Back to Journals » International Journal of Nanomedicine » Volume 13
Nitric oxide-releasing nanoparticles improve doxorubicin anticancer activity
Authors Alimoradi H, Greish K ![]() , Barzegar-Fallah A, Alshaibani L, Pittalà V
, Barzegar-Fallah A, Alshaibani L, Pittalà V ![]()
Received 11 September 2018
Accepted for publication 27 October 2018
Published 20 November 2018 Volume 2018:13 Pages 7771—7787
DOI https://doi.org/10.2147/IJN.S187089
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Thomas Webster
Houman Alimoradi,1,* Khaled Greish,2,3,* Anita Barzegar-Fallah,1 Lama Alshaibani,2 Valeria Pittalà4
1Department of Pharmacology and Toxicology, University of Otago, Dunedin, New Zealand; 2College of Medicine and Medical Sciences, Department of Molecular Medicine, and Nanomedicine Unit, Princess Al-Jawhara Centre for Molecular Medicine and Inherited Disorders, Arabian Gulf University, Manama, Kingdom of Bahrain; 3Department of Oncology, Faculty of Medicine, Suez Canal University, Ismailia, Egypt; 4Department of Drug Science, University of Catania, Catania, Italy
*These authors contributed equally to this work
Purpose: Anticancer drug delivery systems are often limited by hurdles, such as off-target distribution, slow cellular internalization, limited lysosomal escape, and drug resistance. To overcome these limitations, we have developed a stable nitric oxide (NO)-releasing nanoparticle (polystyrene-maleic acid [SMA]-tert-dodecane S-nitrosothiol [tDodSNO]) with the aim of enhancing the anticancer properties of doxorubicin (Dox) and a Dox-loaded nanoparticle (SMA-Dox) carrier.
Materials and methods: Effects of SMA-tDodSNO and/or in combination with Dox or SMA-Dox on cell viability, apoptosis, mitochondrial membrane potential, lysosomal membrane permeability, tumor tissue, and tumor growth were studied using in vitro and in vivo model of triple-negative breast cancer (TNBC). In addition, the concentrations of SMA-Dox and Dox in combination with SMA-tDodSNO were measured in cells and tumor tissues.
Results: Combination of SMA-tDodSNO and Dox synergistically decreased cell viability and induced apoptosis in 4T1 (TNBC cells). Incubation of 4T1 cells with SMA-tDodSNO (40 µM) significantly enhanced the cellular uptake of SMA-Dox and increased Dox concentration in the cells resulting in a twofold increase (P<0.001). Lysosomal membrane integrity, evaluated by acridine orange (AO) staining, was impaired by 40 µM SMA-tDodSNO (P<0.05 vs control) and when combined with SMA-Dox, this effect was significantly potentiated (P<0.001 vs SMA-Dox). Subcutaneous administration of SMA-tDodSNO (1 mg/kg) to xenografted mice bearing 4T1 cells showed that SMA-tDodSNO alone caused a twofold decrease in the tumor size compared to the control group. SMA-tDodSNO in combination with SMA-Dox resulted in a statistically significant 4.7-fold reduction in the tumor volume (P<0.001 vs control), without causing significant toxicity as monitored through body weight loss.
Conclusion: Taken together, these results suggest that SMA-tDodSNO can be used as a successful strategy to increase the efficacy of Dox and SMA-Dox in a model of TNBC.
Keywords: biologic barriers, nanoparticles, nitric oxide, doxorubicin, synergistic cytotoxicity, SMA-tDodSNO
Introduction
Targeted antitumor therapies supported by nanotechnology have emerged as attractive strategies to overcome the lack of selectivity of traditional chemotherapeutic drugs. Described as “a royal gate for targeted anticancer nanomedicines”,1 the enhanced permeability and retention (EPR) effect of macromolecules is the main cause of passive targeting of various long-lasting pharmaceutical nanocarriers larger than 7 nm in solid tumors.2 The EPR effect in tumors arises from the insufficient formation of tight junctions in the endothelium, leaky vasculature, slow venous return, and poor lymphatic clearance.3 Animal studies have shown that nanocarriers can achieve three to ten times higher concentration of drugs at the tumor site with a concomitant reduction of off-target side effects.2 However, in clinical settings, the EPR effect does not always guarantee an efficient targeting of tumor tissues due to the heterogeneity of tumor cells, microenvironment, and several barriers preventing them reaching their targets in cancer cells.4,5 In fact, nanoparticles (NPs) to reach the target should overcome the extracellular matrix (ECM), which is a semisolid barrier consisting of an interconnected network of collagen fibers intermingled with proteins. The higher rigidity of the ECM in most tumors compared to normal tissues hinders NP diffusion.6 In addition, NPs cannot simply enter cells via diffusion; they are internalized through endocytic process, which is time and energy consuming, and this endocytic process can prevent the absorption of NPs carrying drugs acting inside the cells.7 Finally, the payload efflux from the cells and multidrug resistance are major hurdles in the treatment of many cancers.8 Besides the restricted uptake, the confinement of NPs in low-pH endosomal or lysosomal compartments and the reduced blood supply, leading to the presence of hypoxic areas in the central region of the tumor and in metastasis, can also negatively impact the efficacy of the treatments.9 Strategies to enhance the EPR effect, aiming at increasing NPs’ selective tumor targeting, include the modulation of the tumor microenvironment. Local administration of nitric oxide (NO) donors to tumors with the aim of increasing the blood flow has been tested clinically with favorable outcome.10 However, limited NO payloads, short NO half-life, and the lack of organ or tissue specificity have restricted the utilization of NO in the area of anticancer management, despite their potential antitumoral effects.11
The aim of this paper is to investigate the effects of a stable NO-releasing compound loaded into styrene maleic acid nanomicelles on the anticancer properties of doxorubicin (Dox) in 4T1, a triple-negative breast cancer (TNBC) model. To this extent, we identified the NO-releasing tert-dodecane S-nitrosothiol (tDodSNO) as a stable NO-donor and encapsulated it into polystyrene-maleic acid (SMA) to formulate SMA-tDodSNO NPs. tDodSNO showed superior stability and NO release profile compared to commercially available NO donors (such as S-nitroso-N-acetylpenicillamine, S-nitrosoglutathione, and sodium nitroprusside) most probably due to its bulkiness and hydrophobicity.12,13 However, its hydrophobicity may limit therapeutic applications. Encapsulation of tDodSNO into SMA makes it water soluble and physically protects it from premature tDodSNO breakdown as well as trans-nitrosation reactions with proteins and thiols, thereby inhibiting unspecific NO release. In addition, as NO plays an important role in vascular tone and permeability3 and controls the cellular trafficking process of endocytosis and exocytosis,14 on this basis we hypothesized that SMA-tDodSNO can increase the efficacy of Dox in vitro and a Dox-loaded NP (SMA-Dox) formulation in vivo. Finally, therapeutic advantages of the new combination were tested in 4T1 syngeneic animal model of TNBC.
Materials and methods
Dox was purchased from Lancrix Chemicals Ltd. (Shanghai, China). Polystyrene-co-maleic anhydride terminated with cumene (SMA) as a polymeric mixture with an average molecular weight of 1,600 Da and all other chemicals were obtained from Sigma-Aldrich Co. (St Louis, MO, USA). MTT and all cell culture reagents were purchased from Thermo Fisher Scientific (Waltham, MA, USA). tDodSNO was synthesized as described before.12
Synthesis of SMA-tDodSNO and SMA-Dox
SMA-tDodSNO and SMA-Dox were prepared according to the previously reported method.15 Briefly, to create optimized loading of tDodSNO, or Dox in SMA NPs, escalating milligram ratios of the compounds dissolved in 2 mL DMSO were added dropwise to 10 mL of a vigorously stirring solution of SMA (10 mg/mL) at pH =5.0, followed by addition of 80 mg EDAC, and solubilized in 2 mL of deionized water. During the addition of EDAC, the pH of the solution was maintained at 5 for 10 minutes, and then it was increased by NaOH 1 M to reach pH 11. Upon the appearance of a clear solution, the pH was adjusted back to 7.4 and purified using ultrafiltration system Pellicon XL filter 10 kDa (Merck Millipore, Billerica, MA, USA). The final concentrated product was freeze-dried to get the SMA-tDodSNO and SMA-Dox powder. To measure the loading of tDodSNO or Dox in the NPs, known weights of the NPs were dissolved in methanol, and the concentrations of SMA-tDodSNO and SMA-Dox were calculated using HPLC and UV-Vis spectroscopy at 480 nm, respectively.
NPs size and zeta potential
NP size, dispersity, and zeta potential were quantified by dynamic and electrophoretic light scattering of 8 mg/mL solutions of the NPs at room temperature using a Malvern Zeta Sizer ZEN3600 (Malvern Instruments, Malvern, UK), and repeated in triplicate.
Drug release from SMA-tDodSNO and SMA-Dox
The release of the payloads from the NPs was measured using a semipermeable dialysis membrane (14 kDa molecular weight cutoff, Sigma-Aldrich Co.) as detailed before.15 The concentration of tDodSNO in the solutions was measured using a BM-20Alite Prominence HPLC system (Shimadzu Corporation, Kyoto, Japan) on a reversed-phase Gemini 3 μm C18, 110A, 150×2 mm column (Phenomenex Gemini-NX, North Shore City, Auckland, New Zealand) at 25°C. The mobile phase was methanol containing 0.3% trifluoroacetic acid, and the flow rate was 0.65 mL/min. A diode array detector was set at 341 nm and linked to Chromeleon software for data analysis. tDodSNO peak was detected at 341 nm with the retention time of 4.3 minutes.
In vitro studies
Mouse 4T1 mammary carcinoma cells were used for in vitro studies. This cell line is one of the most aggressive phenotypes of cancer cells exhibiting aggressive phenotype similar to human TNBC.16 Mouse 4T1 cells were purchased from ATCC, were cultured in RPMI-1640 medium supplemented with 5% heat-inactivated FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin (Thermo Fisher Scientific) and maintained in a humidified atmosphere with 5% CO2 and 95% air at 37°C.
Cell viability assay and synergistic toxicity of SMA-tDodSNO and Dox
4T1 cells were seeded at 5,000 cells/well into 96-well plates and allowed to adhere overnight. Cells were then treated with various concentrations of SMA-tDodSNO (1–100 μM) and/or Dox (0.001–5 μM) and incubated for 48 hours. The medium was removed and 0.4 mg/mL MTT dissolved in media was added for 3 hours. After removing medium, the residual crystals were dissolved in 100 μL DMSO and cell viability was calculated at ƛ=550 nm.
Cell cycle analysis
Two lakh 4T1 cells/well were seeded in six-well plates and allowed to adhere overnight prior to treatment with different concentrations of Dox (0.1 μM) and SMA-tDodSNO (10 and 40 μM). Forty-eight hours post treatment, cells were harvested, washed in cold PBS, followed by fixation with 4°C chilled 70% ethanol for 30 minutes. The fixed cells were washed twice with phosphate-citrate buffer (Na2HPO4 192 mM, citric acid 4 mM), treated with 50 μL of 100 μg/mL RNase A, and finally stained with 200 μL of 50 μg/mL propidium iodide (PI) at 4°C. DNA content and percentage of cells in the different phases of the cell cycle were determined using FACScan flow cytometer (Becton Dickinson) and FlowJo software (TREE STAR, Inc., San Carlos, CA, USA). For all flow cytometry analysis, a minimum of 10,000 cells/sample were acquired and gated to remove cell debris and doublets as shown in Figure S1. The main population of cells was separated from possible noises (side scatter area vs forward scatter area [FSC-A]), then single cells were selected using forward scatter height vs FSC-A plot (Figure S1).
Apoptosis assay
Double fluorescence staining with Annexin V-FITC/PI was used to determine the percentage of cells actively undergoing apoptosis.17 The cells were seeded and treated for 48 hours, as mentioned above. Cells were harvested, centrifuged at 2,000 rpm for 3 minutes (at 4°C), washed in cold PBS, and resuspended in 100 μL per tube of Annexin V binding buffer (HEPES: 0.1 M, NaCl 1.4 M, CaCl2 25 mM, pH 7.4). Two microliters per tube of Annexin V conjugated Alexa Fluor 488 (Alexa 488) was added to the cell solution and incubated at room temperature for 15 minutes, protected from light. Four hundred microliters of the binding buffer and 5 μL per tube of 50 μg/mL PI were then added to the solution and kept on ice. The resulting samples were analyzed by FACScan. Annexin V-positive, PI-negative cells (Annexin V+/PI−), found in the lower right quadrant of the FACS dot plot, indicated early apoptotic population; Annexin V+/PI+ cells, found in the upper right quadrant of the histogram, represented either late apoptotic or secondary necrotic populations.18
Analysis of mitochondrial membrane potential
Changes in mitochondrial membrane potential were evaluated using the mitochondrial marker tetramethyl-rhodamine ethyl ester (TMRE).19,20 Following SMA-tDodSNO (10 and 40 μM) and/or Dox (0.1 μM) treatments, cells were harvested by trypsinization, centrifuged at 2,000 rpm for 3 minutes, and resuspended in 500 μL of media. Then, 2 μL of 10 μM TMRE was added to the suspensions of cells and incubated for 20 minutes at 37°C, protected from light, washed with PBS, and resuspended in media. The resulting samples were analyzed by FACScan cytometer.
Effect of SMA-tDodSNO on the cellular uptake of SMA-Dox and Dox
To elucidate whether SMA-tDodSNO affects the cellular uptake of SMA-Dox, 4T1 cells in six-well plates (200,000 cells/well) were treated with SMA-tDodSNO (10 or 40 μM) and/or SMA-Dox (1 μM) in medium and incubated for 4 hours. The cells were then washed three times with prewarmed fresh media and the fluorescent intensity of Dox in the cells was quantified by the Nikon Inverted Ti-E microscope (Coherent Instruments, Adelaide, Australia) and by flow cytometry. To evaluate the effect of SMA-tDodSNO on the Dox intracellular concentration, 4T1 cells were treated with SMA-tDodSNO (10 and 40 μM) and Dox (0.1 μM) and incubated for 48 hours, then the cellular concentration of Dox was quantified.
Determination of lysosomal membrane permeability
To evaluate whether the lysosomal membrane permeability was affected by SMA-tDodSNO treatment, acridine orange (AO) staining was used. 4T1 cells plated in six-well plates (200,000 cells/well) were treated in the medium for 4 hours with SMA-tDodSNO (10 and 40 μM) and/or SMA-Dox (1 μM), stained with 2 μL of solution (dissolved in double distilled water [ddW] at a concentration of 1 mg/mL) for 15 minutes at 37°C and imaged by fluorescent microscopy. For flow cytometry analysis, after incubation with AO, the cells were removed from the plate with trypsin, collected in RPMI, then resuspended in the medium and analyzed by the FACScan.
In vivo studies
All procedures involving the use of animals were conducted according to animal care policy of the Arabian Gulf University (AGU) and approved by the Research and Ethics Committee, AGU. To this end, BLAB/c female mice, 6 weeks old, weighing 22.4±2.6 g, were housed in the laboratory animal care facility of the AGU, under a standardized condition of 12 hours light and dark cycle. 4T1 cells (2×106 cell) were implanted subcutaneously in the dorsal skin (right and left sides) of 6-week-old male BALB/c mice. When tumors reached 100 mm3 in size, mice were injected with their respective test drugs. The in vivo study includes two sets of experiments. A) Evaluation of the influence of local administration of SMA-tDodSNO on SMA-Dox accumulation in tumor. To this extent, the xenografted mice were treated with either SMA-Dox or SMA-Dox+SMA-tDodSNO (four mice/group). In this experiment, SMA-Dox was injected intravenously via the tail at the dose of 5 mg/kg, and 1 mg/kg SMA-tDodSNO was injected intratumorally. Twenty four hours later, the mice were euthanized and tumors were excised and preserved in liquid nitrogen. B) Evaluation of the antitumor properties of SMA-tDodSNO alone and in combination with SMA-Dox. To this extent, the mice (5/group) were randomly distributed into four groups: 1) SMA-Dox, 2) SMA-tDodSNO, 3) SMA-Dox+SMA-tDodSNO, and 4) control group. The day of initial administration was set as day 0. At day 0 and day 8 of the study, 5 mg/kg SMA-Dox intravenous and/or SMA-tDodSNO 1 mg/kg intratumorally were injected. The tumor volume was measured by manual caliber; the volume was estimated by using the formula V (mm3) = ((transverse section)2 × (longitudinal cross-section)/2). Tumor volumes were normalized by using the initial tumor volume. The body weight of mice was measured and normalized daily for 14 days as an index of acute toxicity. Tumor tissues were thawed and cut into small pieces. For each 1 g of the tissue, 10 mL of 33% HCl in ethanol was added and the suspension was heated to 70°C in an oven for 30 minutes. The mixture was homogenized followed by sonication (for five intervals), vortex, and centrifugation (for three times). Finally, the supernatant was isolated from the pellet, and using a UV-VIS spectrometer, the absorbance of Dox was measured at 480 nm.
Statistical analysis and quantification of synergism
The results were presented as the mean and SD of the mean. Half-maximal inhibitory concentrations (IC50s) for cytotoxicity were interpolated from semilogarithmic plots of concentration–response curves using OriginPro 8 software. To quantify the interaction of the combinations (synergism and antagonism), the methodology of Chou and Talay was applied using CompuSyn 1.01 software (ComboSyn, Inc., Paramus, NJ, USA).21 The results were expressed as a combination index (CI), where CI <1, CI =1, and CI >1 were considered to indicate synergistic, additive, and antagonistic effects, respectively.21 Statistical analysis was performed by GraphPad Prism, and all comparisons were carried out by a one-way ANOVA followed by the Bonferroni post hoc or independent sample t-test. The differences were considered significant if P-value <0.05.
Results
Synthesis and characterization of SMA-tDodSNO and SMA-Dox
A summary of the characteristics of synthesized NPs, SMA-tDodSNO, and SMA-Dox, is reported in Table 1. The loading of the correspondent active principle in SMA-tDodSNO and SMA-Dox were 21.1±1.4 and 13.1%±2.6% (w/w), respectively. SMA-tDodSNO and SMA-Dox had a mean diameter of 227±37 and 34.3±8.2 nm, respectively. Measured polydispersity index for SMA-tDodSNO and SMA-Dox was 0.211±0.024 and 0.26±0.15, respectively. The surface charge of both micellar systems in ddW was essentially neutral, with a mean zeta potential of −0.001±0.032 mV for tDodSNO and +0.15±0.14 mV for SMA-Dox (Table 1). In PBS (pH =7.4), the NPs were stable and limited release rate was detected within 6 hours (SMA-tDodSNO: 8.46%±4.12% and SMA-Dox: 3.22%±1.96%).
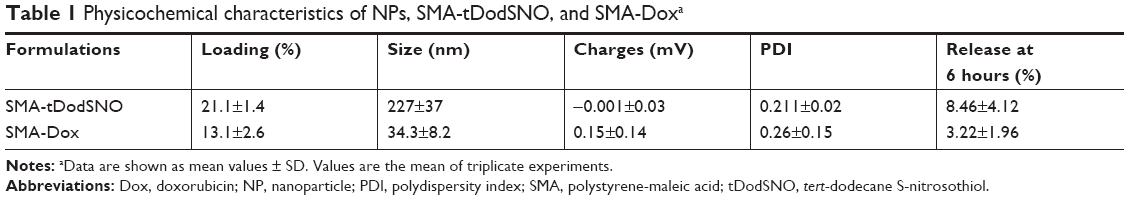 | Table 1 Physicochemical characteristics of NPs, SMA-tDodSNO, and SMA-Doxa |
Effect of the SMA-tDodSNO and Dox treatments on cell viability
Incubation of 4T1 cells with SMA-tDodSNO for 48 hours over a range of different concentrations showed a concentration-dependent decrease in cell survival, with an IC50 calculated from the concentration–response curve of 56.7±4.9 μM (Figure 1A). The combination of SMA-tDodSNO and Dox potentiated the cytotoxicity of Dox with a consequent IC50 shift from 205±34 to 1.79±0.7 nM (P<0.001) when 60 μM of SMA-tDodSNO was added to the cells (Figure 1B). Chou-Talalay analysis of the treatments showed a CI <1, indicating a synergistic interaction between SMA-tDodSNO and Dox on 4T1 cell survival (Figure 1C). Apart from the lowest concentrations (Dox 0.01 μM and SMA-tDodSNO 10 μM), all other doses combination showed a synergistic effect.
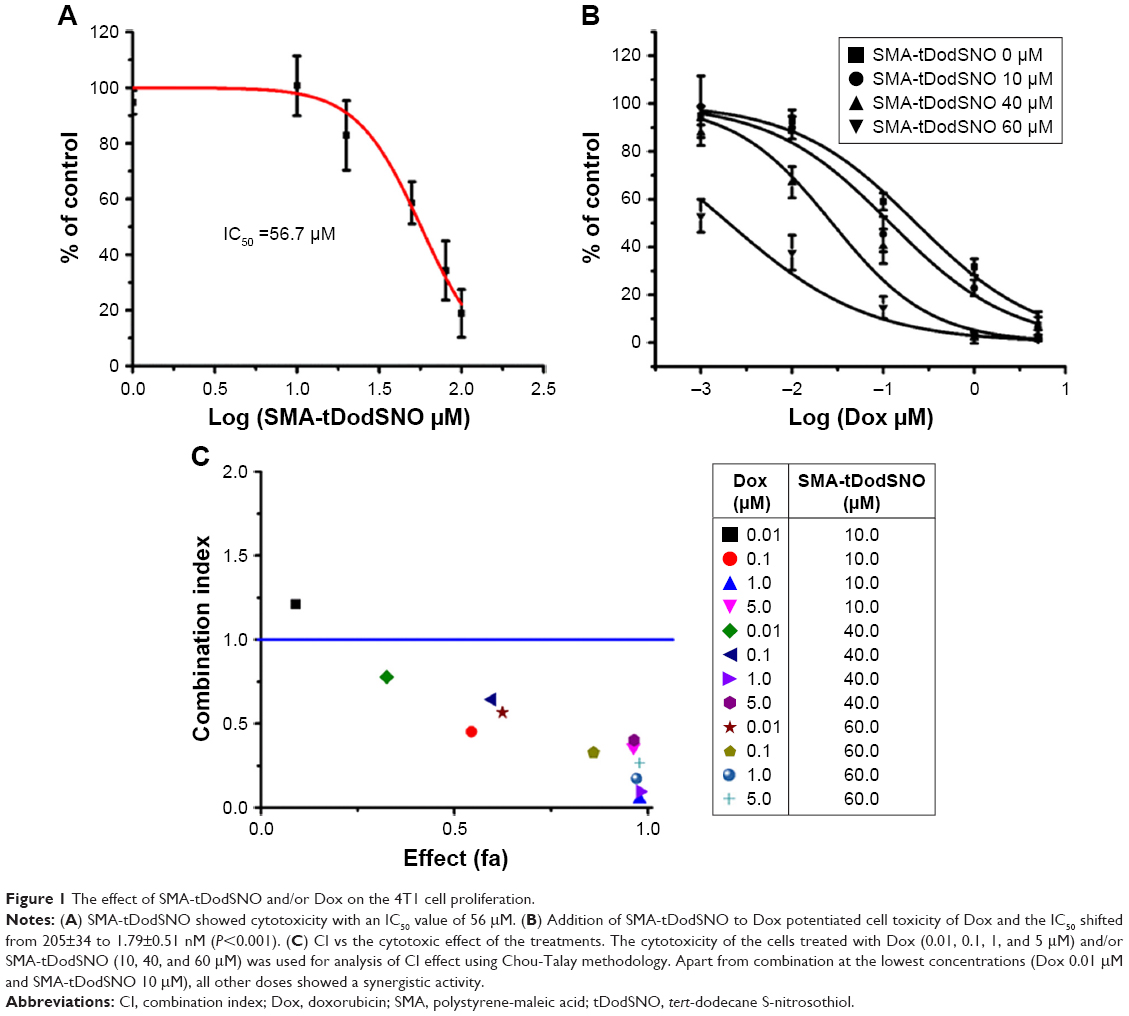 | Figure 1 The effect of SMA-tDodSNO and/or Dox on the 4T1 cell proliferation. |
Effect of the SMA-tDodSNO and Dox treatments on cell cycle
To evaluate the mechanisms by which the different treatments inhibited 4T1 cell proliferation, cell cycle distribution was analyzed by flow cytometry. The identification and gating of single cells by standard biparametric dot plot are given in Figure S1. Both Dox and 40 μM SMA-tDodSNO significantly increased the population of cells in the subG1 phase (P<0.05 and P<0.001, respectively) compared with untreated cells (Figure 2). When a combination of SMA-tDodSNO and Dox was used, the cells population in subG1 phase was 2.8-fold more than only Dox-treated cells (P<0.001) and was significantly more than SMA-tDodSNO-treated cells (P<0.05).
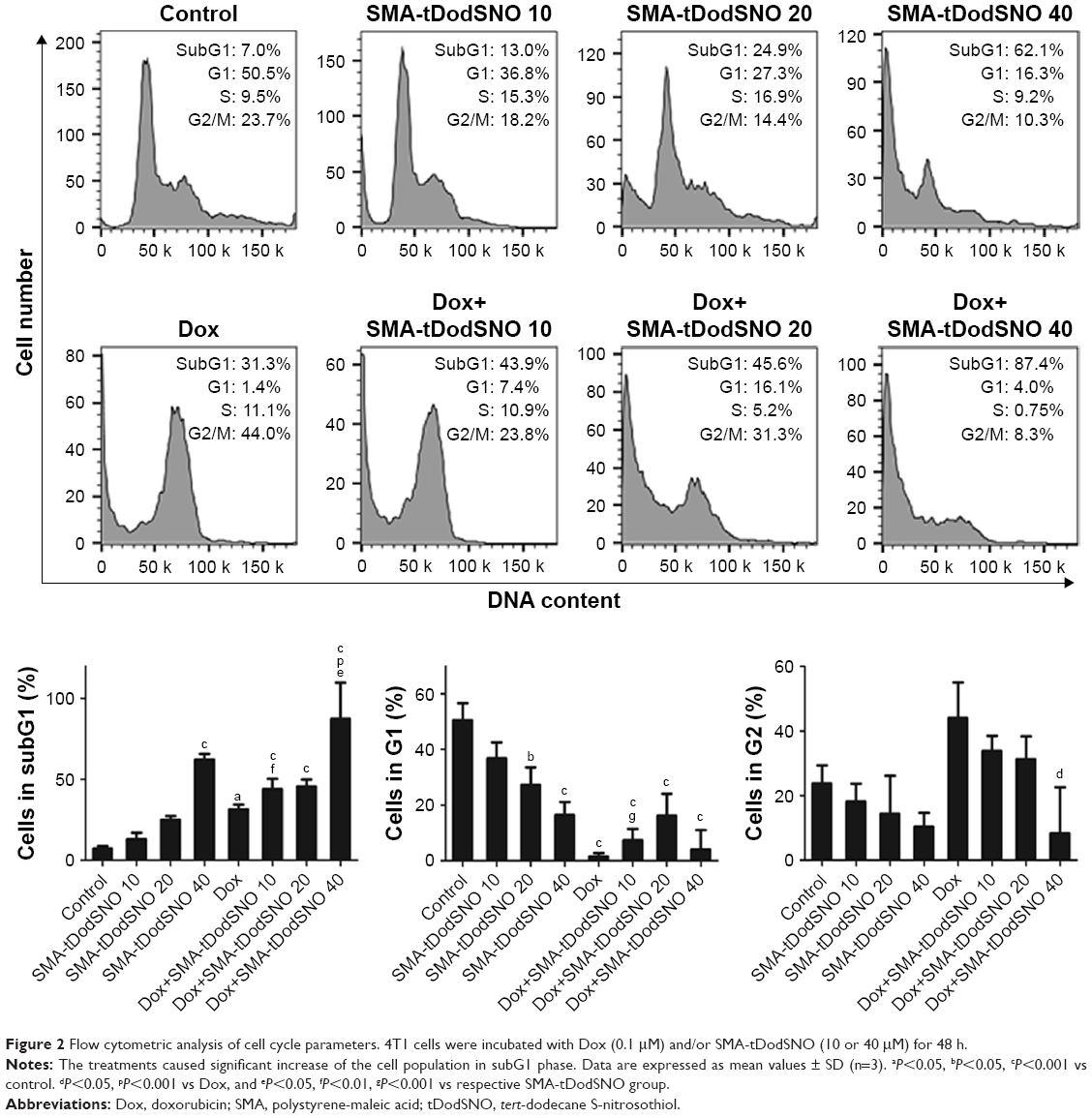 | Figure 2 Flow cytometric analysis of cell cycle parameters. 4T1 cells were incubated with Dox (0.1 μM) and/or SMA-tDodSNO (10 or 40 μM) for 48 h. |
Effect of the SMA-tDodSNO and Dox treatments on apoptosis
The Annexin V and PI double labeling assay was used to determine18 the extent of apoptosis following the treatments and the results are summarized in Figure 3. SMA-tDodSNO at concentrations of 10 and 40 μM decreased the percentage of viable cells (Annexin V−/PI−) to approximately 48.8%±8.7% and 33.9%±3.8% (P<0.001 vs control). Upon SMA-tDodSNO treatments, cells shifted to preapoptotic (33%±8.4% and 47.1%±3.7%; 9- and 13-fold greater than that of control, respectively, P<0.001) and necrotic states (15.9%±1.6% and 14.6%±1.4%, respectively; Figure 3). Similarly, Dox (0.1 μM) caused a significant reduction of viable cells to 38%±8.7% (P<0.05) and enhancement in the population of apoptotic (8.5-fold, P<0.001 vs control) and necrotic (3.3-fold, P<0.01 vs control). When the cells were treated concurrently with Dox and SMA-tDodSNO (40 μM), the percentage of the alive cells dropped to 21.7%±3.9%, which was significantly less than control (P<0.001) and either treatment alone (P<0.05). In addition, the population of apoptotic cells was significantly greater compared to Dox-treated cells (P<0.05; Figure 3).
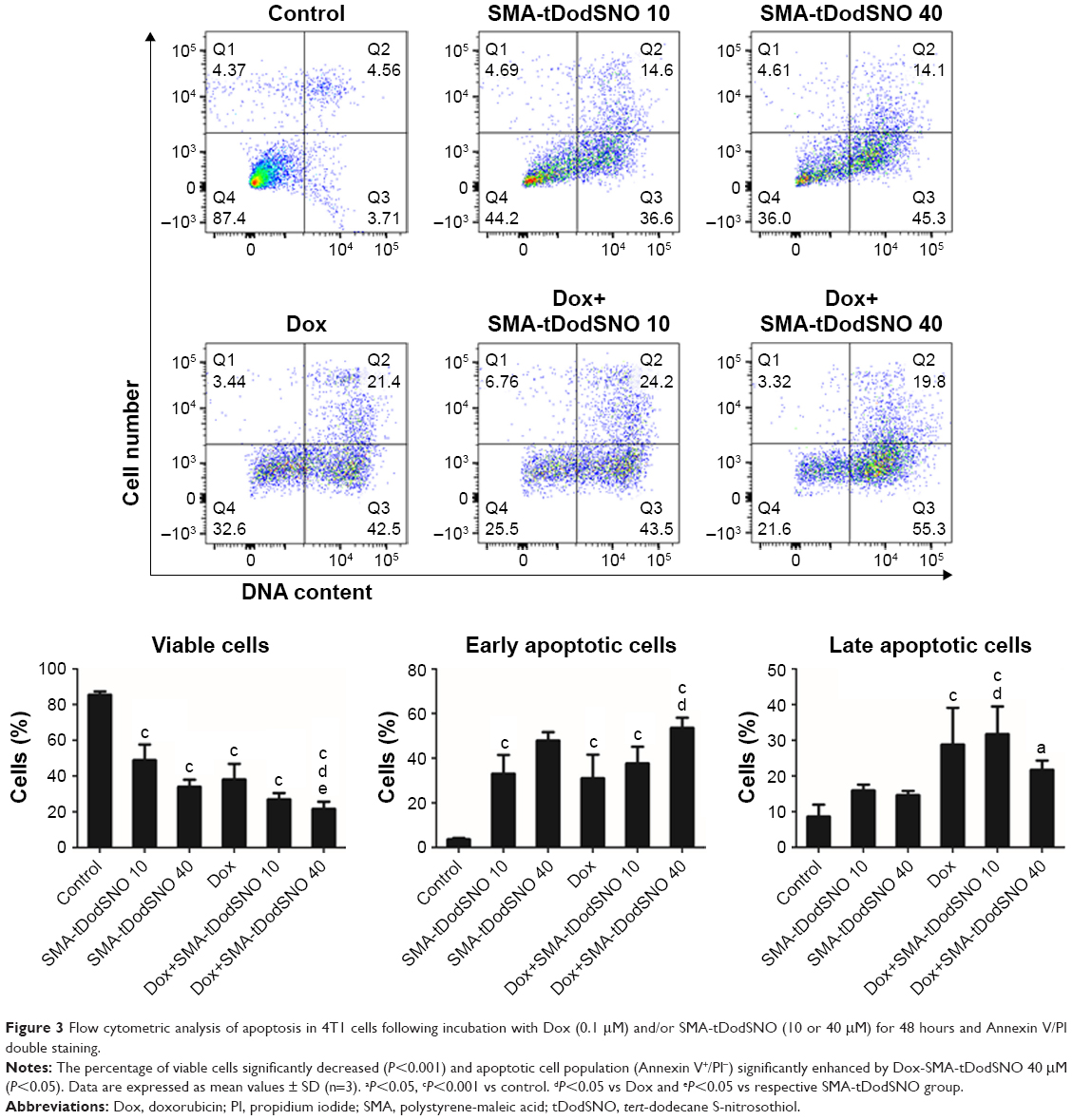 | Figure 3 Flow cytometric analysis of apoptosis in 4T1 cells following incubation with Dox (0.1 μM) and/or SMA-tDodSNO (10 or 40 μM) for 48 hours and Annexin V/PI double staining. |
Role of mitochondria in the cytotoxicity of SMA-tDodSNO and Dox treatments
Mitochondrial dysfunction has been reported as an early indicator of Dox and NO-induced apoptosis,22,23 hence the effect of SMA-tDodSNO and Dox was evaluated on mitochondrial membrane potential. The TMRE histograms (Figure 4) show that both SMA-tDodSNO and Dox significantly shifted the fluorescence peak to the left. The percentage of TMRE+ cells in the SMA-tDodSNO (40 μM)- and Dox (0.1 μM)-treated samples were 16.9±9.5 and 35.8±3.2, respectively, which were significantly different from the control 86.5±5.6 (P<0.05) indicating a loss in mitochondrial membrane potential.24 Combination treatment shifted the peak further to the left, and the population of TMRE+ cells was four times less than Dox-treated cells. However, the decrease in the fluorescent unit was not statistically significant compared to SMA-tDodSNO (40 μM)-treated cells.
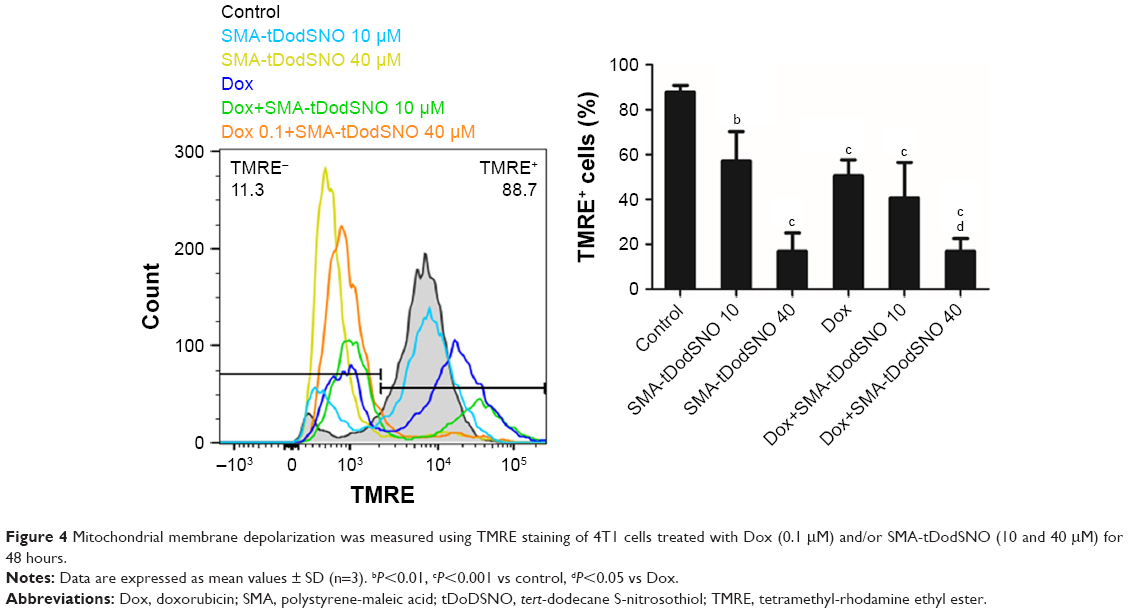 | Figure 4 Mitochondrial membrane depolarization was measured using TMRE staining of 4T1 cells treated with Dox (0.1 μM) and/or SMA-tDodSNO (10 and 40 μM) for 48 hours. |
Effect of SMA-tDodSNO on the endocytosis of SMA-Dox
Due to the size of SMA-Dox (mean diameter =34 nm), it cannot be internalized through diffusion, and their internalization depends on endocytosis.25 To investigate the facilitating role of SMA-tDodSNO in SMA-Dox uptake, we measured the fluorescent imaging of the cells. 4T1 cells were treated with either SMA-Dox or SMA-Dox+SMA-tDodSNO for 4 hours and, as reported in Figure 5, SMA-tDodSNO enhances the cellular uptake of SMA-Dox. Quantification of the fluorescent intensity showed that the addition of 40 μM of SMA-tDodSNO to equimolar concentration of NPs was about twofold higher than in the cells treated with SMA-Dox alone (from 48.6±5.3 to 79±10.1, P<0.05) (Figure 5). Higher number of cells uptaking Dox accompanied the higher intracellular concentrations after 48 hours (Figure 6). The percentage of cell population with high fluorescent intensity of Dox was significantly enhanced when cotreated with SMA-tDodSNO (P<0.001) from 38.3±4.9 (Dox-treated cells) to 72.5±4.0 (Dox+SMA-tDodSNO 10 μM) and 89.1±2.3 (Dox+SMA-tDodSNO 40 μM; Figure 6).
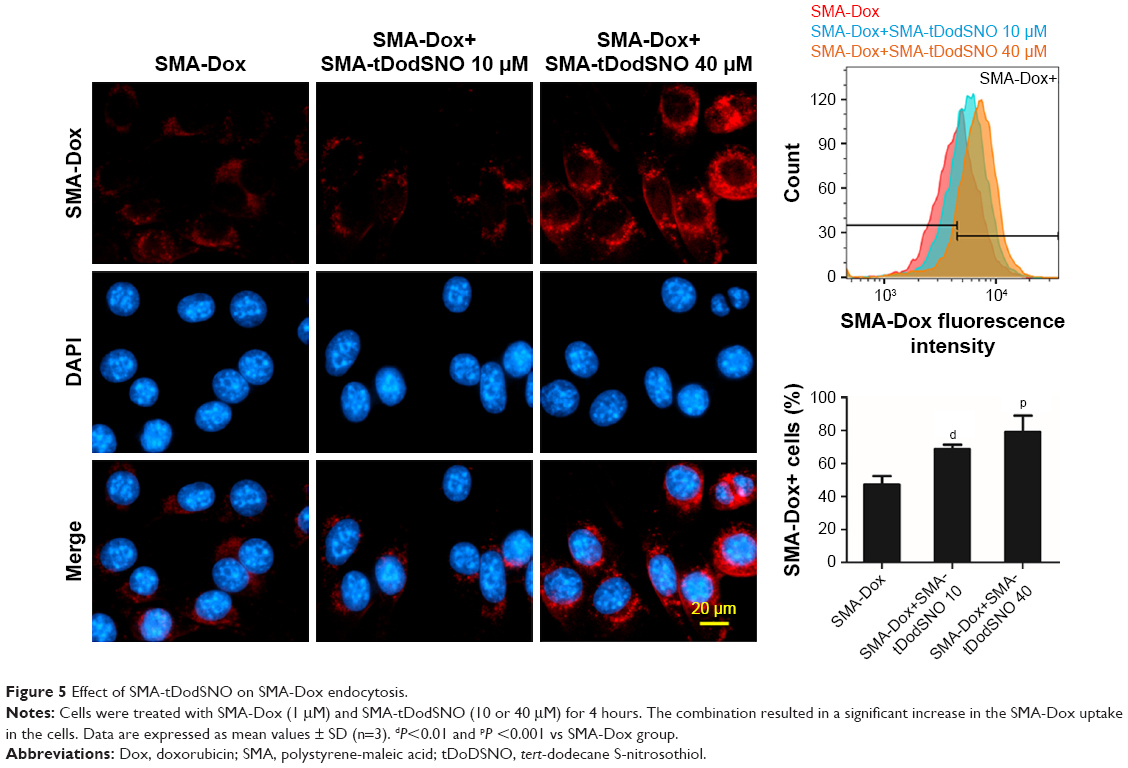 | Figure 5 Effect of SMA-tDodSNO on SMA-Dox endocytosis. |
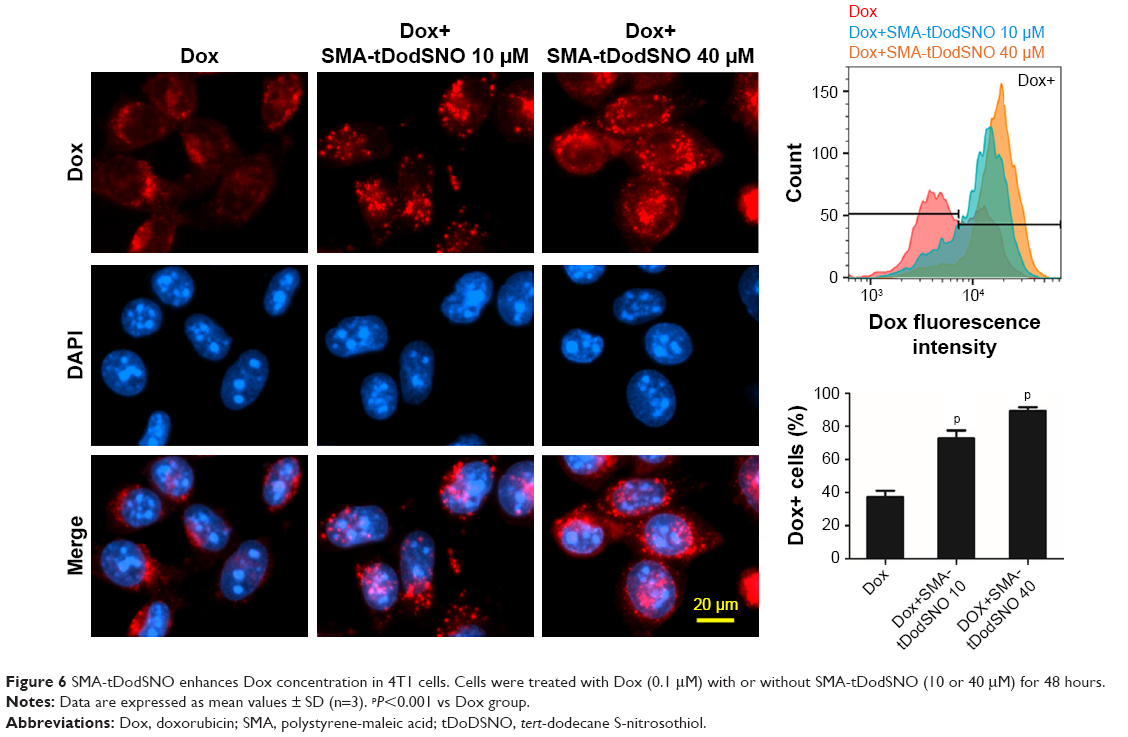 | Figure 6 SMA-tDodSNO enhances Dox concentration in 4T1 cells. Cells were treated with Dox (0.1 μM) with or without SMA-tDodSNO (10 or 40 μM) for 48 hours. |
Effect of SMA-tDodSNO on lysosome membrane permeability
AO is a lipophilic fluorescent dye that is extensively used to stain acidic vesicular organelles including autolysosomes.26 It readily diffuses into the cell compartments, and in an acidic pH of lysosomes is protonated and sequestered. The emission spectrum of protonated AO has a longer wavelength (AO is green and protonated AO is red), hence the intensity of the red fluorescence is proportional to the degree of acidity and/or the volume of the cellular acidic compartment.27 Flow cytometry analysis was used to investigate the effect of SMA-tDodSNO treatment on lysosome membrane permeability. Results showed the difference in the percentage of the cell population with a high intensity of AO (AO+ cells). AO was significantly decreased when the cells were incubated with 40 μM SMA-tDodSNO for 4 hours (P<0.05 vs control; Figure 7). Four-hour incubation with SMA-Dox (1 μM) did not affect the lysosomal membrane permeability. However, the combination of SMA-tDodSNO and SMA-Dox resulted in a significant decrease in the population of AO+ cells compared to control and either treatment alone (Figure 7). In addition, as reported in Figure S2, Chou-Talalay analysis of combined SMA-tDodSNO (10 and 40 μM)- and Dox (1 μM)-treated cells for 48 hours showed a synergistic reduction in the population of AO+ cells (CI <1, Figure S2). The highest effect was observed at Dox 1 μM and SMA-tDodSNO 40 μM. In control cells, fluorescence is limited and evenly disbursed throughout the cell. While in the SMA-tDodSNO-treated cells, the organelles are larger in size and localized in few parts of the cells (Figure 8).
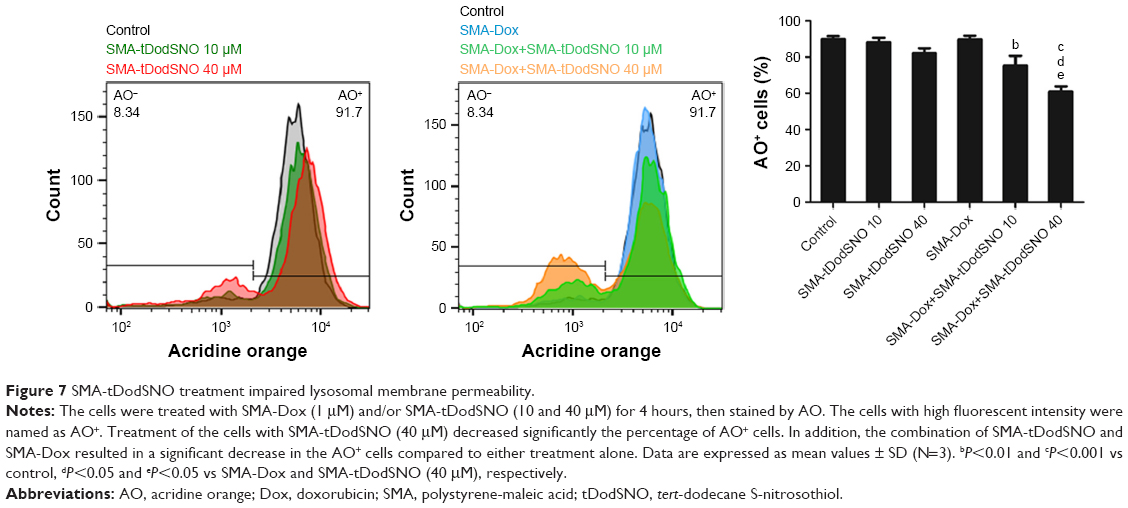 | Figure 7 SMA-tDodSNO treatment impaired lysosomal membrane permeability. |
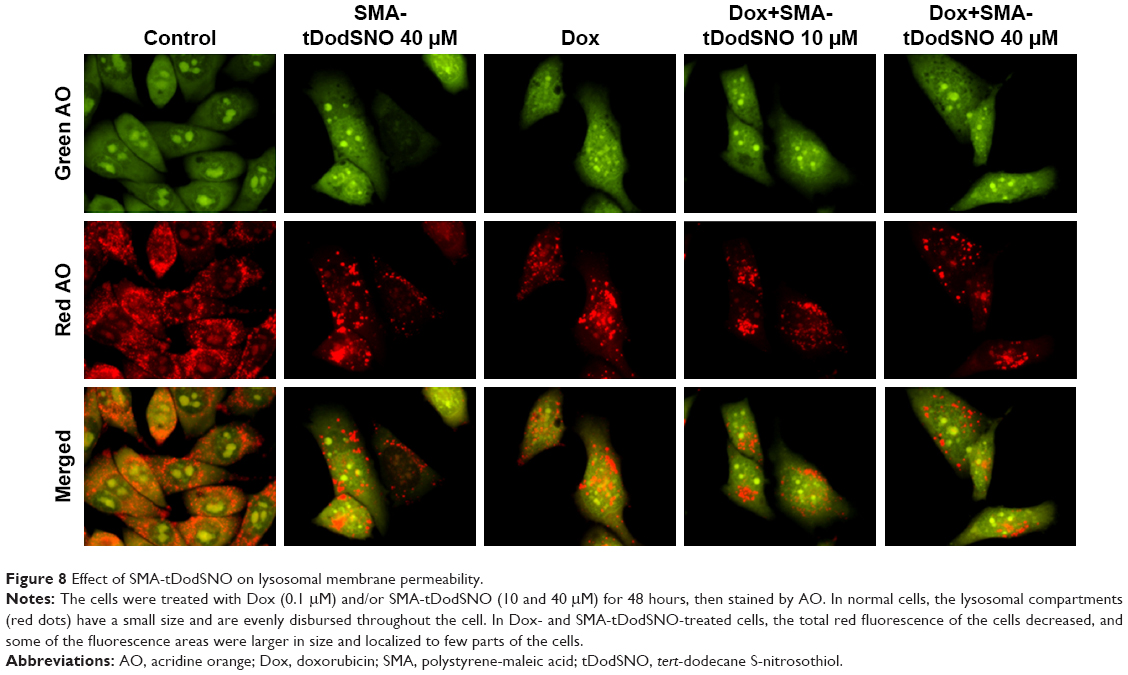 | Figure 8 Effect of SMA-tDodSNO on lysosomal membrane permeability. |
Effect of SMA-tDodSNO and SMA-Dox on concentrations in tumor tissues, tumor growth, and animals’ weight
We measured the concentration of SMA-Dox in tumor tissue upon treatment alone or in combination with SMA-tDodSNO. Local administration of SMA-tDodSNO (1 mg/kg) increased the tumor concentration of SMA-Dox 24 hours after the injections (1.65-fold of SMA-Dox alone); however, the difference did not reach statistical significance (Figure 9). The effect of the treatments on tumor growth was measured in vivo in a xenograft model of TNBC. After 17 days of tumor inoculation, the control group and SMA-Dox-treated mice had equal rate of tumor progression (5.6-fold increase in the size compared to the day of drug injection D0) indicating that SMA-Dox on this tumor model at 5 mg/kg concentration was not effective (Figure 10A). Interestingly, SMA-tDodSNO alone resulted in a twofold reduction of the tumor size compared to the control group (244% vs 561% for control and 569% for SMA-Dox alone). Combined treatment of SMA-Dox and SMA-tDodSNO significantly arrested the tumor growth for the first 6 days of treatment (80% vs 375% for control; P<0.05) and at day 10 post injection, tumor volume was three times less than in the mice treated with SMA-Dox alone (195% vs 569% for SMA-Dox group; Figure 10A); however, the difference was not statistically significant (vs either SMA-tDodSNO or SMA-Dox alone). General toxicity in mice following the SMA-tDodSNO was assessed through the experiment. The treatments did not result in any significant weight loss over the study duration indicating the safety of the combination (Figure 10B).
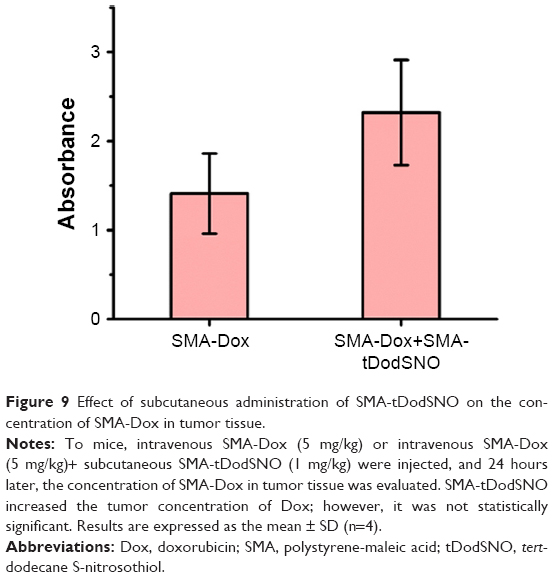 | Figure 9 Effect of subcutaneous administration of SMA-tDodSNO on the concentration of SMA-Dox in tumor tissue. |
 | Figure 10 Effect of SMA-tDodSNO and SMA-Dox on tumor growth. |
Discussion
Targeted anticancer therapies using NP-based drug carriers have emerged as an appealing approach to overcome the limitations associated with conventional drugs such as poor pharmacokinetics resulting in dose-limiting side-effects. Few Nano-based drugs reached clinical applications as in the case of Dox, where improvements in tumor targeting and decreased cardiotoxicity resulted in the Food and Drug Administration approval of Doxil® (Doxorubicin liposomal) for the treatment of Kaposi’s sarcoma.28 However, efficacious responses to Dox-loaded NPs (as well as other anticancer NPs) in tumors can be impaired by reticuloendothelial clearance, blood flow limitations, cellular internalization, escape from endosomal and lysosomal compartments, and drug efflux pumps.29,30
Recent experimental studies have shown that combination of NO donor drugs and Dox or Dox-loaded NPs potentiate the anticancer effects of Dox.31–33 However, currently available NO donors are unstable and possess limited NO payloads; therefore, they cannot be used for targeting tumors.11 In addition, the mechanism by which NO donors improve the anticancer effects of Dox is not yet fully investigated.
Here, we report that SMA-tDodSNO in combination with free Dox or SMA-Dox improves their anticancer properties. Combination treatments reduce cell viability and significantly enhanced early and late apoptosis in 4T1 cancer cells when compared to control as demonstrated by cell cycle analysis. Quantification of the cell population in the subG1 phase has been known as an easy way to score apoptotic events.18 Upon incubation with a combination of SMA-tDodSNO (40 μM) and Dox (0.1 μM) for 48 hours, the majority of the cell population (87%) were in the subG1 phase. Annexin V/PI assay showed that around 80% of the treated cells were not stained by PI (PI−) indicating that they were alive.34 Hence, it can be concluded that combined treatment of SMA-tDodSNO and Dox arrests the cells in SubG1 phase, which then results in apoptosis. In addition, as an early indicator of apoptosis, we studied the mitochondrial dysfunction.22,23 Treatments with SMA-tDodSNO and/or Dox resulted in a significant depolarization of mitochondrial membrane, shown by the decrease in the TMRE fluorescence. This effect was potentiated when cells were treated with both treatments concurrently. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) can significantly affect mitochondrial membrane permeability and guide cells to apoptosis and necrosis.35 ROS and RNS generation plays important role in the cytotoxicity induced by Dox and NO donors.36,37 Dox increases mitochondrial generation of O2.−.38 In addition, the reaction of NO with other free radicals such as O2.− produces highly cytotoxic free radicals such as ONOO−.39 Therefore, combination of NO donor and Dox probably facilitates the production of such highly reactive species that cause damage at key intracellular sites and theoretically can reduce resistance to Dox.
Combination of SMA-Dox and SMA-tDodSNO in vitro remarkably enhanced the intracellular concentration of SMA-Dox within 4 hours compared to the SMA-Dox-treated cells. Cellular uptake of NPs occurs through endocytosis;40 therefore, the enhanced SMA-Dox uptake could be due to the facilitated endocytosis. This is supported by earlier reports that showed that NO accelerates endocytosis by S-nitrosylation of dynamin, a GTPase enzyme that has a critical role in endocytic membrane fission events,41 and increases its ability to hydrolyze GTP and cleave the endocytic vesicle free from the cell membrane.42 Hence, in our model, NO-releasing NPs, such as SMA-tDodSNO, can be used to enhance membrane penetration of NPs. In addition, when SMA-tDodSNO was added to Dox, a twofold higher concentration of Dox in the cells was detected.43 The increased intracellular concentration of Dox is in agreement with previously reported studies and seems to be responsible for the synergistic cytotoxicity of NO donor drugs and Dox when used in combination.40,44
One limitation related to the intracellular delivery of NPs is the escape from endosomal and lysosomal compartments, as low-pH environment and enzymes are proved to be detrimental to nanodelivery systems cargo.29 Therefore, we measured AO staining as a measure of lysosomal membrane integrity. The loss of lysosomal membrane integrity can be measured as the loss of red fluorescence of AO.45,46 SMA-tDodSNO significantly decreased the percentage of the cell population with a high intensity of red fluorescence. In addition, when it combined with SMA-Dox or free Dox, its effect on lysosomal compartment was significantly potentiated. The effects of NO on lysosomes have been poorly investigated so far. Sarkar et al have shown that NO exhibits complex inhibitory effects on the autophagy process.47,48 Following the loss of lysosome membrane integrity, the lysosomal cysteine proteases, cathepsins, penetrate into the cytoplasm and activate necrosis and apoptosis.49,50 Our results suggest that the another possible mechanism for the synergistic anticancer activity of Dox and SMA-tDodSNO might be their synergistic destabilization of lysosomal compartments. This is in agreement with a recent study that has shown that supplementation of Dox with lysosome inhibitors such as bafilomycin A1 and chloroquine provided a more efficient anticancer effect in hepatic cancer cells by increasing lysosomal membrane permeability.51
Subcutaneous administration of SMA-tDodSNO (1 mg/mL) did not induce any sign of toxicity in mice and when administered with SMA-Dox, it increased the concentration of Dox in tumor tissue, even though the difference was not statistically significant. This is in agreement with earlier reports that showed that NO donors52–55 and other vasodilators like sildenafil56 enhance EPR effects in tumor tissues. In addition, our findings are in agreement with earlier reports showing that the NO donors inhibited drug resistance by decreasing the expression of P-glycoproteins in CT26 cell line.31
Treatments with SMA-tDodSNO in vivo resulted in a twofold decrease in tumor size, which supports the in vitro apoptotic and anticancer properties of the NPs. Herein, we hypothesize that the antitumor effects of SMA-tDodSNO could be due to the generation of nitrosative and oxidative stress in the cancer cells.57,58 As expected, the highest antitumor effects were observed when SMA-tDodSNO and SMA-Dox were injected concurrently. It has been shown that NO sensitizes cancer cells to subtoxic chemo-immuno-cytotoxic agents by its direct inhibition of nuclear factor kappa B (NF-κΒ) and Snail (the NF-κΒ target gene).59 Previous studies in 4T1 TNBC model using the free Dox at the same dose equivalence of SMA-Dox in this study failed to show any significant anticancer activity.56,60
Finally, in Figure 11, we schematically showed our hypothesis explaining the potential beneficial effects of SMA-tDodSNO on the antitumor properties of Dox and SMA-Dox. SMA-tDodSNO could enhance blood supply and vessel permeability in tumor tissues, which results in the accumulation of SMA-Dox. In addition, SMA-tDodSNO can help to overcome some of the biologic barriers that hinder the delivery of SMA-Dox. Uptake studies showed that SMA-tDodSNO that increases the intracellular concentration of SMA-Dox may be favoring their endosomal escape. Attenuating the drug efflux from the cells or increasing its intracellular concentration with consequent generation of RNS and ROS would help to prevent drug resistance and improve the efficacy of anticancer drugs.
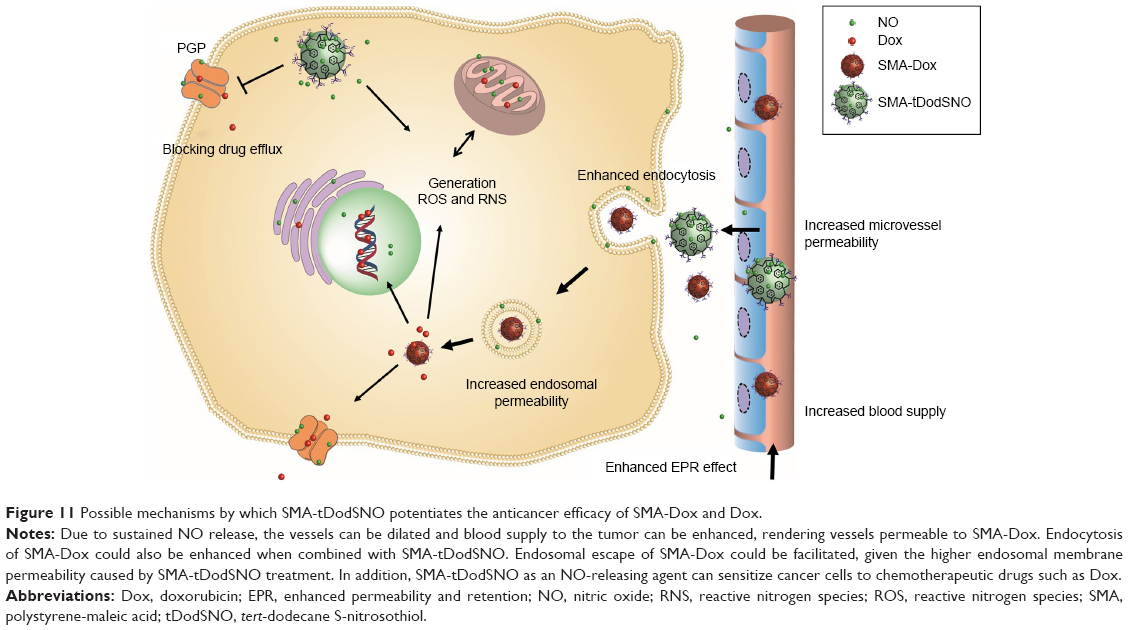 | Figure 11 Possible mechanisms by which SMA-tDodSNO potentiates the anticancer efficacy of SMA-Dox and Dox. |
Conclusion
In this work, we demonstrated that SMA-tDodSNO as a novel NO-releasing NP synergistically increased anticancer properties of Dox-loaded NPs through enhanced EPR effect, compromising endosomal membrane integrity, enhancing intracellular concentrations, and augmenting the overall drug concentrations in tumor tissues. Our work suggests that stable NO donor NPs such as SMA-tDodSNO can be used to overcome the biologic barriers, which hinder the efficacy of anticancer NPs.
Acknowledgments
The authors would like to thank Dr Greg Giles (Otago University) for all the generous support he provided throughout this work and Prof Sarah Hook (Otago University) for the use of FACScan flow cytometer.
Author contributions
HA designed and conducted in vitro experiments, analyzed the data, and wrote the draft of the manuscript; KG conceived the idea, acquired the fund, supervised the work, designed and conducted the in vivo work, and contributed to writing the manuscript; AB-F conducted the flow cytometry work and analyzed its data; LA contributed to in vivo work; VP contributed to in vivo work and to writing and revising the manuscript. All authors contributed toward data analysis, drafting and revising the paper, gave approval for the final version to be published and agree to be accountable for all aspects of the work.
Disclosure
The authors report no conflicts of interest in this work.
References
Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J Drug Target. 2007;15(7–8):457–464. | ||
Maeda H, Nakamura H, Fang J. The EPR effect for macromolecular drug delivery to solid tumors: improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo. Adv Drug Deliv Rev. 2013;65(1):71–79. | ||
Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65(1–2):271–284. | ||
Alimoradi H, Matikonda SS, Gamble AB, Giles GI, Greish K. Hypoxia responsive drug delivery systems in tumor therapy. Curr Pharm Des. 2016;22(19):2808–2820. | ||
Barua S, Mitragotri S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: a review of current status and future prospects. Nano Today. 2014;9(2):223–243. | ||
Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60(9):2497–2503. | ||
Bareford LM, Swaan PW. Endocytic mechanisms for targeted drug delivery. Adv Drug Deliv Rev. 2007;59(8):748–758. | ||
Yin Q, Shen J, Zhang Z, Yu H, Li Y. Reversal of multidrug resistance by stimuli-responsive drug delivery systems for therapy of tumor. Adv Drug Deliv Rev. 2013;65(13–14):1699–1715. | ||
Selby LI, Cortez-Jugo CM, Such GK, Johnston APR. Nanoescapology: progress toward understanding the endosomal escape of polymeric nanoparticles. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2017;9(5):e1452. | ||
Kumar V, Hong SY, Maciag AE, et al. Stabilization of the nitric oxide (NO) prodrugs and anticancer leads, PABA/NO and double JS-K, through incorporation into PEG-protected nanoparticles. Mol Pharm. 2010;7(1):291–298. | ||
Miller MR, Megson IL. Recent developments in nitric oxide donor drugs. Br J Pharmacol. 2007;151(3):305–321. | ||
Giles NM, Kumari S, Gang BP, Yuen CW, Billaud EM, Giles GI. The molecular design of S-nitrosothiols as photodynamic agents for controlled nitric oxide release. Chem Biol Drug Des. 2012;80(3):471–478. | ||
Kumari S, Sammut IA, Giles GI. The design of nitric oxide donor drugs: s-nitrosothiol tDodSNO is a superior photoactivated donor in comparison to GSNO and SNAP. Eur J Pharmacol. 2014;737:168–176. | ||
Lowenstein CJ. Nitric oxide regulation of protein trafficking in the cardiovascular system. Cardiovasc Res. 2007;75(2):240–246. | ||
Greish K, Sawa T, Fang J, Akaike T, Maeda H. SMA-doxorubicin, a new polymeric micellar drug for effective targeting to solid tumours. J Control Release. 2004;97(2):219–230. | ||
Kaur P, Nagaraja GM, Zheng H, et al. A mouse model for triple-negative breast cancer tumor-initiating cells (TNBC-TICs) exhibits similar aggressive phenotype to the human disease. BMC Cancer. 2012;12:120. | ||
Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods. 1995;184(1):39–51. | ||
Rello-Varona S, Herrero-Martín D, López-Alemany R, Muñoz-Pinedo C, Tirado OM. “(Not) all (dead) things share the same breath”: identification of cell death mechanisms in anticancer therapy. Cancer Res. 2015;75(6):913–917. | ||
Loew LM, Carrington W, Tuft RA, Fay FS. Physiological cytosolic Ca2+ transients evoke concurrent mitochondrial depolarizations. Proc Natl Acad Sci U S A. 1994;91(26):12579–12583. | ||
Farkas DL, Wei MD, Febbroriello P, Carson JH, Loew LM. Simultaneous imaging of cell and mitochondrial membrane potentials. Biophys J. 1989;56(6):1053–1069. | ||
Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70(2):440–446. | ||
Green PS, Leeuwenburgh C. Mitochondrial dysfunction is an early indicator of doxorubicin-induced apoptosis. Biochim Biophys Acta. 2002;1588(1):94–101. | ||
Doutheil J, Althausen S, Treiman M, Paschen W. Effect of nitric oxide on endoplasmic reticulum calcium homeostasis, protein synthesis and energy metabolism. Cell Calcium. 2000;27(2):107–115. | ||
Boss O, Samec S, Kühne F, et al. Uncoupling protein-3 expression in rodent skeletal muscle is modulated by food intake but not by changes in environmental temperature. J Biol Chem. 1998;273(1):5–8. | ||
Zhang S, Li J, Lykotrafitis G, Bao G, Suresh S. Size-dependent endocytosis of nanoparticles. Adv Mater. 2009;21:419–424. | ||
Pierzyńska-Mach A, Janowski PA, Dobrucki JW. Evaluation of acridine orange, LysoTracker Red, and quinacrine as fluorescent probes for long-term tracking of acidic vesicles. Cytometry A. 2014;85(8):729–737. | ||
Traganos F, Darzynkiewicz Z. Lysosomal proton pump activity: supravital cell staining with acridine orange differentiates leukocyte subpopulations. Methods Cell Biol. 1994;41:185–194. | ||
Barenholz Y. Doxil® – the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. | ||
Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–951. | ||
Riganti C, Miraglia E, Viarisio D, et al. Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux. Cancer Res. 2005;65(2):516–525. | ||
Tan L, Huang R, Li X, Liu S, Shen YM. Controllable release of nitric oxide and doxorubicin from engineered nanospheres for synergistic tumor therapy. Acta Biomater. 2017;57:498–510. | ||
Song Q, Tan S, Zhuang X, et al. Nitric oxide releasing d-α-tocopheryl polyethylene glycol succinate for enhancing antitumor activity of doxorubicin. Mol Pharm. 2014;11(11):4118–4129. | ||
Kashfi K, Rigas B. Molecular targets of nitric-oxide-donating aspirin in cancer. Biochem Soc Trans. 2005;33(Pt 4):701–704. | ||
Rieger AM, Nelson KL, Konowalchuk JD, Barreda DR. Modified annexin V/propidium iodide apoptosis assay for accurate assessment of cell death. J Vis Exp. 2011;50:2597. | ||
Wink DA, Mitchell JB. Chemical biology of nitric oxide: insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med. 1998;25(4–5):434–456. | ||
Sen S, Kawahara B, Chaudhuri G. Mitochondrial-associated nitric oxide synthase activity inhibits cytochrome c oxidase: implications for breast cancer. Free Radic Biol Med. 2013;57:210–220. | ||
Mimnaugh EG, Trush MA, Bhatnagar M, Gram TE. Enhancement of reactive oxygen-dependent mitochondrial membrane lipid peroxidation by the anticancer drug adriamycin. Biochem Pharmacol. 1985;34(6):847–856. | ||
Goormaghtigh E, Pollakis G, Ruysschaert JM. Mitochondrial membrane modifications induced by adriamycin-mediated electron transport. Biochem Pharmacol. 1983;32(5):889–893. | ||
Brown GC. Nitric oxide and mitochondrial respiration. Biochim Biophys Acta. 1999;1411(2–3):351–369. | ||
Riganti C, Miraglia E, Viarisio D, et al. Nitric oxide reverts the resistance to doxorubicin in human colon cancer cells by inhibiting the drug efflux. Cancer Res. 2005;65(2):516–525. | ||
Ferguson SM, De Camilli P. Dynamin, a membrane-remodelling GTPase. Nat Rev Mol Cell Biol. 2012;13(2):75–88. | ||
Wang G, Moniri NH, Ozawa K, Stamler JS, Daaka Y. Nitric oxide regulates endocytosis by S-nitrosylation of dynamin. Proc Natl Acad Sci U S A. 2006;103(5):1295–1300. | ||
Evans CJ, Phillips RM, Jones PF, et al. A mathematical model of doxorubicin penetration through multicellular layers. J Theor Biol. 2009;257(4):598–608. | ||
Kim J, Yung BC, Kim WJ, Chen X. Combination of nitric oxide and drug delivery systems: tools for overcoming drug resistance in chemotherapy. J Control Release. 2017;263:223–230. | ||
Petersen NH, Olsen OD, Groth-Pedersen L, et al. Transformation-associated changes in sphingolipid metabolism sensitize cells to lysosomal cell death induced by inhibitors of acid sphingomyelinase. Cancer Cell. 2013;24(3):379–393. | ||
Kirkegaard T, Roth AG, Petersen NH, et al. Hsp70 stabilizes lysosomes and reverts Niemann-Pick disease-associated lysosomal pathology. Nature. 2010;463(7280):549–553. | ||
Sarkar S, Korolchuk VI, Renna M, et al. Complex inhibitory effects of nitric oxide on autophagy. Mol Cell. 2011;43(1):19–32. | ||
Yu H, Xiao Y, Jin L. A lysosome-targetable and two-photon fluorescent probe for monitoring endogenous and exogenous nitric oxide in living cells. J Am Chem Soc. 2012;134(42):17486–17489. | ||
Repnik U, Stoka V, Turk V, Turk B. Lysosomes and lysosomal cathepsins in cell death. Biochim Biophys Acta. 2012;1824(1):22–33. | ||
Kroemer G, Jäättelä M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5(11):886–897. | ||
Li Y, Sun Y, Jing L, et al. Lysosome inhibitors enhance the chemotherapeutic activity of doxorubicin in HepG2 cells. Chemotherapy. 2017;62(2):85–93. | ||
Kinoshita R, Ishima Y, Chuang VTG, et al. Improved anticancer effects of albumin-bound paclitaxel nanoparticle via augmentation of EPR effect and albumin-protein interactions using S-nitrosated human serum albumin dimer. Biomaterials. 2017;140:162–169. | ||
Maeda H. Macromolecular therapeutics in cancer treatment: the EPR effect and beyond. J Control Release. 2012;164(2):138–144. | ||
Ishima Y, Inoue A, Fang J, et al. Poly-S-nitrosated human albumin enhances the antitumor and antimetastasis effect of bevacizumab, partly by inhibiting autophagy through the generation of nitric oxide. Cancer Sci. 2015;106(2):194–200. | ||
Ishima Y. Albumin-based nitric oxide traffic system for the treatment of intractable cancers. Biol Pharm Bull. 2017;40(2):128–134. | ||
Greish K, Fateel M, Abdelghany S, Rachel N, Alimoradi H, Bakhiet M, et al. Sildenafil citrate improves the delivery and anticancer activity of doxorubicin formulations in a mouse model of breast cancer. J Drug Target. 2018;26(7):610–615. | ||
Choudhari SK, Chaudhary M, Bagde S, Gadbail AR, Joshi V. Nitric oxide and cancer: a review. World J Surg Oncol. 2013;11:118. | ||
Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2002;160(3):985–1000. | ||
Bonavida B, Garban H. Nitric oxide-mediated sensitization of resistant tumor cells to apoptosis by chemo-immunotherapeutics. Redox Biol. 2015;6:486–494. | ||
Greish K, Taurin S, Morsy MA. The effect of adjuvant therapy with TNF-α on animal model of triple-negative breast cancer. Ther Deliv. 2018;9(5):333–342. |
Supplementary materials
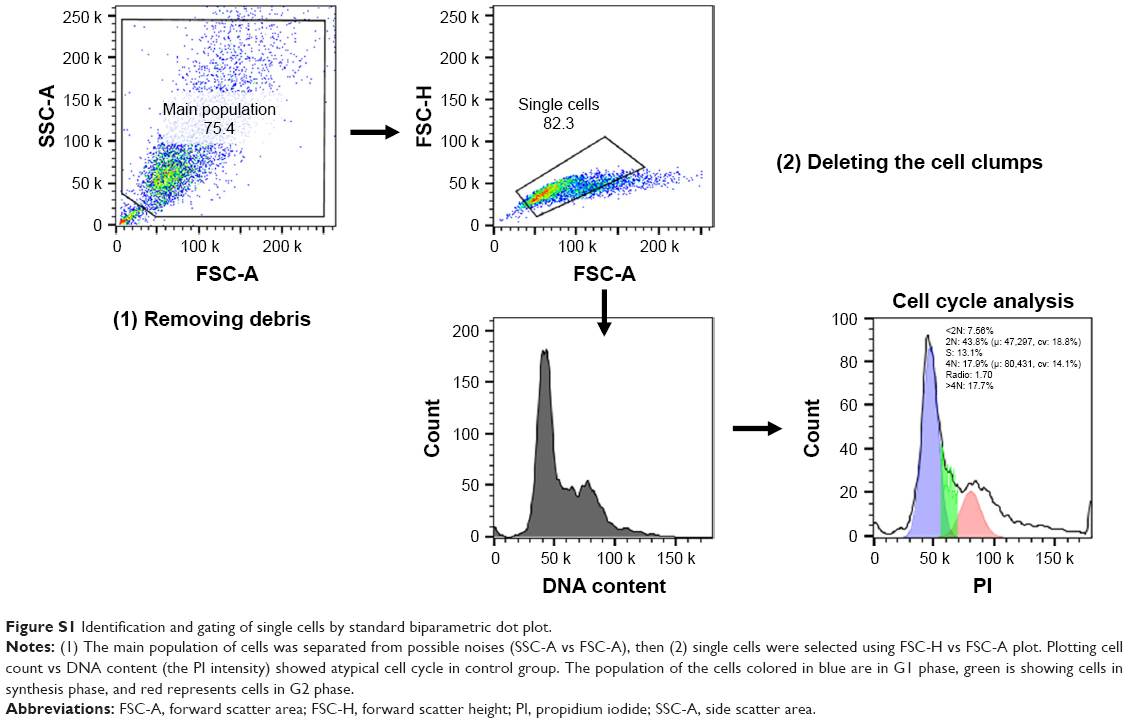 | Figure S1 Identification and gating of single cells by standard biparametric dot plot. |
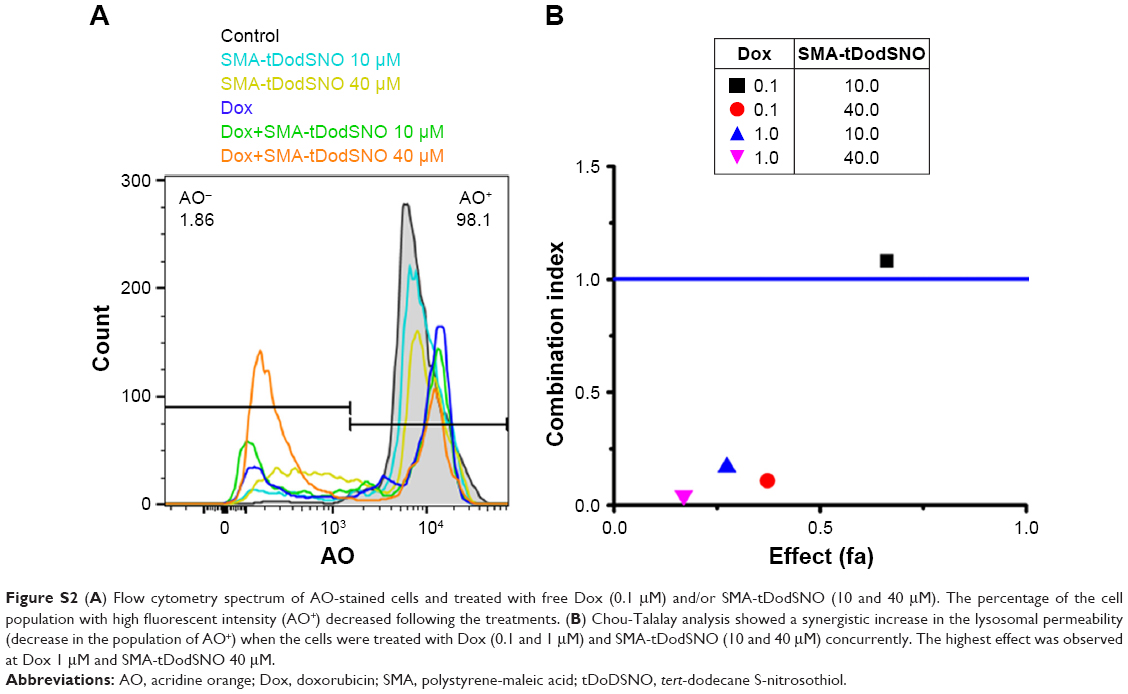 | Figure S2 (A) Flow cytometry spectrum of AO-stained cells and treated with free Dox (0.1 μM) and/or SMA-tDodSNO (10 and 40 μM). The percentage of the cell population with high fluorescent intensity (AO+) decreased following the treatments. (B) Chou-Talalay analysis showed a synergistic increase in the lysosomal permeability (decrease in the population of AO+) when the cells were treated with Dox (0.1 and 1 μM) and SMA-tDodSNO (10 and 40 μM) concurrently. The highest effect was observed at Dox 1 μM and SMA-tDodSNO 40 μM. |
 © 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2018 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.