-FITC was further analyzed by confocal microscopy. As shown in Figure 3D, the uniform fluorescence throughout the whole cells suggested no Rq aggregation inside the cytoplasm, implying that the translocation of Rq may not be dependent on endocytosis or macropinocytosis pathway, which could cause obvious fluorescence aggregation in vesicles and lysosomes.40–42
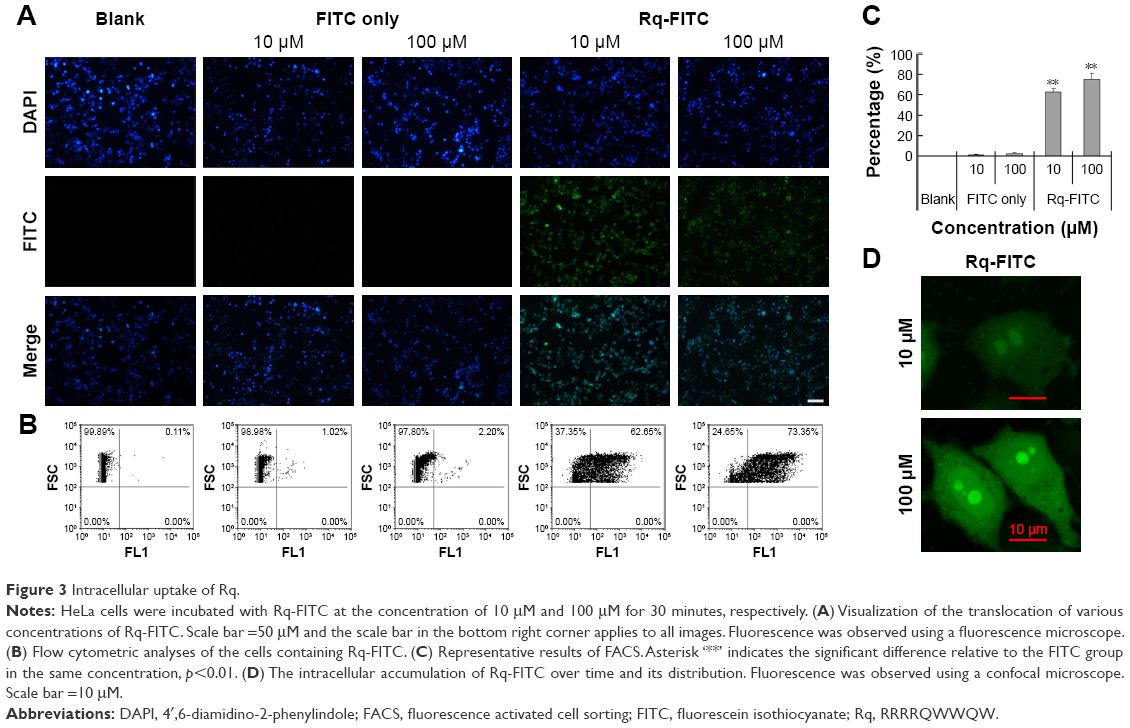 | Figure 3 Intracellular uptake of Rq. |
Cell viability after treating with Rq/PTX
CPP-mediated drug delivery is largely dependent on incubation time, concentration, and cargo molecule.40,43 To evaluate the potential therapeutic effect of the Rq/PTX system, the cytotoxicity of cells treated with Rq/PTX was determined using the MTT assay. We first investigated the effect of Rq and PTX concentrations on cell viability. As shown in Figure 4A, in the presence of 1,470 ng/mL PTX, 10 μM was the minimum concentration of Rq which efficiently delivered PTX into cells and further led to cell death. Not surprisingly, the cell death rate increased with increasing PTX concentration, reaching the maximum at 1,470 ng/mL (Figure 4B). We further determined the effect of incubation time on cell viability. In comparison with the free PTX group, the cell viability was effectively inhibited (p=0.004) after 30 seconds of treatment (Figure 4C), reduced by half to around 51.03%±9.85% (p=0.001) after 5 minutes of treatment, and further declined to around 33.65%±5.48% (p=0.001) after 30 minutes of treatment with Rq/PTX. All these experimental results indicated that the intracellular delivery rate of PTX was facilitated by Rq. The delivery rate of PTX assisted by Rq was not only much faster than free PTX but also much more rapid than the previous CPPs system.26,43 Furthermore, the cytotoxicity of the Rq/PTX complex to the other cell types was investigated. A dramatic drop in the cell viability of tumor cells but not normal cells was observed (Figure S2), revealing that Rq/PTX led to much more damage to tumor cells than normal cells.
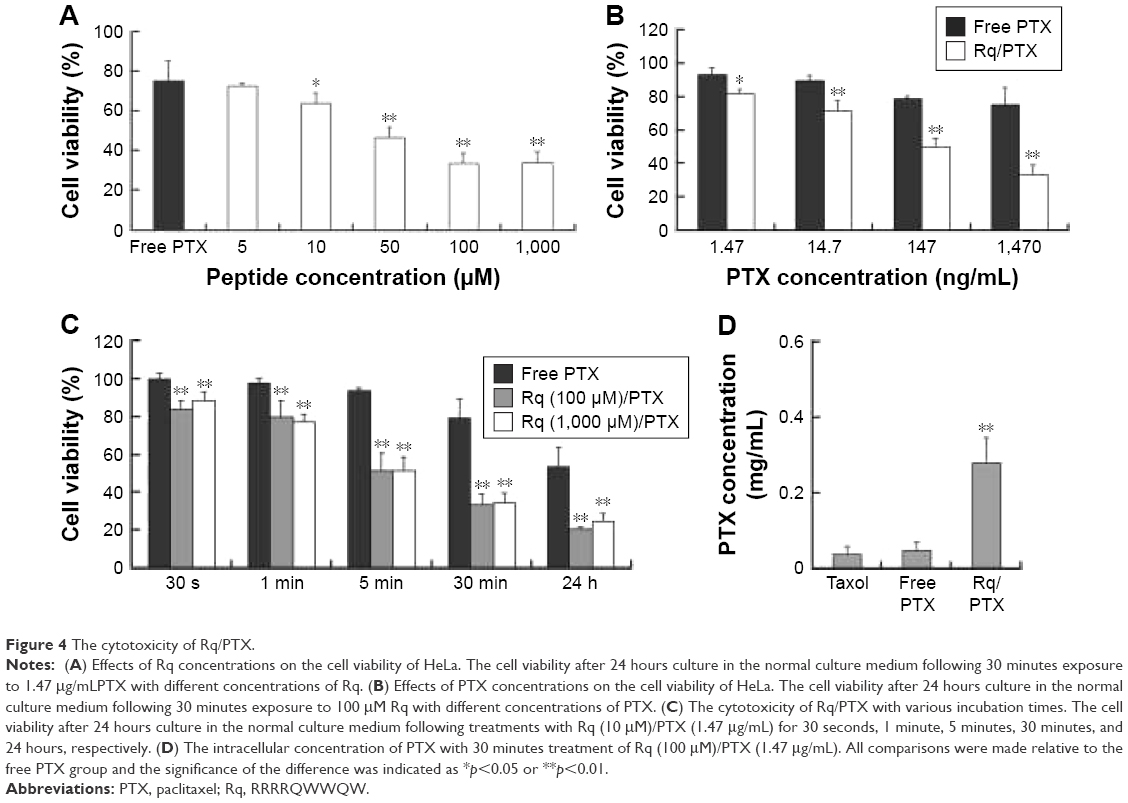 | Figure 4 The cytotoxicity of Rq/PTX. |
PTX accumulation inside cells
To evaluate the cellular uptake of PTX, the intracellular concentration of PTX delivered by Rq was determined by HPLC. It has been reported that the intracellular PTX concentration can accumulate to 0.206 mg/mL after 20 hours of incubation with 1 μM (0.854 μg/mL) PTX,44 around hundreds of times greater than the extracellular PTX concentration.44–46 Here, as shown in Figure 4D, the intracellular PTX concentration of the Rq/PTX group was nearly seven-fold greater than that of the free PTX group, reaching 0.279±0.068 mg/mL (p=0.001) after 30 minutes incubation with Rq/PTX, indicating that the uptake of PTX was mainly mediated by Rq.
Microtube analysis
It has been reported that treatment with PTX results in cell microtubule mass increase.44 Therefore, the microtubule morphology of HeLa cells after treatment with free PTX and Rq/PTX for 30 minutes was detected using the immunofluorescence method. Compared with the untreated group, more green color in cells was observed and the cells became much larger after treating with Rq/PTX (Figure 5A). The greater fluorescence intensity for the Rq/PTX group than the free PTX group (Figure 5B) indicated an increase in the microtube mass after the Rq/PTX treatment,47 implicating that Rq/PTX could efficiently stabilize the microtubule polymer and protect it from disassembly.
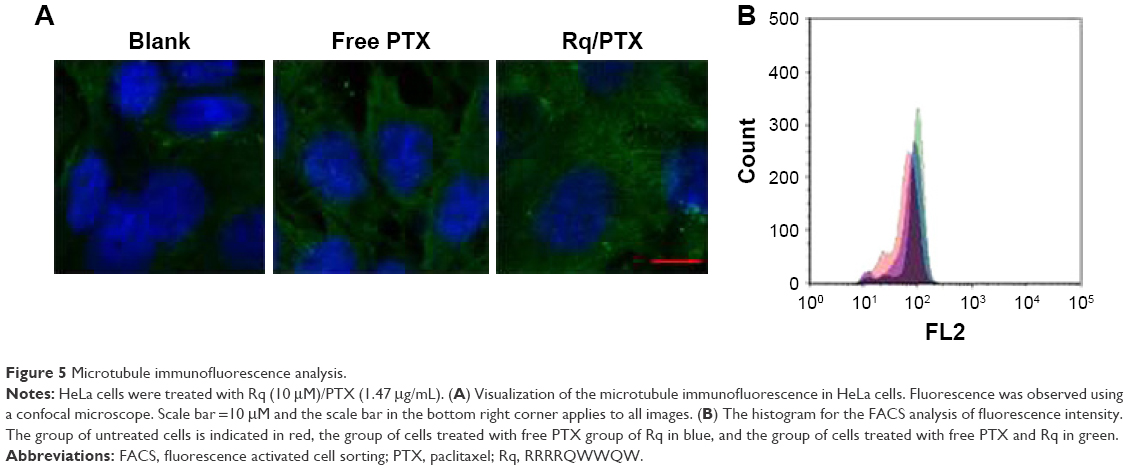 | Figure 5 Microtubule immunofluorescence analysis. |
Translocation pathway of Rq/PTX
The internalization pathway of CPP/cargo appears to not only be responsible for the intracellular distribution of CPP/cargo but also affect its release after penetration.48,49 Although there has been no conclusion of CPP translocation mechanism, CPP translocation usually relates to direct penetration and the endocytosis-mediated entry.26,48,49 Usually, the direct translocation based on the strong electrostatic interaction between the positively charged CPPs and negatively charged membrane is an energy-independent process.50,51 In contrast, endocytosis, which is an energy dependent pathway, can be effectively inhibited at 4°C by the low energy state of cell and NaN3/2-D-G by ATP depletion.50,51 Several researches have reported that some positively charged peptides can directly penetrate into cells at a concentration of 10 μM.50,51 Ziegler et al found that TAT uptake is related to heparin and polarized endocytotic flow at a concentration of 100 μM.52 Thus, here, the uptake mechanism of Rq/PTX was investigated at the concentration of 10 and 100 μM Rq. As shown in Figure 6, cell viability was not significantly affected by the low temperature and the presence of ATP inhibitors, suggesting that the Rq/PTX uptake is not energy-dependent but is direct translocation related.53 No significant change in the cell viability (Figure 6), in the presence of inhibitor of clathrin-dependent endocytosis (CPZ) and inhibitor of lipid raft/caveolae-mediated endocytosis (Nys), further confirmed that clathrin-dependent endocytosis and lipid raft/caveolae-dependent endocytosis were not involved in Rq/PTX intracellular delivery.
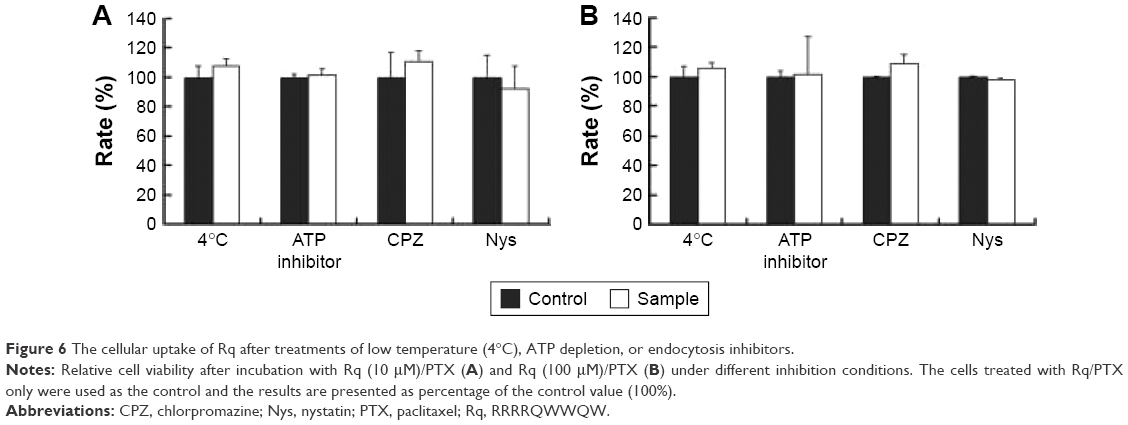 | Figure 6 The cellular uptake of Rq after treatments of low temperature (4°C), ATP depletion, or endocytosis inhibitors. |
In vivo experiments
The plasma concentrations of PTX were measured at 30 minutes and 1 day after injection. As shown in Figure 7A, regarding PTX clearance, the PTX concentration in plasma sharply decreased after 24 hours injection of free PTX, consistent with the results of a previous study.54 In contrast, <30% drop in the PTX concentration for the Rq/PTX group suggested that the PTX clearance in plasma can be slowed down by the presence of Rq. The PTX distribution in tissue is illustrated in Figure 7B. In the free PTX group, PTX accumulated in liver, kidney, and heart when free PTX was injected, in agreement with a previous study.55 In contrast, the PTX level was significantly raised in kidney (p=0.001), heart (p=0.001), and lung (p=0.001) for the Rq/PTX group, still much lower than the micelles delivery system (about 1 μg/mg) and similar to Taxol through intravenous injection in previous studies,56,57 implicating that Rq/PTX was a safe delivery system. In addition, the high acidic environment of tumor cells in comparison to normal cells may be one of the reasons for a dramatic elevation (p=0.001) in the PTX level in tumor for the Rq/PTX group.58 Under a natural physiological condition, the tumor growth was effectively suppressed after treatment with Rq/PTX (about 47.69±7.24 mm3), at a much smaller scale than the Taxol group (about 105.11±11.05 mm3, p=0.004) and the free PTX group (about 134.44±21.40 mm3, p=0.002) (Figures 7C and S3). These results suggested that Rq/PTX has stronger inhibition effects on HeLa tumor growth than Taxol and free PTX. There was no significant weight loss for all tested groups (Figure 7D). Histological examination results also exhibited no obvious damage to the major organs after treating with Rq/PTX, which were mainly filled with abundant cells (Figure 7E). All these experimental results indicated that Rq/PTX had low systemic toxicity and was more biocompatible than Taxol, and implicated that the presence of Rq could lead to the accumulation of PTX in vivo,59 further resulting in the suppression of tumor growth.
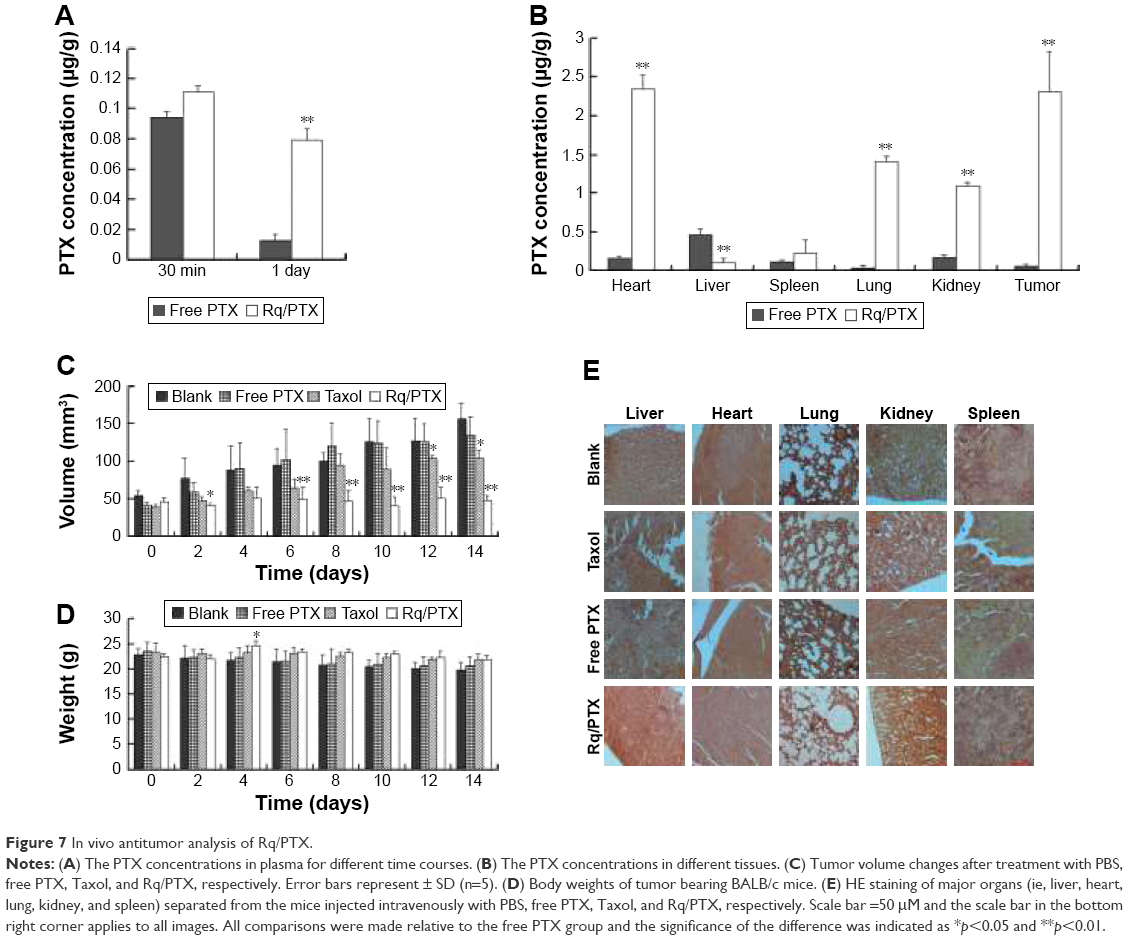 | Figure 7 In vivo antitumor analysis of Rq/PTX. |
Conclusion
A novel CPP, RRRRQWWQW (Rq), was developed based on molecular simulations to deliver PTX into HeLa cells via noncovalent interaction. PTX is efficiently delivered into HeLa cells with the help of Rq through energy-independent pathway and not by endocytosis. The noncovalent interaction between PTX and the CPP allows fast release of PTX once CPP/PTX is inside cells through direct translocation. In vivo animal experiments show that the tumor is successfully suppressed by Rq/PTX through a tail vein injection. This drug delivery system offers a new avenue for the development of advanced drug delivery systems for anticancer therapy.
Acknowledgments
This research was supported by the National Natural Science Foundation of China (21506230 and 21576266). The authors wish to thank the peer reviewers and editors for giving us constructive suggestions which have helped us to improve the quality of the paper.
Disclosure
The authors report no conflicts of interest in this work.
References
Wang TH, Wang HS, Soong YK. Paclitaxel-induced cell death: where the cell cycle and apoptosis come together. Cancer. 2000;88(11):2619–2628. | ||
Rowinsky EK. Clinical pharmacology of Taxol. J Natl Cancer Inst Monogr. 1993;15(15):25–37. | ||
Du AW, Lu H, Stenzel M. Stabilization of paclitaxel-conjugated micelles by cross-linking with cystamine compromises the antitumor effects against two- and three-dimensional tumor cellular models. Mol Pharm. 2016;13(11):3648–3659. | ||
Surapaneni MS, Das SK, Das NG. Designing paclitaxel drug delivery systems aimed at improved patient outcomes: current status and challenges. ISRN Pharmacol. 2012;2012(4):623139–623143. | ||
Yoshizawa Y, Kono Y, Ogawara KI, Kimura T, Higaki K. PEG liposomalization of paclitaxel improved its in vivo disposition and anti-tumor efficacy. Int J Pharm. 2011;412(1–2):132–141. | ||
Chang H, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomedicine. 2012;7:49–60. | ||
Kim SC, Kim DW, Shim YH, et al. In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release. 2001;72(1–3):191–202. | ||
Kim TY, Kim DW, Chung JY, et al. Phase I and pharmacokinetic study of Genexol-PM, a Cremophor-free, polymeric micelle-formulated paclitaxel, in patients with advanced malignancies. Clin Cancer Res. 2004;10(11):3708–3716. | ||
Win KY, Feng SS. In vitro and in vivo studies on vitamin E TPGS-emulsified poly(D,L-lactic-co-glycolic acid) nanoparticles for paclitaxel formulation. Biomaterials. 2006;27(10):2285–2291. | ||
Gan CW, Chien S, Feng SS. Nanomedicine: enhancement of chemotherapeutical efficacy of docetaxel by using a biodegradable nanoparticle formulation. Curr Pharm. 2010;16(21):2308–2320. | ||
Wang Y, Xin D, Liu K, Zhu M, Xiang J. Heparin-paclitaxel conjugates as drug delivery system: synthesis, self-assembly property, drug release, and antitumor activity. Bioconjug Chem. 2009;20(12):2214–2221. | ||
Ran N, Lee YM, Kim J, et al. Poly-cyclodextrin and poly-paclitaxel nano-assembly for anticancer therapy. Nat Commun. 2014;5(6183):3702–3714. | ||
Soga O, Nostrum CFV, Fens M, et al. Thermosensitive and biodegradable polymeric micelles for paclitaxel delivery. J Control Release. 2005;103(2):341–353. | ||
Li G, Liu J, Pang Y, et al. Polymeric micelles with water-insoluble drug as hydrophobic moiety for drug delivery. Biomacromolecules. 2011;12(6):2016–2026. | ||
Osada K, Cabral H, Mochida Y, et al. Bioactive polymeric metallosomes self-assembled through block copolymer–metal complexation. J Am Chem Soc. 2012;134(32):13172–13175. | ||
Ulbrich K, Holá K, Šubr V, Bakandritsos A, Tuček J, Zbořil R. Targeted drug delivery with polymers and magnetic nanoparticles: covalent and noncovalent approaches, release control, and clinical studies. Chem Rev. 2016;116(9):5338–5431. | ||
Kakinoki A, Kaneo Y, Tanaka T, Hosokawa Y. Synthesis and evaluation of water-soluble poly(vinyl alcohol)-paclitaxel conjugate as a macromolecular prodrug. Biol Pharm. 2008;31(5):963–969. | ||
Moktan S, Ryppa C, Kratz F, Raucher D. A thermally responsive biopolymer conjugated to an acid-sensitive derivative of paclitaxel stabilizes microtubules, arrests cell cycle, and induces apoptosis. Invest New Drugs. 2012;30(1):236–248. | ||
Choi JS, Jo BW. Enhanced paclitaxel bioavailability after oral administration of pegylated paclitaxel prodrug for oral delivery in rats. Int J Pharm. 2004;280(1–2):221–227. | ||
Ding J, Chen L, Xiao C, Chen L, Zhuang X, Chen X. Noncovalent interaction-assisted polymeric micelles for controlled drug delivery. Chem Commun. 2014;50(77):11274–11290. | ||
Schüttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. 2004;60(Pt 8):1355–1363. | ||
Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem. 2010;31(2):455–461. | ||
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–1718. | ||
Paál K, Müller J, Hegedûs L. High affinity binding of paclitaxel to human serum albumin. Eur J Biochem. 2001;268(7):2187–2191. | ||
Yan XL, Khor E, Lim LY. Chitosan-alginate films prepared with chitosans of different molecular weights. J Biomed Mater Res. 2001;58(4):358–365. | ||
Richard JP, Melikov K, Vives E, et al. Cell-penetrating peptides. A reevaluation of the mechanism of cellular uptake. J Biol Chem. 2003;278(1):585–590. | ||
Wender PA, Mitchell DJ, Pattabiraman K, Pelkey ET, Steinman L, Rothbard JB. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc Natl Acad Sci U S A. 2000;97(24):13003–13008. | ||
Mitchell DJ, Kim DT, Steinman L, Fathman CG, Rothbard JB. Polyarginine enters cells more efficiently than other polycationic homopolymers. J Pept Res. 2000;56(5):318–325. | ||
Furuhata M, Kawakami H, Toma K, Hattori Y, Maitani Y. Intracellular delivery of proteins in complexes with oligoarginine-modified liposomes and the effect of oligoarginine length. Bioconjug Chem. 2006;17(4):935–942. | ||
Wei Y, Li C, Zhang L, Xu X. Design of novel cell penetrating peptides for the delivery of trehalose into mammalian cells. Biochim Biophys Acta. 2014;1838(7):1911–1920. | ||
Alves NJ, Stimple SD, Handlogten MW, Ashley JD, Kiziltepe T, Bilgicer B. Small-molecule-based affinity chromatography method for antibody purification via nucleotide binding site targeting. Anal Chem. 2012;84(18):7721–7728. | ||
Chiappori F, Merelli I, Milanesi L, Marabotti A. Static and dynamic interactions between GALK enzyme and known inhibitors: guidelines to design new drugs for galactosemic patients. Eur J Med Chem. 2013;63(5):423–434. | ||
Belting M. Heparan sulfate proteoglycan as a plasma membrane carrier. Trends Biochem Sci. 2003;28(3):145–151. | ||
Novikov FN, Stroganov OV, Khaliullin IG, et al. Molecular modeling of different substrate-binding modes and their role in penicillin acylase catalysis. FEBS J. 2013;280(1):115–126. | ||
Saar K, Lindgren M, Hansen M, et al. Cell-penetrating peptides: a comparative membrane toxicity study. Anal Biochem. 2008;345(1):55–65. | ||
Gautam A, Nanda JS, Samuel JS, et al. Topical delivery of protein and peptide using novel cell penetrating peptide IMT-P8. Sci Rep. 2016;6:26278–26288. | ||
Kim S, Hyun S, Lee Y, Yan L, Yu J. Non-hemolytic cell penetrating peptides: site specific introduction of glutamine and lysine residues into the α-helical peptide causes deletion of its direct membrane disrupting ability but retention of its cell penetrating ability. Biomacromolecules. 2016;17(9):3007–3015. | ||
Oh D, Shirazi AN, Northup K, et al. Enhanced cellular uptake of short polyarginine peptides through fatty acylation and cyclization. Mol Pharm. 2014;11(8):2845–2854. | ||
Nan YH, Sunghaeng L, Hakjun K, Songyub S. Mammalian cell toxicity and candidacidal mechanism of Arg- or Lys-containing Trp-rich model antimicrobial peptides and their D-enantiomeric peptides. Peptides. 2010;31(10):1826–1831. | ||
El-Andaloussi S, Järver P, Johansson HJ, Langel U. Cargo-dependent cytotoxicity and delivery efficacy of cell-penetrating peptides: a comparative study. Biochem J. 2007;407(Pt 2):285–292. | ||
Shang L, Nienhaus K, Jiang X, et al. Nanoparticle interactions with live cells: quantitative fluorescence microscopy of nanoparticle size effects. J Nanotechnol. 2014;5(1):2388–2397. | ||
Commisso C, Davidson SM, Soydaner-Azeloglu RG, et al. Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature. 2013;497(7451):633–637. | ||
Dawei X, Derek D, Linhai J, Samways DSK, He D. Designed filamentous cell penetrating peptides: probing supramolecular structure-dependent membrane activity and transfection efficiency. Chem Commun. 2015;51(59):11757–11760. | ||
Jordan MA, Wendell K, Gardiner S, Derry WB, Copp H, Wilson L. Mitotic block induced in HeLa cells by low concentrations of paclitaxel (Taxol) results in abnormal mitotic exit and apoptotic cell death. Cancer Res. 1996;56(4):816–825. | ||
Jordan MA, Toso RJ, Thrower D, Wilson L. Mechanism of mitotic block and inhibition of cell proliferation by Taxol at low concentrations. Proc Natl Acad Sci U S A. 1993;90(20):9552–9556. | ||
Zasadil LM, Andersen KA, Yeum D, et al. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med. 2014;6(229):229–243. | ||
Carazo-Salas RE, Gruss OJ, Mattaj IW, Karsenti E. Ran-GTP coordinates regulation of microtubule nucleation and dynamics during mitotic-spindle assembly. Nat Cell Biol. 2001;3(3):228–234. | ||
Patel LN, Zaro JL, Shen WC. Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharm Res. 2007;24(11):1977–1992. | ||
Teravetisyan G, Tünnemann G, Nowak D, et al. Cell entry of arginine-rich peptides is independent of endocytosis. J Biol Chem. 2009;284(6):3370–3378. | ||
Hillaireau H, Couvreur P. Nanocarriers’ entry into the cell: relevance to drug delivery. Cell Mol Life Sci. 2009;66(17):2873–2896. | ||
Futaki S, Nakase I, Tadokoro A, Takeuchi T, Jones AT. Arginine-rich peptides and their internalization mechanisms. Biochem Soc Trans. 2007;35(Pt 4):784–787. | ||
Ziegler A, Nervi P, Dürrenberger M, Seelig J. The cationic cell-penetrating peptide CPP(TAT) derived from the HIV-1 protein TAT is rapidly transported into living fibroblasts: optical, biophysical, and metabolic evidence. Biochemistry. 2005;44(1):138–148. | ||
Fonseca SB, Pereira MP, Kelley SO. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv Drug Deliv Rev. 2009;61(11):953–964. | ||
Hamaguchi T, Matsumura Y, Suzuki M, et al. NK105, a paclitaxel-incorporating micellar nanoparticle formulation, can extend in vivo antitumour activity and reduce the neurotoxicity of paclitaxel. Br J Cancer. 2005;92(7):1240–1246. | ||
Sparreboom A, Van TO, Nooijen WJ, Beijnen JH. Tissue distribution, metabolism and excretion of paclitaxel in mice. Anticancer Drugs. 1996;7(1):78–86. | ||
Wang F, Shen Y, Xu X, et al. Selective tissue distribution and long circulation endowed by paclitaxel loaded PEGylated poly(ε-caprolactone-co-L-lactide) micelles leading to improved anti-tumor effects and low systematic toxicity. Int J Pharm. 2013;456(1):101–112. | ||
Soma D, Kitayama J, Ishigami H, Kaisaki S, Nagawa H. Different tissue distribution of paclitaxel with intravenous and intraperitoneal administration. J Surg Res. 2009;155(1):142–146. | ||
Cheng H, Zhu JY, Xu XD, et al. Activable cell-penetrating peptide (ACPP) conjugated prodrug for tumor targeted drug delivery. ACS Appl Mater Interfaces. 2015;7(29):16061–16069. | ||
Su J, Sun H, Meng Q, et al. Long circulation red-blood-cell-mimetic nanoparticles with peptide-enhanced tumor penetration for simultaneously inhibiting growth and lung metastasis of breast cancer. Adv Funct Mater. 2016;26(8):1243–1252. |
Supplementary materials
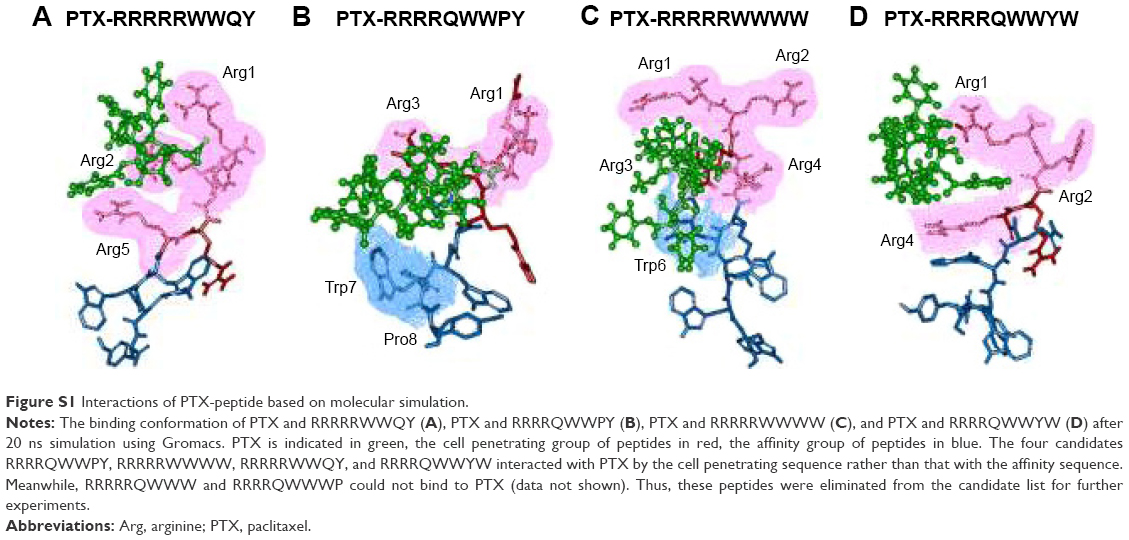 | Figure S1 Interactions of PTX-peptide based on molecular simulation. |
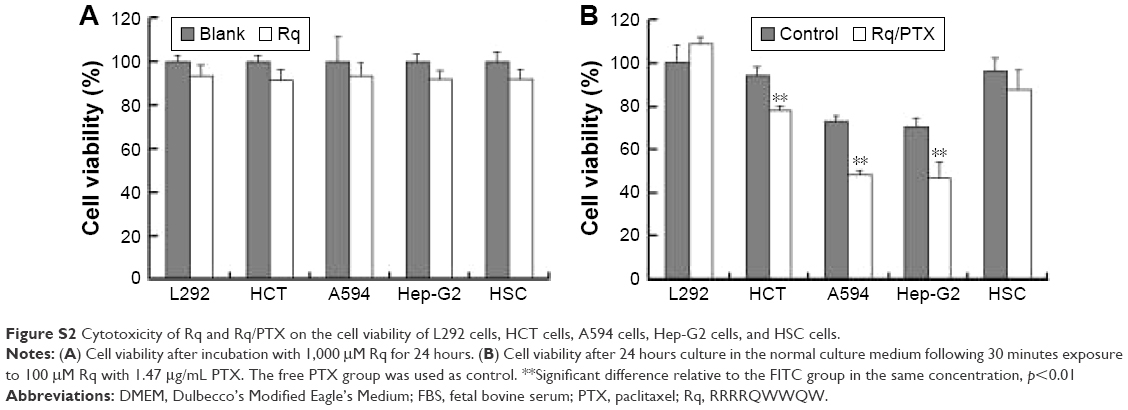 | Figure S2 Cytotoxicity of Rq and Rq/PTX on the cell viability of L292 cells, HCT cells, A594 cells, Hep-G2 cells, and HSC cells. |
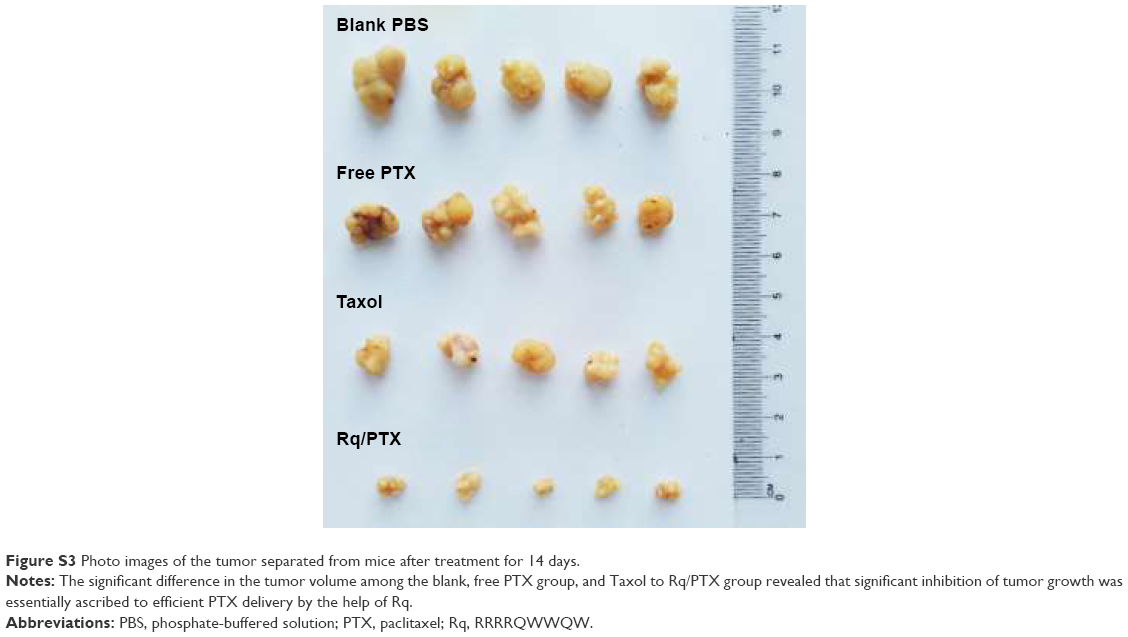 | Figure S3 Photo images of the tumor separated from mice after treatment for 14 days. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.