Back to Journals » Diabetes, Metabolic Syndrome and Obesity » Volume 12
NLRP3 inflammasome promotes diabetes-induced endothelial inflammation and atherosclerosis
Authors Wan Z, Fan Y, Liu X, Xue J, Han Z, Zhu C, Wang X
Received 5 July 2019
Accepted for publication 26 August 2019
Published 20 September 2019 Volume 2019:12 Pages 1931—1942
DOI https://doi.org/10.2147/DMSO.S222053
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Prof. Dr. Juei-Tang Cheng
Zhaofei Wan,1 Yan Fan,2 Xiaojun Liu,1 Jiahong Xue,1 Zhenhua Han,1 Canzhan Zhu,1 Xinhong Wang1
1Department of Cardiovascular Medicine, Second Affiliated Hospital of Medical College, Xi’an Jiaotong University, Xi’an, Shaanxi, People’s Republic of China; 2Department of Cardiovascular Medicine, Gansu Provincial Hospital, Lanzhou, People’s Republic of China
Correspondence: Xinhong Wang
Department of Cardiovascular Medicine, Second Affiliated Hospital of Medical College, Xi’an Jiaotong University, 157 Xiwu Road, Xi’an, Shaanxi 710004, People’s Republic of China
Tel +86 298 632 0430
Email [email protected]
Background: NLRP3 inflammasome can be activated by high glucose and links inflammation and metabolic disease. This study aimed to investigate the role of NLRP3 inflammasome in hyperglycemia-induced endothelial inflammation and diabetic atherosclerosis.
Methods: NLRP3 levels in peripheral blood mononuclear cell (PBMC) and plasma IL-1β level were measured in diabetes patients. The activation of NLPR3 was detected in diabetic ApoE−/− mice and human umbilical vein endothelial cells (HUVECs).
Results: Compared with healthy controls, NLRP3 expression levels in PBMC and plasma IL-1β level were significantly higher in diabetes patients but considerably decreased after lifestyle interventions and medicine. Moreover, carotid atherosclerosis was significantly related to plasma IL-1β level in diabetes patients. In diabetic atherosclerosis mouse model, NLRP3 knockdown suppressed NLRP3 inflammasome activation, inhibited the expression of adhesion molecules ICAM-1 and VCAM-1 in intima, reduced atherosclerosis and stabilized atherosclerotic plaque. In vitro, the expression of NLRP3 inflammasome components and the secretion of IL-1β were augmented by high glucose in HUVECs. Moreover, either high glucose or IL-1β promoted the expression of adhesion molecules, which were suppressed by NLRP3 knockdown or IL-1β receptor antagonist.
Conclusion: These findings provide novel insights into pathological mechanisms of diabetic atherosclerosis and have potential therapeutic implications for cardiovascular complications in diabetes.
Keywords: NLRP3 inflammasome, diabetes, atherosclerosis, inflammation
Introduction
The number of diabetic patients has dramatically increased, making diabetics one of the major threats to public health. Cardiovascular complications associated with pervasive and serious atherosclerosis are the most common cause of morbidity and mortality in diabetic patients.1 Although the mechanisms that accelerate diabetic atherosclerosis are still poorly understood, inflammatory response has been involved in all phases of atherosclerosis.2
IL-1β is a major proinflammatory cytokine that induces the generation of other inflammatory mediators, thus instigating a self-enlarging cytokine network.3 NLRP3 inflammasome is a platform for IL-1β production and links inflammation and metabolic disease.4 NLRP3 inflammasome plays a central role in insulin resistance and diabetes.5 Recent evidence has demonstrated that NLRP3 inflammasome participates in the initiation and progression of atherosclerosis.6,7 It is notable that ROS mediates NLRP3 inflammasome activation and contributes to hemodynamic-induced endothelial inflammation and atherosclerosis.8,9
Increased oxidative stress has been detected in diabetic model mice and diabetes patients. Hyperglycemia drives ROS production by mitochondrial electron transport chain, glucose autoxidation and NADPH oxidase.10 Moreover, NLRP3 inflammasome could be activated by high glucose in several types of cells.11–14 Therefore, we hypothesized that NLRP3 inflammasome is involved in hyperglycemia-induced atherosclerosis in diabetes condition. In this study, we performed both in vitro and in vivo experiments to show that the activation of NLRP3 inflammasome mediated high glucose-induced endothelial inflammation and diabetic atherosclerosis.
Materials and methods
Subjects
Fifty-five newly-diagnosed type 2 diabetes mellitus (T2DM) patients without any medication and 20 generally healthy controls with similar age and gender distributions were consecutively recruited. The study complied with the Declaration of Helsinki and was approved by the Ethics Committee of Second Affiliated Hospital of Medical College, Xi’an Jiaotong University, and written informed consent was obtained from all subjects. Definition of T2DM was the following: fasting plasma glucose levels were ≥7 mmol/L, HbA1c levels were ≥6.5%, plasma glucose levels after 2 hrs were ≥11.1 mmol/L or a random plasma glucose level was ≥11.1 mmol/L. Fifty-five T2DM patients were arranged to receive lifestyle interventions and medicine, and finally, 50 of them were analyzed because 5 T2DM patients did not finish the whole procedure. Exclusion criteria included advanced liver disease, renal failure, cancer, valvular heart disease, severe heart failure, stroke, atrial fibrillation, peripheral arterial disease and other vascular diseases. Carotid atherosclerosis of 50 T2DM patients was evaluated by standard B-mode ultrasound examination with the use of a 13.0 MHz multifrequency linear array transducer and a high-resolution ultrasound system (GE Vingmed Ultrasound AS, Horten, Norway). Intima-media thickness (IMT) was calculated as the mean maximal IMT of the six segments of the carotid artery. Two independent operators performed the ultrasonography blindly.
Venous blood samples
Fasting venous blood samples of all subjects were collected in the morning in EDTA vacutainer tubes. Fasting venous blood samples of T2DM patients were collected again when HbA1C decreased to below 6.5% after lifestyle interventions and medicine. The plasma samples were centrifuged at 3000 rpm for 20 mins at 4°C and then kept frozen at −80°C until analysis.
Isolation of peripheral blood mononuclear cell (PMBC)
PBMC was isolated by Ficoll standard density gradient centrifugation. The upper layer containing PBMC was harvested and washed with Hank’s balanced salt solution and then with PBS.
Animal experiments
Animal experiment protocols were approved by the Animal Use and Care Committee of Xi’an JiaoTong University. The experimental procedures were carried out in accordance with the Guide for the Care and Use of Laboratory Animals of National Institutes of Health revised in 2011. ApoE−/− mice were bred at the experimental animal center with pathogen-free, temperature-controlled facility at 25°C with a 12-hr light/12-hr dark cycle. All mice were fed normal chow diet until 6 weeks of age and then changed to western diet containing 21% fat and 0.15% cholesterol. Diabetes was induced in ApoE−/− mice by two daily intraperitoneal injections of streptozotocin (100 mg/kg/day; Sigma-Aldrich, St. Louis, MO, USA).15 Ten days postinjection, the survived animals with blood glucose >11 mmol/L were defined as diabetes mellitus model.16 Diabetic ApoE−/− mice were subsequently randomized to 3 groups to be injected via the tail vein with 2.1×107 transduction units of NLRP3 shRNA lentivirus, scrambled shRNA lentivirus (Santa Cruz Biotechnology) or saline twice a week for 4 weeks. At 22 weeks, all mice were sacrificed to collect a blood sample and the aorta under general anesthesia with isoflurane.
Analysis of atherosclerotic lesions in aortic root of mice
The aorta root was dissected, embedded and cut into sections at 6 μm. The first section was harvested when the three aortic valve cusps became visible in the lumen of the aorta, and every seventh section was harvested on one slide (10 sections per slide). Lipids were detected using oil-red O staining as previously described.17 Section images were analyzed by an Olympus BX51 imaging system (Olympus, Tokyo, Japan) and quantified with Image-Pro Plus 6.0 software. The atherosclerotic plaque was expressed as a percentage of the total area of the aorta. The protocol of plaque collagen staining was performed as described previously.18 Atherosclerotic plaque collagen was stained by Sirius red and the sections were analyzed by microscopy.
Cell culture
Primary human umbilical vein endothelial cells (HUVECs) were isolated as described.19 The experiment was approved by the Ethics Committee of Second Affiliated Hospital of Medical College, Xi’an Jiaotong University. HUVECs were cultured in M199 medium (Gibco, Invitrogen) supplemented with 15% fetal bovine serum (Gibco, Invitrogen), 3 ng/mL endothelial cells growth factor (Sigma) and 100 U/mL penicillin-streptomycin at 37°C. HUVECs were treated with IL-1β (Sino Biological Inc., North Wales, PA, USA) at 5–20 ng/mL with or without IL-1β receptor antagonist Anakinra (Kineret, Amgen, USA) at 5 μg/mL for 12–48 hrs. Cell viability was assessed by trypan blue exclusion. HUVECs at approximately 50–70% confluence were transfected with NLRP3 shRNA, scrambled shRNA or copGFP control lentiviral particles (Santa Cruz Biotechnology) in 5 μg/mL polybrene/complete medium for 12 hrs.
Real-time PCR
Intimal RNA was isolated from the aorta as reported previously.20 TRIzol reagent (Invitrogen) was used to extract total RNA from intima and HUVECs. First-strand cDNA was synthesized from total RNA (2 mg) using RevertAidTM First Strand cDNA Synthesis Kit (Fermentas). qRT-PCR was performed to detect mRNA levels of NLRP3, Caspase-1, ASC, IL-1β, ICAM-1 and VCAM-1 using SYBR Premix Ex TaqTM (Takara). The primer sequences are shown in Table 1.
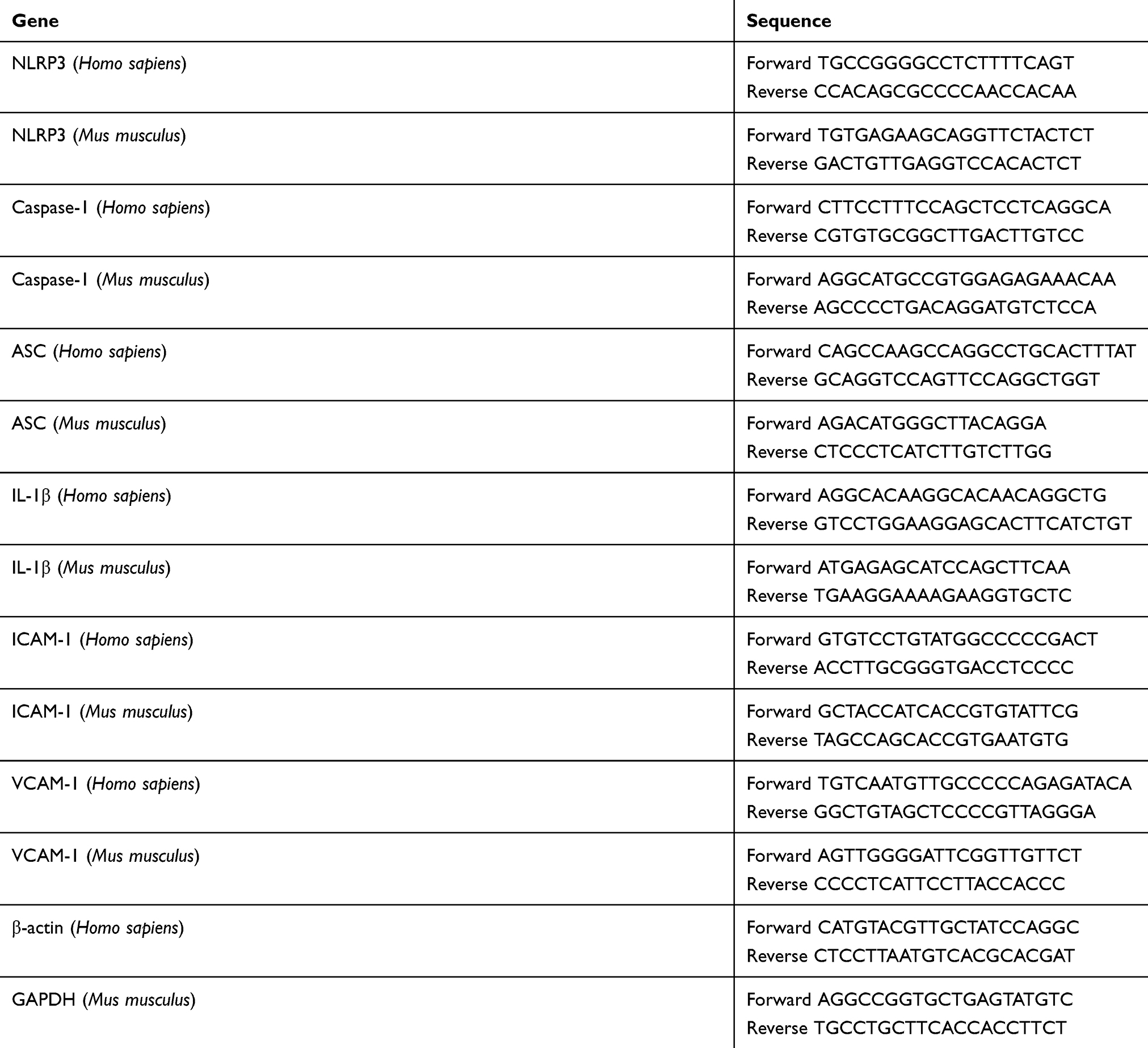 |
Table 1 Primers used in this study |
Western blot analysis
Total proteins were extracted, and equal amounts of protein lysate extracts were separated by 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride membranes. Nonspecific binding was blocked with 5% skim milk for 2 hrs at room temperature. Membranes were incubated overnight at 4°C with primary antibodies for NLRP3 (Cell Signaling Technology), Caspase-1 (Cell Signaling Technology), ASC, ICAM-1 and VCAM-1 (Santa Cruz Biotechnology), washed and then incubated for 2 hrs at room temperature with horseradish peroxide-conjugated anti-rabbit or anti-mouse secondary antibodies (Santa Cruz Biotechnology). Blots were visualized with enhanced chemiluminescent substrate (Pierce Biotechnology). Relative quantities of proteins were determined with a densitometer.
Enzyme-linked immunosorbent assay
IL-1β in cell culture supernatant and the plasma of the mice and the subjects were assayed with ELISA kit (Tera Bio, Guangzhou, China) following the manufacturer’s protocols.
Statistical analysis
Data were collected and analyzed with SPSS 13.0 (SPSS Inc., Chicago, IL, USA). Quantitative variables were expressed as mean ± standard deviation. Categorical variables were expressed as frequency and percentage. Kolmogorov–Smirnov test was used to assess normal distribution of quantitative variables. The Student t-test or ANOVA was used to analyze the differences among groups. Two-tailed P<0.05 was considered significant.
Results
Characteristics of the subjects
The demographic and clinical characteristics of the subjects are shown in Table 2. Body mass index and fasting blood sugar in patients with T2DM were higher than those in generally healthy controls, but were notably decreased after lifestyle interventions and medicine.
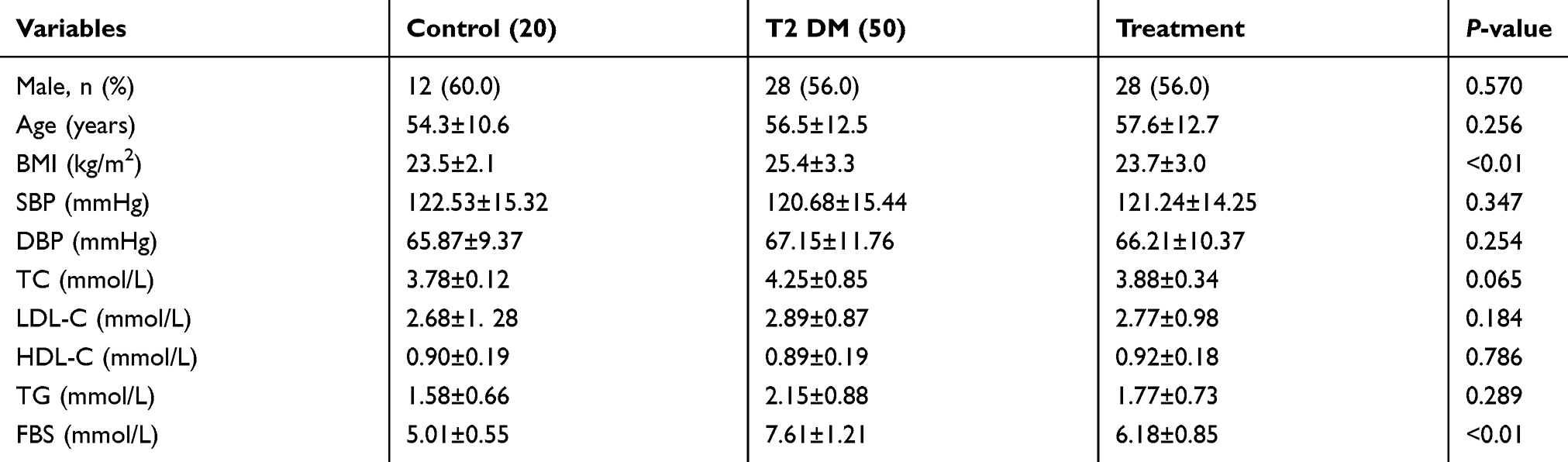 |
Table 2 The baseline characteristics of the subjects |
Activated NLRP3 inflammasome and elevated plasma IL-1β were related to atherosclerosis in diabetic patients
Compared with healthy controls, NLRP3 protein level (Figure 1A) and NLRP3 mRNA level (Figure 1B) in PBMC and plasma IL-1β level (Figure 1C) were significantly higher in DM patients at admission, but they decreased after lifestyle interventions and medicine. Moreover, when T2DM patients were divided into 3 subgroups according to tertiles of plasma IL-1β level, IMT increased significantly with the increase in plasma IL-1β level (Tertile 1, 0.88±0.04 mm; Tertile 2, 1.59±0.12 mm; and Tertile 3, 2.03±0.18 mm, P<0.01, Figure 1D).
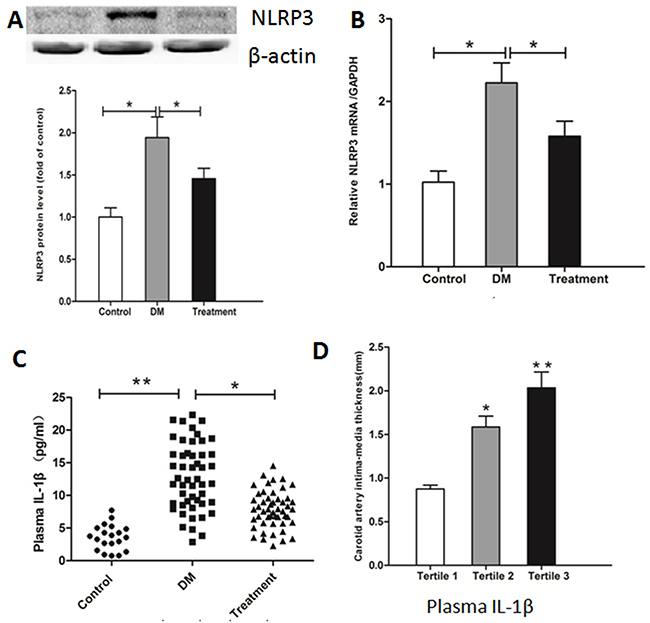 |
Figure 1 Comparison of NLRP3 expression and plasma IL-1β level in healthy controls and diabetic patients. PBMCs were isolated, and NLRP3 protein level (A) and mRNA level (B) were determined by Western blotting and real-time PCR. Plasma IL-1β was assayed with ELISA (C). Carotid atherosclerosis of diabetic patients was evaluated by standard B-mode ultrasound (D). Data represent mean ± SD. *P<0.05, **P<0.01 vs controls and treatment or vs tertile 1. |
NLRP3 knockdown reduced diabetic atherosclerosis and stabilized the atherosclerotic plaque
As shown in Figure 2A and C, aortic root atherosclerotic plaque area percentage was significantly increased in diabetic ApoE−/− mice compared to ApoE−/− mice (43.46±4.25% vs 22.34±2.24%; P<0.01), but NLRP3 knockdown notably reduced atherosclerotic plaque of diabetic ApoE−/− mice (25.36±2.43%; P<0.01). Moreover, NLRP3 knockdown drastically increased plaque collagen content percentage (ApoE−/− mice, 15.45±4.5%; diabetic ApoE−/− mice 17.72±3.5%; diabetic ApoE−/− mice with scrambled shRNA, 16.58±3.5%; diabetic ApoE−/− mice with NLRP3 shRNA, 27.61±4.4%; P<0.01) (Figure 2B and D). These results indicated that atherosclerotic lesions were accelerated in diabetic ApoE−/− mice but NLRP3 knockdown alleviated atherosclerotic lesions and stabilized the plaques.
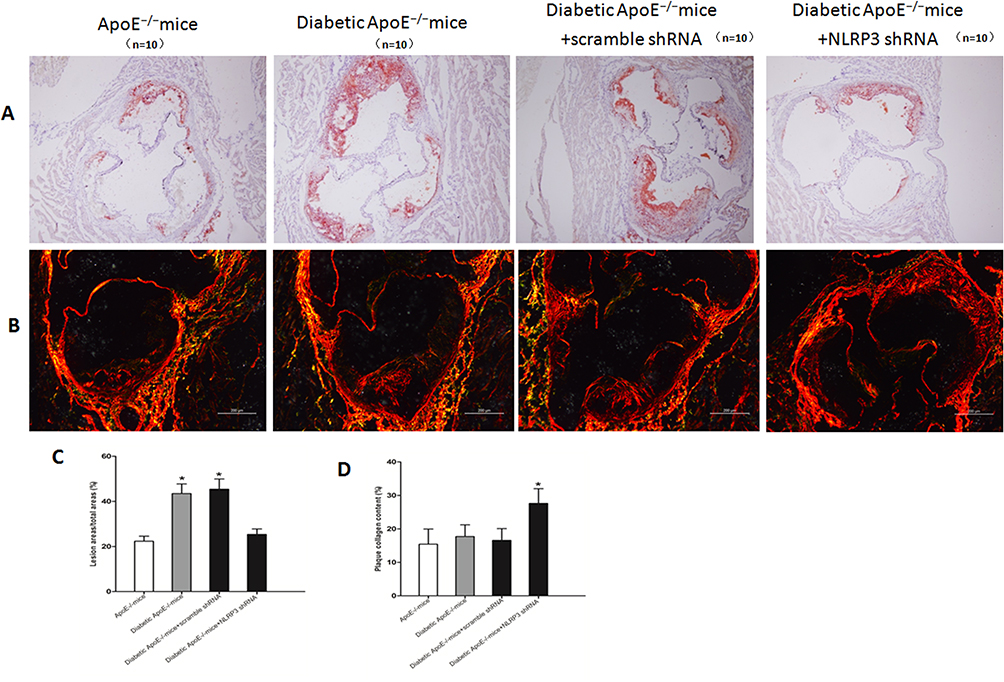 |
Figure 2 NLRP3 knockdown reduced diabetic atherosclerosis and stabilized the atherosclerotic plaque. (A and C) Quantitative analysis of aortic root atherosclerosis lesion in ApoE−/− mice, diabetic ApoE−/− mice and diabetic ApoE−/− mice treated with NLRP3 shRNA or vehicle. Red Oil O staining, original magnification: 100×, bar: 200 μm. (B and D) Quantitative analysis of collagen content of plaques in ApoE−/− mice, diabetic ApoE−/− mice and diabetic ApoE−/− mice treated with NLRP3 shRNA or vehicle. Sirius Red staining, original magnification: 140×, bar: 200 μm. Data are mean ± SD. *P<0.05 vs ApoE−/− mice, diabetic ApoE−/− mice and diabetic ApoE−/− mice treated with vehicle. |
NLRP3 knockdown suppressed NLRP3 inflammasome and improved endothelial inflammation in diabetic ApoE−/− mice
The expression levels of NLRP3 inflammasome components NLRP3, Caspase-1 and ASC, and adhesion molecules including ICAM-1 and VCAM-1 in the endothelium of diabetic ApoE−/− mice were significantly higher than those in ApoE−/− mice, but NLRP3 knockdown suppressed their expression and improved endothelial inflammation in diabetic ApoE−/− mice (Figure 3A and C). NLRP3 knockdown significantly decreased serum IL-1β level of diabetic ApoE−/− mice (ApoE−/− mice 126.64±10.75 pg/mL; diabetic ApoE−/− mice 184.39±29.58 pg/mL; NLRP3 shRNA diabetic ApoE−/− mice 129.76±9.97 pg/mL; scrambled shRNA diabetic ApoE−/− mice 178.83±30.43 pg/mL; P<0.01) (Figure 3B).
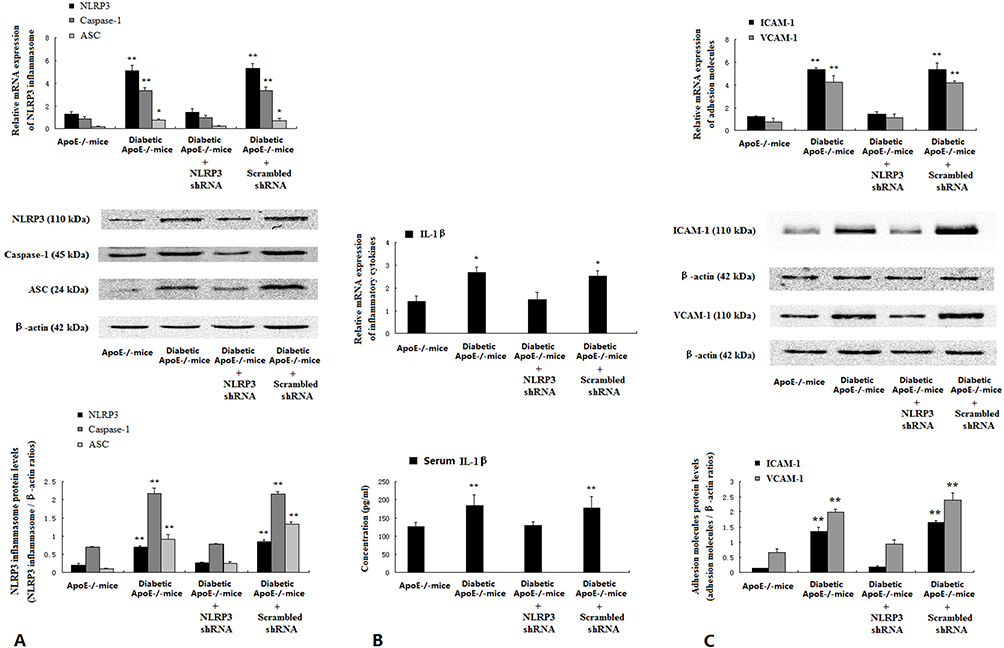 |
Figure 3 NLRP3 knockdown suppressed NLRP3 inflammasome and improved endothelial inflammation in diabetic ApoE−/− mice. Intima was separated from media and adventitia of thoracic aorta of the mice. Protein and mRNA expression of NLRP3 components NLRP3, Caspase-1 and ASC (A), and adhesion molecules ICAM-1 and VCAM-1 (C) were assessed by Western blotting and real-time PCR. Plasma IL-1β was assayed with ELISA (B). Data are mean ± SD (n=5). *P<0.05, **P<0.01 vs ApoE−/− mice and diabetic ApoE−/− mice treated with NLRP3 shRNA. |
High glucose activated NLRP3 inflammasome in HUVECs
D-glucose increased mRNA and protein levels of NLRP3 inflammasome components NLRP3, Caspase-1 and ASC in HUVECs in a dose- and time-dependent manner (Figure 4A and B). D-glucose also increased IL-1β levels in supernatants of HUVECs in a time- and dose-dependent manner (Figure 4C and D).
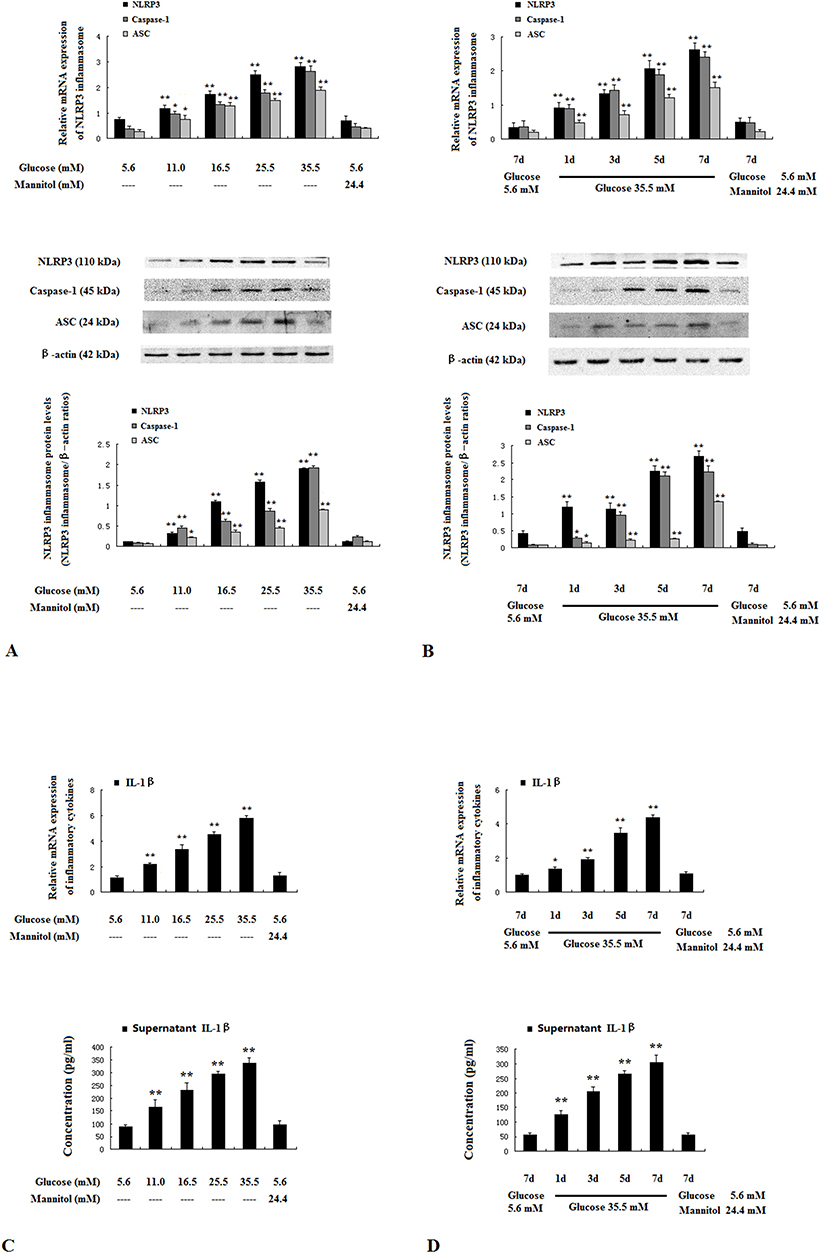 |
Figure 4 High glucose activated NLRP3 inflammasome in HUVECs. HUVECs were incubated for 7 days with 5.6–35.5 mM glucose, or 5.6 mM glucose with 24.4 mM mannitol (A and C). HUVECs were incubated for 1–7 days with 35.5 mM glucose, 5.6 mM glucose or 5.6 mM glucose with 24.4 mM mannitol (B and D). The mRNA and protein expressions of NLRP3 inflammasome components NLRP3, Caspase-1 and ASC (A and B) were determined by real-time PCR and Western blotting. IL-1β in supernatants was assayed with ELISA (C and D). Data are mean ± SD (n=5). *P<0.05, **P<0.01 vs controls (5.6 mM glucose or 5.6 mM glucose with 24.4 mM mannitol for 7 days). |
NLRP3 knockdown decreased the secretion of IL-1β in HUVECs
HUVECs were transfected with NLRP3 shRNA, scrambled shRNA or copGFP control lentiviral particles, only NLRP3 shRNA lentiviral particles significantly inhibited NLRP3 mRNA and protein expression (Figure 5A). Notably, IL-1β secretion decreased significantly in HUVECs treated with NLRP3 shRNA lentiviral particles (Figure 5B).
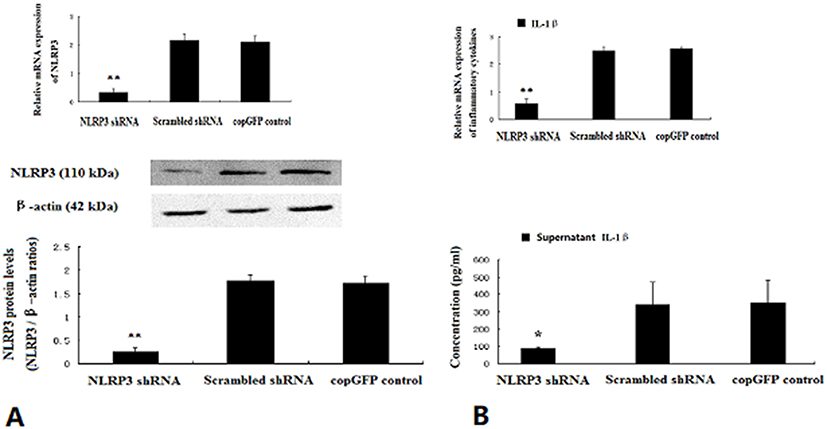 |
Figure 5 NLRP3 knockdown inhibited the secretion of IL-1β in HUVECs. HUVECs exposed to 35.5 mM glucose at approximately 50–70% confluence were infected with NLRP3 shRNA, scrambled shRNA or copGFP control lentiviral particles for 12 hrs. The mRNA and protein levels of NLRP3 were determined by real-time PCR and Western blotting 24 hrs later (A). IL-1β in supernatants was assayed with ELISA (B). Data are mean ± SD (n=5). *P<0.05, **P<0.01 vs scrambled shRNA or copGFP control. |
IL-1β upregulated the expression of adhesion molecules in HUVECs
To elucidate the role of IL-1β in endothelial inflammation, we treated HUVECs with IL-1β and found that mRNA and protein levels of ICAM-1 and VCAM-1 were markedly enhanced by IL-1βin a dose- and time-dependent manner (Figure 6A and B).
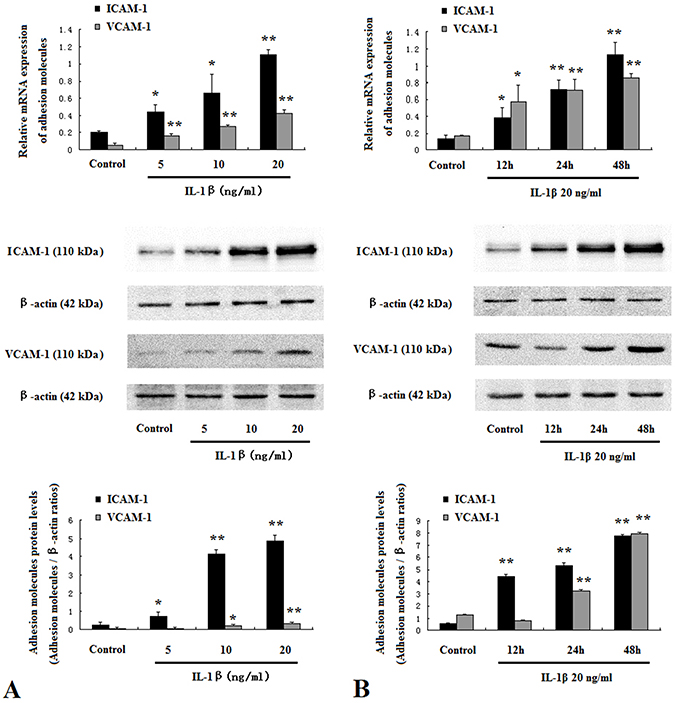 |
Figure 6 IL-1β promoted adhesion molecules expression in HUVECs. HUVECs were treated with IL-1β at 5 ng/mL to 20 ng/mL for 48 hrs (A). HUVECs were treated with IL-1β at 20 ng/mL for 12–48 hrs (B). Protein and mRNA expressions of ICAM-1 and VCAM-1 were assessed by Western blotting and real-time PCR. Data are mean ± SD (n=5). *P<0.05, **P<0.01 vs control. |
NLRP3 knockdown or IL-1β receptor antagonist suppressed adhesion molecules expression enhanced by high glucose in HUVECs
Compared to normal glucose group, the expression of adhesion molecules ICAM-1 and VCAM-1 was considerably promoted by high glucose (Figure 7A and B). However, NLRP3 knockdown or anakinra, IL-1β receptor antagonist, significantly decreased the expression of ICAM-1 and VCAM-1 induced by high glucose (Figure 7A and B).
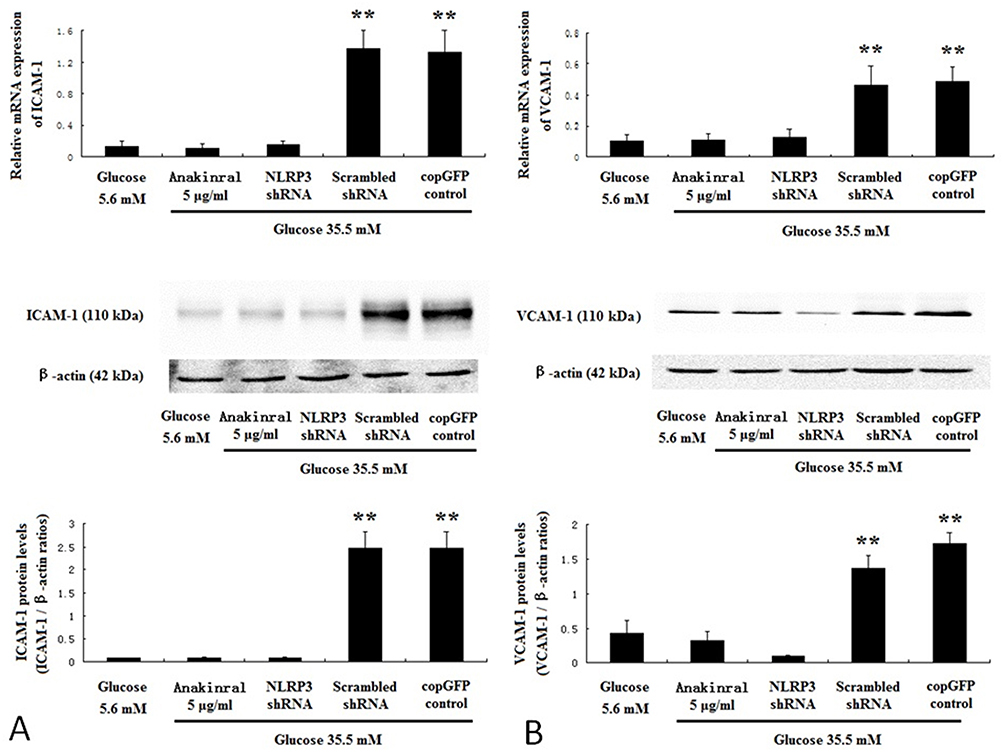 |
Figure 7 NLRP3 knockdown or IL-1β receptor antagonist suppressed adhesion molecules expression induced by high glucose in HUVECs. HUVECs were infected with NLRP3 shRNA, scrambled shRNA or copGFP control lentiviral particles, and incubated for 7 days with 35.5 mM glucose or 35.5 mM glucose with 5 μg/mL anakinra. HUVECs incubated for 7 days with 5.6 mM glucose served as control. The mRNA and protein expressions of ICAM-1 (A) and VCAM-1 (B) were determined by real-time PCR and Western blotting. Data are mean ± SD (n=5). **P<0.01 vs scrambled shRNA or copGFP control. |
Discussion
Diabetes is a main risk factor for the initiation and progression of atherosclerosis.21 Interestingly, recent studies have suggested that the activation of NLRP3 inflammasome is implicated in metabolic diseases such as atherosclerosis and diabetes.22–24 Therefore, the present study aimed to explore the role of NLRP3 inflammasome in diabetic atherosclerosis. We found that NLRP3 inflammasome activation was augmented in diabetic patients and diabetic ApoE−/− mice, and NLRP3 knockdown alleviated endothelial inflammation, reduced atherosclerotic lesion burden and increased plaque collagen content in a mouse model of diabetic atherosclerosis. Moreover, high glucose induced the activation of NLRP3 inflammasome and the upregulation of adhesion molecules in HUVECs, while NLRP3 knockdown or IL-1β receptor antagonist suppressed adhesion molecular expression induced by high glucose in HUVECs. These findings provide novel insights into the pathological mechanisms that directly link NLRP3 inflammasome to diabetic atherosclerosis.
IL-1β is proposed as the index of NLRP3 inflammasome activation. IL-1β is an important proinflammatory cytokine and plays an important role in atherosclerosis.6 As expected, we found that carotid atherosclerosis was significantly related to plasma IL-1β level in diabetic patients. Lee et al reported that compared with healthy subjects, the monocytes exhibited high expression of inflammasome components and high serum levels of IL-1β in newly identified untreated type 2 diabetic patients.25 Consistent with the finding, we found that the NLRP3 inflammasome activation was elevated in PBMC from newly diagnosed untreated diabetic patients than from generally healthy subjects. After lifestyle interventions and medicine, HbA1c decreased to less than 6.5% in diabetic patients and NLRP3 inflammasome activation was significantly attenuated. Similarly, it was reported that NLRP3 inflammasome activation was augmented in adipose tissue from an obese mouse model and alleviated after calorie restriction.26 Furthermore, we found that the expression of NLRP3 inflammasome components NLRP3, Caspase-1 and ASC in the intima and plasma IL-1β level were higher in diabetic ApoE−/− mice than in ApoE−/− model mice. The main difference between diabetic ApoE−/− mice and ApoE−/− mice is hyperglycemia. Indeed, previous studies demonstrated that high glucose could activate NLRP3 inflammasome.11–14 Using HUVECs as in vitro cell model, we found that the expression of NLRP3 inflammasome components and IL-1β secretion were augmented by high glucose in a dose- and time-dependent manner.
It is well known that adhesion molecules facilitate monocyte-endothelial cell interaction and play a key role in atherosclerosis.27–29 In the present study, the expression levels of adhesion molecules in the intima from diabetic ApoE−/− mice were higher than those from ApoE−/− mice. In vitro, high glucose could promote adhesion molecules expression in HUVECs. Previous studies showed that adhesion molecules expression could be enhanced by IL-1β.30,31 In this study, IL-1β receptor antagonist or NLRP3 knockdown could significantly suppress adhesion molecules expression enhanced by high glucose in HUVECs. More importantly, NLRP3 knockdown alleviated the intima inflammation in diabetic ApoE−/− mice. Taken together, these results suggest that hyperglycemia causes intima inflammation via the activation of NLRP3 inflammasome and the production of IL-1β. Our finding was partly consistent with a recent study reporting that anagliptin ameliorated high glucose-induced endothelial dysfunction via the suppression of NLRP3 inflammasome activation mediated by SIRT1.32
In conclusion, the present study demonstrated that hyperglycemia-induced NLRP3 inflammasome activation mediated diabetic atherosclerosis by causing endothelial cell inflammation. These findings provide novel insights into pathological mechanisms of diabetic atherosclerosis and have potential therapeutic implications for cardiovascular complications in diabetes.
Acknowledgment
This study was supported by Natural Science Funds of Shaanxi Province (No. 2016JM8059).
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tomkin GH, Owens D. Diabetes and dyslipidemia: characterizing lipoprotein metabolism. Diabetes Metab Syndr Obes. 2017;10:333–343. doi:10.2147/DMSO.S115855
2. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. 2011;17(11):1410–1422. doi:10.1038/nm.2538
3. Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev. 2008;223:20–38. doi:10.1111/j.1600-065X.2008.00624.x
4. De Nardo D, Latz E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol. 2011;32(8):373–379. doi:10.1016/j.it.2011.05.004
5. Stienstra R, van Diepen JA, Tack CJ, et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc Natl Acad Sci USA. 2011;108(37):15324–15329. doi:10.1073/pnas.1100255108
6. Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464(7293):1357–1361. doi:10.1038/nature08940
7. He J, Yang Y, Peng DQ. Monosodium urate (MSU) crystals increase gout associated coronary heart disease (CHD) risk through the activation of NLRP3 inflammasome. Int J Cardiol. 2012;160(1):72–73. doi:10.1016/j.ijcard.2012.05.083
8. Xiao H, Lu M, Lin TY, et al. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic-induced atherosclerosis susceptibility. Circulation. 2013;128(6):632–642. doi:10.1161/CIRCULATIONAHA.113.002714
9. Tschopp J, Schroder K. NLRP3 inflammasome activation: the convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10(3):210–215. doi:10.1038/nri2725
10. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107(9):1058–1070. doi:10.1161/CIRCRESAHA.110.223545
11. Feng H, Gu J, Gou F, et al. High glucose and lipopolysaccharide prime NLRP3 inflammasome via ROS/TXNIP pathway in mesangial cells. J Diabetes Res. 2016;2016:6973175. doi:10.1155/2016/6973175
12. Shi H, Zhang Z, Wang X, et al. Inhibition of autophagy induces IL-1β release from ARPE-19 cells via ROS mediated NLRP3 inflammasome activation under high glucose stress. Biochem Biophys Res Commun. 2015;463(4):1071–1076. doi:10.1016/j.bbrc.2015.06.116
13. Ward R, Ergul A. Relationship of endothelin-1 and NLRP3 inflammasome activation in HT22 hippocampal cells in diabetes. Life Sci. 2016;159:97–103. doi:10.1016/j.lfs.2016.02.043
14. Wu J, Li X, Zhu G, Zhang Y, He M, Zhang J. The role of Resveratrol-induced mitophagy/autophagy in peritoneal mesothelial cells inflammatory injury via NLRP3 inflammasome activation triggered by mitochondrial ROS. Exp Cell Res. 2016;341(1):42–53. doi:10.1016/j.yexcr.2016.01.014
15. Takeda Y, Fujita Y, Honjo J, et al. Reduction of both beta cell death and alpha cell proliferation by dipeptidyl peptidase-4 inhibition in a streptozotocin-induced model of diabetes in mice. Diabetologia. 2012;55(2):404–412. doi:10.1007/s00125-011-2365-4
16. Terasaki M, Nagashima M, Nohtomi K, et al. Preventive effect of dipeptidyl peptidase-4 inhibitor on atherosclerosis is mainly attributable to incretin’s actions in nondiabetic and diabetic apolipoprotein E-null mice. PLoS One. 2013;8(8):e70933. doi:10.1371/journal.pone.0070933
17. Nakano K, Egashira K, Ohtani K, et al. Azelnidipine has anti-atherosclerotic effects independent of its blood pressure-lowering actions in monkeys and mice. Atherosclerosis. 2008;196(1):172–179. doi:10.1016/j.atherosclerosis.2007.03.036
18. Tian Y, Chen T, Wu Y, et al. Pioglitazone stabilizes atherosclerotic plaque by regulating the Th17/Treg balance in AMPK-dependent mechanisms. Cardiovasc Diabetol. 2017;16(1):140. doi:10.1186/s12933-017-0624-5
19. Wang X, Liu W, Wu Y, et al. C-reactive protein reduces protein S-nitrosylation in endothelial cells. Mol Cell Biochem. 2013;375(1–2):131–138. doi:10.1007/s11010-012-1535-0
20. Sun X, Icli B, Wara AK, et al. MicroRNA-181b regulates NF-κB-mediated vascular inflammation. J Clin Invest. 2012;122(6):1973–1990. doi:10.1172/JCI61495
21. Robinson JG, Davidson MH. Can we cure atherosclerosis? Rev Cardiovasc Med. 2018;19(S1):S20–S24.
22. Sharma A, Tate M, Mathew G, Vince JE, Ritchie RH, de Haan JB. Oxidative stress and NLRP3-inflammasome activity as significant drivers of diabetic cardiovascular complications: therapeutic implications. Front Physiol. 2018;9:114. doi:10.3389/fphys.2018.00114
23. Lee J, Wan J, Lee L, Peng C, Xie H, Lee C. Study of the NLRP3 inflammasome component genes and downstream cytokines in patients with type 2 diabetes mellitus with carotid atherosclerosis. Lipids Health Dis. 2017;16(1):217. doi:10.1186/s12944-017-0565-8
24. Wu M, Han W, Song S, et al. NLRP3 deficiency ameliorates renal inflammation and fibrosis in diabetic mice. Mol Cell Endocrinol. 2018;478:115–125. doi:10.1016/j.mce.2018.08.002
25. Lee HM, Kim JJ, Kim HJ, Shong M, Ku BJ, Jo EK. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62(1):194–204. doi:10.2337/db12-0863
26. Vandanmagsar B, Youm YH, Ravussin A, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–188. doi:10.1038/nm.2279
27. Gaudreault N, Kumar N, Posada JM, et al. ApoE suppresses atherosclerosis by reducing lipid accumulation in circulating monocytes and the expression of inflammatory molecules on monocytes and vascular endothelium. Arterioscler Thromb Vasc Biol. 2012;32(2):264–272. doi:10.1161/ATVBAHA.111.238964
28. Gower RM, Wu H, Foster GA, et al. CD11c/CD18 expression is upregulated on blood monocytes during hypertriglyceridemia and enhances adhesion to vascular cell adhesion molecule-1. Arterioscler Thromb Vasc Biol. 2011;31(1):160–166. doi:10.1161/ATVBAHA.110.215434
29. Wu H, Gower RM, Wang H, et al. Functional role of CD11c+ monocytes in atherogenesis associated with hypercholesterolemia. Circulation. 2009;119(20):2708–2717. doi:10.1161/CIRCULATIONAHA.108.823740
30. Verginelli F, Adesso L, Limon I, et al. Activation of an endothelial Notch1-Jagged1 circuit induces VCAM1 expression, an effect amplified by interleukin-1beta. Oncotarget. 2015;6(41):43216–43229. doi:10.18632/oncotarget.6456
31. Chang WC, Chen CH, Lee MF, et al. Chlorogenic acid attenuates adhesion molecules upregulation in IL-1beta-treated endothelial cells. Eur J Nutr. 2010;49(5):267–275. doi:10.1007/s00394-009-0083-1
32. Jiang T, Jiang D, Zhang L, Ding M, Zhou H. Anagliptin ameliorates high glucose-induced endothelial dysfunction via suppression of NLRP3 inflammasome activation mediated by SIRT1. Mol Immunol. 2019;107:54–60. doi:10.1016/j.molimm.2019.01.006
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.