Back to Journals » Drug Design, Development and Therapy » Volume 15
No Relevant Pharmacokinetic Drug–Drug Interaction Between the Sodium-Glucose Co-Transporter-2 Inhibitor Empagliflozin and Lobeglitazone, a Peroxisome Proliferator-Activated Receptor-γ Agonist, in Healthy Subjects
Authors Kim YK ![]() , Hwang JG
, Hwang JG ![]() , Park MK
, Park MK ![]()
Received 15 January 2021
Accepted for publication 14 April 2021
Published 28 April 2021 Volume 2021:15 Pages 1725—1734
DOI https://doi.org/10.2147/DDDT.S302215
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Qiongyu Guo
Yu Kyong Kim,1,* Jun Gi Hwang,1,* Min Kyu Park1,2
1Department of Clinical Pharmacology and Therapeutics, Chungbuk National University College of Medicine and Hospital, Cheongju, Republic of Korea; 2Department of Pharmacology and Clinical Pharmacology, Dong-A University College of Medicine, Dong-A University Hospital, Busan, Republic of Korea
*These authors contributed equally to this work
Correspondence: Min Kyu Park
Department of Clinical Pharmacology and Therapeutics, Chungbuk National University College of Medicine and Hospital, 776, 1 Sunhwan-ro, Seowon-gu, Cheongju-si, Chungcheongbuk-do, 28644, Republic of Korea
Tel +82 43 269 8708
Fax +82 43 269 8724
Email [email protected]
Purpose: Combination therapy with insulin-independent sodium-glucose cotransporter 2 inhibitors and thiazolidinedione drugs, such as lobeglitazone, has been reported to elicit potential additive efficacy in glycemic control in type 2 diabetes mellitus. This study was conducted to evaluate the pharmacokinetic (PK) drug–drug interactions between empagliflozin and lobeglitazone in healthy subjects.
Subjects and Methods: A randomized, open-label, multiple-dose study was conducted in 30 healthy subjects using a three-treatment, six-sequence, three-way crossover design. Subjects received one of the following treatments once daily for 5 days in each period: 25 mg empagliflozin, 0.5 mg lobeglitazone sulfate, or a combination. Serial blood sampling before every dose and up to 24 h after the last dose was performed during each treatment period. The PK parameters were estimated using noncompartmental methods with the plasma empagliflozin and lobeglitazone concentrations. The absence of a PK interaction was construed as the 90% confidence interval (90% CI) of maximum concentration at steady state (Cmax,ss) and area under the concentration-time curve over the dosing interval (AUCtau) for combination therapy-to-monotherapy ratios within the limits of 0.80– 1.25.
Results: The steady-state plasma empagliflozin and lobeglitazone concentration-time profiles of combination therapy and monotherapy were comparable in the 25 subjects who completed the study. Coadministration of empagliflozin with lobeglitazone did not affect empagliflozin PK (with 90% CIs of 0.956– 1.150 and 0.945– 1.133 for Cmax,ss and AUCtau, respectively). Likewise, empagliflozin did not affect lobeglitazone Cmax,ss or AUCtau (with 90% CIs of 0.869– 0.995 and 0.851– 1.018, respectively). All treatment groups tolerated mild adverse events well.
Conclusion: The lack of PK interactions between lobeglitazone and empagliflozin in combination therapy, along with their good tolerability, indicates that the two drugs can be coadministered without dose adjustment.
Trial Registration Number: NCT02854748, Registered on August 7, 2016.
Keywords: clinical trial, antidiabetic drug, pharmacokinetic interaction, thiazolidinedione, type 2 diabetes
Introduction
A global increase in the prevalence of diabetes mellitus has been heightened by a growing number of metabolic risk factors such as obesity.1–3 As obesity and metabolic syndromes are related to various complications, such as cardiovascular risk, the use of an integrative patient-centered treatment algorithm is crucial in the management of diabetes.4–8 The most recent Standards of Medical Care in Diabetes (2020) recommends metformin and comprehensive lifestyle modification as the first-line therapy for type II diabetes mellitus (T2DM).5 However, more than 40% of newly diagnosed T2DM patients are nonresponders to metformin monotherapy; hence, the use of dual and triple oral glucose-lowering therapies is suggested when the hemoglobin A1C (HbA1C) level remains above the individualized target after 3 months of treatment.5,6,9
Due to each of the hypoglycemic agents having strengths such as efficacy, cardiovascular benefit, minimal hypoglycemia risk, and body weight reduction, choosing the most appropriate combination of pharmacotherapy ultimately leads to successful glycemic control.4 The sodium-glucose cotransporter 2 (SGLT-2) inhibitor class of drugs is known for its potential benefit in cardiorenal protection and weight loss.10–12 Empagliflozin, a selective SGLT-2 inhibitor, exerts its blood glucose level-lowering effect by inducing glucosuria.13 After rapid absorption following oral administration, empagliflozin is primarily metabolized via Phase II conjugation reactions and is excreted in the urine and feces.14
The thiazolidinedione (TZD) class of drugs is reported to be associated with a positive effect on reducing macrovascular risks.15 As a novel peroxisome proliferator-activated receptor-γ agonist with a TZD moiety, lobeglitazone elicits its antihyperglycemic effect by directly improving the survival and function of pancreatic beta cells.16,17 Lobeglitazone is rapidly absorbed into the systemic circulation and is then cleared in a monoexponential manner with a negligible amount excreted in the urine.18,19 SGLT-2 inhibitors and TZDs not only have different mechanisms of action in regulating blood glucose levels but also have a relatively low risk of hypoglycemia, and their synergistic effect on attenuation of the early phase of diabetic nephropathy progression further supports combination therapy as one of the preferred treatment options in the management of T2DM.20 Therefore, a thorough evaluation of drug–drug interactions for expected concomitant drug candidates is crucial even without an index perpetrator or substrate drug.21
Among the SGLT-2 inhibitors and TZDs approved by the Korea Ministry of Food and Drug Safety, empagliflozin and lobeglitazone have yet to be evaluated for pharmacokinetic interactions. In this study, we performed a concomitant-use clinical investigation of potential pharmacokinetic drug–drug interactions between empagliflozin and lobeglitazone in healthy subjects.
Subjects and Methods
The Investigational Drug, Ethics Approval, and Consent to Participate
In order to maximize the probability of identifying a drug–drug interaction, the maximum dose in the shortest dosing interval for empagliflozin and lobeglitazone sulfate, 25 mg and 0.5 mg, respectively, were coadministered or administered separately for 5 days for the assessment of pharmacokinetic interaction and tolerability in healthy male subjects who were eligible for study participation per the inclusion and exclusion criteria of the study.17 Healthy candidates were defined based on clinical assessments such as physical examination performed at the time of screening and clinical laboratory test results. All subjects provided informed consent before participation in any procedure.
The study protocol, including the informed consent form, was approved by the Dong-A University Hospital Institutional Review Board and conducted at the Clinical Trials Center, Dong-A University Hospital, Busan, Korea (ClinicalTrials.gov registry no.: NCT02854748). The study was conducted under the principles stipulated in the Declaration of Helsinki as amended in October 2013 (Fortaleza, Brazil) and the International Council on Harmonization Good Clinical Practice Guideline.22,23
Study Design
Thirty eligible subjects between the ages of 19 and 45 years (inclusive) were randomized for participation in an open-label, multiple-dose, 3-period, 6-sequence crossover study (Figure 1). Five subjects were allocated per sequence, with at least 7 days of washout in between the periods. The investigational drugs were orally administered with 150 mL of water as once-daily doses for 5 days under fasting conditions. All subjects visited the clinical trial center for the first 3 days of dosing in each period, and they were restricted from consuming any food for at least 8 h to maintain an empty stomach before each dosing. On the fourth day of dosing for each period, the subjects were admitted to the clinical trial center in the evening and remained fasted for at least 10 h until the next day for the last dosing and pharmacokinetic blood sampling. All concomitant medications or foods containing caffeine, grapefruit, or alcohol and smoking were prohibited throughout the entire study duration.
 |
Figure 1 Study design. E: empagliflozin 25 mg once daily for 5 days, L: lobeglitazone sulfate 0.5 mg once daily for 5 days. Subjects were randomly assigned to one of the indicated crossover treatment sequences (Sequences A - E) on the day before the investigational drug administration (−1 day), and each period was separated by at least 7 days of washout. |
For the pharmacokinetic profiling of lobeglitazone and empagliflozin, blood samples (4–6 mL) were serially collected in tubes containing ethylenediaminetetraacetic acid (EDTA) before dosing and at 0.33, 0.67, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, and 24 h after the fifth dosing in each period. The samples were centrifuged at 2000 × g for 10 minutes at 4°C. The plasma was separated and aliquoted to be stored below −70°C until the time of concentration analysis.
Determination of Empagliflozin Plasma Concentration
Plasma concentrations of empagliflozin were assayed with a validated method using a Waters ACQUITY ultra-performance liquid chromatography (UPLC) system from Waters (Milford, MA, USA) equipped with a Waters ACQUITY UPLC®BEH C18 column (1.7 μm, 2.1×50 mm) and Waters Micromass XevoTM TQ-S mass spectrometer. The positive ion electrospray method in multiple reaction monitoring (MRM) mode was implemented for quantification. MRM transitions of m/z = 468.15 → 71.15 and 426.15 → 167.10 were monitored for empagliflozin and dapagliflozin (internal standard, ISTD), respectively.
An empagliflozin stock solution of 500 μg/mL was prepared with 10.3 mg empagliflozin in 20% methanol. Where necessary, the samples were diluted with blank human plasma to bring the concentration within the range of calibration. The calibration standards were then prepared by spiking blank plasma samples with known concentrations of empagliflozin ranging from 5.00 to 1000 ng/mL. The calibration curve of empagliflozin was linear within these ranges with a correlation coefficient (r2) value of 0.9990. The accuracy of the quality control samples during the sample analysis of empagliflozin, expressed as the percentage deviation of the mean from the theoretical concentration (%DMT), for within-run and between-run accuracy ranges were −10.6–1.1% and −2.1–3.7%, respectively. The relative standard deviations (%RSD) of the within-run and between-run precision ranges were 1.1–4.8% and 3.4–5.6%, respectively.
Determination of Lobeglitazone Plasma Concentration
A separate validated method from empagliflozin to assess the lobeglitazone plasma concentration was implemented using a Waters ACQUITY UPLC system from Waters (Milford, MA, USA) equipped with a Waters ACQUITY UPLC®BEH C18 column (1.7 μm, 2.1×50 mm) and a Waters Micromass Quattro PremierTM XE mass spectrometer. For lobeglitazone and lobeglitazone-d3 (ISTD), the monitored MRM transitions were m/z = 481.40 → 258.40 and 484.40 → 261.40, respectively.
Lobeglitazone sulfate (12.4 mg) was dissolved in 70% methanol to prepare a stock solution of 100 μg/mL. The calibration standards were then prepared by spiking blank plasma samples with known amounts of lobeglitazone ranging from 1.00 to 250 ng/mL. The calibration curve with lobeglitazone ranging from 1.00 to 250 ng/mL in human plasma was linear with an r2 value of 0.9984. The accuracy (%DMT) of lobeglitazone quality control samples was −7.0–9.0% for within-run accuracy and −4.5–−1.3% for between-run accuracy. The %RSD values were 1.7–6.6% for within-run precision and 2.4–9.1% for between-run precision.
Pharmacokinetic Drug Interaction Evaluation
To evaluate the pharmacokinetic (PK) parameters of empagliflozin and lobeglitazone, the plasma concentrations of each analyte with time after dose were analyzed. The parameters subjected to PK evaluation were as follows: the peak concentration (Cmax,ss) and the time to reach peak concentration after the fifth dose (Tmax,ss), the area under the concentration-time curve from the time of the fifth dosing to the dosing interval (AUCtau), the terminal half-life after the fifth dose (t1/2,ss) and the trough concentration before each dose (Ctrough). The AUCtau was calculated using the linear up/log down method. The actual observed concentrations and times were used to estimate the Cmax,ss and Tmax,ss of empagliflozin and lobeglitazone. t1/2,ss was calculated as ln(2)/λz, where λz stands for the elimination rate constant calculated using linear regression of the log-linear portion of the plasma concentration-time curve.
The PK parameters were estimated using Phoenix® WinNonlin® (version 6.4, Certara USA Inc., NJ, USA). For evaluation of the drug interaction between empagliflozin and lobeglitazone, a mixed effect model was applied to the exponentiated geometric mean differences and the corresponding 90% confidence intervals (CIs) of Cmax,ss and AUCtau. If the 90% CIs of these parameters were within the range of 0.80–1.25, it was concluded that there was an absence of a PK interaction between empagliflozin and lobeglitazone.24,25 All statistical analyses were performed using SPSS® Statistics (version 22.0, IBM, Armonk, NY, USA).
Tolerability Assessment
Tolerability assessments, including vital signs (systolic and diastolic blood pressures and pulse rate), physical examinations, and adverse events, were conducted from the administration of the investigational drug until the end of the clinical study. Information on adverse events was collected via nonleading open questioning and individual interviews of each subject. Where required, concomitant medications were provided for the treatment of adverse events, and further administration of the investigational drugs was discontinued.
Results
Subjects
Thirty-one subjects were randomized and included in the demographic characteristics as the full analysis set. One subject withdrew consent before the investigational drug administration; the remaining 30 participants who received the investigational drug at least once were subjected to tolerability assessment. Among these subjects, a total of 25 subjects completed the study and were included in the PK evaluation (Figure 2).
 |
Figure 2 Subject disposition. |
The mean ± standard deviation of age, height, weight, and BMI were 26.5 ± 4.8 years, 176.0 ± 6.1 cm, 73.1 ± 9.2 kg, and 23.6 ± 2.7 kg/m2, respectively, for the 31 study participants. There were no statistical differences across the sequence groups in terms of demographic distribution with P-values for age, height, weight and BMI of 0.2116, 0.6774, 0.4282 and 0.4616, respectively.
Effects of Lobeglitazone on the Pharmacokinetic Properties of Empagliflozin
The mean plasma empagliflozin concentration-time curves after multiple oral administrations with and without lobeglitazone were almost superimposable in healthy subjects (Figure 3A). The Tmax,ss ranges remained constant regardless of the presence of lobeglitazone (0.67–4.00 h) along with similar t1/2,ss of approximately 8 h (Figure 3A and Table 1).
 |
Figure 3 Mean plasma concentration versus time curves after multiple oral doses for 5 days in healthy subjects (N=25) on linear scales; (A) empagliflozin 25 mg once daily and (B) lobeglitazone 0.5 mg once daily. Inset shows the semi-log scale plots. |
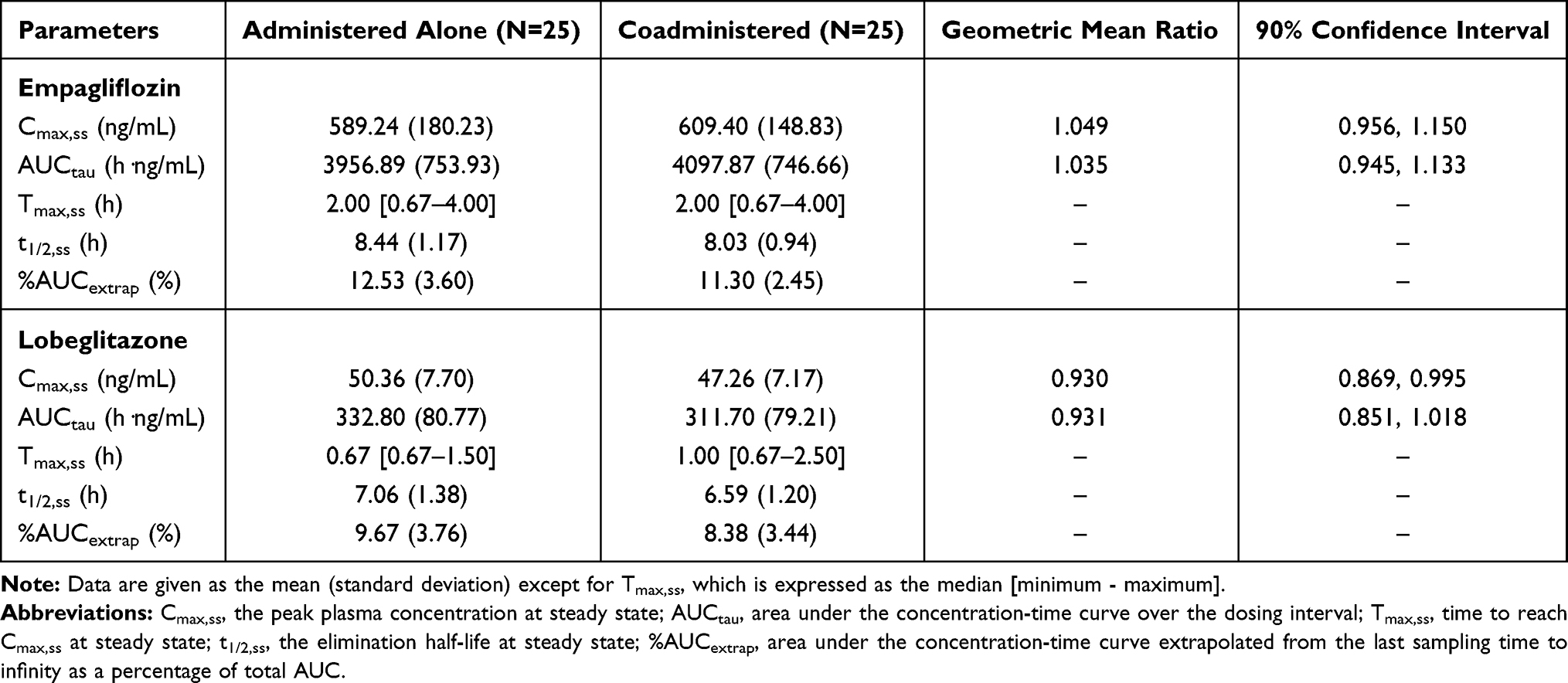 |
Table 1 Pharmacokinetic Parameters of Empagliflozin and Lobeglitazone After Multiple Oral Administrations in Healthy Subjects |
After the fifth oral dose of empagliflozin alone and in combination with lobeglitazone, the systemic exposure to empagliflozin reached a steady state, with the calculated P-values for the Ctrough on Day 5 and Day 4 being 0.723 and 0.602, respectively. The point estimates and the 90% CI of the ln-transformed empagliflozin Cmax,ss and AUCtau between empagliflozin alone and in combination with lobeglitazone fell entirely within the bioequivalence criteria of 0.80 and 1.25 (Table 1, Figure 4A and B). This indicates that multiple administrations of lobeglitazone have no effect on the pharmacokinetics of empagliflozin in healthy subjects.
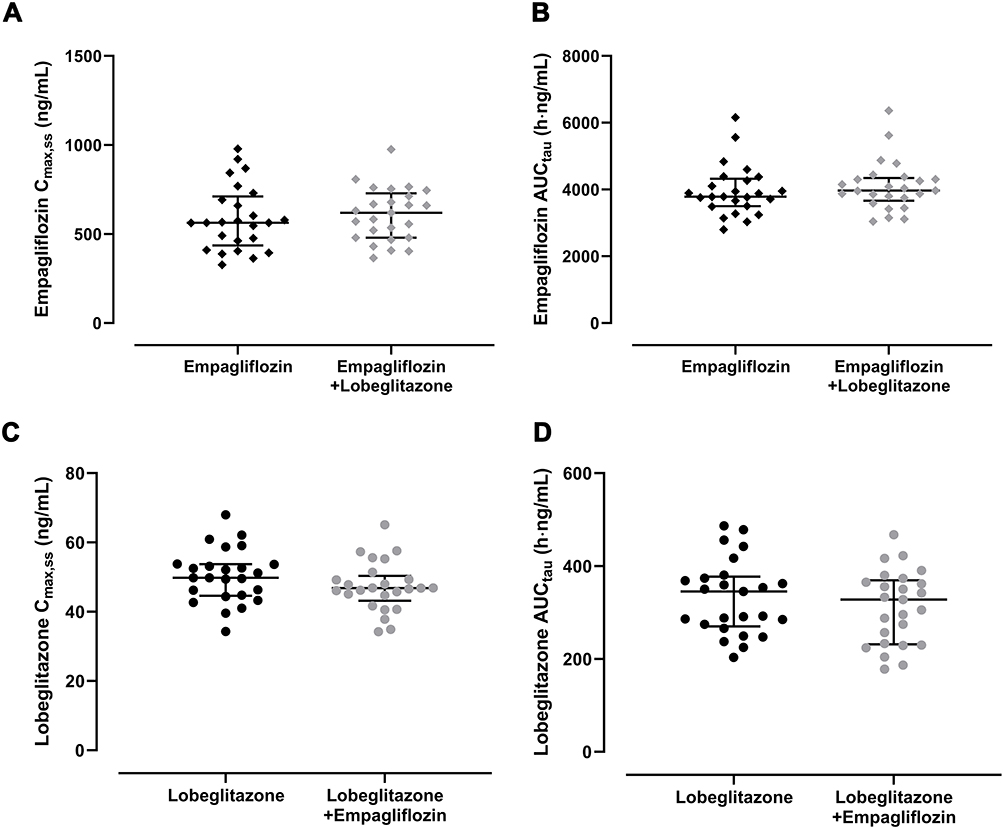 |
Figure 4 Pharmacokinetic parameters Cmax,ss (A and C) and AUCtau (B and D) after multiple oral doses for 5 days in healthy subjects; (A and B) empagliflozin 25 mg once daily and (C and D) lobeglitazone 0.5 mg once daily. The three horizontal lines represent the median (middle), the first quartile (bottom), and the third quartile (top). |
Effects of Empagliflozin on Lobeglitazone Pharmacokinetic Properties
Similar to those of empagliflozin, the mean plasma lobeglitazone concentration-time curves after multiple oral administrations alone and in combination with empagliflozin were similar in healthy subjects, as presented in Figure 3B. The Tmax,ss reached before 2.5 h in both treatment groups, with a t1/2,ss of approximately 7 h (Figure 3B and Table 1).
Lobeglitazone reached steady state before the fifth oral dose alone and in combination with empagliflozin, with the calculated P-values for the Ctrough on Day 4 and Day 3 being 0.293 and 0.453, respectively. The point estimates of the geometric mean ratios and the 90% CI of lobeglitazone Cmax,ss and AUCtau for coadministration of lobeglitazone and empagliflozin compared with lobeglitazone alone were also within the criteria of 0.80 to 1.25 (Figure 4C and D); hence, multiple administrations of empagliflozin did not affect the pharmacokinetics of lobeglitazone in healthy subjects (Table 1).
Tolerability
Among the thirty subjects who received the investigational drug at least once during the study, three adverse events observed in three subjects were considered to be possibly related to the investigational drug administration: nausea, skin exfoliation, and contact dermatitis (Table 2). The subject with contact dermatitis who received lobeglitazone discontinued the study due to the concomitant therapy required for the treatment of the adverse event.
 |
Table 2 Summary of Investigational Drug-Related Adverse Events |
All adverse events were transient and mild in intensity, with no clinically significant findings in other tolerability parameters, such as the laboratory test results, vital signs, and physical examination after multiple administrations of empagliflozin and lobeglitazone in healthy subjects.
Discussion
As a concomitant-use study to assess the pharmacokinetic drug–drug interaction between the two major classes of oral T2DM medications, a multiple-dose crossover design was implemented in this study. The 24-hour blood sampling schedule was sufficient to estimate the exposures of the investigational drugs, with the mean percentage of AUC extrapolated from the last sampling point to infinity for all treatment groups ranging from 8.38% to 12.53% (Table 1). Furthermore, at least 7 days of washout between the periods allowed both empagliflozin and lobeglitazone to be excreted to the lowest level of quantification at predose sampling in the second period. Some of the subjects were overweight in accordance with the BMI categories of over 25 kg/m2 (inclusive), however, there was no statistically significant discrepancies in pharmacokinetic parameters between the overweight and normal weight groups, for both empagliflozin and lobeglitazone (data not shown).
Lobeglitazone, a peroxisome proliferator-activated receptor-γ agonist, stimulates lipogenic activities in adipocytes, thus improving insulin suppression of lipolysis and insulin sensitivity.26–29 Due to its highly protein-bound properties after oral administration, lobeglitazone is mainly metabolized by cytochrome P450 enzymes such as CYP1A2, 2C9 and 2C19, and only a minimal amount is excreted unchanged in the urine.18,30 However, as a sodium-glucose cotransporter-2 inhibitor, the primary route of empagliflozin metabolism in humans is glucuronidation by UGT1A3, 1A8, 1A9 and 2B7 isozymes, and empagliflozin is eliminated in feces and urine as an unchanged parent drug in similar portions.14 Empagliflozin exerts its hypoglycemic effect by reducing renal tubular glucose reabsorption, promoting urinary glucose excretion without impacting the 24-h urine volume.31,32 As expected by the nonoverlapping metabolic pathways, the probability of pharmacokinetic drug interaction after coadministration of empagliflozin and lobeglitazone is low, hence there were no relevant interactions (Table 1 and Figure 3). The empagliflozin and lobeglitazone AUCtau and Cmax,ss were consistent with those previously reported, regardless of the presence of one another.18,33–35 The investigational drug-related adverse events were all within the ranges of known adverse events, and neither the frequency nor the severity were altered when the two investigational drugs were exposed to their highest extent at steady state (Table 2).
This finding is of considerable importance to the real-life clinical setting, as up to 50% of T2DM patients on standard first-line metformin therapy are nonresponders who require alternative or additional hypoglycemic agents with other mechanisms of action.9 Combinations of oral hypoglycemic agents as initial therapy are also advantageous in bringing out synergistic effects on glycemic target achievement and even β-cell preservation when used concomitantly with the thiazolidinedione class of drugs.36–38
This study has potential limitations. Firstly, the current findings are difficult to extrapolate to other drugs sharing the same DDI properties as per the purpose of a concomitant-use study with a non-index drug. However, relevant concomitant medications such as add-on drug therapies or treatments for common co-morbidities in the clinical practice setting are subjected for evaluation of any changes in the drug’s exposure.21 Secondly, as with all exploratory clinical studies including only healthy young men, the representativeness of the study results is limited. Although this study reports the findings from healthy male subjects instead of the indicated group of T2DM patients, neither lobeglitazone nor empagliflozin showed clinically relevant differences in the pharmacokinetic characteristics by sex.19,39 Nonetheless, cautious interpretation of the results and thorough monitoring are compulsory in practice.
Conclusions
The combination of lobeglitazone and empagliflozin did not induce any PK interaction at steady state with good tolerability, predicting no need for dose adjustment when using the two drugs to treat hyperglycemia in T2DM patients.
Abbreviations
AUC, area under the plasma concentration-time curve; AUCtau, area under the plasma concentration-time curve during a dosage interval; Cmax,ss, maximum steady-state plasma concentration during a dosage interval; Ctrough, trough plasma concentration measured at the end of a dosing interval at steady state taken directly before next administration; %DMT, percentage deviation of the mean from the theoretical concentration; EDTA, ethylenediaminetetraacetic acid; HbA1C, hemoglobin A1C; ISTD, internal standard; MRM, multiple reaction monitoring; PK, pharmacokinetic; RSD, relative standard deviations; SGLT-2, sodium-glucose cotransporter 2; t1/2,ss, terminal half-life; T2DM, type 2 diabetes mellitus; Tmax,ss, time to reach maximum plasma concentration following drug administration at steady state; TZD, thiazolidinedione; UPLC, ultra-performance liquid chromatography.
Data Sharing Statement
The dataset supporting the conclusions of this article is included in the article.
Ethics Approval and Informed Consent
Prior to the study conduct, the clinical study protocol was approved by the Dong-A University Hospital Institutional Review Board. The study was conducted in subjects who have provided a written informed consent, under the principles stipulated in the Declaration of Helsinki and the International Council on Harmonization Good Clinical Practice Guideline.22,23
Funding
This study was sponsored by Chong Kun Dang Pharm.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–1053. doi:10.2337/diacare.27.5.1047
2. Yoon KH, Lee JH, Kim JW, et al. Epidemic obesity and type 2 diabetes in Asia. Lancet. 2006;368(9548):1681–1688. doi:10.1016/S0140-6736(06)69703-1
3. Barnes AS. The epidemic of obesity and diabetes: trends and treatments. Tex Heart Inst J. 2011;38(2):142–144.
4. Kim MK, Ko SH, Kim BY, et al. 2019 clinical practice guidelines for type 2 diabetes mellitus in Korea. Diabetes Metab J. 2019;43(4):398–406. doi:10.4093/dmj.2019.0137
5. American Diabetes Association. Standards of medical care in diabetes-2020 abridged for primary care providers. Clin Diabetes. 2020;38(1):10–38. doi:10.2337/cd20-as01
6. Doyle-Delgado K, Chamberlain JJ, Shubrook JH, Skolnik N, Trujillo J. Pharmacologic approaches to glycemic treatment of type 2 diabetes: synopsis of the 2020 American diabetes association’s standards of medical care in diabetes clinical guideline. Ann Intern Med. 2020;173(10):813–821. doi:10.7326/M20-2470
7. Garber AJ, Handelsman Y, Grunberger G, et al. Consensus statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the comprehensive type 2 diabetes management algorithm - 2020 executive summary. Endocr Pract. 2020;26(1):107–139.
8. Sano M. A paradigm shift in the treatment of type 2 diabetes and heart failure. J Atheroscler Thromb. 2020;27(8):727–731. doi:10.5551/jat.RV17042
9. Rashid M, Shahzad M, Mahmood S, Khan K. Variability in the therapeutic response of Metformin treatment in patients with type 2 diabetes mellitus. Pak J Med Sci. 2019;35(1):71–76. doi:10.12669/pjms.35.1.100
10. Scheen AJ. Cardiovascular and renal protection with sodium-glucose cotransporter type 2 inhibitors: new paradigm in type 2 diabetes management … and potentially beyond. Ann Transl Med. 2019;7(Suppl 3):S132. doi:10.21037/atm.2019.05.82
11. Pereira MJ, Eriksson JW. Emerging role of SGLT-2 inhibitors for the treatment of obesity. Drugs. 2019;79(3):219–230. doi:10.1007/s40265-019-1057-0
12. Minze MG, Will KJ, Terrell BT, Black RL, Irons BK. Benefits of SGLT2 inhibitors beyond glycemic control - a focus on metabolic, cardiovascular and renal outcomes. Curr Diabetes Rev. 2018;14(6):509–517. doi:10.2174/1573399813666170816142351
13. Frampton JE. Empagliflozin: a review in type 2 diabetes. Drugs. 2018;78(10):1037–1048. doi:10.1007/s40265-018-0937-z
14. Ingelheim B. Jardiance® (Empagliflozin) Prescribing Information. CT USA: US Food and Drug Administration; 2016.
15. Erdmann E, Charbonnel B, Wilcox R. Thiazolidinediones and cardiovascular risk-a question of balance. Curr Cardiol Rev. 2009;5(3):155–165. doi:10.2174/157340309788970333
16. Kwon MJ, Lee YJ, Jung HS, et al. The direct effect of lobeglitazone, a new thiazolidinedione, on pancreatic beta cells: a comparison with other thiazolidinediones. Diabetes Res Clin Pract. 2019;151:209–223. doi:10.1016/j.diabres.2019.04.006
17. Pharm CKD. Duvie Tab. (Lobeglitazone Sulfate) Prescribing Information. Republic of Korea: Ministry of Food and Drug Safety; 2013.
18. Kim JW, Kim JR, Yi S, et al. Tolerability and pharmacokinetics of lobeglitazone (CKD-501), a peroxisome proliferator-activated receptor-gamma agonist: a single- and multiple-dose, double-blind, randomized control study in healthy male Korean subjects. Clin Ther. 2011;33(11):1819–1830. doi:10.1016/j.clinthera.2011.09.023
19. Park MK, Kim TE, Kim J, et al. Tolerability and pharmacokinetics of lobeglitazone, a novel peroxisome proliferator-activated receptor-gamma agonist, after a single oral administration in healthy female subjects. Clin Drug Investig. 2014;34(7):467–474. doi:10.1007/s40261-014-0197-y
20. Han E, Shin E, Kim G, et al. Combining SGLT2 inhibition with a thiazolidinedione additively attenuate the very early phase of diabetic nephropathy progression in type 2 diabetes mellitus. Front Endocrinol (Lausanne). 2018;9:412. doi:10.3389/fendo.2018.00412
21. U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drugs Evaluation Research. Guidance for Industry: Clinical Drug Interaction Studies - Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions. Maryland: US Food and Drug Administration; 2020.
22. World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191–2194. doi:10.1001/jama.2013.281053
23. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. Guideline for good clinical practice E6 (R2). J Eur Med Agency. 2016.
24. Zhang L, Reynolds KS, Zhao P, Huang SM. Drug interactions evaluation: an integrated part of risk assessment of therapeutics. Toxicol Appl Pharmacol. 2010;243(2):134–145. doi:10.1016/j.taap.2009.12.016
25. U.S. Department of Health and Human Services, Food and Drug Administration, Research CfDE. Guidance for Industry: Statistical Approaches to Establishing Bioequivalence. Maryland: US Food and Drug Administration; 2001.
26. Chiarelli F, Di Marzio D. Peroxisome proliferator-activated receptor-gamma agonists and diabetes: current evidence and future perspectives. Vasc Health Risk Manag. 2008;4(2):297–304. doi:10.2147/VHRM.S993
27. Bermudez V, Finol F, Parra N, et al. PPAR-γ agonists and their role in type 2 diabetes mellitus management. Am J Ther. 2010;17(3):274–283. doi:10.1097/MJT.0b013e3181c08081
28. De Vos P, Lefebvre AM, Miller SG, et al. Thiazolidinediones repress ob gene expression in rodents via activation of peroxisome proliferator-activated receptor gamma. J Clin Invest. 1996;98(4):1004–1009. doi:10.1172/JCI118860
29. Oakes ND, Thalen PG, Jacinto SM, Ljung B. Thiazolidinediones increase plasma-adipose tissue FFA exchange capacity and enhance insulin-mediated control of systemic FFA availability. Diabetes. 2001;50(5):1158–1165. doi:10.2337/diabetes.50.5.1158
30. Lee JH, Noh CK, Yim CS, et al. Kinetics of the absorption, distribution, metabolism, and excretion of lobeglitazone, a novel activator of peroxisome proliferator-activated receptor gamma in rats. J Pharm Sci. 2015;104(9):3049–3059. doi:10.1002/jps.24378
31. Hsia DS, Grove O, Cefalu WT. An update on sodium-glucose co-transporter-2 inhibitors for the treatment of diabetes mellitus. Curr Opin Endocrinol Diabetes Obes. 2017;24(1):73–79. doi:10.1097/MED.0000000000000311
32. Scheen AJ. Pharmacokinetic and pharmacodynamic profile of empagliflozin, a sodium glucose co-transporter 2 inhibitor. Clin Pharmacokinet. 2014;53(3):213–225. doi:10.1007/s40262-013-0126-x
33. Sarashina A, Koiwai K, Seman LJ, et al. Safety, tolerability, pharmacokinetics and pharmacodynamics of single doses of empagliflozin, a sodium glucose cotransporter 2 (SGLT2) inhibitor, in healthy Japanese subjects. Drug Metab Pharmacokinet. 2013;28(3):213–219. doi:10.2133/dmpk.DMPK-12-RG-082
34. Seman L, Macha S, Nehmiz G, et al. Empagliflozin (BI 10773), a potent and selective SGLT2 inhibitor, induces dose-dependent glucosuria in healthy subjects. Clin Pharmacol Drug Dev. 2013;2(2):152–161. doi:10.1002/cpdd.16
35. Heise T, Seman L, Macha S, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of multiple rising doses of empagliflozin in patients with type 2 diabetes mellitus. Diabetes Ther. 2013;4(2):331–345. doi:10.1007/s13300-013-0030-2
36. Cahn A, Cefalu WT. Clinical considerations for use of initial combination therapy in type 2 diabetes. Diabetes Care. 2016;39(Suppl 2):S137–S145. doi:10.2337/dcS15-3007
37. Defronzo RA, Tripathy D, Schwenke DC, et al. Prevention of diabetes with pioglitazone in ACT NOW: physiologic correlates. Diabetes. 2013;62(11):3920–3926. doi:10.2337/db13-0265
38. Kahn SE, Haffner SM, Heise MA, et al. Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med. 2006;355(23):2427–2443. doi:10.1056/NEJMoa066224
39. Baron KT, Macha S, Broedl UC, Nock V, Retlich S, Riggs M. Population pharmacokinetics and exposure-response (efficacy and safety/tolerability) of empagliflozin in patients with type 2 diabetes. Diabetes Ther. 2016;7(3):455–471. doi:10.1007/s13300-016-0174-y
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.