Back to Journals » Pharmacogenomics and Personalized Medicine » Volume 14
Non-Classical 21-Hydroxylase Deficiency: Analysis of a Mutant Gene in a Uyghur Family and Literature Review
Authors Maimaitiming J, Amuti G, TuHuTi A, Chen Y, Song XX, Wang J, Alimu A, Zhang K, Abudounaiyimu M, Jiang J, Wang XL, Guo YY
Received 22 December 2020
Accepted for publication 2 March 2021
Published 7 April 2021 Volume 2021:14 Pages 409—416
DOI https://doi.org/10.2147/PGPM.S297607
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Martin H Bluth
Jimilanmu Maimaitiming,1 Guli Amuti,1 AiHeMaiTiJiang TuHuTi,1 Yuan Chen,1 Xiang-Xin Song,1 Jing Wang,1 Adila Alimu,1 Kaidi Zhang,1 Munila Abudounaiyimu,1 Jun Jiang,2 Xin-Ling Wang,1 Yan-Ying Guo1
1People’ s Hospital of Xinjiang Uygur Autonomous Region, Urumqi, 830001, People’s Republic of China; 2Key Laboratory of Genome Science and Information, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, 10029, People’s Republic of China
Correspondence: Yan-Ying Guo; Xin-Ling Wang
Department of Endocrinology, People’s Hospital of Xinjiang Uygur Autonomous Region, No. 91 of Tianchi Road, Tianshan District, Urumqi, 830001, People’s Republic of China
Tel +86 991 8563546
Fax +86 991 8563607
Email [email protected]; [email protected]
Objective: The gene mutation and clinical characteristics of a patient with non-classical 21-hydroxylase deficiency and his family were analyzed.
Methods: A patient was diagnosed with non-classical 21-hydroxylase deficiency in the Department of Endocrinology of People’s Hospital of Xinjiang Uygur Autonomous Region in December 2016. The clinical data and related gene-sequencing results were analyzed. The detected mutations were verified in nine members of the family.
Results: Gene-sequencing results revealed that the proband and the other three members of the family (proband, proband’s mother’s younger brother and the proband’s mother’s younger brother’s younger daughter, and proband’s second elder sister) shared the following mutations: Ile173Asn, Ile237Asn, Val238Glu, Met240Lys, Val282Leu, Leu308Phefs*6, Gln319Ter, Arg357Trp, and Arg484Profs. The Val282Leu mutation was heterozygous in the proband’s mother’s younger brother’s younger daughter, but homozygous in the other three individuals. The father of the proband, the elder brother of the father of the proband, the third younger brother of the father of the proband, and the elder sister of the proband all carried only the Val282Leu mutation.
Conclusion: Val282Leu is the gene responsible for non-classical 21-hydroxylase deficiency. Screening for this gene in the offspring of patients with non-classical 21-hydroxylase deficiency may help to identify cases early.
Keywords: congenital adrenal hyperplasia, non-classical 21-hydroxylase deficiency, CYP21A2 gene, gene mutation
Introduction
Congenital adrenal hyperplasia (CAH) is a disorder of adrenal corticosteroid synthesis caused by the mutation of key genes. The most common type of CAH is 21-hydroxylase deficiency (21-OHD), which accounts for 90–95% of all cases and follows a pattern of autosomal recessive inheritance. The clinical type of 21-OHD can be classified according to the activity of the residual enzyme caused by the gene mutation as either classical or non-classical. Classical CAH can be divided into salt-wasting or simple virilizing.1 Non-classical CAH is also known as delayed CAH, and its prevalence in the population is 0.001%,2 although it may be as high as 6% in some ethnic groups and women with a medical disorder that produces excessive hair growth.3 Non-classical 21-OHD is a type of 21-OHD that exhibits a low decrease in enzyme activity, and the symptoms are non-specific. Therefore, clinicians lack understanding of the disease. In the present study, one patient with non-classical 21-OHD and his nine family members were genetically sequenced, and the screened genes were analyzed.
Materials and Methods
Subjects
The subjects of this study were a Uyghur family, consisting of one patient and nine other family members, with non-classical 21-OHD. Clinical data and serum specimens were collected. This study was conducted in accordance with the declaration of Helsinki. This study was conducted with approval from People’s Hospital of Xinjiang Uygur Autonomous Region (KY2019051515). Written informed consent was obtained from the participants.
Molecular Biological Detection
In full-length sequencing of the CYP21A2 gene, the point mutation type of the CYP21A2 gene was detected by direct sequencing. According to related literature,12,16–21 specific upstream and downstream primers of CYP21A2, CYP21A1P, and chimeric genes were designed and synthesized based on the difference in sequences of the real gene CYP21A2 and pseudogene CYP21A1P. Full-length sequencing of amplified 3.4 Kb DNA was conducted and compared with the CYP21A2 standard reference sequence GenBank M12792.1 to detect point mutations in the full length of the CYP21A2 gene.
Results
Clinical Data
The proband was a married 29-year-old man, and his main symptom was a previous history of hyperlipidemia. The proband required physical examination in the hospital due to premature ejaculation. Examination of sex hormone levels revealed elevated progesterone levels. At birth, the external genitals of the patient were normal, and there was no evidence of symptoms, such as low cry, reduced activity, or weakness. At the age of 12 years, the patient’s Adam’s apple was protruded and his beard appeared. At the age of 13 years, changes in his voice occurred, his penis and testes increased in size, and spermatorrhea and morning erection occurred.
Physical Examination
The patient was 176 cm tall and weighed 82 kg. His blood pressure measured 137/70 mmHg. His facial features appeared normal, and there was no abnormality in the facial midline. The patient had an Adam’s apple, but no thyromegaly. The length of the penis was approximately 8 cm, and the volume of bilateral testes was approximately 15 mL.
Laboratory Examination
No abnormalities were found in routine blood, urine, and fecal tests. Serum liver, renal and thyroid function, and blood ion levels all appeared normal. Levels of adrenocorticotropic hormone (ACTH), cortisol and sex hormones in the patient at different time points are presented in Table 1.
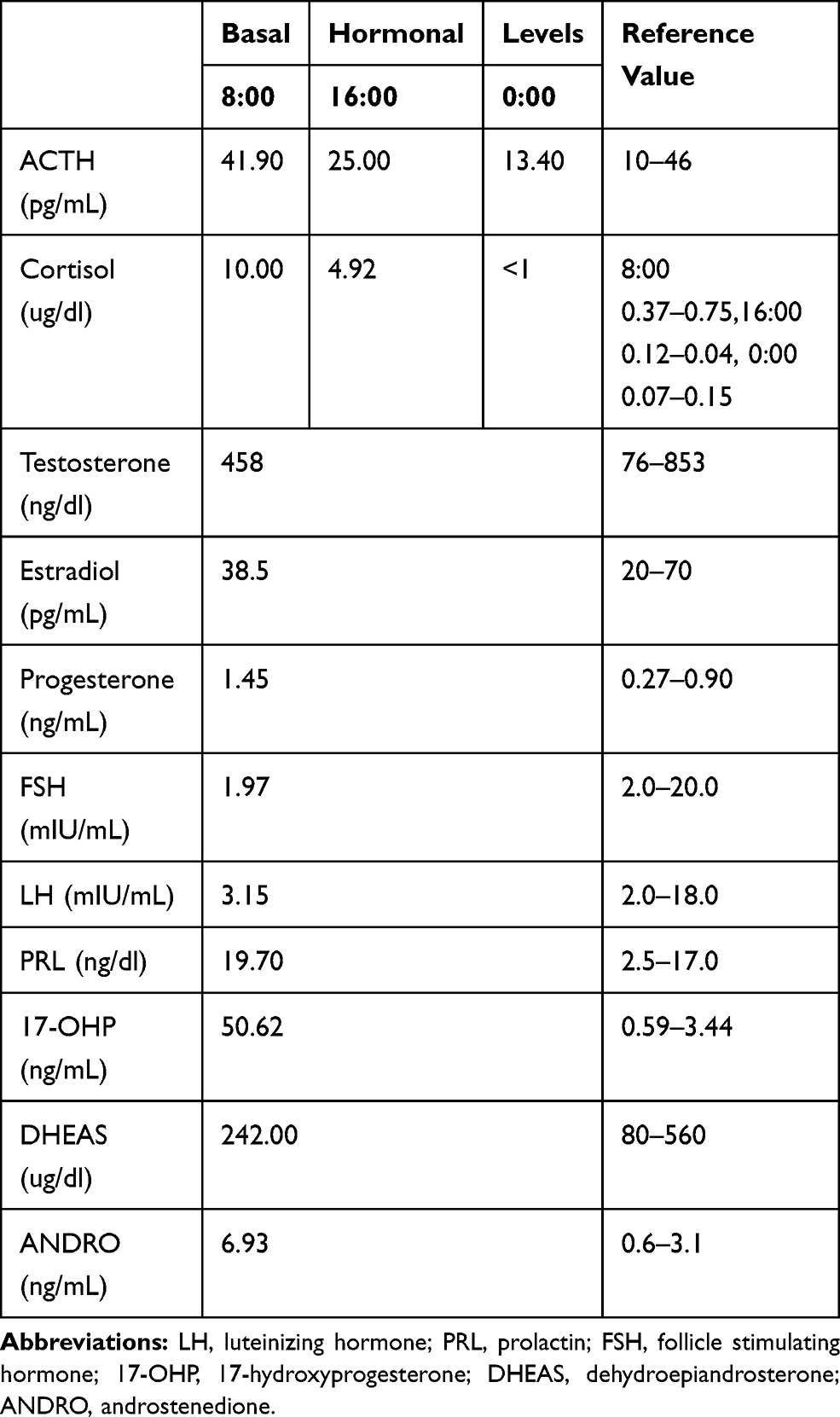 |
Table 1 ACTH, Cortisol and Basal Hormonal Levels of Patient at Different Time Points |
Imaging Results
Adrenal CT suggested thickening of the left adrenal joint. No abnormalities were found after type-B ultrasonography of the genitals and urinary tract.
Family Mapping
The patient’s father was living and proved to be a carrier of the mutation, but the patient’s mother was deceased. Both parents were of Uyghur ethnicity. Theirs was not a consanguineous marriage, and there was no family history of the disease (Figure 1).
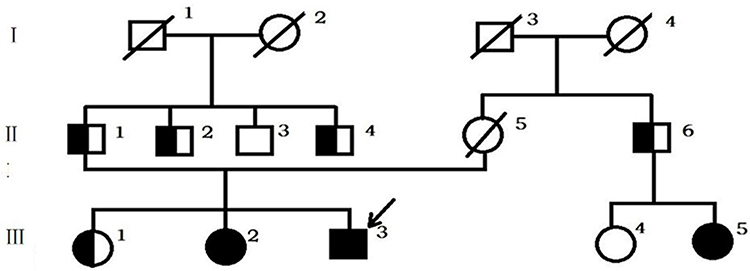 |
Figure 1 Genealogical tree of Sardinian family: full black circle or square = Val282Leu homozygous patients; black and white circle or square = Val282Leu heterozygous subjects; full white circle or square = wild-type subjects. |
Values of 17-Hydroxyprogesterone and Free Testosterone in the Family Members
Hormone levels in various family members are presented in Table 2. Some of the results were out of normal range.
 |
Table 2 Levels of 17 Hydroxyprogesterone and Free Testosterone in Family Members |
DNA Sequencing Results
Results of DNA sequencing are given in Figure 2. A total of 10 blood samples were collected from the patient and his family (members I1, I2, I3, I4 and II4 died before study). Serum DNA was extracted, and exons on the CYP21A2 gene associated with 21-OHD were sequenced by PCR. The method for the amplification of the CYP21A2 gene can be found in Tables 3–5. These results were compared with the CYP21A2 gene sequence. The results revealed that the proband, the proband’s mother’s younger brother, the proband’s mother’s younger brother’s younger daughter, and the proband’s second elder sister shared the following mutations: Ile173Asn, Ile237Asn, Val238Glu, Met240Lys, Val282Leu, Leu308Phefs*6, Gln319Ter, Arg357Trp, and Arg484Profs. The Val282Leu mutation was heterozygous in the proband’s mother’s younger brother, and homozygous in the other three members. The father of the proband, the first younger brother of the father of the proband, the third younger brother of the father of the proband, and the elder sister of the proband all carried only one mutation, namely Val282Leu. No abnormalities were detected in related fragments in other family members. The results of the gene sequencing in the proband are presented in Table 6, and the peak shape diagram of the gene sequencing is shown in Figure 2. Ile173Asn, Ile237Asn, Val238Glu, Met240Lys, Arg357Trp, and Val282Leu were missense mutations, Leu308Phefs*6 was an insertion mutation, Arg484Profs was a deletion mutation, and Gln319Ter was a nonsense mutation.
 |
Table 3 Primers for PCR Amplification and Sequencing of CYP21 Gene |
 |
Table 4 Amplification Protocol |
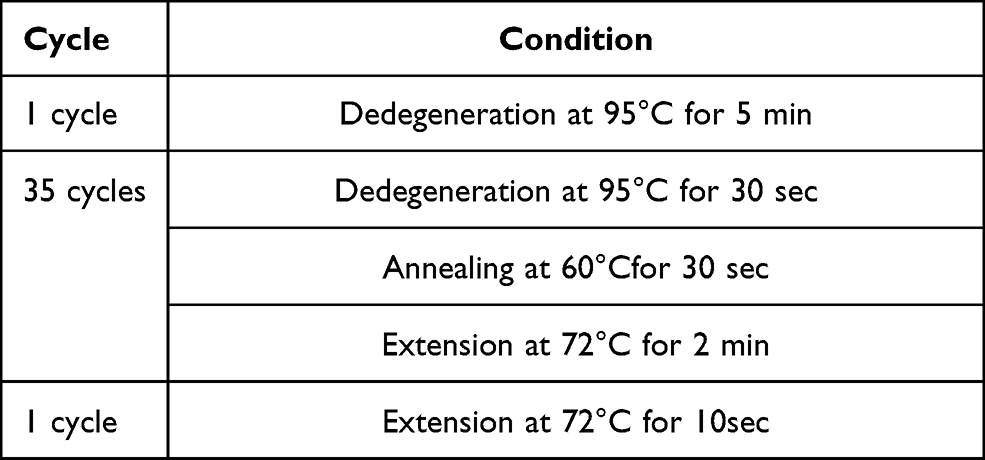 |
Table 5 Reaction Condition |
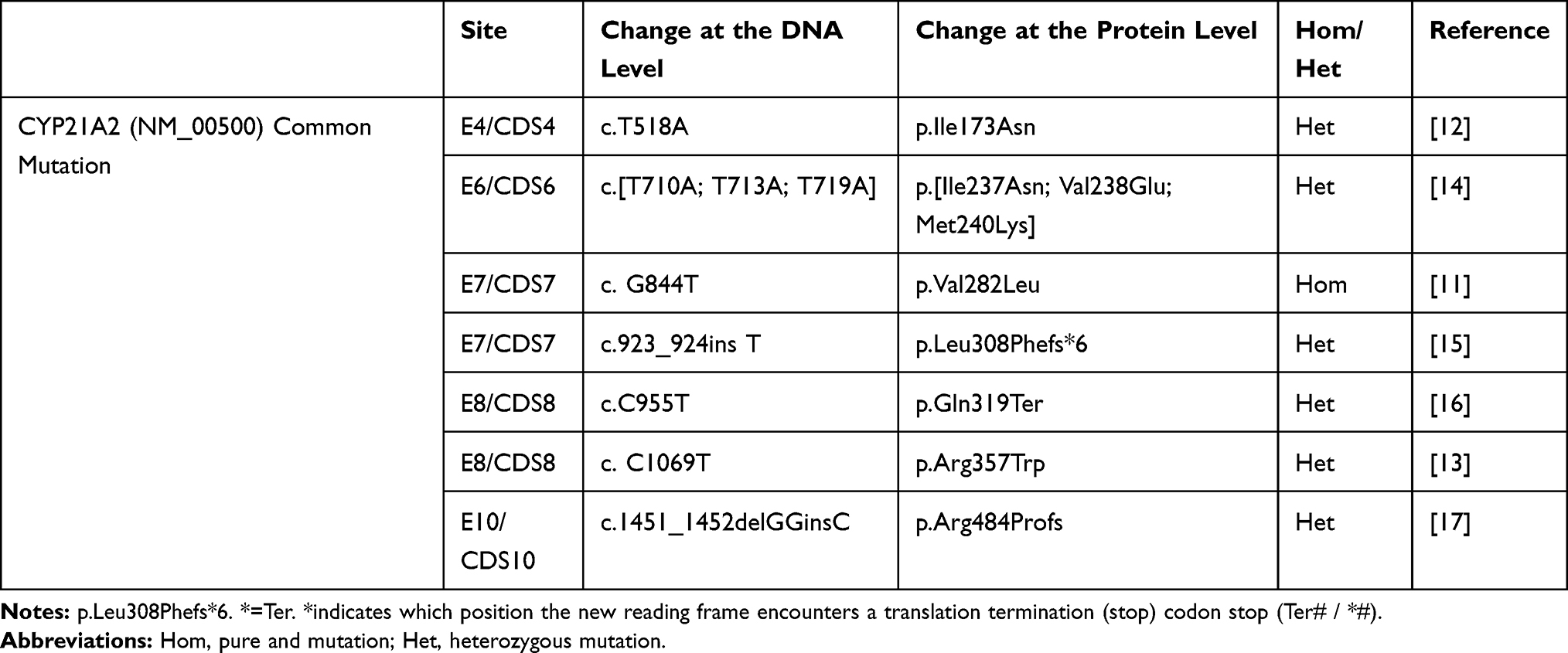 |
Table 6 Proband Gene Sequencing |
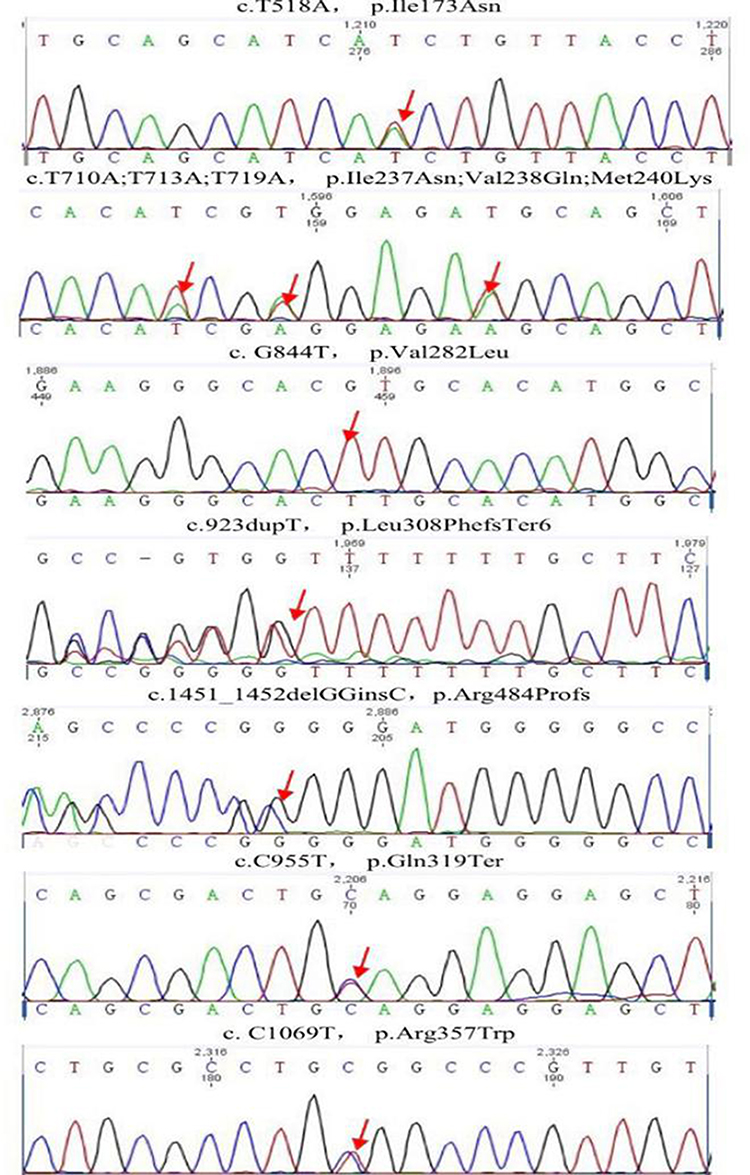 |
Figure 2 Proband gene sequencing: the red arrows indicate the sites of genetic mutation. |
Discussion
There are two CYP21 genes in humans, CYP21A2, which is active, and the inactive CYP21 pseudogene (CYP21A1P), and these are located in the short arm of chromosome 6 (6p21.3). The CYP21A2 gene is highly homologous to its pseudogene. It is arranged in a series of exons and introns, and the homogeneity of these is 98% and 96%, respectively. This leads to similarities in its DNA structure.4 The 21-OHD disease-causing gene CYP21A is one of the most polymorphic genes in humans. Typically, a patient with non-classical CAH retains 20–50% of residual 21-hydroxylase activity.5 This causes an increase in serum androgen and 17-hydroxyprogesterone, which has little effect on the synthesis of cortisol. Levels of serum cortisol and aldosterone remain in the normal range. Therefore, these clinical symptoms are non-specific.
In the present study, nine mutations were detected at seven sites in the CYP21A2 gene coding region in the proband and his relatives. Currently, more than 200 CYP21A2 mutations have been reported (http://www.hgmd.cf.ac.uk). The mutations carried by the proband were nonsense mutations, all have been previously reported, and no new mutation site was found. However, this is the first time that a patient carrying nine mutations has been reported.
This disease follows an autosomal recessive pattern of inheritance, in which both parents are carriers of the relevant gene. The proband and two other family members all carried the homozygous mutation Val282Leu. In patients with non-classical 21-OHD, the majority of mutations are caused by intergenic recombination, including deletion and conversion, between real genes and pseudogenes. Some 95% of mutant genes are recombinant, but only 5% arise from spontaneous mutations rather than gene conversion events.6 As the patient’s mother had already died, her genes could not be sequenced. However, the patient’s father only carried Val282Leu. It was speculated that the patient’s mother also carried Val282Leu and that the other mutations were also derived from her.
Tajima et al reported that the most common genotypic mutation of non-classical 21-OHD patients is P30L in Japanese patients7 and V281L in Caucasian patients. These account for 45–60% of all mutations,8 although the rate for V281L is as high as 80% in Jewish patients.9 Peking Union Medical College Hospital reports that the most common genotypic mutation of non-classical 21-OHD patients is P30L, which occurs at a rate of 37.5% in Chinese (Han nationality) individuals, while the rate for V281L is 25%, which is similar to that seen in Japanese patients.10 A previous study reported that in 20 Uyghur children from Xinjiang, mutations were most common in I172N, while P30L mutations were not detected.11 We also failed to find any evidence of P30L mutations in this study.
The V281L mutation occurs when the 281st amino acid codon, GTG, is replaced by TTG, and the normal codon of valine is replaced by a codon of leucine or glycine, causing a missense mutation. This mutation reduces the activity of the enzyme to 20–50% of normal levels. This mutation is a genetic marker of non-classic 21-OHD associated with HLA-B14 and DR1.12 V281L is the most common mutation in non-classical CAH patients, accounting for 73–87% of the mutations observed in this group. Very few patients with this mutation develop classic CAH.13–15
The I173N missense mutation arises when the 173th normal codon of ATC is replaced by AAC. It is usually detected in the CYP21 pseudogene, which may be transformed into the CYP21B gene, although DNA expression is unaltered. Furthermore, a partial ability to synthesize aldosterone is retained, although 21-hydroxylase activity is reduced by a factor of 10. However, 17-hydroxylase activity is retained. This mutation usually manifests as simple virilizing CAH, but it can also be detected in patients with the salt-wasting variant of the disease. This phenotypic difference may be caused by individual differences in the synthesis of the mutated enzyme or differences in catabolism or excretion rate.16,17
J.-H. Choi et al reported that three consecutive mutations were detected in the same exon in a female patient with CAH: Ile237Asn, Val238Glu and Met240Lys. This cluster of mutations induces complete loss of 21-hydroxylase activity. The clinical phenotype should be salt-wasting CAH.18 The insertion mutation Leu308Phefs*6 results in complete loss of enzyme activity, which can lead to salt-wasting CAH.19 The nonsense mutation Gln319Ter, located in exon 8, replaces the glutamine encoded by CAG with TAG. This causes mRNA transcription to be terminated early, and the subsequent absence of the heme binding region of the P450 polypeptide leads to loss of enzyme activity. This mutation is more common in salt-wasting and simple virilizing CAH patients.20 Chiou reported that the mutation Arg357Trp was detected in patients with simple virilizing CAH. This nonsense mutation is located at the substrate binding site of the enzyme of P450C and causes the failure of enzyme substrate transformation. Although enzymatic activity is partially retained, the loss of enzyme activity is significantly lower than that of 172N. This nonsense mutation does not affect the expression of DNA, but instead causes a defect in the enzyme.17 The Arg484Profs mutation has been detected in a patient with salt-wasting CAH. This frameshift mutation induces the predicted protein to be longer than the normal protein by 45 amino acids. This may seriously disrupt enzyme activity, causing classical CAH.21
CAH genotypes show good correlation with clinical phenotypes, especially in salt-wasting CAH and non-classical CAH. However, a few studies have reported that the clinical phenotypes of salt-wasting or simple virilizing CAH are characterized by non-classical CAH.19 Two-thirds of patients with non-classical CAH carry a severe mutation in at least one of the two alleles. However, the clinical picture in these patients is not markedly different to patients with less serious mutations in both alleles.22
The average age at diagnosis of male patients with non-classical CAH is 24.5 years, and most patients have no obvious signs of disease at diagnosis. Indeed, 51% of patients are detected only when family sequencing is carried out. Symptomatic patients are identified by early pubic hair (29%), hirsutism or acne (11%), infertility (2%), early or delayed adolescence (4%), and height problems (15%).14 In our study, the proband was diagnosed at 29 years of age. There were no obvious clinical signs or symptoms in the patient himself or in members of his family. However, elevated levels of progesterone were detected during physical examination. The proband and several other relatives carried the V281L mutation, and all displayed the clinical phenotype of non-classical CAH. The 2010 clinical practice guidelines of the Endocrine Society recommend that genotyping should be used to validate a diagnosis and to enable genetic counseling to be offered. It is recommended that all patients affected by CAH should be genotyped. Long-term follow-up of the offspring of women with non-classical CAH showed that while no growth abnormalities or intellectual deficits were seen in these children, there was an increased incidence of both classical and non-classical CAH.23–25 Therefore, family members of patients should undergo genetic screening.
Conclusion
In the present study, we found mutations in Val282Leu in several family members. This mutation was heterozygous in one individual and homozygous in another three. In four family members, this was the only mutation found. Genetic screening in patients with CAH patients should lead to a better understanding of the mutations responsible for 21-OHD.
Data Sharing Statement
The datasets used and/or analysed during the current study available from the corresponding author, Yan-Ying Guo ([email protected]), on reasonable request.
Ethics Approval and Consent to Participate
This study was conducted in accordance with the declaration of Helsinki. This study was conducted with approval from People’s Hospital of Xinjiang Uygur Autonomous Region (KY2019051515). Written informed consent was obtained from the participants.
Consent for Publication
Obtained from the participants.
Acknowledgments
Thanks for the support from The Xinjiang Uygur Autonomous Region science and technology basic conditions platform construction project (PT1601).
Funding
The study was supported by Special Scientific Research Project of Young Medical Science and Technology talents in Health and Health of Autonomous region(WJWY-201813) and Scientific research project of special training plan for scientific and technological talents of ethnic minorities in Xinjiang (2019D03022).
Disclosure
The authors report no conflicts of interest in this work.
References
1. New MI. An update of congenital adrenal hyperplasia. Ann N Y Acad Sci. 2004;1038(1):14–43. doi:10.1196/annals.1315.009
2. Speiser PW, Dupont B, Rubinstein P, et al. High frequency of nonclassical steroid 21-hydroxylase deficiency. Am J Hum Genet. 1985;37(4):650–667.
3. Kuttenn F, Couillin P, Girard F, et al. Late-onset adrenal hyperplasia in hirsutism. N Engl J Med. 1985;313(4):224–231. doi:10.1056/NEJM198507253130404
4. Speiser PW, New MI, Pc W. white, Molecular genetic analysis of nonclassic steroid 21-hydroxylase deficiency associated with HLA-B14, DR1. N Engl J Med. 1988;319:19–23.
5. Amor M, Parker KL, Globerman H, New MI, White PC. Mutation in the CYP21B gene (Ile-172—-Asn) causes steroid 21-hydroxylase deficiency. Proc Natl Acad Sci U S A. 1988;85(5):1600–1604. doi:10.1073/pnas.85.5.1600
6. Chiou SH, Hu MC, Chung BC. A missense mutation at Ile172—-Asn or Arg356—-Trp causes steroid 21-hydroxylase deficiency. J Biol Chem. 1990;265(6):3549–3552. doi:10.1016/S0021-9258(19)39804-7
7. Choi JH, Jin HY, Lee BH, et al. Clinical phenotype and mutation spectrum of the CYP21A2 gene in patients with steroid 21-hydroxylase deficiency. Exp Clin Endocrinol Diabetes. 2012;120(01):23–27. doi:10.1055/s-0031-1287789
8. Speiser PW, Dupont J, Zhu D, et al. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. 1992;90(2):584–595. doi:10.1172/JCI115897
9. Globerman H, Amor M, Parker KL, New MI, White PC. Nonsense mutation causing steroid 21-Hydroxylase deficiency. J Clin Invest. 1988;82(1):139–144. doi:10.1172/JCI113562
10. Wedell A, Ritzén EM, Haglund-Stengler B, Luthman H. Steroid 21-hydroxylase deficiency: three additional mutated alleles and establishment of phenotype-genotype relationships of common mutations. Proc Natl Acad Sci U S A. 1992;89(15):7232–7236. doi:10.1073/pnas.89.15.7232
11. Krone N, Arlt W. Genetics of congenital adrenal hyperplasia. Best Pract Res Clin Endocrinol Metab. 2009;23(2):181–192. doi:10.1016/j.beem.2008.10.014
12. Tusie-Luna MT, Traktman P, White PC. White, Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J Biol Chem. 1990;265:20916–20922.
13. Lee HH. CYP21 mutations and congenital adrenal hyperplasia. Clin Genet. 2001;59:293–301.
14. Tajima T, Fujieda K, Nakae J, Mikami A, Cutler GB
15. Balsamo A, Cacciari E, Baldazzi L, et al. CYP21 analysis and phenotype/genotype relationship in the screened population of the Italian Emilia-Romagna region. Clin Endocrinol (Oxf). 2000;53(1):117–125. doi:10.1046/j.1365-2265.2000.01048.x
16. Weintrob N, Brautbar C, Pertzelan A,et al. Genotype-phenotype associations in non-classical steroid 21-hydroxylase deficiency. Clin endocrinol (Oxf). 2000;53(1):117–125.
17. Zhang B, Lu L, Lu Z. Molecular diagnosis of Chinese patients with 21-hydroxylase deficiency and analysis of genotype–phenotype correlations. J Int Med Res. 2017;45(2):481–492. doi:10.1177/0300060516685204
18. Li J, Luo YF. Genotypes and phenotypes in Uygur children with 21-hydroxylase deficiency in Xinjiang, China. Zhongguo Dang Dai Er Ke Za Zhi. 2016;18(2):141–146.
19. New MI, Abraham M, Gonzalez B, et al. Genotype-phenotype correlation in 1507 families with congenital adrenal hyperplasia owing to 21-hydroxylase deficiency. Proc Natl Acad Sci U S A. 2013;110(7):2611–2616. doi:10.1073/pnas.1300057110
20. Livadas S, Dracopoulou M, Dastamani A, et al. The spectrum of clinical, hormonal and molecular findings in 280 individuals with nonclassical congenital adrenal hyperplasia caused by mutations of the CYP21A2 gene. Clin endocrinol (Oxf). 2015;82(4):543–549. doi:10.1111/cen.12543
21. Bidet M, Bellanné-Chantelot C, Galand-Portier MB, et al. Clinical and molecular characterization of a cohort of 161 unrelated women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency and 330 family members. J Clin Endocrinol Metab. 2009;94(5):1570–1578. doi:10.1210/jc.2008-1582
22. Savaş-Erdeve Ş, Çetinkaya S, Abalı ZY, et al. Clinical, biochemical and genetic features with nonclassical 21-hydroxylase deficiency and final height. J Pediatr Endocrinol Metab. 2017;30(7):759–766. doi:10.1515/jpem-2017-0088
23. Hagenfeldt K, Janson PO, Holmdahl G, et al. Fertility and pregnancy outcome in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum Reprod. 2008;23:1607–1613.
24. Bidet M, Bellanné-Chantelot C, Galand-Portier MB, et al. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2010;95(3):1182–1190. doi:10.1210/jc.2009-1383
25. Moran C, Azziz R, Weintrob N, et al. Reproductive outcome of women with 21-hydroxylase-deficient nonclassic adrenal hyperplasia. J Clin Endocrinol Metab. 2006;91(9):3451–3456. doi:10.1210/jc.2006-0062
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.