Back to Journals » Journal of Inflammation Research » Volume 19
NHWD-870, a Potent BET Inhibitor, Ameliorated Endotoxemia-Induced Hepatic Inflammation via Suppression of BRD4-STAT1 Signaling and Macrophage M1 Polarization
Authors Zhou N ![]() , Tan B
, Tan B ![]() , Jiang Y, Wang H, Peng W, Yin M, Fu L, Peng S
, Jiang Y, Wang H, Peng W, Yin M, Fu L, Peng S
Received 18 February 2026
Accepted for publication 28 May 2026
Published 6 June 2026 Volume 2026:19 598139
DOI https://doi.org/10.2147/JIR.S598139
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 5
Editor who approved publication: Dr Anh Ngo
Nianqi Zhou,1 Bin Tan,1 Ying Jiang,1 Huiwen Wang,2 Wenting Peng,1 Mingzhu Yin,3 Lei Fu,1,* Shifang Peng1,*
1Department of Infectious Diseases, Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China; 2Infection Control Center, Xiangya Hospital, Central South University, Changsha, Hunan, People’s Republic of China; 3Clinical Research Center, Medical Pathology Center, Cancer Early Detection and Treatment Center and Translational Medicine Research Center, Chongqing University Three Gorges Hospital, Chongqing University, Chongqing, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Lei Fu; Shifang Peng, Email [email protected]; [email protected]
Background: Sepsis remains a leading cause of mortality in intensive care units, with endotoxemia-induced hepatic inflammation being a major contributor to multiorgan failure. Bromodomain and extraterminal domain (BET) proteins are key epigenetic regulators of inflammation. While the BET inhibitor JQ1 shows protective effects in sepsis models, its toxicity limits translational application. NHWD-870, a next-generation BET inhibitor with improved potency and safety, has not been evaluated in septic liver injury.
Methods: Eight-week-old male C57BL/6 mice were challenged with lipopolysaccharide (LPS) to induce endotoxemia and treated with NHWD-870 1 hour before LPS challenge. Survival rate was assessed and 0.5mg/kg was selected as the optimal dose. Liver injury and inflammation were assessed by histopathology, serum biochemistry, ELISA, RT-qPCR, and immunohistochemistry. In vitro, primary mouse hepatocytes (PMHs) and bone marrow-derived macrophages (BMDMs) were stimulated with LPS to examine NHWD-870 effects on cytokine production and macrophage polarization.
Results: NHWD-870 improved survival in lethal LPS challenge and attenuated LPS induced ALT/AST elevations and hepatic inflammatory infiltration. Serum and hepatic IL6, TNFα, and MCP1 were reduced after NHWD-870; IL6 suppression at 12 h exceeded JQ1. NHWD-870 decreased hepatic macrophage and neutrophil infiltration and no obvious histopathological abnormalities were observed in the major organs at the active dose. In vitro, NHWD-870 dose dependently reduced LPS induced cytokine expression in PMH and BMDM and inhibited macrophage M1 polarization (CD86, iNOS). BRD4 and STAT1 expression increased after LPS; NHWD 870 suppressed both.
Conclusion: NHWD-870 ameliorates endotoxemia-associated hepatic inflammation and improves survival in mice, at least in part via suppression of BRD4-STAT1 signaling and inhibition of macrophage M1 polarization. Further preclinical development is warranted.
Keywords: sepsis, hepatic inflammation, NHWD-870, bromodomain protein 4, M1 polarization
Introduction
Sepsis is a life-threatening condition characterized by infection-induced dysregulated host responses and multiple organ dysfunction.1,2 Recent global estimates indicated that sepsis remained a major public health burden worldwide. In 2021, approximately 166 million sepsis cases and 21.4 million sepsis-related deaths were reported globally, accounting for 31.5% of total deaths,3,4 with mortality rising again during 2020–2021 after a previous decline from 1990 to 2019.5 These epidemiological data underscore that, despite advances in antimicrobial therapy and critical care management, sepsis remains a highly lethal syndrome and highlights the urgent need for novel therapeutic strategies.
Among the affected organs, the liver is both a target and an amplifier of systemic inflammation during sepsis. Sepsis- associated liver injury (SALI) is commonly manifested by elevated transaminases levels and inflammatory cell infiltration in hepatic tissue. Liver dysfunction during sepsis correlates strongly with adverse prognosis, with mortality rates exceeding 60% in severe cases.6,7 The liver plays a pivotal role in sepsis and endotoxemia because of its continuous exposure to circulating pathogens and microbial products through the portal and systemic circulation.8–11 Under physiological conditions, hepatocytes and resident Kupffer cells (KCs) coordinate immune tolerance and pathogen clearance.12 However, during sepsis, this homeostatic balance was disrupted, leading to excessive production of inflammatory cytokines and chemokines, recruitment of immune cells, and progressive hepatic injury.13
Mechanistically, both hepatocytes and macrophages actively contribute to hepatic inflammation through reciprocal activation.14,15 Lipopolysaccharide (LPS)-stimulated hepatocytes release chemokines such as MCP-1 that recruit macrophages, whereas activated macrophages secrete proinflammatory mediators that further aggravate hepatocyte injury.16 This hepatocyte-macrophage crosstalk forms a self-amplifying inflammatory loop that sustained tissue damage. Macrophages exhibit remarkable plasticity, polarizing into M1-like (proinflammatory) or M2-like (anti-inflammatory) states.17 M1 macrophages secrete IL-6, TNFα, and reactive oxygen species, promoting pathogen clearance but also collateral tissue damage when excessively activated.18–20 Accumulating evidence suggests that disruption of the M1/M2 balance is a critical determinant of sepsis severity and organ dysfunction. Importantly, signal transducer and activator of transcription 1 (STAT1) is a key transcription factor involved in inflammatory gene transcription and macrophage M1 polarization, and dysregulated STAT1 activation has been implicated in exaggerated inflammatory responses during sepsis.21,22
Epigenetic regulation, particularly histone acetylation, is a key mechanism for transcriptional reprogramming of many immune-related genes during sepsis.23,24 Bromodomain and extra-terminal domain (BET) proteins—BRD2, BRD3, BRD4, and BRDT—function as epigenetic readers of acetylated lysine residues and facilitate transcriptional activation of proinflammatory genes. Among them, BRD4 is the most extensively studied and has been shown to promote transcription of NF-κB-dependent inflammatory genes while regulating macrophage activation and inflammatory signaling pathways, including STAT1-associated transcriptional programs.25,26 Preclinical studies showed that BET inhibitors such as JQ1 and I-BET exert protective anti-inflammatory effects in models of sepsis, colitis, and cardiac injury.27–31 However, the clinical translation of first-generation BET inhibitors remains limited due to their short half-life, modest potency, and dose-limiting systemic toxicity.32
NHWD-870 is a newly developed, high-affinity BET inhibitor demonstrating ~50-fold higher potency and improved safety compared with JQ1.33 In recent years, NHWD-870 has shown strong efficacy in preclinical studies of small cell lung cancer,34 melanoma,35,36 osteosarcoma,37 and psoriasis,38 and is undergoing clinical trials in primary pulmonary NUT carcinoma.39,40 There exists no relevant investigation conducted in its protective role in endotoxemia-induced hepatic inflammation. Therefore, in this study, we hypothesized that NHWD-870 may attenuate LPS-induced hepatic inflammation in a preclinical model and further investigated the underlying mechanism.
Materials and Methods
Animals and Treatment
All animal studies were approved by the Institutional Animal Care and Use Committee of Xiangya Hospital, Central South University (No. CSU-2023-0034) and were conducted in accordance with the ARRIVE guidelines and the institutional guidelines for the care and use of laboratory animals. Eight-week-old male C57BL/6 mice were purchased from Hunan SJA Laboratory Animal Co., Ltd. The approval number SYXK 2023–0006 and the animal license number SCXK 2021–0002. All mice were maintained under SPF conditions and housed with a 12 h light/dark cycle, at 22 ± 2°C and 50 ± 10% humidity in standard ventilated cages. Adaptive feeding for one week was conducted before the start of the formal experimental randomization.
For the survival experiment, mice were challenged with a lethal dose of LPS (15 mg/kg, i.p., Escherichia coli (O111:B4), Sigma-Aldrich, Germany) and given 0.125mg/kg (n=6), 0.25mg/kg (n=6), 0.5mg/kg (n=6), 1mg/kg (n=6) NHWD-870 (i.p.) or an equal amount of vehicle (n=5), respectively, and monitored for survival over 120 h. Mice were monitored every 12 hours for survival and humane endpoints throughout the observation period.
For the prophylactic pre-treatment experiment, mice were challenged with a lower dose of LPS (7.5 mg/kg, i.p.) and with 0.5mg/kg NHWD-870 or an equal volume of vehicle, and euthanized at 12 h or 24 h for tissue collection (n=5 per group). All NHWD-870 was administered 1 hour before LPS challenge, similar to the method commonly used for other BETi in previous studies, establishing a prophylactic pre-treatment model rather than a post-onset therapeutic model.31,41,42 All in vivo experiments were conducted in three independent replicates (biological replicates performed on separate occasions), with data pooled for analysis.
LPS was dissolved in saline, and NHWD-870 was dissolved in 2%DMSO solvent system (vehicle). Mice were anesthetized by 1% sodium pentobarbital solution, which was carried out in accordance with American Veterinary Medical Association. Blood samples were obtained by retro-orbital bleeding under anesthesia and serum was collected and immediately stored at −80°C until analysis. A portion of the liver was fixed in 4% paraformaldehyde (PFA) for histopathology, and the remaining liver tissue was rapidly frozen in liquid nitrogen for further use.
Animal monitoring and humane endpoints: All experimental animals were monitored every 6 hours after LPS injection for signs of severe distress requiring euthanasia (severe lethargy, inability to stand, respiratory distress, or weight loss >20%). Animals meeting these humane endpoint criteria were immediately euthanized (pentobarbital overdose or cervical dislocation). Survival was recorded as the primary endpoint at 120 hours. Expected or unexpected adverse events were reported in the results.
Isolation and Culture of Mouse Primary Hepatocytes (PMHs) and Bone Marrow-Derived Macrophages (BMDMs)
PMHs and BMDMs were isolated using collagenase perfusion and standard differentiation protocols. Briefly, liver tissue of the anesthetized mice was perfused and digested with perfusion buffer and enzyme buffer respectively. The digested liver tissue was filtered and live hepatocytes were separated with Percoll (Solarbio, China). Then, carefully separated the lower limbs, and flushed out the bone marrow using DMEM (Gibco, USA). After precipitation and red blood cells lysis, mouse BMDMs were obtained and cultured in DMEM complete medium containing 20 ng/mL M-CSF (Sino Biological, China). Cells were stimulated with LPS (100 ng/mL) with or without NHWD-870 (1.25~10 nM).
Histological and Immunofluorescence Assays
4% PFA-fixed liver tissues were embedded in paraffin and sectioned (5 µm). Hematoxylin–eosin (H&E, Servicebio, China), immunohistochemistry (IHC), and immunofluorescence (IF) were performed using antibodies against F4/80 (Proteintech, China), MPO (Aifang Bio, China), and CD86 (Bioss, China). IHC and IF staining were performed as previously described.43 Briefly, antigen retrieval was achieved by heating Tris-EDTA buffer in a microwave oven for 6 min 3 times. After incubation with primary antibodies at 4°C overnight, tissue sections were incubated with appropriate secondary antibodies at 37°C for 1 h. IF samples were then incubated with DAPI (Servicebio, China) for nuclear staining, while IHC samples were incubated with hematoxylin for nuclear staining. For each liver specimen (n = 5 per group), 10 random fields were analyzed per section, and the positive area was quantified using ImageJ by a blinded investigator.
RNA Extraction and RT-qPCR
Total RNA and proteins were extracted from tissues or cultured cells. RNA was extracted using the Trizol (Takara, Japan) and dissolved in DEPC water (Biosharp, China). Reverse transcription kit was from Yeasen, China. The enzyme used for RT-qPCR was from TOROIVD, China. Relative mRNA expression of cytokines (IL-6, TNF-α, MCP-1), macrophage markers (CD86, iNOS), and signaling molecules (BRD4, STAT1) were quantified by the 2−ΔΔCt method, normalized to 18S rRNA. The primer sequences are shown in Supplementary Table 1.
Western Blot Analysis
Mouse liver tissue was treated with RIPA (Ecotop, China) and protease inhibitors and phosphatase inhibitors (NCM Biotech, China) to obtain total liver protein. Protein concentration was measured using a BCA protein assay kit (Thermo Scientific, German). Proteins were separated by 8% SDS-PAGE according to concentration and transferred to a 0.45 μm PVDF membrane (MILLIPORE, USA). The membrane was blocked with 5% skim milk for 1 h, and primary antibodies were added and incubated overnight at 4°C. After washing with TBST, the membrane was incubated with HRP-conjugated secondary antibodies for 1 h. Enhanced chemiluminescence system (ECL) was used for detection. Protein levels were normalized to β-actin and quantified using Image J 1.51 (Bethesda, USA) (n = 5 per group). Western blot imaging was performed using the Bio-Rad ChemiDOCTM XRS+ system, with images captured using the Image Lab 6.0.1 software in the Chemi Hi Sensitivity mode and automatic exposure settings. No further adjustments were made to the contrast of the images. Antibodies used in WB were mouse ACTB antibody (SantaCruz, USA), mouse BRD4 antibody (Abcam, USA), mouse STAT1 antibody (ZEN-BIOSCIENCE, China).
Enzyme-Linked Immunosorbent Assay (ELISA)
Mouse IL6 Precoated ELISA Kit (Cat:1210602), and Mouse TNF-α Precoated ELISA Kit (Cat:1217202) were purchased from Dakewe, China, and MCP1 ELISA kits were purchased from RenjieBio, China and operated according to the instructions.
Biochemistry
Mouse serum liver function was analyzed by the Laboratory Department of Xiangya Hospital, Central South University using Beckman Coulter AU5800 automatic biochemical analyzer.
Statistical Analysis
Statistical analysis and drawing were performed using Graphpad prism 9.0. The data were tested for normality using a QQ graph. Data were presented as mean ± SD. Two-group comparisons used Student’s t-test; multiple groups were analyzed by one-way ANOVA with Tukey’s post hoc test. Survival rate was analyzed by Kaplan–Meier, and Log rank tests were used to compare differences. p < 0.05 was considered statistically significant.
Results
Endotoxemia Liver Inflammation Model Was Successfully Established
We constructed an LPS-induced endotoxemia model at different time points (Figure 1A). The modeling group was given an intraperitoneal injection of 7.5 mg/kg LPS while the control group was injected with an equal volume of saline. Mice were euthanized at 12 h and 24 h after LPS challenge. Hepatic injury was more pronounced at 12 h, with evident hepatocyte swelling, disorganized lobular architecture, and inflammatory infiltration (Figure 1B). At the same time, serum liver function results showed that after LPS administration, the levels of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were significantly increased (Figure 1C and D). ELISA (Figure 1E–G) and RT-qPCR (Figure 1H–J) results showed that the expression levels of cytokine IL6, TNFα, and chemokine MCP1 in the serum and liver tissue were significantly increased in LPS group. The above results confirmed successful sepsis liver injury model establishment.
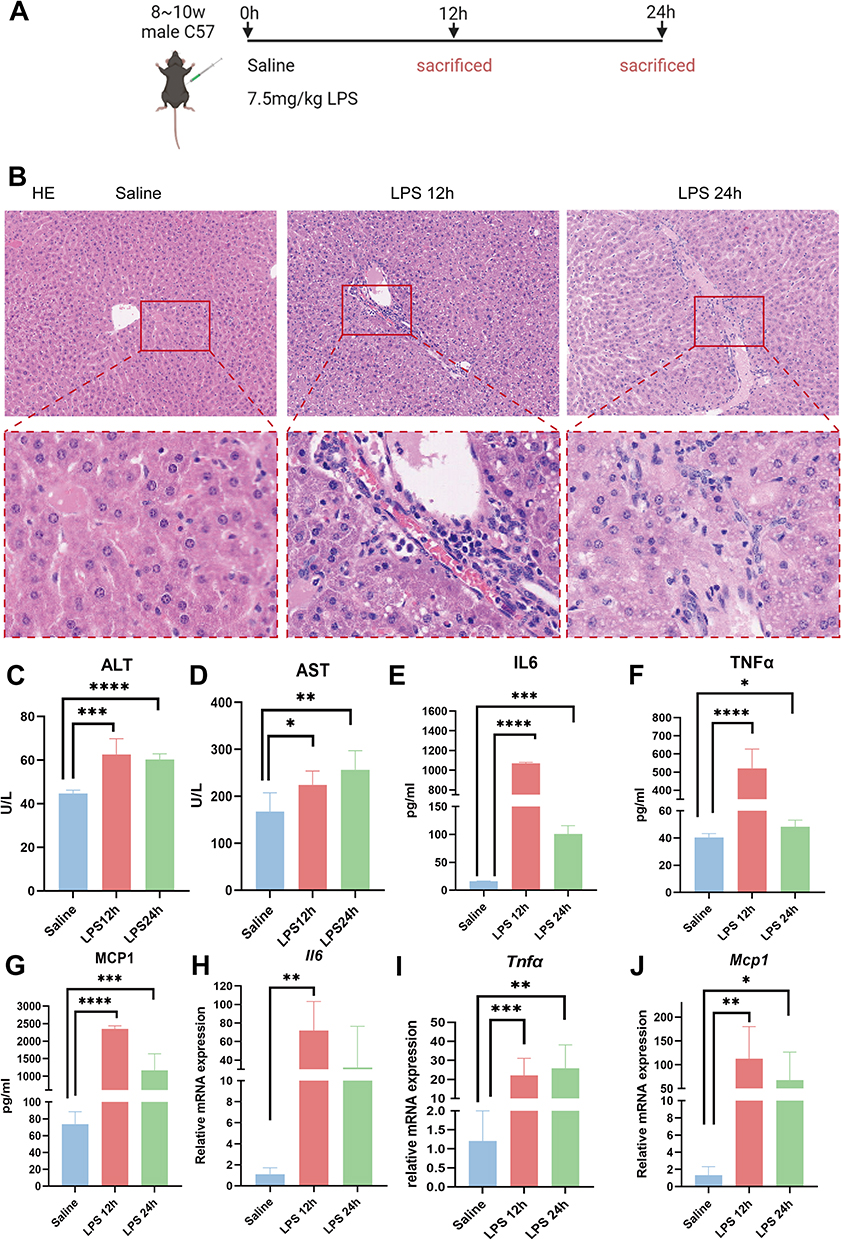 |
Figure 1 Endotoxemia liver inflammation model was successfully established. (A) Schematic diagram of LPS modeling (7.5mg/kg). (B) HE staining of liver tissue (magnification 100×). (C and D) Levels of serum ALT(C) and AST(D). (E–G) ELISA was used to detect the levels of serum IL6 (E), TNFα (F), and MCP1 (G). (H–J) RT-qPCR was used to detect the mRNA expression of IL6 (H), TNFα(I), and MCP1 (J) in the liver tissues of mice. Mice were divided into 3 groups: Saline (n=5), LPS 12h (n=5), LPS 24h (n=5). The data are presented as mean ± SD. (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). |
NHWD-870 Improved Survival and Alleviated Liver Injury in Mice with Endotoxemia
In lethal LPS challenge, NHWD-870 increased survival, and 0.5mg/kg conferred the optimal balance of efficacy and tolerability and was used for mechanistic studies (Figure 2A and B). Further, we selected JQ1, a classic BET inhibitor, as a positive control and challenged mice with LPS injection (Figure 2C). NHWD-870 (0.5 mg/kg) significantly reduced serum ALT at 12 and 24h and AST at 24h compared with vehicle; JQ1 showed smaller effects at the doses tested (Figure 2D and E). HE staining showed that the immune cells infiltration of liver tissue and the swelling of hepatocytes were significantly reduced after treatment with both NHWD-870 and JQ1 (Figure 2F). NHWD-870 reduced serum IL6, TNFα and MCP1 at 12h and 24h (Figure 2G–I); the magnitude of IL6 suppression at 12h exceeded that of JQ1. Hepatic mRNA levels mirrored serum cytokine reductions (Figure 2J–L).
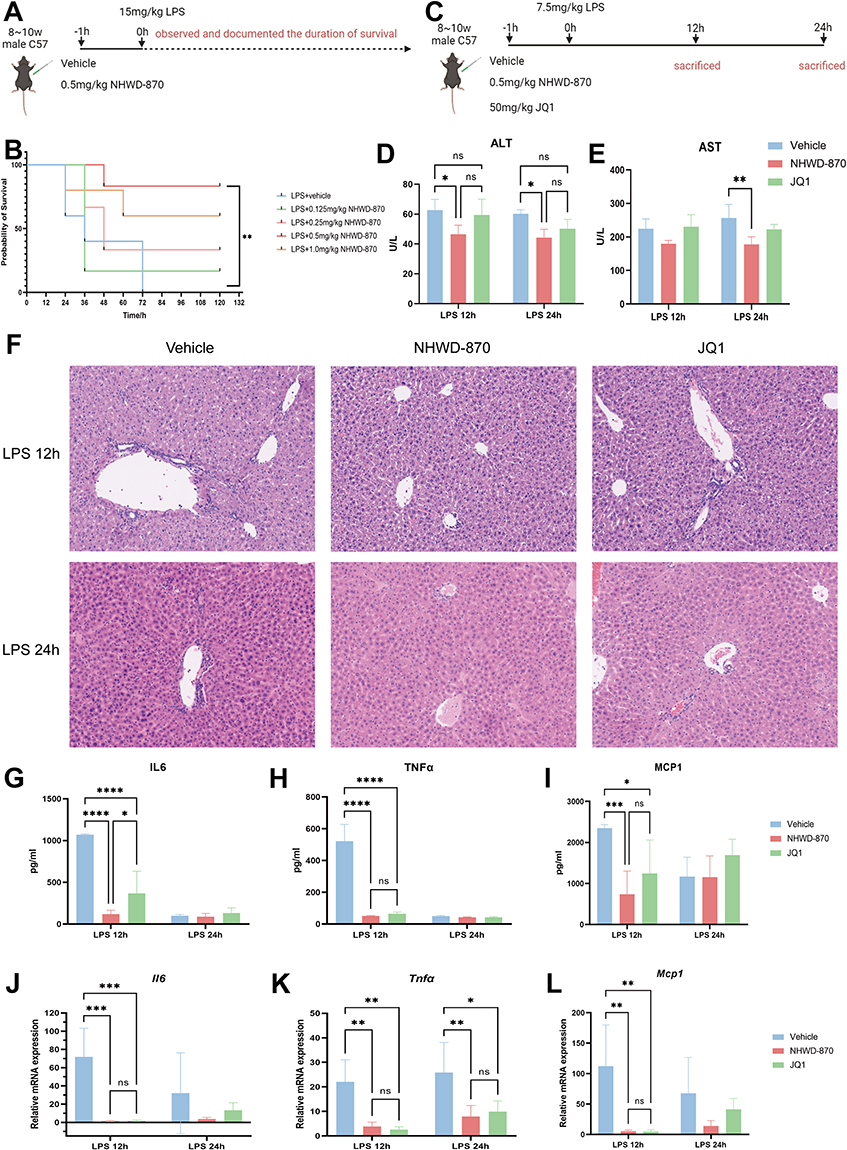 |
Figure 2 NWD-870 improved the survival rate and liver inflammation of mice with endotoxemia. (A) Schematic diagram of the lethal model of endotoxemia. (B) Survival analysis of mice treated with NHWD-870. Mice were divided into five groups as following: LPS (15mg/kg) +vehicle (n=5), LPS+0.125mg/kg NHWD-870 (n=6), LPS+0.25mg/kg NHWD-870 (n=6), LPS+0.5mg/kg NHWD-870 (n=6), LPS+1.0mg/kg NHWD-870 (n=5). (C) Schematic diagram of NHWD-870 treatment of endotoxemia liver inflammation model. (D and E) Serum levels of ALT and AST. (F) HE staining of liver tissue (Magnification 100×). (G-I) ELISA detection of IL6 (G), TNFα (H), and MCP1(I) expression levels in mouse serum. (J-L) RT-qPCR detection of IL6 (J), TNFα (K), and MCP1 (L) mRNA expression levels in mouse liver tissue after NHWD-870 intervention. Each time point of endotoxemia model were divided into 3 groups: vehicle (n=5), NHWD-870 (n=5), JQ1 (n=5). (A and B) showed survival study using 15 mg/kg LPS (lethal dose); (C–L) showed mechanistic study using 7.5 mg/kg LPS (sublethal dose) to enable tissue collection at 12h and 24h. Different doses were selected based on distinct experimental objectives as detailed in Methods. Data are presented as mean ± SD. (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Abbreviations, ALT, alanine aminotransferase; AST, aspartate aminotransferase; HE, hematoxylin-eosin staining; ELISA, enzyme-linked immunosorbent assay; RT-qPCR, real-time quantitative reverse transcription PCR. |
NHWD-870 Reduced Inflammatory Cell Infiltration Without Causing Organ Toxicity
Macrophages and neutrophils are the earliest and most important immune cells that respond to bacterial endotoxin-induced liver inflammation, and their infiltration degree is related to inflammatory damage. Quantitative analysis of IHC-positive area confirmed that NHWD-870 significantly reduced hepatic infiltration of macrophages (F4/80⁺) and neutrophils (MPO⁺) compared with vehicle (Figure 3A-D). Histological examination of heart, kidney, spleen, and lung revealed no pathological damage, indicating favorable safety (Figure 3E). ALT and AST showed no significant difference compared with vehicle group also indirectly confirmed this point (Figure 3F and G). Serum IL6 and TNFα were unchanged after NHWD-870 alone, while MCP1 decreased slightly, suggesting selective immunomodulation rather than systemic suppression (Figure 3H–J).
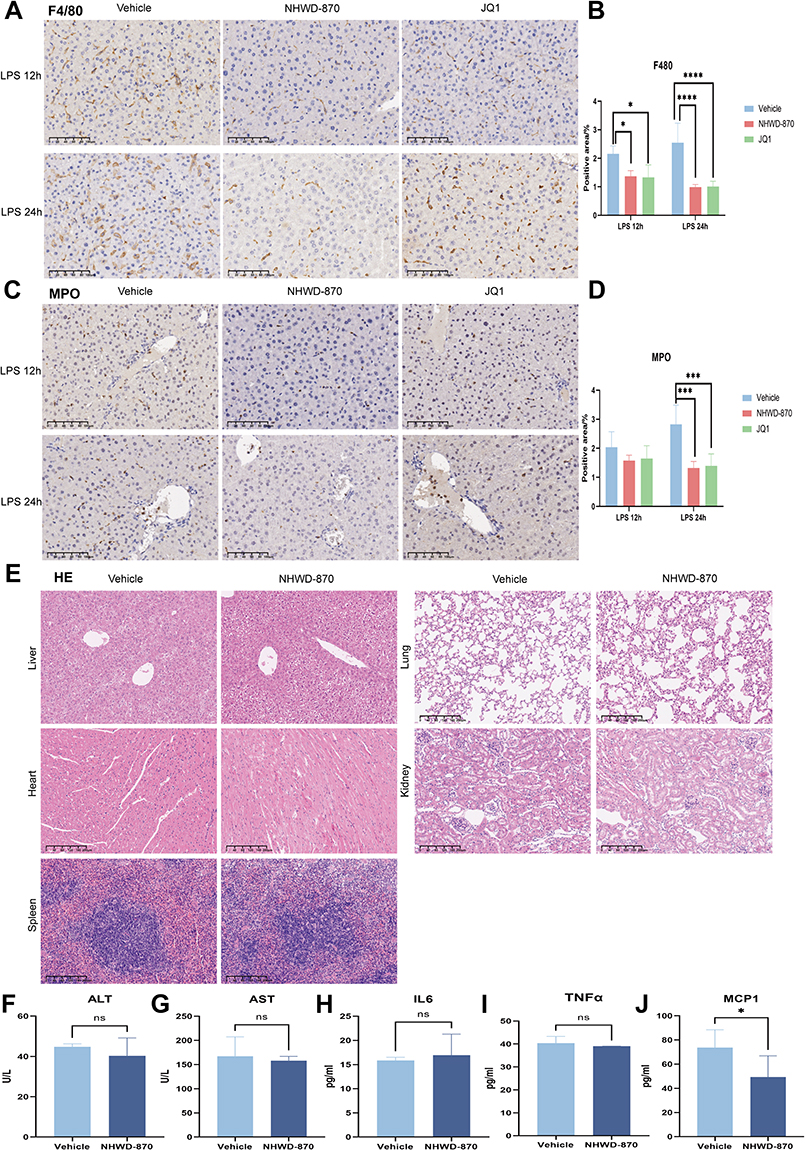 |
Figure 3 Effects of NHWD-870 on inflammatory cell infiltration in endotoxemia mice and its effects on various organs. (A) IHC detection of F4/80 expression in liver tissues. (scale bar 100μm) (B) Statistics of the positive area of F4/80 IHC staining. (C) IHC detection of MPO expression in liver tissues. (scale bar 100μm) (D) Statistics of the positive area of MPO IHC staining. IHC positive areas are brown-yellow. Each time point of endotoxemia model were divided into 3 groups: vehicle (n=5), NHWD-870 (n=5), JQ1 (n=5). (E) HE staining of NHWD-870 treatment on the heart, liver, spleen, kidney, and liver morphology of mice. (Magnification 100×) (F-G): ALT (F) and AST (G) levels after NHWD-870 intervention. (H-J) ELISA detection of IL6 (H), TNFα (I), and MCP1 (J) levels in mouse serum. Vehicle group (n=5); NHWD-870 group (n=5). Data are presented as mean ± standard deviation. (*p<0.05, ***p<0.001, ****p<0.0001). Abbreviations: IHC, Immunohistochemical; ALT, alanine aminotransferase; AST, aspartate aminotransferase; HE, hematoxylin-eosin staining; ELISA, enzyme-linked immunosorbent assay. |
NHWD-870 Suppressed Inflammatory Response in Hepatocytes and Macrophages
In vitro, LPS stimulation was constructed to test NHWD-870 directly or indirectly ameliorating hepatocytes and macrophages inflammation. In both hepatocytes (Figure 4A–C) and macrophages (Figure 4D–F), IL6, TNFα and MCP1 dramatically increased after LPS challenge and NHWD-870 ameliorated this response in a dose-dependent manner (Figure 4A–F). When hepatocytes were incubated with supernatants from LPS-activated macrophages, cytokine expression further increased, but this effect was reversed by NHWD-870, confirming its dual inhibition of macrophage and hepatocyte inflammatory signaling (Figure 4G–I).
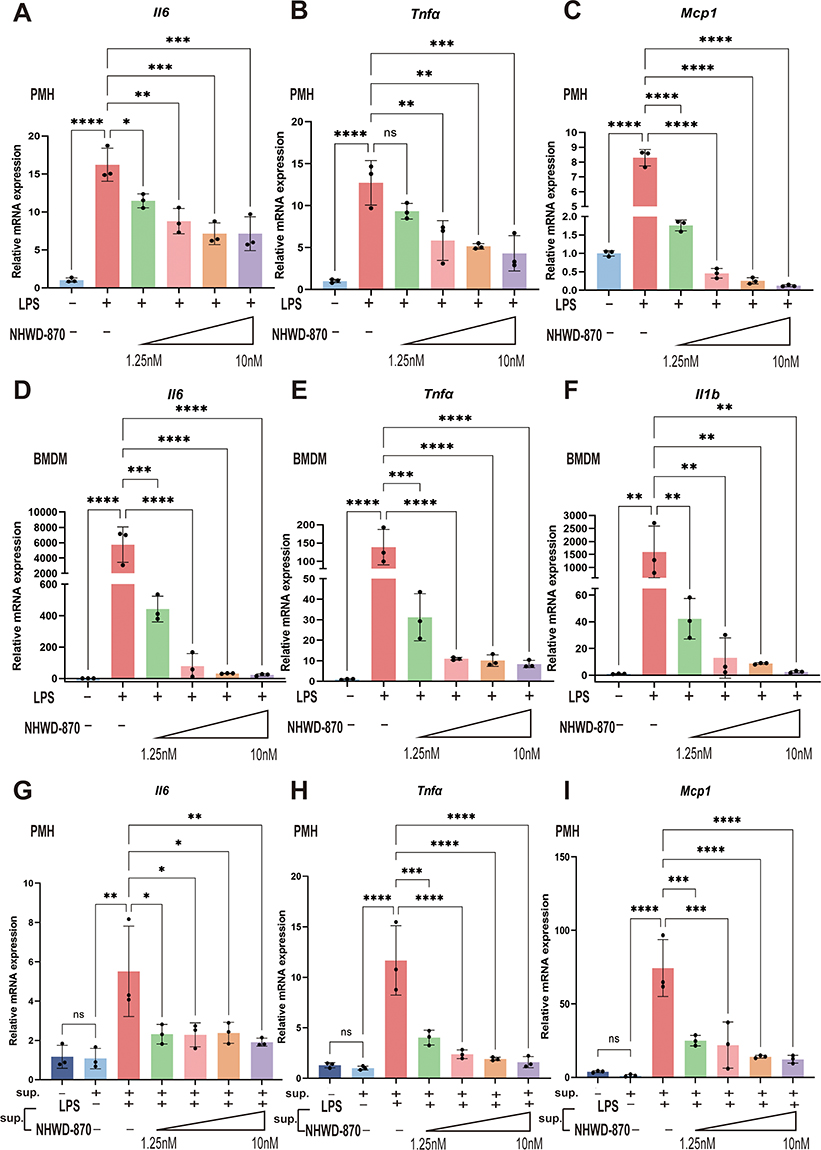 |
Figure 4 NHWD-870 reduced the inflammatory response of PMHs and BMDMs. (A–C) Expression levels of inflammatory factors and chemokines after NHWD-870 intervention in PMHs. (D–F) Expression levels of inflammatory factors and chemokines after NHWD-870 intervention in BMDMs. (Figure A-F legends: The first line indicates whether LPS was given, and the second line indicates whether NHWD-870 was given and its concentration, which are 1.25nM, 2.5nM, 5nM, 10nM, respectively) (G–I) Expression levels of inflammatory factors and chemokines after BMDMs supernatant was used to intervene in PMHs. The first line indicates whether BMDMs supernatant was given for intervention, and the second line indicates the components in macrophage supernatant. All results were obtained by RT-qPCR, and n=3 per group. Data are presented as mean ± standard deviation. (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). Abbreviations: RT-qPCR, real-time quantitative PCR; PMHs, primary mouse hepatocytes; BMDMs, bone marrow-derived macrophages; sup., supernatant. |
NHWD-870 Inhibited Macrophage M1 Polarization
LPS has been widely recognized to promote the polarization of M1-like macrophages, and triggered robust expression of CD86 and inducible nitric oxide synthase (iNOS). To determine whether BET inhibition affects macrophage polarization, we examined M1-like macrophage markers CD86 and iNOS. Both markers were markedly increased after LPS stimulation and were significantly suppressed by NHWD-870 at the mRNA level in macrophages and liver tissue (Figure 5A–D). Immunofluorescence staining confirmed reduced hepatic CD86 expression, indicating that NHWD-870 prevented excessive M1 macrophage activation during endotoxemia (Figure 5E and F).
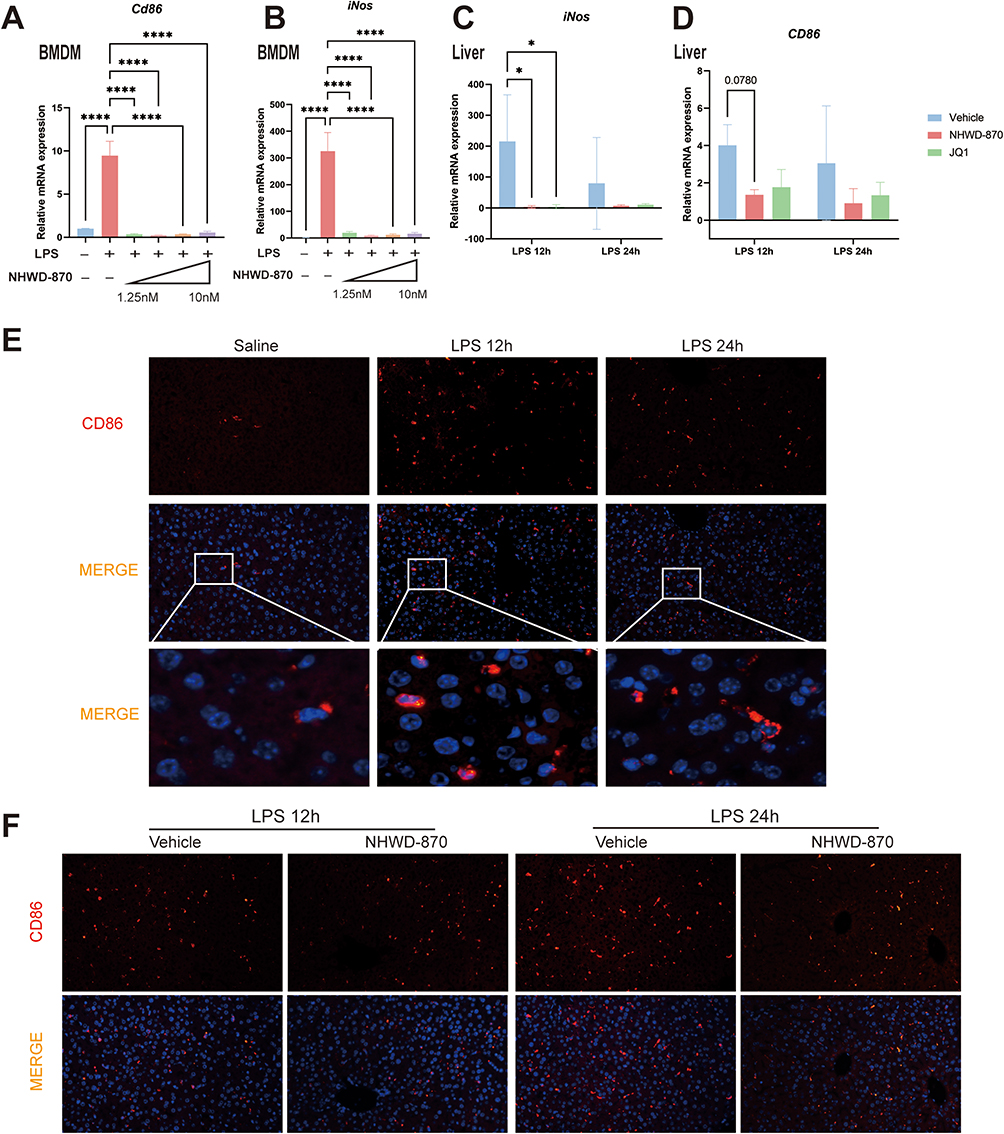 |
Figure 5 NHWD-870 inhibited macrophage M1 polarization. (A and B) RT-qPCR detection of BMDMs CD86 (A) and iNOS (B) mRNA expression levels (n=3 in each group). (C and D) RT-qPCR detection of CD86 (C) and iNOS (D) mRNA expression levels in mouse liver tissue after NHWD-870 intervention (n=5 in each group). (E and F) CD86 immunofluorescence staining of liver tissue in each group (marked in red), and nucleus were marked with DAPI (blue). Data are presented as mean ± standard deviation. (*p<0.05, ****p<0.0001). Abbreviation: iNOS, inducible nitric oxide synthase. |
NHWD-870 Downregulated BRD4-STAT1 Signaling in Endotoxemia
In BET family, the most widely studied member is BRD4. Given that BRD4 could promotes the development of various diseases, including sepsis or endotoxemia, by regulating gene transcription. Previous studies by our group found that BRD4 expressed in human hepatocytes and macrophages43 and we confirmed this finding in mouse liver tissue (Figure 6A and B). Given that M1-like macrophage polarization is closely related to STAT1, we explored whether NHWD-870 mediated this pathway. The results showed that LPS stimulation upregulated BRD4 and STAT1 expression in parallel (Figure 6C–G), and their levels were positively correlated at both mRNA and protein levels (Figure 6H and I). Meanwhile, NHWD-870 treatment markedly suppressed STAT1 expression, suggesting inhibition of this axis underlies its anti-inflammatory effects (Figure 6J–L).
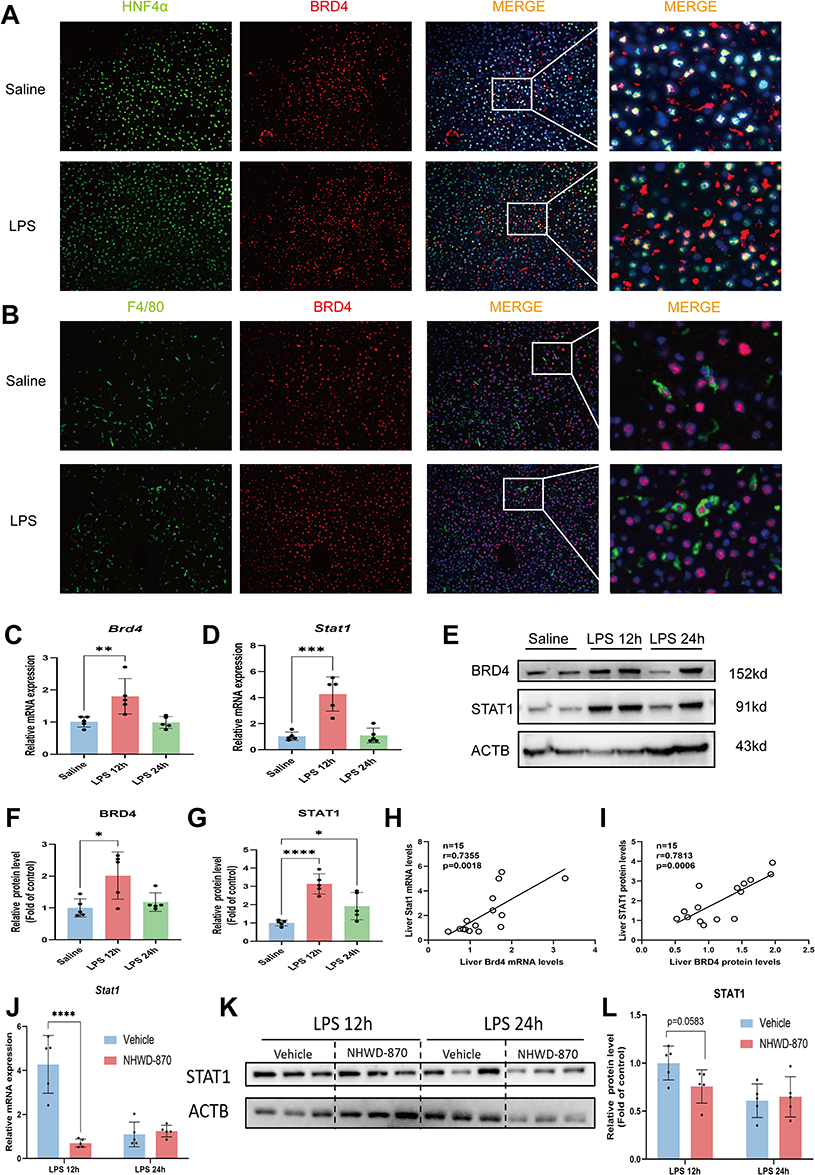 |
Figure 6 STAT1 expression was highly correlated with BRD4 and significantly inhibited by NHWD-870. (A) Immunofluorescence colocalization of hepatocyte markers (HNF4α, green label) and BRD4 (red label) in both saline and LPS group, and the cell nuclei were labeled with DAPI (blue). (B) Immunofluorescence colocalization of macrophage markers (F4/80, green label) and BRD4 (red label) in the saline group and LPS group, and the cell nuclei were labeled with DAPI (blue). Immunofluorescence images were magnified at 200×, n=3 in each group. (C) RT-qPCR was used to detect the expression of BRD4 mRNA in mouse liver tissue (n=5 in each group). (D) RT-qPCR was used to detect the expression of STAT1 mRNA in mouse liver tissue (n=5 in each group). (E–G) Western Blotting was used to detect the expression of BRD4 and STAT1 protein (E) and the statistical chart (F and G) (n=5 in each group). (H and I) Correlation analysis of STAT1 and BRD4 expression at mRNA (H) and protein (I) levels (n=15). (J–L) RT-qPCR (J) and Western Blotting (K and L) were used to detect the expression levels of STAT1 mRNA and protein in liver tissue after NHWD-870 intervention (n=5 in each group). Data are presented as mean ± standard deviation. (*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001). |
These findings suggest that NHWD-870 suppresses endotoxemia-induced hepatic inflammation by inhibiting the BRD4-STAT1 axis and macrophage M1 polarization, as summarized in Figure 7.
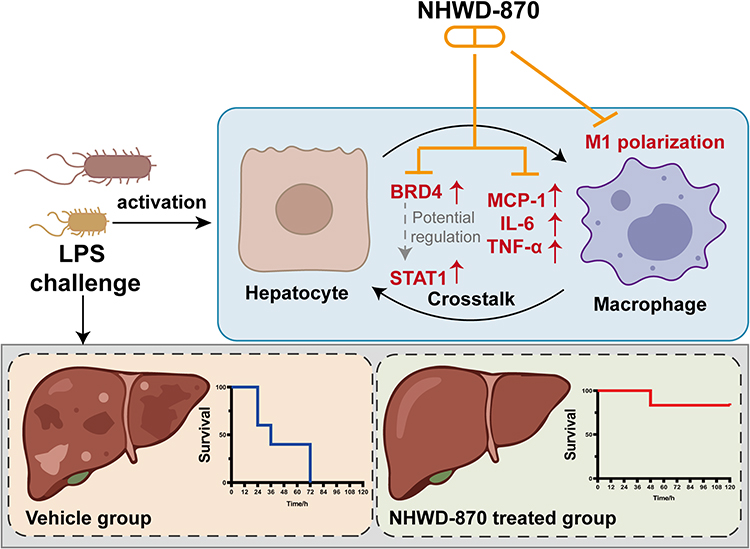 |
Figure 7 Schematic illustration of the protective effects of NHWD-870 in endotoxemia-induced hepatic inflammation. LPS challenge activates hepatocytes and macrophages, leading to hepatocyte-macrophage crosstalk, upregulation of BRD4 and STAT1, increased production of proinflammatory mediators (MCP-1, IL-6, and TNF-α), and promotion of macrophage M1 polarization. NHWD-870, a BET inhibitor, suppresses the BRD4-STAT1 signaling axis, attenuates macrophage M1 polarization, reduces inflammatory mediator production, and ultimately ameliorates endotoxemia-induced liver injury and improves survival. The lower panels show the survival outcomes in the vehicle group and the NHWD-870-treated group. |
Discussion
In the present study, we demonstrated that NHWD-870 significantly attenuated endotoxemia-induced hepatic inflammation, improved survival, and alleviated liver injury in LPS-challenged mice. Mechanistically, NHWD-870 suppressed macrophage M1 polarization and reduced activation of the BRD4-STAT1 signaling axis, suggesting that BET inhibition may represent a promising preclinical strategy for sepsis-related liver injury.
Although substantial advances have been made in antimicrobial therapy and critical care management, sepsis remains a major cause of morbidity and mortality worldwide.3,5 Current treatment strategies primarily rely on infection control, hemodynamic support, and organ function preservation, whereas targeted therapies directed against dysregulated host inflammatory responses remain under active investigation.44,45 Therefore, identifying novel molecular targets involved in sepsis-associated immune dysregulation is of considerable translational interest.
BET proteins have emerged as critical epigenetic regulators of inflammatory gene transcription, and increasing evidence suggests that pharmacological inhibition of BET signaling exerts protective effects in multiple inflammatory disease models.46–49 Previous studies have shown that classical BET inhibitors such as JQ1 and I-BET attenuate inflammatory injury in models of sepsis, acute liver injury,42 cardiac dysfunction,27 and colitis.41 JQ1 has been demonstrated to attenuate NLRP3 inflammasome priming and activation, thereby reducing caspase-1 maturation and subsequent interleukin-1β (IL-1β) and IL-18 release in LPS-challenged macrophages.50–52 Furthermore, BRD4 inhibition suppresses gasdermin D (GSDMD) expression, the key executioner of pyroptotic cell death, providing cytoprotection in inflammatory tissue injury, supporting the concept that BET inhibition may mitigate inflammation induced by bacterial pathogens and their products.50,53 However, the clinical translation of first-generation BET inhibitors has been hindered by limitations including short half-life, modest potency, and dose-limiting toxicity. NHWD-870, a newly developed BET inhibitor, has demonstrated superior potency and improved pharmacologic properties compared with conventional BET inhibitors.33–36,54 Our findings further expand its anti-inflammatory potential by showing that NHWD-870 effectively protects against endotoxemia-induced hepatic injury.
The liver serves as both an immune sensor and effector organ during sepsis, continuously exposed to circulating pathogens and endotoxins.55,56 Upon LPS stimulation, hepatocytes and macrophages release inflammatory cytokines and chemokines that recruit additional immune cells and amplify hepatic inflammation. In agreement with this concept, our data showed that NHWD-870 reduced hepatic inflammatory cell infiltration, improved histopathological injury, and decreased serum liver injury markers, suggesting broad protective effects on both hepatic parenchymal cells and immune-mediated inflammatory responses.
Mechanistically, BRD4 is one of the most extensively studied BET family members and has been implicated in regulating inflammatory transcriptional programs.57 Recent studies have suggested that BRD4 contributes to macrophage polarization partly through modulation of STAT1-dependent transcriptional activity.58 STAT1 is a critical transcription factor involved in inflammatory signaling and macrophage M1 polarization, and excessive STAT1 activation has been associated with exaggerated inflammatory injury in sepsis-related models.59,60 In the present study, we observed a strong positive correlation between BRD4 and STAT1 expression following LPS challenge, both of which were significantly suppressed by NHWD-870 treatment. These findings suggest that NHWD-870 may inhibit proinflammatory gene transcription by suppressing BRD4-STAT1 signaling, thereby attenuating macrophage M1 polarization and downstream cytokine release. However, the observed correlation between BRD4 and STAT1 expression, while supportive of a functional axis, does not establish causation. BRD4 may regulate STAT1 transcriptionally, or both may be co-regulated by upstream signals. Alternatively, reduced STAT1 may reflect diminished cytokine signaling secondary to BET inhibition rather than direct BRD4-mediated transcriptional control.
Interestingly, NHWD-870 markedly suppressed the early surge of IL-6 and TNF-α following LPS exposure (12h), whereas its effects on circulating cytokine levels became less pronounced at later measured time points (24h). This observation may reflect the transient nature of cytokine release in acute endotoxemia models. Nevertheless, histological analyses demonstrated sustained reductions in hepatic inflammatory infiltration and hepatocellular injury, suggesting that NHWD-870 exerts both early immunomodulatory and persistent tissue-protective effects.
While the current study demonstrates that a single dose of NHWD-870 provides significant survival benefit, the observed waning of anti-inflammatory effects at 24h suggests that multiple-dose regimens or sustained-release formulations may be necessary for prolonged protection. Future studies will systematically evaluate the pharmacokinetic-pharmacodynamic relationship and optimize dosing intervals to maintain therapeutic efficacy.
Compared with JQ1, NHWD-870 achieved comparable or superior anti-inflammatory efficacy at substantially lower doses (0.5mg/kg versus 50mg/kg) in our experimental setting, highlighting its potential pharmacologic advantage. In addition, no obvious histopathological abnormalities or major biochemical toxicity were observed in principal organs at the active dose, supporting the preliminary safety of NHWD-870 under the conditions tested.
Several limitations of this study should be acknowledged. First, NHWD-870 was administered prophylactically before LPS challenge; therefore, whether similar protective effects can be achieved with therapeutic post-treatment remains to be determined. The prophylactic administration used in this study was designed to establish proof-of-concept for NHWD-870’s protective efficacy and elucidate underlying mechanisms. While therapeutic post-treatment models would better simulate clinical scenarios, they require prior knowledge of effective dosing windows. Our findings provide the necessary foundation for future studies evaluating NHWD-870 in therapeutic settings. Second, although our findings suggest that BRD4 inhibition is a major mechanism underlying the observed protective effects, the potential contribution of other BET family members cannot be excluded. Third, while the observed correlation between BRD4 and STAT1 supports involvement of this signaling axis, direct mechanistic validation through genetic knockdown, chromatin immunoprecipitation, or rescue experiments is required to establish causality.
Collectively, our findings provide preclinical evidence that NHWD-870 attenuates endotoxemia-induced hepatic inflammation by suppressing macrophage activation and inhibiting BRD4-STAT1 signaling. These results support further investigation of NHWD-870 as a potential therapeutic strategy for sepsis-associated organ injury. However, the clinical translation practice of NHWD-870 in the future still requires efficacy verification; target confirmation; efficacy and safety assessment of multiple organ dysfunction other than liver injury; pharmacokinetics-pharmacodynamic optimization and other steps.
Conclusions
NHWD-870 ameliorates endotoxemia-induced hepatic inflammation by inhibiting macrophage M1 polarization and suppressing BRD4-STAT1 signaling, while direct genetic mechanistic confirmation and broader organ protection assessment representing critical next steps toward clinical translation.
Institutional Review Board Statement
All animal studies were approved by the Institutional Animal Care and Use Committee of Xiangya Hospital, Central South University (No. CSU-2023-0034) and conducted per ARRIVE guidelines. The approval number SYXK 2023-0006 and the animal license number SCXK 2021-0002.
Data Sharing Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Acknowledgments
We thank Prof. Yin et al for kindly providing NHWD-870 for this study. The compound was synthesized and characterized by Prof. Yin’s laboratory as previously described. No independent chemical validation or batch-specific quality control was performed in the present study. Prof. Yin was not involved in data collection, analysis, or interpretation.
Author Contributions
Nianqi Zhou: Methodology, Investigation, Data curation, Formal analysis, Visualization, Writing – original draft, Writing – review & editing. Bin Tan: Methodology, Investigation, Data curation, Validation, Writing – original draft. Ying Jiang: Methodology, Investigation, Data curation, Validation, Writing – review & editing. Huiwen Wang: Methodology, Investigation, Data curation, Validation, Writing – review & editing. Wenting Peng: Methodology, Investigation, Data curation, Validation, Writing – review & editing. Mingzhu Yin: Resources, Writing – review & editing. Lei Fu: Conceptualization, Supervision, Project administration, Funding acquisition, Writing – review & editing. Shifang Peng: Conceptualization, Supervision, Project administration, Funding acquisition, Writing – review & editing.
All authors gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
This research was funded by National Natural Science Foundation of China (No. 82570747) and (NO.82170640); Natural Science Foundation of Hunan Province (NO.2022JJ30954) and (NO.2025JJ50599).
Disclosure
The authors declare no conflicts of interest related to this publication.
References
1. Meyer NJ, Prescott HC. Sepsis and septic shock. N Engl J Med. 2024;391(22):2133–16. doi:10.1056/NEJMra2403213
2. Singer M, Deutschman CS, Seymour CW, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA. 2016;315(8):801–810. doi:10.1001/jama.2016.0287
3. Gray AP, Chung E, Hsu RL. Global, regional, and national sepsis incidence and mortality, 1990–2021: a systematic analysis. Lancet Glob Health. 2025;13(12):e2013–e2026. doi:10.1016/s2214-109x(25)00356-0
4. Rudd KE, Johnson SC, Agesa KM, et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the global burden of disease study. Lancet. 2020;395(10219):200–211. doi:10.1016/s0140-6736(19)32989-7
5. Prescott HC, Antonelli M, Alhazzani W, et al. Surviving sepsis campaign: international guidelines for management of sepsis and septic shock 2026. Intensive Care Med. 2026. doi:10.1007/s00134-026-08361-1
6. Cui L, Bao J, Yu C, et al. Development of a nomogram for predicting 90-day mortality in patients with sepsis-associated liver injury. Sci Rep. 2023;13(1):3662. doi:10.1038/s41598-023-30235-5
7. Lu Y, Shi Y, Wu Q, et al. An overview of drug delivery nanosystems for sepsis-related liver injury treatment. Int J Nanomed. 2023;18:765–779. doi:10.2147/ijn.S394802
8. Wang J, Tao X, Liu Z, et al. Noncoding RNAs in sepsis-associated acute liver injury: roles, mechanisms, and therapeutic applications. Pharmacol Res. 2025;212:107596. doi:10.1016/j.phrs.2025.107596
9. Moura FA, Siqueira A. Gut-liver axis in sepsis-associated liver injury: epidemiology, challenges and clinical practice. World J Gastroenterol. 2025;31(1):99987. doi:10.3748/wjg.v31.i1.99987
10. Schnabl B, Brenner DA. Interactions between the intestinal microbiome and liver diseases. Gastroenterology. 2014;146(6):1513–1524. doi:10.1053/j.gastro.2014.01.020
11. Protzer U, Maini MK, Knolle PA. Living in the liver: hepatic infections. Nat Rev Immunol. 2012;12(3):201–213. doi:10.1038/nri3169
12. Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14(10):996–1006. doi:10.1038/ni.2691
13. Heymann F, Tacke F. Immunology in the liver--from homeostasis to disease. Nat Rev Gastroenterol Hepatol. 2016;13(2):88–110. doi:10.1038/nrgastro.2015.200
14. Nesseler N, Launey Y, Aninat C, Morel F, Mallédant Y, Seguin P. Clinical review: the liver in sepsis. Crit Care. 2012;16(5):235. doi:10.1186/cc11381
15. Strnad P, Tacke F, Koch A, Trautwein C. Liver - guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol. 2017;14(1):55–66. doi:10.1038/nrgastro.2016.168
16. Sun X, Wu J, Liu L, et al. Transcriptional switch of hepatocytes initiates macrophage recruitment and T-cell suppression in endotoxemia. J Hepatol. 2022;77(2):436–452. doi:10.1016/j.jhep.2022.02.028
17. Araujo David B, Andreata F, Blériot C, Ginhoux F, Kubes P, Iannacone M. Kupffer cells in liver homeostasis and disease: from immune sentinels to metabolic gatekeepers. Nat Rev Immunol. 2026;8. doi:10.1038/s41577-026-01288-0
18. Wan Y, Wang T, Wang K, Huo Q, Cheng X. Ailanthone ameliorates CCl(4)-induced liver fibrosis by targeting PKM2-mediated macrophage M1 polarization and glycolytic reprogramming. Int Immunopharmacol. 2026;179(179):116640. doi:10.1016/j.intimp.2026.116640
19. Jiang S, Meng J, Wang Q, Zou Y, Mou Y. Glucose modulates macrophage polarization via glycating adsorbed albumin on titanium. Mater Today Bio. 2026;38:103047. doi:10.1016/j.mtbio.2026.103047
20. Hao M, Xiang K, Zhang K, et al. Bee sting-shaped microneedles for accelerating Achilles tendinopathy healing via enhanced regulating macrophage polarization. J Nanobiotechnology. 2026;24(1). doi:10.1186/s12951-026-04364-8
21. Xu Q, Liu X, Heng H, et al. Cytokine-driven PANoptosis of alveolar macrophages mediated by STAT1 underlies acute lung injury in hypervirulent Klebsiella pneumoniae infection. mBio. 2026:e0395825. doi:10.1128/mbio.03958-25
22. Liu M, Han Y, Xie B, et al. Legumain restrains granuloma formation by inhibiting mTORC1/STAT1-mediated m1 macrophage polarization in sarcoidosis. Adv Sci. 2026:e20635. doi:10.1002/advs.202520635
23. Leentjens J, Kox M, van der Hoeven JG, Netea MG, Pickkers P. Immunotherapy for the adjunctive treatment of sepsis: from immunosuppression to immunostimulation. Time for a paradigm change? Am J Respir Crit Care Med. 2013;187(12):1287–1293. doi:10.1164/rccm.201301-0036CP
24. Carson WF, Cavassani KA, Dou Y, Kunkel SL. Epigenetic regulation of immune cell functions during post-septic immunosuppression. Epigenetics. 2011;6(3):273–283. doi:10.4161/epi.6.3.14017
25. Etoh K, Araki H, Koga T, et al. Citrate metabolism controls the senescent microenvironment via the remodeling of pro-inflammatory enhancers. Cell Rep. 2024;43(8):114496. doi:10.1016/j.celrep.2024.114496
26. Filippakopoulos P, Picaud S, Mangos M, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149(1):214–231. doi:10.1016/j.cell.2012.02.013
27. Li W, Shen X, Feng S, et al. BRD4 inhibition by JQ1 protects against LPS-induced cardiac dysfunction by inhibiting activation of NLRP3 inflammasomes. Mol Biol Rep. 2022;49(9):8197–8207. doi:10.1007/s11033-022-07377-2
28. Wasiak S, Gilham D, Daze E, et al. Epigenetic modulation by apabetalone counters cytokine-driven acute phase response in vitro, in mice and in patients with cardiovascular disease. Cardiovasc Ther. 2020;2020:9397109. doi:10.1155/2020/9397109
29. Jahagirdar R, Attwell S, Marusic S, et al. RVX-297, a BET bromodomain inhibitor, has therapeutic effects in preclinical models of acute inflammation and autoimmune disease. Mol Pharmacol. 2017;92(6):694–706. doi:10.1124/mol.117.110379
30. Seal J, Lamotte Y, Donche F, et al. Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of I-BET151 (GSK1210151A). Bioorg Med Chem Lett. 2012;22(8):2968–2972. doi:10.1016/j.bmcl.2012.02.041
31. Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468(7327):1119–1123. doi:10.1038/nature09589
32. Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Ann Oncol. 2017;28(8):1776–1787. doi:10.1093/annonc/mdx157
33. Yin M, Guo Y, Hu R, et al. Potent BRD4 inhibitor suppresses cancer cell-macrophage interaction. Nat Commun. 2020;11(1):1833. doi:10.1038/s41467-020-15290-0
34. Ma L, Zhang X, Xu S, et al. NHWD-870 suppresses tumor proliferation via the BRD4/STRADA/CCND1 axis in small cell lung cancer. Lung Cancer. 2025;210:108841. doi:10.1016/j.lungcan.2025.108841
35. Wang W, Li X, Hu R, et al. BET inhibitor in combination with BCG vaccine enhances antitumor efficacy and orchestrates T cell reprogramming for melanoma. Cell Rep Med. 2025;6(3):101995. doi:10.1016/j.xcrm.2025.101995
36. Hu R, Hou H, Li Y, et al. Combined BET and MEK Inhibition synergistically suppresses melanoma by targeting YAP1. Theranostics. 2024;14(2):593–607. doi:10.7150/thno.85437
37. Ma Y, Lai P, Sha Z, et al. TME-responsive nanocomposite hydrogel with targeted capacity for enhanced synergistic chemoimmunotherapy of MYC-amplified osteosarcoma. Bioact Mater. 2025;47:83–99. doi:10.1016/j.bioactmat.2025.01.006
38. Jin L, Dong L, Pei S, et al. A BET inhibitor, NHWD-870, can downregulate dendritic cells maturation via the IRF7-mediated signaling pathway to ameliorate imiquimod-induced psoriasis-like murine skin inflammation. Eur J Pharmacol. 2024;968:176382. doi:10.1016/j.ejphar.2024.176382
39. Ye Z, Li X, Zhang M, et al. Complete response to BET inhibitor in primary pulmonary NUT carcinoma with single-cell sequencing-based analysis: a case report. JTO Clin Res Rep. 2025;6(10):100885. doi:10.1016/j.jtocrr.2025.100885
40. Cao J, Liu Y, Lu C. Primary thyroid nuclear protein in testis carcinoma: a case report and literature review. Gland Surg. 2024;13(6):1116–1125. doi:10.21037/gs-24-77
41. Chen L, Zhong X, Cao W, et al. JQ1 as a BRD4 inhibitor blocks inflammatory pyroptosis-related acute colon injury induced by LPS. Front Immunol. 2021;12:609319. doi:10.3389/fimmu.2021.609319
42. Qian Z, Shuying W, Ranran D. Inhibitory effects of JQ1 on listeria monocytogenes-induced acute liver injury by blocking BRD4/RIPK1 axis. Biomed Pharmacother. 2020;125:109818. doi:10.1016/j.biopha.2020.109818
43. Wu C, Cheng D, Peng Y, et al. Hepatic BRD4 is upregulated in liver fibrosis of various etiologies and positively correlated to fibrotic severity. Front Med. 2021;8:683506. doi:10.3389/fmed.2021.683506
44. Antcliffe DB, Burrell A, Boyle AJ, Gordon AC, McAuley DF, Silversides J. Sepsis subphenotypes, theragnostics and personalized sepsis care. Intensive Care Med. 2025;51(4):756–768. doi:10.1007/s00134-025-07873-6
45. Durand F, Kellum JA, Nadim MK. Fluid resuscitation in patients with cirrhosis and sepsis: a multidisciplinary perspective. J Hepatol. 2023;79(1):240–246. doi:10.1016/j.jhep.2023.02.024
46. Matuszewska M, Cieślik M, Sulejczak D, Wilkaniec A, Czapski GA. BET protein inhibitor JQ1 reduces inflammationand hippocampal amyloid-β level without altering Tau phosphorylation in LPS-challenged adult wild-type mice. Brain Res. 2026;1884:150318. doi:10.1016/j.brainres.2026.150318
47. Li S, Liu DX. Interaction of coronavirus E protein with BRD2 plays important regulatory roles in viral replication and induction of pro-inflammatory response. J Virol. 2026;100(3):e0220125. doi:10.1128/jvi.02201-25
48. Kumar R, Neidemire-Colley L, Garfinkle EA, et al. BET inhibition blunts antibody production and macrophage-mediated fibrosis to restore lung function in murine cGVHD. Blood. 2026. doi:10.1182/blood.2025031983
49. Jiang F, Li H, Sun Q, et al. Dual / bifunctional targeting of bromodomain and extra-terminal (BET) proteins: expanding the paradigm of epigenetic drug discovery. Eur J Med Chem. 2026;305:118524. doi:10.1016/j.ejmech.2025.118524
50. Li X, Wang Z, Zhang T, et al. Aloperine mitigates cigarette smoke-induced inflammation and pyroptosis by inhibiting the BRD4/NLRP3/GSDMD pathway in chronic obstructive pulmonary disease. Toxicol Appl Pharmacol. 2026;507:117704. doi:10.1016/j.taap.2025.117704
51. Weng Y, Zhang Y, Dong W, et al. Acetylation reader BRD4-driven TXNIP transcription enhances NLRP3 inflammasome activation in PCOS. Cell Mol Life Sci. 2025;82(1):427. doi:10.1007/s00018-025-05925-0
52. Wang L, Gong W. Bromodomain protein 4 inhibitor JQ-1 alleviates hepatic ischemia-reperfusion injury by blocking the NLRP3/caspase-1 pathway. World J Emerg Med. 2025;16(4):340–347. doi:10.5847/wjem.j.1920-8642.2025.081
53. Chen F, Li S, Liu M, et al. Targeting BRD4 mitigates hepatocellular lipotoxicity by suppressing the NLRP3 inflammasome activation and GSDMD-mediated hepatocyte pyroptosis. Cell Mol Life Sci. 2024;81(1):295. doi:10.1007/s00018-024-05328-7
54. Liao M, Long K, Dong L, et al. Targeting CLEC4E in immunosuppressive tumour-associated macrophages via BET inhibition. Clin Transl Med. 2025;15(10):e70505. doi:10.1002/ctm2.70505
55. Vijayasimha M, Srikanth M, Jayarajan D, Babu L, Malinidevi M. Aminotransferase-to-platelet ratio index for sepsis-associated liver dysfunction: promising triage signal but phenotype confounding and implementation calibration must be intensive care unit ready. Indian J Crit Care Med. 2026;30(2):177–178. doi:10.5005/jp-journals-10071-25122
56. Fei M, Xu Y, Jin P, Wang Y, Zhou M. Mitochondrial dysfunction in sepsis-induced liver injury: from pathophysiology to preclinical therapeutic targets. J Transl Med. 2025;23(1):1339. doi:10.1186/s12967-025-07369-3
57. Wang N, Wu R, Tang D, Kang R. The BET family in immunity and disease. Signal Transduct Target Ther. 2021;6(1):23. doi:10.1038/s41392-020-00384-4
58. Wang A, Kang X, Wang J, Zhang S. IFIH1/IRF1/STAT1 promotes sepsis associated inflammatory lung injury via activating macrophage M1 polarization. Int Immunopharmacol. 2023;114:109478. doi:10.1016/j.intimp.2022.109478
59. Zhu B, Sun C, Luo D, et al. Coptisine improves liver inflammation in sepsis by regulating STAT1/IRF1/GPX4 signaling-mediated kupffer cells ferroptosis. Phytother Res. 2025;39(9):4308–4326. doi:10.1002/ptr.70063
60. Xiao X, Li JX, Li HH, Teng F. ACE2 alleviates sepsis-induced cardiomyopathy through inhibiting M1 macrophage via NF-κB/STAT1 signals. Cell Biol Toxicol. 2024;40(1):82. doi:10.1007/s10565-024-09923-z
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.