Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
Niosomal carriers enhance oral bioavailability of carvedilol: effects of bile salt-enriched vesicles and carrier surface charge
Authors Arzani G, Haeri A, Daeihamed M, Bakhtiari-Kaboutaraki H, Dadashzadeh S
Received 16 March 2015
Accepted for publication 29 May 2015
Published 29 July 2015 Volume 2015:10(1) Pages 4797—4813
DOI https://doi.org/10.2147/IJN.S84703
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Thomas Webster
Gelareh Arzani,1 Azadeh Haeri,1 Marjan Daeihamed,1 Hamid Bakhtiari-Kaboutaraki,1 Simin Dadashzadeh1,2
1Department of Pharmaceutics, Faculty of Pharmacy, 2Pharmaceutical Sciences Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran
Abstract: Carvedilol (CRV) is an antihypertensive drug with both alpha and beta receptor blocking activity used to preclude angina and cardiac arrhythmias. To overcome the low, variable oral bioavailability of CRV, niosomal formulations were prepared and characterized: plain niosomes (without bile salts), bile salt-enriched niosomes (bilosomes containing various percentages of sodium cholate or sodium taurocholate), and charged niosomes (negative, containing dicetyl phosphate and positive, containing hexadecyl trimethyl ammonium bromide). All formulations were characterized in terms of encapsulation efficiency, size, zeta potential, release profile, stability, and morphology. Various formulations were administered orally to ten groups of Wistar rats (n=6 per group). The plasma levels of CRV were measured by a validated high-performance liquid chromatography (HPLC) method and pharmacokinetic properties of different formulations were characterized. Contribution of lymphatic transport to the oral bioavailability of niosomes was also investigated using a chylomicron flow-blocking approach. Of the bile salt-enriched vesicles examined, bilosomes containing 20% sodium cholate (F2) and 30% sodium taurocholate (F5) appeared to give the greatest enhancement of intestinal absorption. The relative bioavailability of F2 and F5 formulations to the suspension was estimated to be 1.84 and 1.64, respectively. With regard to charged niosomes, the peak plasma concentrations (Cmax) of CRV for positively (F7) and negatively charged formulations (F10) were approximately 2.3- and 1.7-fold higher than after a suspension. Bioavailability studies also revealed a significant increase in extent of drug absorption from charged vesicles. Tissue histology revealed no signs of inflammation or damage. The study proved that the type and concentration of bile salts as well as carrier surface charge had great influences on oral bioavailability of niosomes. Blocking the lymphatic absorption pathway significantly reduced oral bioavailability of CRV niosomes. Overall twofold enhancement in bioavailability in comparison with drug suspension confers the potential of niosomes as suitable carriers for improved oral delivery of CRV.
Keywords: niosomes, bile salts, surface charge, bioavailability, oral delivery, lymphatic transport
Introduction
Oral delivery is the most convenient route of drug administration, especially for chronic illness, with high patient compliance, ease of administration, cost-effectiveness, and other benefits.1 Although high oral bioavailability is highly desirable, many important drugs in the clinic suffer from poor oral bioavailability and highly variable exposure. This can be due to various factors, including low solubility, limited permeability, first-pass metabolism and drug efflux.2
To reach the bloodstream, a drug should first dissolve in the gastrointestinal (GI) fluid. Thus, dissolution may be the rate-limiting step in oral administration of poorly water-soluble drugs,3 resulting in erratic absorption and low oral bioavailability. Intestinal and hepatic first-pass metabolism can also restrict oral bioavailability to a significant extent.4,5 In the efforts to enhance oral bioavailability, various approaches have been employed, such as solid dispersions,6 salt forms,7 nanosizing, and micronization.8 In the last two decades, there has been increased interest in studying colloidal particulate carriers such as liposomes, niosomes, and micelles, as well as polymeric and lipidic nanoparticles. These have shown distinct advantages over conventional dosage forms in oral drug delivery.9 In addition to enhanced solubility and dissolution rates, these carriers provide a powerful means to avoid first-pass metabolism through stimulation of lymphatic transport, leading to improved bioavailability.10,11 In recent years, spheroid vesicular structures formed by self-assembly of nonionic surfactants (niosomes), have received significant attention as drug delivery systems. Although niosomes are similar to liposomes in physical properties and biopharmaceutical functions, they possess advantages, including higher stability, easier handling and storage, and lower cost, making them a promising alternative.12,13 These qualities also make them promising tools in the oral delivery of various therapeutic agents,14,15 as well as in preoral and mucosal immunization.16
By modifying the physicochemical characteristics of nanocarriers, including particle size, elasticity, and surface charge, it may be possible to design vesicles with attributes appropriate for a given application.10,17,18 It has been reported that the biological fate of vesicular systems following oral administration is affected by the inclusion of bile salts and charge-inducing agents.17,19
Bile salts are endogenous detergents used extensively in drug delivery as permeability enhancers, facilitating drug penetration across biological barriers including skin,20 the intestinal wall,21 the blood–brain barrier,22 nasal mucosa,23 and the cornea.24 Liposomes containing bile salts have been claimed to improve the oral bioavailability of some drugs and macromolecules.21,25,26 Although the exact mechanisms of this enhanced absorption have not been determined, it has been proposed that the addition of bile salts to lipid bilayers gives vesicles the ability to withstand disruption by physiological bile salts in the GI tract.27 Although there have been a few studies on oral delivery by bile salt-containing liposomes, to our knowledge, bile salt-enriched niosomes have not been investigated as carriers for oral delivery of poorly water-soluble drugs. This inspired our investigation into the effects of bile salts (sodium cholate [SC] and sodium taurocholate [STC] in various percentages) in niosomal formulations on intestinal absorption of a poorly water-soluble drug.
Another physicochemical characteristic with a prominent role in vesicular interactions with biological membranes is vesicular surface charge. It has been reported that a negative charge on the surface of nanocarriers promotes M-cell uptake.17,28 On the other hand, positively charged nanocarriers create strong electrostatic interactions with negative charges on the cell surface, resulting in enhanced endocytosis (especially clathrin-mediated endocytosis).17,29 Furthermore, endosomal degradation may be prevented by positive surface charge on carriers. Moreover, positive charge facilitates nanostructure transport across mucosal membranes.17
Carvedilol (CRV) is a third-generation β-blocker combining nonselective β- and α-blockade at pharmacologic doses with ion channel blockade at high concentrations.30 CRV has also shown several ancillary activities, including anti-inflammatory, antioxidant, antifibrotic and anti-apoptotic properties. These activities likely contribute to the cardioprotective effects of CRV in hypertension, myocardial infarction (MI), congestive heart failure, and post-MI left ventricular dysfunction.30,31 Low water solubility and extensive pre-systemic metabolism of CRV result in incomplete absorption (~25%) and limited systemic exposure after oral administration.31 Therefore, strategies to enhance the oral bioavailability of CRV are in great demand.
Due to the lack of evidence on the effect of bile salts and surface charge on the oral bioavailability of niosomal formulations, this work was undertaken to investigate the influence of these parameters on oral bioavailability of a poorly water-soluble drug, CRV, from niosomal formulations. Formulation parameters, including entrapment efficiency (EE), vesicle size, zeta potential, morphology, stability, and in vitro release profile, were studied. Furthermore, the in vivo pharmacokinetics of different formulations were assessed in rats to elucidate the effects of bile salts and vesicle surface charge on oral absorption of a poorly water-soluble CRV. To estimate the influence of lymphatic transport on the oral bioavailability of niosomes, in vivo studies were also conducted in chylomicron-blocked rats.
Materials and methods
Materials
CRV (98% purity) was a kind gift from Hakim Pharmaceutical Co. (Tehran, Iran). Span 20, Span 60, Span 80, Tween 80, SC, hexadecyl-trimethyl ammonium bromide (CTAB), chloroform, methyl tertiary butyl ether, HPLC-grade acetonitrile, and HPLC-grade methanol were obtained from Merck (Darmstadt, Germany). Dicetyl phosphate (DCP), STC, cycloheximide, and cholesterol (Chol, purity >98%) were provided by Sigma-Aldrich (St Louis, MO, USA). All other chemicals were of analytical grade or of the best grade available.
Niosome preparation
Different niosomal formulations were prepared by ether injection or conventional thin film hydration method. Briefly, in ether injection, surfactants (Span 60, Span 80, or Span 20), Chol and drug were dissolved in ether. The solution was slowly injected through a fine needle into warm (65°C) phosphate buffered saline (PBS, pH =7.4) rotating at a constant rate. Following removal of ether from the mixture, niosomal vesicles formed. In thin film hydration, surfactants (Span 60, Span 80, or Span 20), Chol, and drug (Table 1) were dissolved in chloroform:methanol (2:1, v/v) in a round-bottomed flask. The total surfactant concentration was varied from 20 to 80 mM during formulation optimization. Organic solvents were then removed under reduced pressure in a rotary evaporator at 65°C for 30 minutes, yielding a thin film on the wall of the flask. The lipid film was maintained under vacuum for at least 2 hours to remove the last traces of organic solvent. The dry thin film was hydrated at a few degrees above the transition temperature of the components with PBS buffer solution for 1.5 hours at a rotation speed of 120 rpm using rotary evaporator (Heidolph, Schwabach, Germany). The resulting suspension was sonicated in a bath for 1 hour. The niosomal dispersion was left at room temperature for 2 hours to ensure thorough hydration and then stored in a refrigerator.
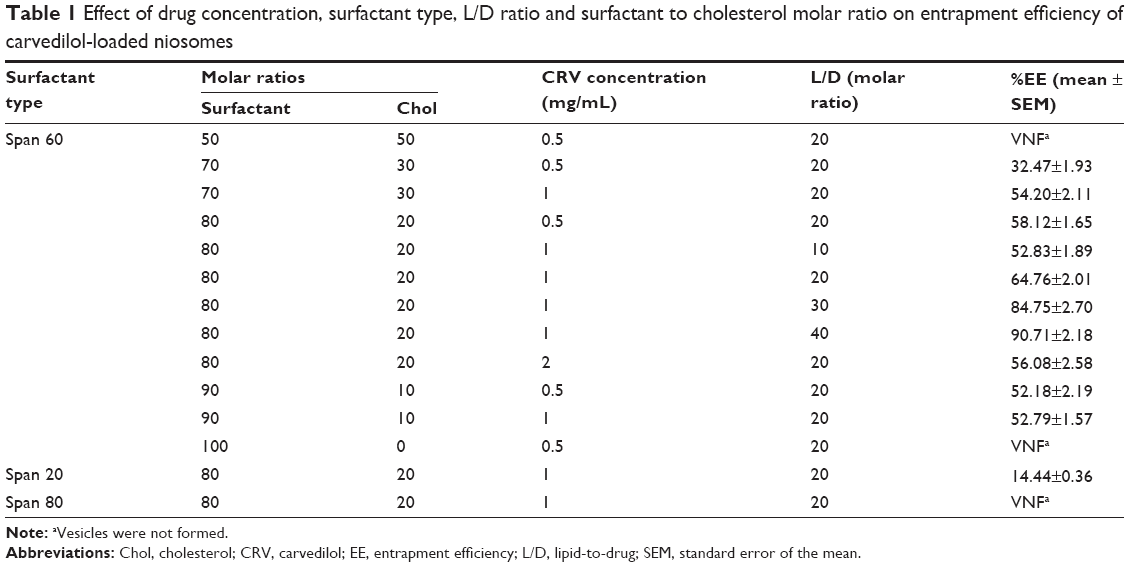 | Table 1 Effect of drug concentration, surfactant type, L/D ratio and surfactant to cholesterol molar ratio on entrapment efficiency of carvedilol-loaded niosomes |
Niosome characterization
Entrapment efficiency (EE), vesicle size, zeta potential, Fourier transform infrared (FTIR) analyses, morphology, stability, and in vitro release profile of the prepared niosomes were investigated.
Entrapment efficiency
To determine CRV content in the niosomal formulations, unentrapped drug was separated from the niosome-retained drug using membrane dialysis method against PBS at 4°C. Afterwards, a small fraction of the dialysate (50 μL) was lysed with isopropanol (950 μL) and further bath-sonicated for 10 minutes. The concentration of CRV in the resulting clear solution was then measured by UV spectrophotometer at the wavelength of 241 nm. The %EE was calculated by the following equation:
|
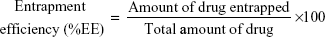
Size distribution and zeta potential
The hydrodynamic size and zeta potential of different niosomal formulations were estimated by dynamic light scattering using a Malvern Zetasizer Nano (Malvern Instruments, Malvern, UK) at 25°C. Just before measurements, samples were diluted with deionized water. Z-average and zeta potential values were presented as the mean ± standard error of the mean (SEM) from triplicate experiments.
Fourier transform infrared spectroscopy
Different samples (pure drug, empty niosomes, and CRV-loaded niosomes) were prepared and infrared spectroscopy analysis was conducted in the transmittance mode on WQF-510 Fourier transform spectrometer (Rayleigh Optics, People’s Republic of China) equipped with a deuterated Lanthanide triglycine sulfate detector and a KBr beam splitter. All spectra were acquired with 4 cm−1 resolution in the region of 4,000–500 cm−1.
Morphology of niosomes
The morphology of niosomes was investigated by atomic force microscopy (AFM). For imaging, the niosomes were diluted with deionized water and 10 μL of diluted niosomes was dripped onto the glass slide, spread thin and air-dried. AFM observations were performed with a Nanowizard II atomic force microscope (JPK Instruments, Berlin, Germany) operating in contact mode under ambient conditions employing low stress silicon nitride cantilevers (AppNano, Mountain View, CA, USA).
In vitro drug release
Release of CRV from niosomal formulations in various release media was studied using dialysis. CRV-loaded niosomes (0.5 mL) were placed into cellulose dialysis membranes (molecular weight cutoff 12,000 Da), which were then transferred to 50 mL of release media (enzyme-free simulated intestinal fluid [SIF] [pH =6.8] containing 0.1% [w/v] Tween 80 to maintain the sink condition and enzyme-free SIF [pH =6.8] containing 0.1% [w/v] Tween 80 and 10 mM STC to simulate bile salt concentrations in the small intestine). The release system was stirred magnetically at 100 rpm and 37°C. At predetermined intervals (0.5, 1, 2, 4, 6, 8, and 10 hours), 1 mL aliquots of release media were removed for HPLC analysis. Samples removed at 0.5, 1, 8, and 10 hours were replenished immediately with the same volume of fresh media, while after sampling of medium at 2, 4, and 6 hours for drug analysis, the whole incubated media were totally replaced with fresh one to maintain perfect sink conditions and increase the driving force for diffusion. Each experiment was carried out in triplicate. To determine drug concentrations, samples were centrifuged at 10,000 rpm for 5 minutes, diluted and injected to the HPLC system.
Niosome stability in 10 mM STC
We tested the ability of CRV-loaded niosomes to retain their physical properties in the presence of STC under perfect sink conditions. Different formulations were placed into pre-warmed enzyme-free release medium containing 10 mM STC and stirred at 100 rpm and 37°C. For each niosomal formulation, a control experiment was carried out in which formulations were incubated in bile salt-free solution. The results were compared to those of bile salt-containing solutions.
Vesicular size distributions, polydispersity index (PDI), and zeta potential were determined before and immediately after 10 hours incubation using a Zetasizer Nano ZS (Malvern Instruments). All experiments were performed in triplicate; results are given as the mean ± SEM.
Drug analysis
Concentration of CRV in the release medium and plasma samples was measured by an HPLC system. The HPLC system (KNAUER, Berlin, Germany) consisted of a WellChrom K-1001 solvent delivery system with an online degasser, a K-1500 solvent organizer, a column oven and a model RF-10 AXL Shimadzu fluorescence detector (excitation, 284 nm; emission, 340 nm). The chromatographic separation was performed on a PerfectSil Target C18 column (150×4.6 mm, 3.5 μm particle size) from MZ-Analysentechnik GmbH (Mainz, Germany), using a mobile phase of methanol–phosphate buffer 20 mM (50:50, v/v, pH 3.5). The column temperature was held at 35°C. The flow rate was set at 1 mL/min and the injection volume was 50 μL. Chromgate HPLC software (Version 317) was used for all chromatographic data calculations. Prior to the analysis, the method was validated according to the International Conference on Harmonization guidelines with respect to selectivity, linearity, precision, accuracy, limit of quantification, and recovery.32
Pharmacokinetics
Animal experiments
Male Wistar rats weighing 200–220 g were obtained from the Pasteur Institute (Tehran, Iran). The rats were kept under controlled temperature (23°C±2°C), humidity (55%±10%), and light (12 hour-light, 12 hour-dark cycles). Before the experiments, the animals were fasted overnight with free access to water. All experiments were approved by the ethics committee for animal experiments at the Shahid Beheshti University of Medical Sciences, Tehran, Iran. The animals were randomly divided into ten groups (six rats in each group). Niosomal formulations (at total lipid and surfactant concentration of about 74 mM) were administered to eight groups, and the two other groups received CRV suspension (in 0.5% w/v sodium carboxymethyl cellulose vehicle) and plain niosomes as controls. Each formulation, equivalent to 8 mg/kg of drug, was administered orally via a gavage tube. Immediately before dosing and then at 0.083, 0.25, 0.5, 1, 1.5, 2, 4, 6, 8, and 10 hours after drug administration, blood samples were withdrawn from the tail vein into heparin-containing tubes and immediately centrifuged at 10,000 rpm for 10 minutes. Plasma samples (100 μL) were then collected and stored at −15°C until HPLC assay.
To prepare plasma samples for the HPLC assay, frozen plasma samples were allowed to thaw at room temperature. The internal standard (cisapride, 35 μL of a 6 μg/mL methanolic solution) was then added to each sample and vortex-mixed for 30 seconds. Afterward, 1 mL of methyl tertiary butyl ether was added to each sample. The mixture was vigorously vortex-mixed for 15 minutes and centrifuged at 10,000 rpm for 10 minutes. The supernatant was separated and dried under a nitrogen stream. The residue was then vortex-mixed with 100 μL of mobile phase for 30 seconds and centrifuged at 10,000 rpm for 2 minutes; finally, 50 μL was injected into the HPLC device.
Assessment of lymphatic transport of niosomes
The intestinal lymphatic transport of niosomes was investigated in a less invasive chylomicron flow blocked rat model.33 Two groups of male Wistar rats, comprising six animals in each group, were fasted overnight and treated with intraperitoneal injection of 3 mg/kg cycloheximide solution (0.6 mg/mL). One hour after the injection, one group received 20% SC-enriched vesicles (F2) and the other one received DCP-containing niosomes (F10) by oral gavage. The amount of each formulation corresponded to 8 mg/kg CRV. The blood samples were collected from tail vein and processed as described above.
Pharmacokinetic analysis
Pharmacokinetic analysis was performed by using noncompartmental method.34 Pharmacokinetic parameters, including maximum plasma concentration (Cmax), time to reach it (Tmax), area under the drug concentration-time curve (AUC) and mean residence time (MRT), were calculated from the plasma concentration–time data.34
Histological evaluation of small intestinal segments
Histological studies were performed to evaluate the possibility of morphological changes and damages caused by niosomes on the intestine. In these experiments, 20% SC-enriched vesicles (F2), DCP-containing niosomes (F10) and PBS were administered to three animal groups by oral gavage and the animals were euthanized after 4 hours. Tissue samples were taken from different sections of the small intestine, fixed in buffered formalin, embedded in paraffin, sectioned at 5 μm and stained with hematoxylin and eosin (H&E) according to standard methods. Photomicrographs were taken with Nikon DS-L2 camera which was connected to the Nikon Eclipse E-200 microscope.
Statistical analysis
All statistical analyses were performed with SPSS version 17 (SPSS Inc., Chicago, IL, USA). Data are shown as the mean ± SEM. Comparison of the formulation and pharmacokinetic parameters was carried out using analysis of variance; P<0.05 was considered statistically significant.
Results and discussion
Niosomes are a milestone in novel drug delivery systems due to their potential to encapsulate drugs, hormones, antigens, and other chemotherapeutic agents and to overcome the instability, insolubility, and rapid degradation of these compounds.35 Their advantages include cost-effectiveness, high stability, and ease of large-scale preparation. Moreover, the use of nonionic surfactants as emulsifiers, wetting agents, solubilizers, and permeability enhancers has led to the exploitation of niosomal vesicles in different areas of drug delivery.12,35 To find a niosomal formulation that increases oral absorption of CRV, various niosomal formulations were prepared and extensively evaluated, including plain niosomes, bilosomes, and charged niosomes.
Preparation of plain niosomes
Niosomes can be prepared using various methods. An appropriate method should be chosen for each drug considering its physicochemical properties.12 Preliminary studies were carried out utilizing ether injection and conventional thin film hydration. A series of formulations was prepared, altering surfactant and Chol content while keeping other factors invariant. Ether injection was not very effective, with low %EE (below 30%). Conventional thin film hydration generally gave an EE approximately three to four times higher. In the next step, to reduce the size of the multilamellar vesicles, various methods including extrusion at a temperature above the phase transition of the surfactants, high frequency probe sonication and bath sonication during hydration were evaluated. Extrusion seemed not to be suitable for size reduction, giving a much lower EE (data not shown). Using probe sonication, the vesicles were unfortunately enlarged after the process ended. Using bath sonication during hydration gave significantly higher drug loading; also, vesicle particle size, and PDI were approximately 400 nm and 0.3, respectively, which are considered suitable for oral delivery.14,15 Therefore, thin film hydration followed by bath-sonication during hydration was employed for CRV niosome preparation.
Factors affecting CRV EE in niosomal formulations
To obtain a niosomal formulation with higher EE, various parameters were optimized, including Chol content (from 0% to 50%), drug concentration (0.5–2 mg/mL niosome), surfactant structure, and lipid-to-drug (L/D) molar ratio (from 10 to 40).
As shown in Table 1, incorporation of Chol into Span 60 niosomes (from 0% to 20%) considerably enhanced CRV EE. However, beyond this amount (from 20% to 50%), a decrease in EE was observed. Similar results were found in our previous studies of liposomal formulations.36,37 The increase in EE can be rationalized by proposing that Chol increases the microviscosity of the membrane by abolishing the gel-to-liquid phase transition of the surfactant bilayer, resulting in a more stable and hydrophobic bilayer38 that retards permeation and prevents leakage of hydrophobic drugs entrapped in the bilayer.39 In contrast, subsequent intercalation of Chol would reduce drug entrapment by competing with the drug for the bilayer, thus preventing incorporation of the amphiphilic or lipophilic drug into the vesicles.
With regard to the influence of drug content, increasing the proportion of drug from 0.5 to 1 mg/mL at two surfactant:Chol molar ratios (80:20 and 70:30) led to a significant increase in EE (P<0.01) (Table 1). This may be due to causing CRV to be entrapped into the niosome bilayers via saturation of the medium with the drug.12 However, a decrease in %EE was observed by the higher level for the drug proportion (2 mg/mL, P<0.01), which is in accordance with published data.39,40 This may be due to the saturation of the drug within the bilayers of niosomes: the excess drug would scatter between the niosomal pellets and precipitate.39,40 This suggests that a constant proportion of Chol and surfactant, creating a certain number of niosomal vesicles, yields limited drug entrapment.
Surfactants, the predominant components of niosomes, play a key role in niosome properties. Therefore, to investigate the effects of surfactant structure on EE, a number of niosomal formulations were prepared, consisting of Span 20, Span 60, or Span 80 with fixed Chol (20%), CRV (1 mg/mL), and L/D molar ratio (=20). As shown in Table 1, of the different types of surfactant used, the highest EE was achieved with Span 60 niosomes. We propose the following explanations: A) Because all Span surfactants have the same head group, Span 60, with a longer alkyl chain (C18), can produce more stable membrane bilayers, yielding higher EE than Span 20 (C12).40,41 Similar results were found for liposomes with longer alkyl chain phospholipids in our previous study.42 Moreover, as shown in Table 1, incorporation of Span 80 nonionic surfactant into niosomes distinctly defects the niosome bilayers, which may be due to the formation of permeable and leaky vesicles promoted by the unsaturated alkyl chain.39 This surfactant may require a considerably higher proportion of Chol for stable vesicle formation.39 B) Among the surfactants examined, Span 60 has the highest transition temperature (Tc =53°C),43 which may account for it yielding the highest EE. C) The hydrophilic–lipophilic balance (HLB) value of a surfactant is a significant factor in drug incorporation. The longer the saturated alkyl chain of a surfactant, the lower its HLB and the higher the EE that can be achieved. It seems reasonable that Span 60 (HLB value =4.7) gave higher drug entrapment than Span 20 (HLB value =8.6).43 Higher EE with Span 60 in niosomes has also been reported for minoxidil40 and colchicine.41
As summarized in Table 1, the L/D molar ratio has an impact on drug loading. Altering the L/D molar ratio from 10 to 40 while keeping other factors invariant augmented the %EE of CRV from 52.8% to 90.7%. It is obvious that increasing the concentration of bilayer-forming materials, which in turn increases the number of vesicles in a given volume, can increase the amount of drug entrapped in the vesicles.44,45 Although enhancement of the L/D molar ratio leads to higher EE, the ratio should be chosen with care because high lipid concentrations are not appropriate for niosome preparation on an industrial scale.
With regard to FTIR spectra (Figure S1), we found that drug loading did not shift or broaden the major peaks of the niosomes. This confirms the absence of major interactions between drug and formulation components.
Overall, as shown in Table 1, CRV-loaded niosomes composed of 80% Span 60 (surfactant), 20% Chol, and 1 mg/mL CRV with L/D molar ratio of 30 (formulation F1, Table 2) gave a suitably high EE. This formulation was therefore selected as the optimal plain niosome and was used for further studies.
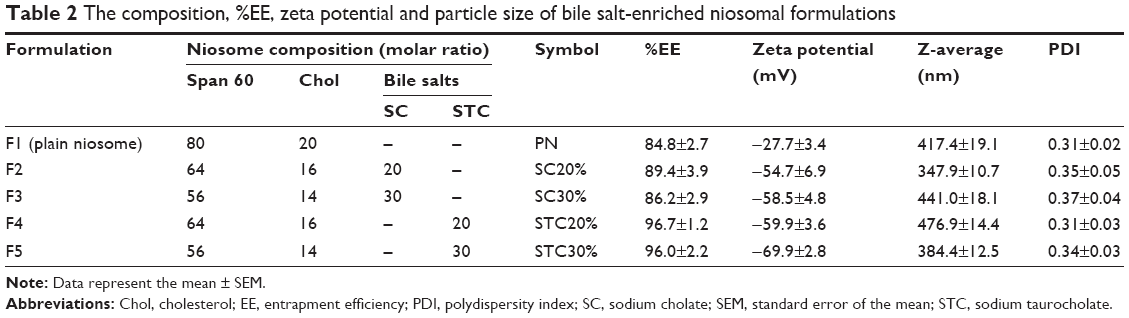 | Table 2 The composition, %EE, zeta potential and particle size of bile salt-enriched niosomal formulations |
Preparation of bile salt-enriched CRV-loaded niosomes (bilosomes)
Bilosome vesicles (Table 2, formulations F2 to F5) were fabricated by adding different types of cholate (20% and 30% of either SC or STC) bile salts to the optimal plain niosomes while keeping the molar ratio of Span 60 and Chol constant at 4:1, similar to plain niosomes. The choice of bile salt percentage was based on previous reports of bile salt-enriched liposomes.26,46 As shown in Table 2, all bilosomes yielded higher EE than plain niosomes (more than 85%). Moreover, almost complete drug entrapment was achieved in formulations containing STC (F4 and F5), which may be due to a more favorable hydrophobic region for lipophilic CRV molecules within vesicle bilayers. Higher drug encapsulation with bile salt-containing vesicles than with conventional carriers has also previously been reported for fenofibrate, a poorly water-soluble drug in liposomal formulations.21
As shown in Figure 1A and B, the plain and bilosome formulations were disperse, almost spherical particles by AFM.
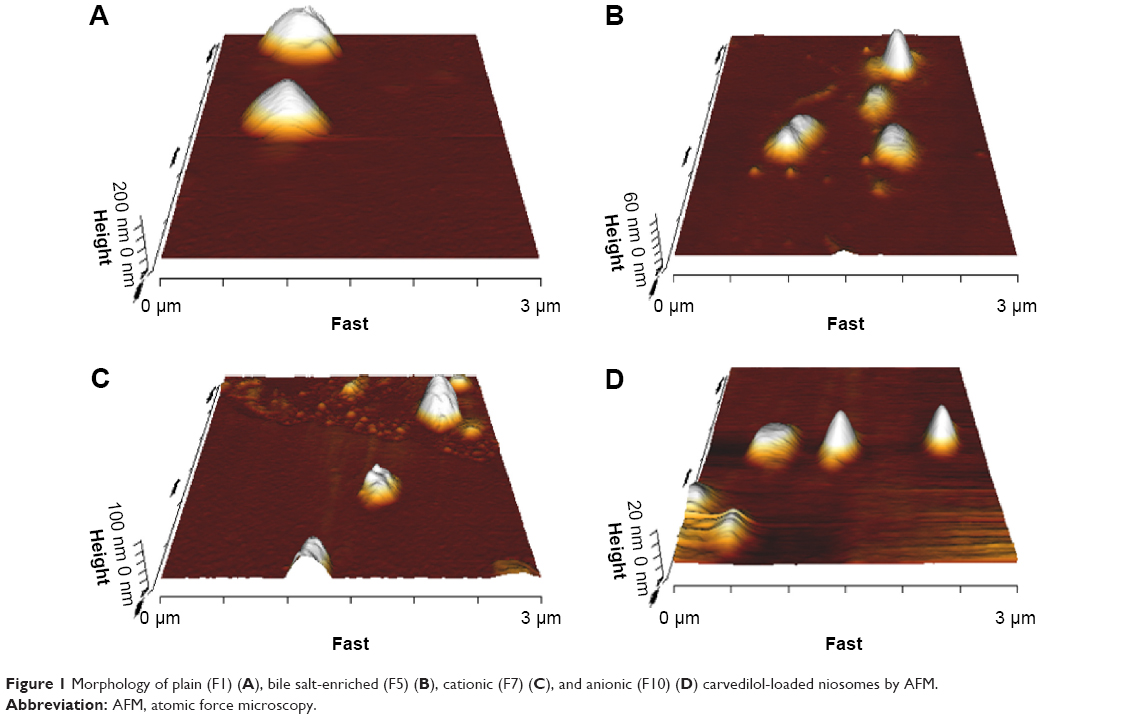 | Figure 1 Morphology of plain (F1) (A), bile salt-enriched (F5) (B), cationic (F7) (C), and anionic (F10) (D) carvedilol-loaded niosomes by AFM. |
Particle size and surface charge of vesicles exert significant influences on stability, release behavior, and biodisposition of nanoparticles. The average particle size of plain niosomes was found to be approximately 417 nm, with a PDI of 0.3 (Table 2). To elucidate the impact of the addition of bile salts to the vesicles, we attempted to keep the mean particle size of the prepared bilosome batches within the approximate range of 350–470 nm, with a narrow size distribution. To achieve this, we verified that less sonication time was required during hydration of bilosomes. This may be due to the effects of bile salts in the vesicle bilayer, which can diminish vesicle surface tension and enhance flexibility.21 Furthermore, zeta potential analysis revealed greater negative surface charge density, due to the incorporation of negatively charged bile salts into the niosomal membranes. As shown in Table 2, STC had a greater effect on membrane surface charge than SC, which is consistent with the observations of Yang et al.47 Despite the absence of bile salts in plain niosomes, a zeta potential of −27.7±3.4 mV was obtained, which may be due to hydroxyl ions adsorbed on the membrane surface, as reported previously.14
Preparation of CRV-loaded niosomes containing charge-inducing agents
By keeping niosome component ratios the same as in the optimal plain niosomes, charged niosomes were prepared by incorporating different proportions of CTAB and DCP as positively and negatively charged agents, respectively. The compositions of the prepared niosomal formulations are shown in Table 3. Among the ratios of charged molecules examined, the addition of 5% CTAB to niosomes was found to adversely affect the formation of vesicles (Table 3). As shown in Table 3, niosomes produced using 10% charged molecules (CTAB 10% [formulation F7] and DCP 10% [formulation F10]) exhibited high EE (>80%) and similar particle size to those of plain niosomes (approximately 400 nm). Based on these results, F7 and F10 formulations were selected for further experiments. Niosomes containing 15% CTAB and 5% DCP were not selected, due to low particle size and low %EE, respectively. The zeta potentials of these optimized negative and positive CRV niosomes were approximately −83 and +56 mV, respectively.
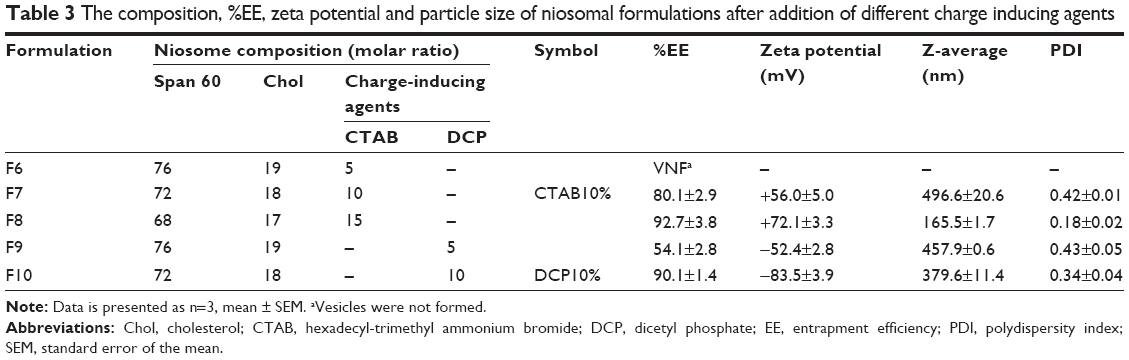 | Table 3 The composition, %EE, zeta potential and particle size of niosomal formulations after addition of different charge inducing agents |
AFM was performed to analyze vesicle morphology. The observations revealed that the selected niosomes were of uniform size and almost spherical shape (Figure 1C and D). However, positively charged niosomes revealed low particle aggregation in a few microscope fields, consistent with a previous report regarding cationic pyrazinamide niosomes.48
In vitro drug release studies
In vitro release studies were carried out to define the influence of bile salt enrichment and charge-inducing agents on the rate of drug release from niosomal carriers. The sink condition was maintained by the addition of Tween 80. Two different release media were examined (SIF containing 0.1% w/v Tween 80 [blank release medium] and SIF containing 0.1% w/v Tween 80 and 10 mM STC [bile salt release medium]).
Figure 2A illustrates the cumulative release of drug from CRV suspension, CRV-loaded plain niosomes (F1), several bilosome formulations, and optimized negatively and positively charged niosomes in blank release medium. Cumulative drug release from F1 (plain niosomes), F2 (SC20%), F3 (SC30%), F4 (STC20%), and F5 (STC30%) in this medium was approximately 75%, 71%, 65%, 91%, and 64%, respectively, within 10 hours. However, CRV suspension gave slower drug release than bilosomes and plain niosomes (P<0.05). Because the particle size of niosomal formulations was much smaller than that of suspensions (mean particle size ≈9.9 μm), the total surface area exposed to release medium is larger for niosomal vesicles than for suspensions. As illustrated in Figure 2, all release profiles were biphasic, showing a primary fast release phase lasting for approximately 2–3 hours, followed by slower release in a more controlled manner. The primary and secondary release phases may be due to diffusion distances for drug molecules in the outer and inner vesicle membranes, respectively. Plain niosomes demonstrated the highest burst release, almost 40% of loaded drug during the first 60 minutes (P<0.05). Considering that the initial release of drug from vesicular structures can be ascribed to desorption of the drug from the surface and outer membrane of the nanoparticles,45 it is likely that most CRV molecules were incorporated near the surface of plain vesicles.
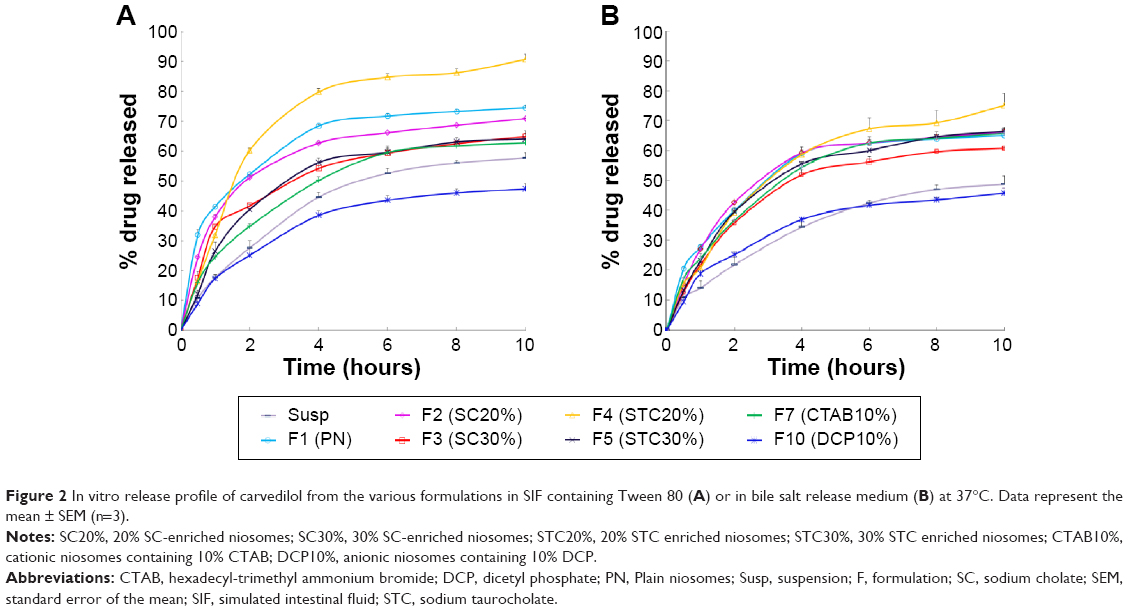 | Figure 2 In vitro release profile of carvedilol from the various formulations in SIF containing Tween 80 (A) or in bile salt release medium (B) at 37°C. Data represent the mean ± SEM (n=3). |
Moreover, CRV release from niosomes was influenced by the surface charge of the carrier. The inclusion of positive and negative charge seemed to slow down drug release in comparison with plain niosomes. This may be because charged molecules stabilize vesicle bilayers, resulting in complete inclusion of drug inside bilayers, reducing the release rate. This is in accordance with the observations of Hasan et al.14 In addition, Figure 2A clearly shows that CTAB formulations released the drug more rapidly than DCP formulations, which may indicate that negatively charged niosomes are more stable.
As shown in Figure 2B, the release behavior of different formulations in bile salt medium resembled that in blank medium (Figure 2A). CRV was released into SIF containing STC in a sustained manner for over 10 hours. Unexpectedly however, the percentage of CRV released from niosomal formulations in bile salt medium was significantly lower than in blank release medium, suggesting that the bile salt (STC) retarded the release of CRV. The exact mechanism of this observation is not clear.
Stability in the presence of bile salts
The stability of the prepared nanocarriers in conditions simulating GI media was investigated in vitro. Solutions of bile acids simulating human intestinal content were prepared by the addition of STC to SIF, giving a final bile salt concentration of 10 mM. To maintain sink conditions, 0.1% w/v Tween 80 was also added.
The influence of bile salts on the stability of CRV-loaded niosomes after incubation is depicted in Figure 3. As shown in Figure 3A, significant decreases (P<0.05) in particle size, approximately 30%, were observed in plain niosomes and F7 (CTAB10%). This suggests that the diffusion of Tween 80 and STC across the dialysis membrane, and their inclusion into niosomal vesicles may affect the vesicles’ physicochemical characteristics. However, there were no substantial differences in particle size between bilosomes and F10 (DCP10%), bearing in mind that inclusion of bile salts and DCP into niosomal vesicles enhances resistance to these simulated GI conditions. In addition, after incubation for 10 hours, negligible change of PDI in bilosomes was observed. This indicates that bile salt enrichment confers stability on the vesicles; PDI changes were statistically significant for F7, F10, and plain niosomes (Figure 3B). Changes were less than 30%; PDI was still in the acceptable range for oral delivery. From the zeta potentials shown in Figure 3C, an increase in negative surface charge occurred, especially for positively charged (F7) and plain niosomes. Considering the components of the solutions, lower zeta potential may be due to the negative charge of STC and the OH groups of Tween 80 diffusing across the dialysis membrane to the vesicles. These data suggest that inclusion of bile salts and DCP into niosomal vesicles may protect them against harsh GI conditions. Although significant changes were observed in plain niosomes and F7, vesicular structures were maintained in the presence of STC and Tween 80; the results were also within the accepted ranges for oral drug delivery.
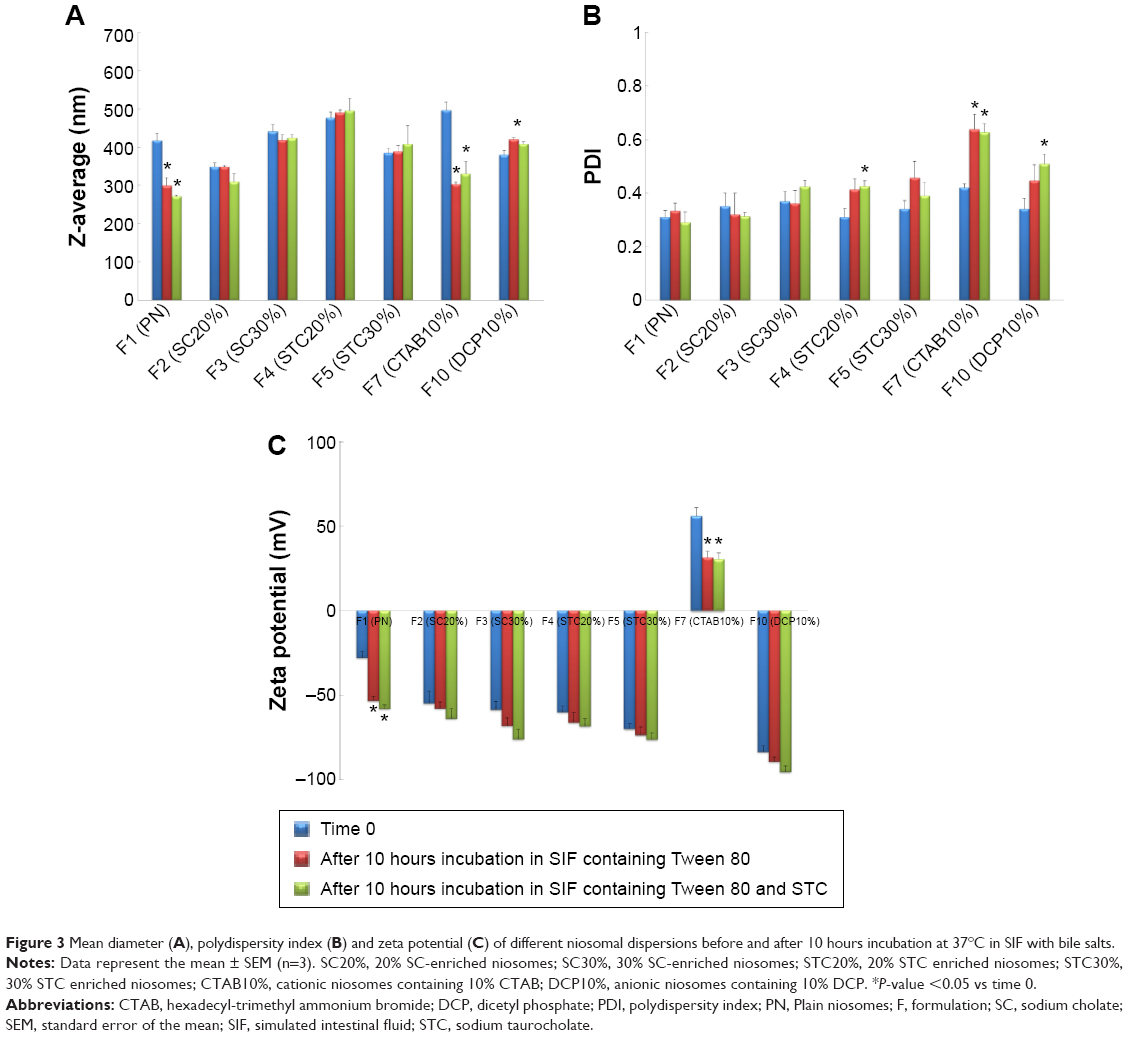 | Figure 3 Mean diameter (A), polydispersity index (B) and zeta potential (C) of different niosomal dispersions before and after 10 hours incubation at 37°C in SIF with bile salts. |
In vivo studies
The results obtained after oral administration of CRV suspension and the different niosome-based formulations are presented in three sections. First, the effects of encapsulating CRV into niosomal carriers on bioavailability are described. Second, the influences of bile salt incorporation into niosome compositions are investigated, followed by the effect of carrier surface charge on oral bioavailability.
The effect of niosome carriers on oral bioavailability of CRV
In general, colloidal particulate systems are believed to enhance the oral bioavailability of Biopharmaceutics Classification System class II drugs, the biopharmaceutical characteristics of which are poor aqueous solubility and high permeability.9 Because dissolution is the rate-limiting step in the absorption of a Biopharmaceutics Classification System class II drug, even a small increase in dissolution rate due to increased surface area can lead to a significant increase in oral absorption.8
The pharmacokinetic parameters of CRV after oral administration of optimal plain niosomes (F1, equivalent to 8 mg/kg of drug) were compared to a suspension containing the same dose as control. Although blood samples were collected until 10 hours for both groups, CRV plasma concentrations were below the quantification limit after 8 hours.
The concentration vs time curves obtained for plasma samples, as well as the relevant pharmacokinetic parameters for plain niosomes and CRV suspension, are given in Figure 4A and Table 4. As summarized in Table 4, the conventional niosome yielded much better absorption, reaching a peak plasma concentration (Cmax) nearly 1.7 times greater than that of the CRV suspension (351.4 vs 208.1 ng/mL), while Tmax and MRT remained nearly unchanged. Moreover, the plain niosomal formulation gave approximately 30% higher AUC0→∞ (680.7±37.9 ng·h/mL) than the orally administered CRV suspension (518.0±22.8 ng·h/mL). Thus, compared with suspension, there appeared to be significantly enhanced oral bioavailability of CRV from the niosomal formulation. These observations were consistent with previous findings that niosomes enhance the oral bioavailability of ganciclovir49 and paclitaxel.50
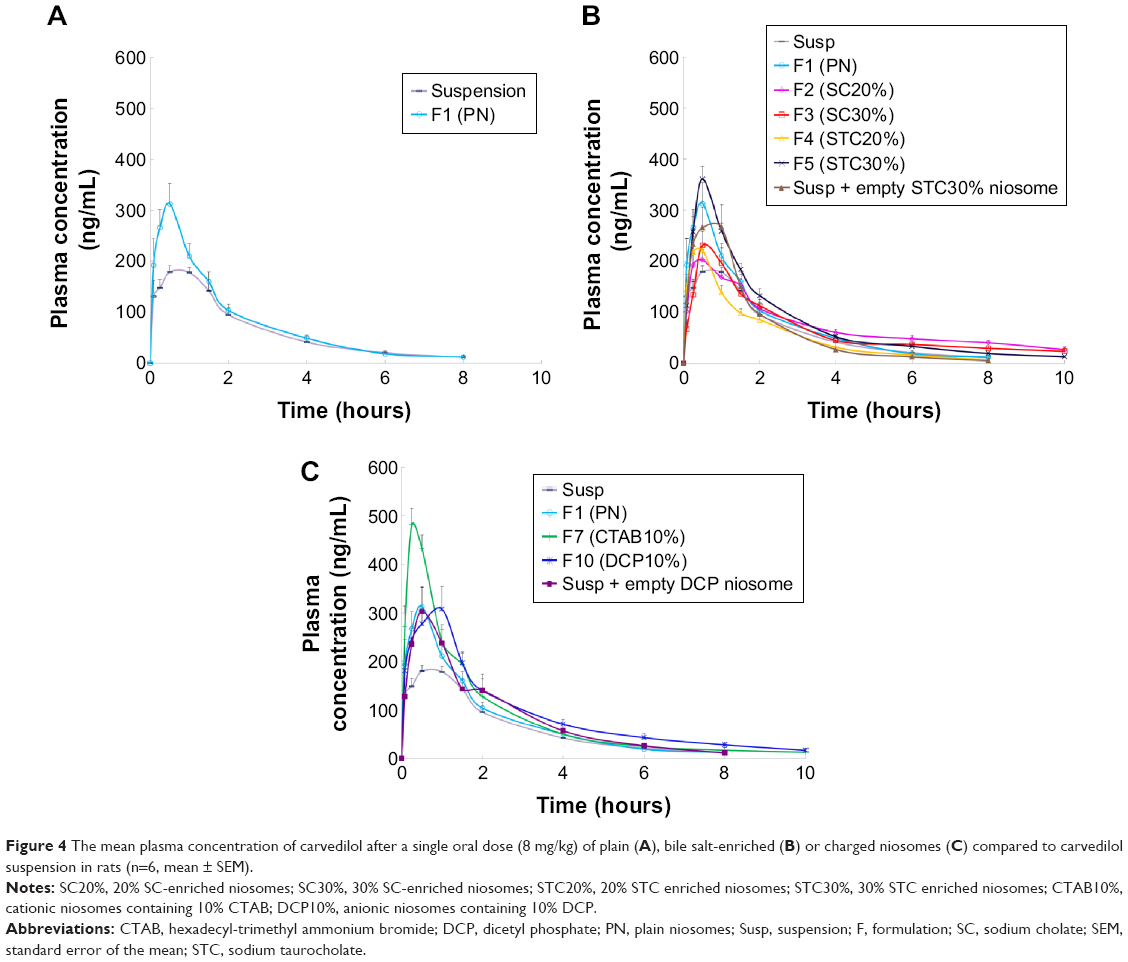 | Figure 4 The mean plasma concentration of carvedilol after a single oral dose (8 mg/kg) of plain (A), bile salt-enriched (B) or charged niosomes (C) compared to carvedilol suspension in rats (n=6, mean ± SEM). |
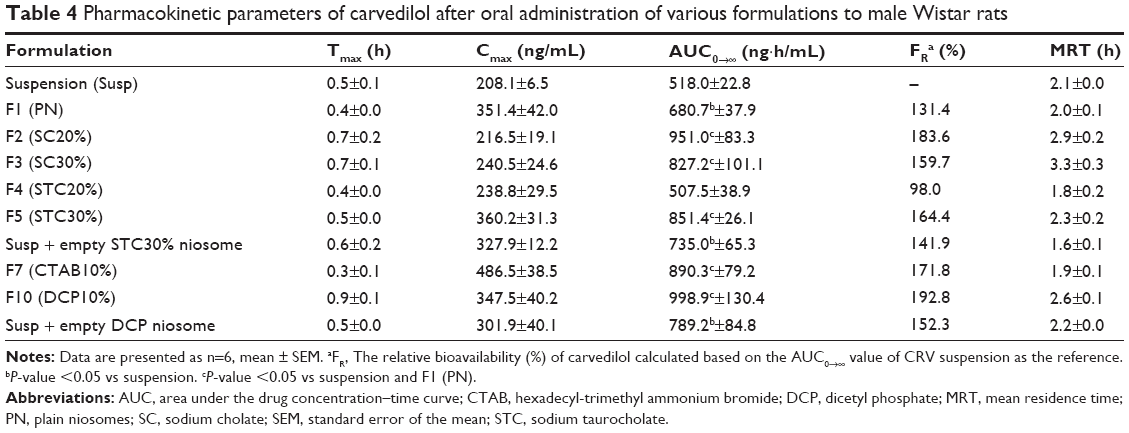 | Table 4 Pharmacokinetic parameters of carvedilol after oral administration of various formulations to male Wistar rats |
Improved CRV absorption and higher plasma drug concentrations after niosomal formulation may be due to the main carrier components (surfactants) acting as penetration enhancers. Others contributors may include increased penetration of mucosa resulting from the lipophilic nature of niosomes, lower vesicular size (approximately 400 nm) and higher dissolution rate/solubility compared to suspensions.49 In addition, the negative surface charge of the conventional CRV niosomes in this study (−27.7±3.4 mV) may promote vesicle uptake and transcytosis via M-cells of Peyer’s patches in the small intestine.17
The effects of bile salt incorporation into niosomes
In the last decade, bile salts have been widely utilized in transdermal drug delivery systems as penetration enhancers, due to their membrane-destabilizing activity.26 Elastic or deformable vesicles generated by the incorporation of an edge activator into transdermal liposomes (Transfersomes®) were first introduced in the early 1990s. It has been widely reported that transfersomes provide greater flexibility and higher penetration of deeper skin layers, allowing them to deliver drugs across the skin better than conventional liposomes.20 Later, in an effort to make vesicles resistant to physiological bile salts in the GI tract, the addition of bile salts to liposome bilayers (bilosomes) was proposed for oral drug delivery.21,26 Considering that bile salts can modify membrane fluidity and permeability and can enhance the oral bioavailability of drugs, this study focused on the incorporation of different amounts of SC and STC into niosomal formulations. To the best of our knowledge, this has not been reported before. In vivo absorption of bile salt-enriched niosomes was compared with CRV suspensions, conventional CRV niosomes, and a mixture of CRV suspension with empty bile salt-containing niosomes. The purpose of this latter control group was to distinguish the effects of drug encapsulation from those of vesicle components (surfactants) on oral bioavailability.
The mean plasma CRV concentration–time curves for bilosomes, and their pharmacokinetic parameters, are shown in Figure 4B and Table 4, respectively. Plasma levels of CRV point to longer detection times (up to 10 hours) for bilosomes compared to suspensions and plain niosomes. A marked increase in plasma CRV was observed 6 and 8 hours after oral administration of bilosomes compared with suspension. As with plain niosomes, comparison of STC bilosomes (F4 and F5) with suspension revealed statistically non-significant differences in MRT and Tmax values. In contrast to F4 and F5 formulations, SC bilosomes (F2 and F3) gave higher MRT, resulting in higher CRV concentrations 6, 8, and 10 hours after oral administration. Moreover, mean Cmax for F5 (360.2±31.3 ng/mL) was higher than for other bilosome formulations (P<0.05). AUC for all evaluated formulations indicated that, except for F4 (20% STC), the incorporation of bile salts into niosomes yielded higher CRV AUC0→∞ than did plain niosomes or especially CRV suspensions (P<0.05). The relative bioavailability of CRV from bile salt-enriched formulations compared to suspension was estimated to be 184%, 160%, and 164% for F2, F3, and F5, respectively (Table 4). In addition, the AUC0→∞ of F2, F3, and F5 was 40%, 22%, and 25% higher than that of plain niosomes, respectively. This indicates significantly enhanced absorption after addition of 20% SC, 30% SC, and 30% STC. Because F4 gave no enhancement in AUC0→∞, niosome formulations do not appear to benefit from incorporation of 20% STC. It can be assumed that 20% SC or 30% STC in lipid bilayers facilitate the vesicle–micelle phase transition in the GI tract and solubilize the poorly-soluble drug in intestinal fluid.51,52 Improved drug absorption may also have resulted from enhanced paracellular or transcellular absorption, most likely due to flexible bilayers for vesicle penetration and to permeation enhancement by bile salts.53 Furthermore, such vesicles may benefit from facilitated uptake by M-cells in Peyer’s patch. There have been reports of higher absorption of liposome-containing bile salts associated with some transmembrane carriers,21 which may also facilitate absorption of niosomes containing bile salts. These mechanisms may yield enhanced oral bioavailability of CRV. Furthermore, when comparing the AUC0→∞ of CRV suspension mixed with drug-free F5 to F5 alone (735.0±65.3 ng·h/mL vs 851.4±26.1 ng·h/mL, respectively), the drug should be incorporated into vesicles to achieve higher oral bioavailability.
Effects of carrier surface charge on oral bioavailability of niosomal CRV
It is widely believed that carrier surface charge has a marked influence on vesicular transport across biological barriers, in vivo fate and biodisposition.17,18 Janga et al54 studied pro-liposomal zaleplon formulations composed of hydrogenated soy phosphatidylcholine and Chol with neutral, positive (stearylamine) or negative (DCP) charge. The positive formulation yielded the highest oral absorption. In contrast, Jung et al28 suggest that anionic nanoparticles with hydrophobic surfaces promote M-cell uptake. In studies of oral delivery by niosomal formulations, cationic14 or anionic55 niosomes were administered to animals. Thus, available evidence on the effect of surface charge on the oral delivery of vesicles is very limited and inconclusive.
In this context, we investigated the influence of surface charge by tailoring niosomes with positive and negative charge-inducing agents. Optimized cationic and anionic niosomes were selected for in vivo studies. To highlight the role of charged molecules, the oral absorption of CRV from each group was compared with those of suspension, plain niosomes and a mixture of suspension and drug-free charged formulation. These various control groups were chosen to rule out other variables that might have influenced the outcome.
Figure 4C shows mean plasma concentration vs time profiles. Relevant pharmacokinetic parameters are given in Table 4. As illustrated in Figure 4C, the F7 (cationic) and F10 (anionic) formulations gave higher plasma concentrations than the CRV suspension (P<0.05). The peak plasma concentration (Cmax) of CRV was 486.5±38.5 ng/mL and 347.5±40.2 ng/mL for the F7 and F10 formulations, respectively, approximately 2.3- and 1.7-fold higher than for the CRV suspension. Furthermore, both charged niosomes gave remarkable increases in absorption (AUC) and relative bioavailability (FR) compared to controls (P<0.05). However, the oral bioavailability of cationic and anionic niosomes did not differ significantly. Overall, charged niosomes appeared to be superior to the conventional formulation regardless of surface charge. Many mechanisms may be involved in the beneficial effects of charged vesicles. The negatively charged proteins in enterocyte membranes possess a negatively charged outer surface.56 Hence, it is assumed that strong electrostatic interactions between negatively charged GI membranes and positively charged carriers selectively increases uptake of cationic carriers. Additionally, it was previously reported that the addition of a positive charge to nanocarriers promotes their uptake by endocytosis, especially clathrin-mediated endocytosis. Moreover, cationic nanocarriers exhibit a tendency to escape endo-lysosomal degradation.17 Absorption of the F7 formulation may benefit from all these mechanisms.
On the other hand, the altered pharmacokinetic parameters (higher MRT and Tmax) and increased oral bioavailability of CRV from the anionic (F10) formulation may result from several factors. Earlier reports of negatively charged nanoparticles indicated greater endocytosis and more uptake via M-cells of Peyer’s patches in intestinal lymphatic tissue.17 Presumably, negative surface charge can be easily recognized by receptors on a variety of cells, including macrophages.57 The increased Tmax of DCP-containing niosomes may also be due to increased absorption via the lymphatic pathway. Overall a 72%–93% increase in relative bioavailability suggests that charged niosomes may improve the oral bioavailability of CRV.
Lymphatic transport of CRV-loaded niosomes
Enhanced lymphatic transport has been suggested to be a potential mechanism for the improved oral bioavailability of niosomal vesicles.50,55 To our knowledge, however, no reports have attempted to quantify the role of lymphatic transport in this improvement.
Two formulations with higher oral bioavailability, F2 (SC20%) and F10 (DCP10%), were chosen to clarify the role of lymphatic transport. The rats were pretreated with cycloheximide one hour prior to oral gavage. Cycloheximide is known to inhibit lymphatic transport through both blockade of chylomicron secretion and inhibition of M-cell phagocytic activity.33,52
The CRV plasma concentration–time profiles and AUC values obtained after oral administration of niosomes to rats pretreated with cycloheximide are shown in Figure 5A and B, respectively.
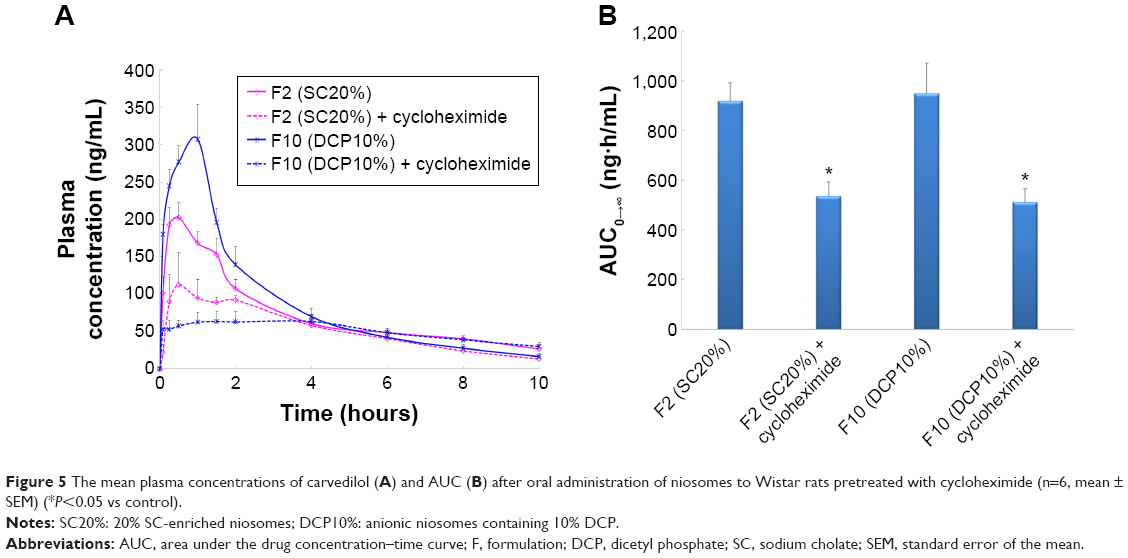 | Figure 5 The mean plasma concentrations of carvedilol (A) and AUC (B) after oral administration of niosomes to Wistar rats pretreated with cycloheximide (n=6, mean ± SEM) (*P<0.05 vs control). |
Following administration of SC-enriched vesicles (F2), the Cmax of cycloheximide treated rats (139.3±26.0 ng/mL) was significantly lower than that of untreated animals (216.5±19.1 ng/mL). In addition, a 1.8-fold decrease was observed in the AUC0–∞ of SC20% niosomes in the chylomicron flow blocked group compared to controls. When intestinal lymphatic transport was selectively blocked, oral administration of DCP-containing niosomes (F10) significantly reduced the plasma concentration of CRV; AUC0–∞ and Cmax were 1.9- and 5.0-fold diminished, respectively. These results suggest that lymphatic transport is the dominant route of absorption for these formulations.
Due to the beneficial impact of intestinal lymphatic transport on the oral bioavailability of hydrophobic drugs with extensive pre-systemic metabolism, DCP- and SC-enriched niosomes may diminish hepatic first-pass extraction and improve the oral absorption of lipophilic agents such as CRV.
Histological evaluation of small intestinal segments
Tissue histological studies were performed to assess pathological changes caused by niosomes in the epithelium of the small intestine (Figure 6). After administration of 20% SC and DCP vesicles, the duodenum, jejunum, and ileum were comparable to the control group in the microscopic appearance of the intestinal epithelium. Inflammatory cells were not observed in these tissue samples. No signs of necrosis were detected. No evidence of damage to villi was found. Although these short-term studies did not show any apparent morphological changes following niosome administration, further studies of chronic administration should be performed to permit conclusions regarding the safety of multiple doses of niosomes.
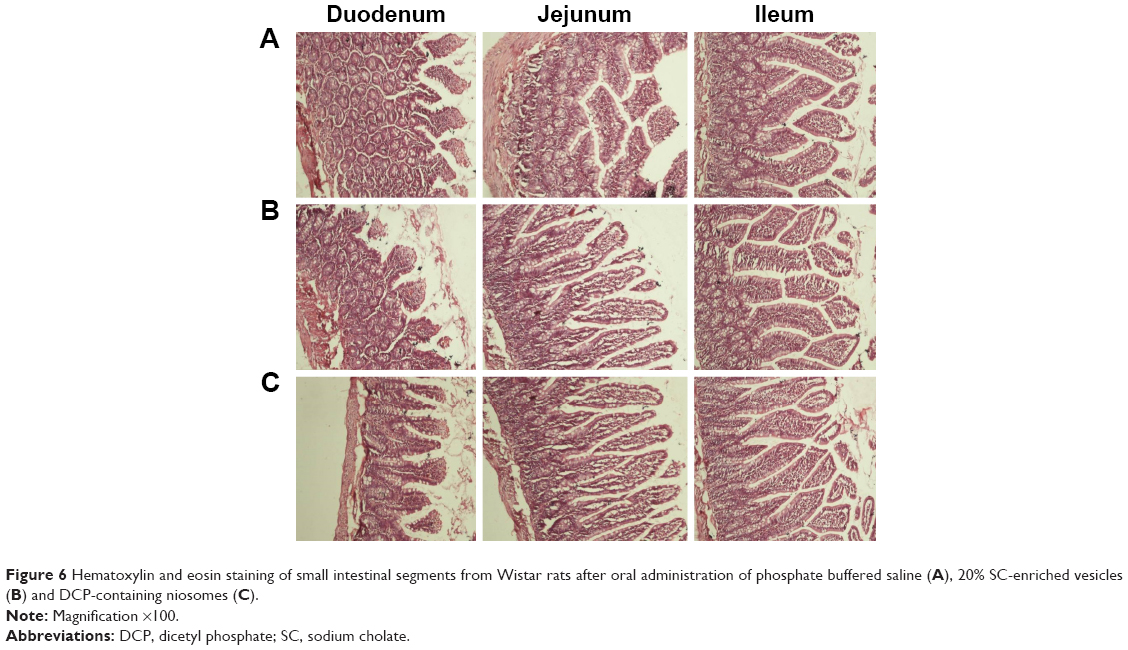 | Figure 6 Hematoxylin and eosin staining of small intestinal segments from Wistar rats after oral administration of phosphate buffered saline (A), 20% SC-enriched vesicles (B) and DCP-containing niosomes (C). |
Conclusion
This study contributes to our understanding of the effect of various niosomal carriers on the oral bioavailability of CRV. Plain niosomes, bile salt-enriched niosomes, and charged niosomes enhanced the oral absorption of CRV. The effects of various formulation parameters (surfactant type, L/D molar ratio, Chol percentage, and drug concentration) on CRV entrapment were evaluated. Particle size, zeta potential, vesicular stability, in vitro release profile, and morphology of these formulations were thoroughly characterized. The optimized niosomes showed adequate %EE and acceptable stability in simulated GI conditions. In vitro release studies revealed sustained delivery of CRV from niosomes over 10 hours. Pharmacokinetic studies showed that incorporation of drug into niosome carriers helped increase plasma levels of CRV compared to a suspension. Moreover, the incorporation of bile salts into vesicles gave enhanced intestinal absorption of CRV. Furthermore, the advantages of combining niosomes and surface charge for improved oral drug delivery were explored. The results of chylomicron flow blocking experiments suggest a major contribution of the lymphatic route to the enhanced oral absorption of CRV conferred by niosomes. Thus, niosomes and suitable bile salts in appropriate amounts, as well as optimal carrier surface charge, may lead to the development of a practical oral formulation of CRV.
Acknowledgments
This work was supported by a grant from Shahid Beheshti University of Medical Sciences. The authors thank Ms Abbasian, Ms Astaraki, and Ms Maghfurian for their valuable technical assistance. The authors express their appreciation to Professor Farshad Shirazi (Shahid Beheshti University of Medical Sciences) for advice with FTIR analyses and Dr Maryam Sotoudeh Anvari (Tehran University of Medical Sciences) for advice with histological evaluation.
Disclosure
The authors report no conflicts of interest in this work.
References
Sastry SV, Nyshadham JR, Fix JA. Recent technological advances in oral drug delivery – a review. Pharm Sci Technolo Today. 2000;3(4): 138–145. | ||
Hu M, Li X, Eds. Barriers to oral bioavailability – an overview. In: Oral Bioavailability: Basic Principles, Advanced Concepts, and Applications, New York: John Wiley & Sons, Inc.; 2011. | ||
Hörter D, Dressman J. Influence of physicochemical properties on dissolution of drugs in the gastrointestinal tract. Adv Drug Deliv Rev. 2001;46(1):75–87. | ||
Back D, Rogers S. Review: first-pass metabolism by the gastrointestinal mucosa. Aliment Pharmacol Ther. 1987;1(5):339–357. | ||
Wilkinson GR. Cytochrome P4503A (CYP3A) metabolism: prediction of in vivo activity in humans. J Pharmacokinet Biopharm. 1996;24(5):475–490. | ||
Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov Today. 2007;12(23):1068–1075. | ||
Huang L-F, Tong W-QT. Impact of solid state properties on developability assessment of drug candidates. Adv Drug Deliv Rev. 2004;56(3):321–334. | ||
Kawabata Y, Wada K, Nakatani M, Yamada S, Onoue S. Formulation design for poorly water-soluble drugs based on biopharmaceutics classification system: basic approaches and practical applications. Int J Pharm. 2011;420(1):1–10. | ||
Zhang L, Wang S, Zhang M, Sun J. Nanocarriers for oral drug delivery. J Drug Target. 2013;21(6):515–527. | ||
Cai Z, Wang Y, Zhu L-J, Liu Z-Q. Nanocarriers: a general strategy for enhancement of oral bioavailability of poorly absorbed or pre-systemically metabolized drugs. Curr Drug Metab. 2010;11(2):197–207. | ||
Nishioka Y, Yoshino H. Lymphatic targeting with nanoparticulate system. Adv Drug Deliv Rev. 2001;47(1):55–64. | ||
Kumar GP, Rajeshwarrao P. Nonionic surfactant vesicular systems for effective drug delivery – an overview. Acta Pharm Sin B. 2011;1(4):208–219. | ||
Moghassemi S, Hadjizadeh A. Nano-niosomes as nanoscale drug delivery systems: an illustrated review. J Control Release. 2014;185:22–36. | ||
Hasan AA, Madkor H, Wageh S. Formulation and evaluation of metformin hydrochloride-loaded niosomes as controlled release drug delivery system. Drug Deliv. 2013;20(3–4):120–126. | ||
Bayindir ZS, Yuksel N. Characterization of niosomes prepared with various nonionic surfactants for paclitaxel oral delivery. J Pharm Sci. 2010;99(4):2049–2060. | ||
Jain S, Vyas S. Mannosylated niosomes as adjuvant-carrier system for oral mucosal immunization. J Liposome Res. 2006;16(4):331–345. | ||
Roger E, Lagarce F, Garcion E, Benoit J-P. Biopharmaceutical parameters to consider in order to alter the fate of nanocarriers after oral delivery. Nanomedicine. 2010;5(2):287–306. | ||
Duan X, Li Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small. 2013;9(9–10):1521–1532. | ||
Huang Y-B, Tsai M-J, Wu P-C, Tsai Y-H, Wu Y-H, Fang J-Y. Elastic liposomes as carriers for oral delivery and the brain distribution of (+)-catechin. J Drug Target. 2011;19(8):709–718. | ||
Cevc G. Transfersomes, liposomes and other lipid suspensions on the skin: permeation enhancement, vesicle penetration, and transdermal drug delivery. Crit Rev Ther Drug Carrier Syst. 1996;13(3–4):257–388. | ||
Chen Y, Lu Y, Chen J, et al. Enhanced bioavailability of the poorly water-soluble drug fenofibrate by using liposomes containing a bile salt. Int J Pharm. 2009;376(1):153–160. | ||
Mikov M, Kevrešan S, Kuhajda K, Jakovljeviæ V, Vasoviæ V. 3a, 7a-Dihydroxy-12-oxo-5b-cholanate as blood-brain barrier permeator. Pol J Pharmacol. 2004;56:367–371. | ||
Bagger MA, Nielsen HW, Bechgaard E. Nasal bioavailability of peptide T in rabbits: absorption enhancement by sodium glycocholate and glycofurol. Eur J Pharm Sci. 2001;14(1):69–74. | ||
Morimoto K, Nakai T, Morisaka K. Evaluation of permeability enhancement of hydrophilic compounds and macromolecular compounds by bile salts through rabbit corneas in-vitro. J Pharm Pharmacol. 1987;39(2):124–126. | ||
Guan P, Lu Y, Qi J, et al. Enhanced oral bioavailability of cyclosporine A by liposomes containing a bile salt. Int J Nanomedicine. 2011;6(965):e974. | ||
Niu M, Lu Y, Hovgaard L, et al. Hypoglycemic activity and oral bioavailability of insulin-loaded liposomes containing bile salts in rats: the effect of cholate type, particle size and administered dose. Eur J Pharm Biopharm. 2012;81(2):265–272. | ||
Conacher M, Alexander J, Brewer JM. Oral immunisation with peptide and protein antigens by formulation in lipid vesicles incorporating bile salts (bilosomes). Vaccine. 2001;19(20):2965–2974. | ||
Jung T, Kamm W, Breitenbach A, Kaiserling E, Xiao J, Kissel T. Biodegradable nanoparticles for oral delivery of peptides: is there a role for polymers to affect mucosal uptake? Eur J Pharm Biopharm. 2000;50(1):147–160. | ||
Harush-Frenkel O, Debotton N, Benita S, Altschuler Y. Targeting of nanoparticles to the clathrin-mediated endocytic pathway. Biochem Biophys Res Commun. 2007;353(1):26–32. | ||
Dulin B, Abraham WT. Pharmacology of carvedilol. Am J Cardiol. 2004;93(9):3–6. | ||
Morgan T. Clinical pharmacokinetics and pharmacodynamics of carvedilol. Clin Pharmacokinet. 1994;26(5):335–346. | ||
ICH Guideline Harmonized Tripartite. “Q2B validation of analytical procedures: methodology.” Fed Regist. 1997;62. | ||
Dahan A, Hoffman A. Evaluation of a chylomicron flow blocking approach to investigate the intestinal lymphatic transport of lipophilic drugs. Eur J Pharm Sci. 2005;24(4):381–388. | ||
Shargel L, Andrew B, Wu-Pong S. Applied Biopharmaceutics and Pharmacokinetics. New York: McGraw-Hill; 2005. | ||
Rajera R, Nagpal K, Singh SK, Mishra DN. Niosomes: a controlled and novel drug delivery system. Biol Pharm Bull. 2011;34(7):945–953. | ||
Haeri A, Sadeghian S, Rabbani S, Anvari MS, Boroumand MA, Dadashzadeh S. Use of remote film loading methodology to entrap sirolimus into liposomes: Preparation, characterization and in vivo efficacy for treatment of restenosis. Int J Pharm. 2011;414(1):16–27. | ||
Bajelan E, Haeri A, Vali AM, Ostad SN, Dadashzadeh S. Co-delivery of doxorubicin and PSC 833 (Valspodar) by stealth nanoliposomes for efficient overcoming of multidrug resistance. J Pharm Pharm Sci. 2012;15(4):568–582. | ||
Manosroi A, Wongtrakul P, Manosroi J, et al. Characterization of vesicles prepared with various non-ionic surfactants mixed with cholesterol. Colloids Surf B Biointerfaces. 2003;30(1):129–138. | ||
Mokhtar M, Sammour OA, Hammad MA, Megrab NA. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int J Pharm. 2008;361(1):104–111. | ||
Balakrishnan P, Shanmugam S, Lee WS, et al. Formulation and in vitro assessment of minoxidil niosomes for enhanced skin delivery. Int J Pharm. 2009;377(1):1–8. | ||
Hao Y, Zhao F, Li N, Yang Y, Li Ka. Studies on a high encapsulation of colchicine by a niosome system. Int J Pharm. 2002;244(1):73–80. | ||
Haeri A, Alinaghian B, Daeihamed M, Dadashzadeh S. Preparation and characterization of stable nanoliposomal formulation of fluoxetine as a potential adjuvant therapy for drug-resistant tumors. Iran J Pharm Res. 2014;13(Suppl):3. | ||
Yoshioka T, Sternberg B, Florence AT. Preparation and properties of vesicles (niosomes) of sorbitan monoesters (Span 20, 40, 60 and 80) and a sorbitan triester (Span 85). Int J Pharm. 1994;105(1):1–6. | ||
Haeri A, Sadeghian S, Rabbani S, Anvari MS, Erfan M, Dadashzadeh S. PEGylated estradiol benzoate liposomes as a potential local vascular delivery system for treatment of restenosis. J Microencapsul. 2012;29(1):83–94. | ||
Haeri A, Sadeghian S, Rabbani S, et al. Sirolimus-loaded stealth colloidal systems attenuate neointimal hyperplasia after balloon injury: a comparison of phospholipid micelles and liposomes. Int J Pharm. 2013;455(1):320–330. | ||
Chu C, Tong S-S, Xu Y, et al. Proliposomes for oral delivery of dehydrosilymarin: preparation and evaluation in vitro and in vivo. Acta Pharmacol Sin. 2011;32(7):973–980. | ||
Yang L, Tucker IG, Østergaard J. Effects of bile salts on propranolol distribution into liposomes studied by capillary electrophoresis. J Pharm Biomed Anal. 2011;56(3):553–559. | ||
El-Ridy MS, Abdelbary A, Nasr EA, et al. Niosomal encapsulation of the antitubercular drug, pyrazinamide. Drug Dev Ind Pharm. 2011;37(9):1110–1118. | ||
Akhter S, Kushwaha S, Warsi MH, et al. Development and evaluation of nanosized niosomal dispersion for oral delivery of Ganciclovir. Drug Dev Ind Pharm. 2012;38(1):84–92. | ||
Sezgin-Bayindir Z, Onay-Besikci A, Vural N, Yuksel N. Niosomes encapsulating paclitaxel for oral bioavailability enhancement: preparation, characterization, pharmacokinetics and biodistribution. J Microencapsul. 2013;30(8):796–804. | ||
Andrieux K, Forte L, Lesieur S, Paternostre M, Ollivon M, Grabielle-Madelmont C. Solubilisation of dipalmitoylphosphatidylcholine bilayers by sodium taurocholate: a model to study the stability of liposomes in the gastrointestinal tract and their mechanism of interaction with a model bile salt. Eur J Pharm Biopharm. 2009;71(2):346–355. | ||
Sun J, Deng Y, Wang S, Cao J, Gao X, Dong X. Liposomes incorporating sodium deoxycholate for hexamethylmelamine (HMM) oral delivery: development, characterization, and in vivo evaluation. Drug Deliv. 2010;17(3):164–170. | ||
Humberstone AJ, Charman WN. Lipid-based vehicles for the oral delivery of poorly water soluble drugs. Adv Drug Deliv Rev. 1997; 25(1):103–128. | ||
Janga KY, Jukanti R, Velpula A, et al. Bioavailability enhancement of zaleplon via proliposomes: Role of surface charge. Eur J Pharm Biopharm. 2012;80(2):347–357. | ||
Jadon PS, Gajbhiye V, Jadon RS, Gajbhiye KR, Ganesh N. Enhanced oral bioavailability of griseofulvin via niosomes. AAPS Pharm Sci Tech. 2009;10(4):1186–1192. | ||
El-Shabouri M. Positively charged nanoparticles for improving the oral bioavailability of cyclosporin-A. Int J Pharm. 2002;249(1):101–108. | ||
Lian T, Ho RJ. Trends and developments in liposome drug delivery systems. J Pharm Sci. 2001;90(6):667–680. |
Supplementary material
 | Figure S1 FTIR spectra of empty niosomes (A), carvedilol-loaded niosomes (B). |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.