Back to Archived Journals » Orphan Drugs: Research and Reviews » Volume 4
New treatments in development for Pseudomonas aeruginosa infections in the lungs of individuals with cystic fibrosis
Authors Akhand S, Pettit R, Gardner T, Anderson G
Received 13 July 2014
Accepted for publication 25 August 2014
Published 24 October 2014 Volume 2014:4 Pages 71—81
DOI https://doi.org/10.2147/ODRR.S50014
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Lise Aagaard
Saeed S Akhand,1 Rebecca S Pettit,2 Tonia E Gardner,3 Gregory G Anderson1
1Department of Biology, Indiana University–Purdue University Indianapolis, 2Riley Hospital for Children, Indiana University Health, 3Department of Pediatrics, Indiana University School of Medicine, Indianapolis, IN, USA
Abstract: In 1989, with the discovery of the genetic mutation causing cystic fibrosis (CF), a cure for this terrible disease seemed just around the corner. Though significant progress has been made to improve the quality of life of CF patients, a remedy for CF is yet to be found. Of great concern, such bacteria as Pseudomonas aeruginosa establish chronic infections that are extremely difficult to eradicate. P. aeruginosa thrives in the CF lung environment, where it protects itself from the host inflammatory defense and contributes immensely to the lung inflammation and tissue destruction seen with the disease. Tackling and eradicating P. aeruginosa infection in the CF lung holds the key to lessen the suffering of these individuals. Unfortunately, current antibiotic regimens are insufficient to eliminate P. aeruginosa from the CF lung. In this review, we address recent drugs that are currently under development to eradicate P. aeruginosa infection in the airways of patients with CF. We discuss the P. aeruginosa infection from the perspective of CF, the shortcomings of current drugs, and the upcoming drugs that are under clinical trials and development. These new drugs and therapies hold much promise for treating recalcitrant P. aeruginosa infections and improving the lives of individuals with CF.
Keywords: cystic fibrosis, orphan drugs, Pseudomonas aeruginosa infection
Introduction
Cystic fibrosis (CF) is an inherited autosomal genetic disorder caused by point mutations in a gene that encodes for the CF transmembrane-conductance regulator (CFTR) protein. Although this lethal genetic disease is most prevalent in people of Central and Northern European ancestry, it also occurs in many other demographic regions. Approximately 30,000 individuals have CF in the US; most are diagnosed by age 2 years.1 While technically considered a rare disease, approximately one in 3,500 and one in 3,700 children are born with CF in the EU and the US, respectively.2,3
Of the more than 1,900 mutations in the CFTR gene that can lead to CF, F508del (deletion of the three-nucleotide codon for phenylalanine at position 508) is by far the most common.4–6 The primary role of CFTR is to function as a chloride channel.7 However, this membrane protein also participates in other regulatory functions like inhibition of sodium transport through the epithelial sodium channel, regulation of adenosine triphosphate channels, regulation of intracellular vesicle transport, regulation of the outwardly rectifying chloride channel, and inhibition of endogenous calcium-activated chloride channels.8–13 These actions lead to excess sodium and water reabsorption, in turn causing dehydration of the airway-surface liquid.14–16 Consequently, airway-cell ciliary beat is impaired and mucociliary clearance becomes severely limited, resulting in excessive amounts of thick and tenacious mucous secretions that obstruct organ ducts and passages.17 Such conditions promote bacterial infection, and some studies suggest that altered chloride concentration in the periciliary layer disrupts the function of important innate antimicrobial molecules.18–20 Moreover, the lack of CFTR function leads to impaired HCO3- transport, resulting in pH falling in airway-surface liquid, which in turn is associated with reduced bacterial killing, as shown in the pig model.21 CFTR also influences immune-cell activity, and in CF, macrophages and neutrophils have been found to be defective in clearing bacterial infection.22 Furthermore, a decrease in T-helper 17 response in CF may correlate with enhanced infection.23 These reactions predispose CF patients to chronic microbial infection. In infancy and early childhood, lungs of CF patients are often colonized or infected with organisms like Staphylococcus aureus and Haemophilus influenzae that may damage the epithelial surfaces.24 However, Pseudomonas aeruginosa eventually becomes the dominant pathogen, and it can persist in the CF lung for decades, despite aggressive antimicrobial therapy.
P. aeruginosa is a Gram-negative, pigment-forming, unipolar motile bacterium. It can adapt to a wide range of conditions, and is thus able to cause numerous different types of infections in a variety of host species.25 This microbe possesses a number of classical virulence factors, including pili, flagella, lipopolysaccharides, and various secreted toxins and degradative enzymes. Additionally, as will be discussed further, P. aeruginosa utilizes an interbacterial communication system called quorum sensing to coordinate expression of these virulence factors. However, the most important trait that makes P. aeruginosa a formidable one for CF is its ability to develop into specialized antibiotic-resistant and immune-resistant aggregates called biofilms.24,26
Biofilms are bacterial communities that are irreversibly attached to living or abiotic surfaces and embedded in an extracellular matrix of exopolysaccharides, protein, and deoxyribonucleic acid (DNA).27,28 The hypersecretion of viscous mucus in the CF airways provides a low oxygen environment, and the presence of DNA and actin promotes P. aeruginosa to switch to the biofilm mode of growth in the CF lung.27,29 Often, biofilm-state P. aeruginosa overproduces the polysaccharide alginate, leading to a “mucoid” phenotype of the bacteria.30 Part of the reason that current antibiotic regimens are not capable of eradicating P. aeruginosa infection in the CF lung is the formation of these biofilms.31,32 Indeed, biofilms are intrinsically recalcitrant, because the matrix inhibits antimicrobial diffusion, the microorganisms grow slowly as a result of limiting nutrients, and specific resistance factors are expressed in biofilm-state bacteria.33
Therefore, treatment strategies for CF lung infections are lacking. However, much progress has been made in recent years toward developing newer and better antimicrobial therapies. In this review, we summarize the current status in the development of drugs against P. aeruginosa infection in the CF airway.
Shortcomings of current therapies
The common classes of antipseudomonal antibiotics currently used in the US include aminoglycosides (ie, amikacin, gentamicin, tobramycin); quinolones (ie, ciprofloxacin, levofloxacin); polymyxins, such as colistin; and β-lactams, including penicillins (ie, piperacillin, ticarcillin), cephalosporins (ie, ceftazidime, cefepime), monobactams (ie, aztreonam), and carbapenems (ie, imipenem, meropenem). Oral azithromycin is also recommended for use in CF patients with chronic P. aeruginosa infection, and it has been shown to exert immunomodulatory activity and inhibit P. aeruginosa signaling.34,35 At first glance, this may seem like a large selection to choose from, but options can quickly become limited, due to a number of bacterial, patient, and logistical issues. In addition to the intrinsic resistance of biofilms, P. aeruginosa possesses an extraordinary ability to acquire additional resistance mechanisms when under selective pressure, through horizontal gene transfer and spontaneous mutation. Moreover, the versatility and large size of the P. aeruginosa genome allows for several resistance mechanisms to be present simultaneously and for cross-resistance to develop. Common resistance mechanisms to aminoglycosides include enzyme modification, such as plasmid-mediated acetylases and adenylases that decrease the binding affinity of the antibiotic for its target in the cell,36 and diminished outer-membrane permeability leading to reduced aminoglycoside accumulation.37 Quinolone resistance occurs by point mutations in the antibiotic’s target enzyme, DNA gyrase, and also by active efflux of the antibiotic out of the cell.38
Numerous β-lactamases representing all four classes (A–D) are produced by P. aeruginosa and contribute significantly to widespread β-lactam resistance patterns. Extended-spectrum β-lactamases of classes A, B, and D have also been found, and confer resistance to many extended-spectrum cephalosporins and aztreonam.39 Constitutive low-level expression of the AmpC β-lactamase (a class C enzyme) leads to penicillin resistance and first- and second-generation cephalosporin resistance, and is not inhibited by β-lactam inhibitors, such as clavulanic acid and sulbactam.39 Third-generation cephalosporin resistance can be seen when this enzyme is overproduced in response to the presence of inducing β-lactams.40
Additionally, the bacterium’s low outer-membrane permeability is an important innate resistance mechanism to several antibiotic classes. Active efflux is another important mechanism of antibiotic resistance seen in P. aeruginosa. This mechanism contributes to resistance against β-lactams, quinolones, and somewhat to aminoglycosides by removing the antibiotic from the cellular space.41
Most alarmingly, any and all of the aforementioned resistance mechanisms may be present simultaneously, leaving clinicians with few or no viable antibiotic options to treat infections. To make matters worse, constant immune and antibiotic pressures lead to the development of small-colony variants – persister cells – that have higher minimum inhibitory concentrations (MICs) and are more likely to survive antibiotic treatment.42
Furthermore, extended use of antibiotics leads to deleterious effects on the patient. Ototoxicity and nephrotoxicity are well-known toxicities associated with the use of aminoglycoside antibiotics, and incidence increases with repeated exposure.36 Renal wasting of electrolytes, such as potassium and magnesium, can also occur if proximal tubule damage has occurred. The most frequently described toxicities associated with colistin use have been nephrotoxicity and neurotoxicity (paresthesias, dizziness, weakness, vertigo, and ataxia) that are dose-dependent and usually reversible with cessation of therapy. Incidence of nephrotoxicity has been estimated at ~20%, though some newer reports have shown lower estimates in the 10%–15% range.43 Neurotoxicity incidence is estimated at 7%; however, the incidence of paresthesias in patients with CF has been reported up to nearly 30%.44 Gastrointestinal side effects, such as nausea, vomiting, and diarrhea, and central nervous system side effects (headaches, dizziness) are the most common complaints seen with fluoroquinolone use. Arthropathy and tendinopathy have also been reported. Headaches, rash, nausea/vomiting, and anemia have been reported with carbapenems. Multiple studies have also found a dose-related increase in the risk for seizure events with imipenem/cilistatin,45 though similar studies have not found the same results with meropenem use.46
Newer/developing therapies
Novel combinations of existing drugs
Inhaled tobramycin (inhaled powder and nebulized solution), inhaled aztreonam, and inhaled colistin are the currently available and utilized treatments for chronic P. aeruginosa infections in CF patients. However, all of these medications have limitations, and many questions about their use remain, perhaps most notably whether cycling two antibiotics (ie, 28 days inhaled tobramycin and 28 days inhaled aztreonam alternating) is better than cycling one inhaled antibiotic (ie, 28 days inhaled tobramycin followed by 28 days of no inhaled antibiotic alternating) (Table 1). There is currently a clinical study investigating this question. The AZLI CAT study is randomizing patients to receive alternating 28-day cycles of inhaled aztreonam or placebo and inhaled tobramycin nebulized solution for three cycles.47 The end points for this study include exacerbation rate and changes in pulmonary function tests. The results of this study may help optimize chronic P. aeruginosa treatment.
 | Table 1 Drugs under clinical trials |
For years, CF patients have been treated with 28-day cycles of inhaled antibiotics. This treatment regimen has been ingrained into practice. However, a new study of inhaled tobramycin powder is challenging this paradigm. The study is comparing two different dosing strategies of inhaled tobramycin powder: once-daily continuous dosing versus twice-daily dosing in cycles of 28 days on and 28 days off.48 The primary end point of the study will be changes in forced expiratory volume in 1 second (FEV1) from baseline, with secondary end points including exacerbation rate. Once daily continuous tobramycin inhalation might be easier for patients to remember, and thus lead to an increased adherence to their inhaled antibiotic regimen.
Both of these studies are investigating the use of existing antipseudomonal antibiotics in new ways to improve treatment and increase compliance. The results of these two trials may impact the treatment of chronic P. aeruginosa in CF patients.
Another question that remains in CF care is about the optimal treatment for the initial eradication of P. aeruginosa. It is clear that an eradication attempt after the first positive P. aeruginosa culture is beneficial to keep the lower airways clear of this microorganism.49 However, the optimal antibiotic eradication strategy has not been established. Many different regimens, including inhaled tobramycin alone, inhaled tobramycin + ciprofloxacin oral, and inhaled colistin + ciprofloxacin oral, have been used for P. aeruginosa eradication.49–51 Most of these regimens have similar eradication success rates. Other strategies for eradication use intravenous (IV) antibiotics instead of inhaled and/or oral antibiotics. More study is needed to determine the optimal treatment regimen to eradicate P. aeruginosa before it colonizes the patient’s lungs.
KB001
KB001 is a pegylated, recombinant anti-Pseudomonas-PcrV antibody Fab’ fragment currently being studied in CF patients (Table 1). KB001, a monoclonal antibody, works by inhibiting the function of the P. aeruginosa type III secretion system (T3SS). T3SS is a protein complex that allows P. aeruginosa to inject specific exotoxins into host cells.52 The T3SS exotoxins add to P. aeruginosa virulence and the inflammatory response to this pathogen in the airways. KB001 blocks T3SS by specifically targeting the PcrV protein, which is a component of the T3SS tip. Importantly, it does not cross-react with mammalian cells.52
A randomized, double blind, placebo-controlled, single-dose study was conducted to determine the pharmacokinetics and safety of KB001 in CF patients >12 years old with FEV1 >40%. Patients on inhaled antibiotics were allowed to participate in the study, but they were not allowed to take inhaled antibiotics during the 28 days of the trial.52 There were two study cohorts. The first cohort was randomized to receive 3 mg/kg KB001, and in the second cohort the KB001 dose was 10 mg/kg. KB001 was administered by a 1-hour IV infusion. Only two patients had infusion-related reactions, which were mild (flushing, pruritus, tachycardia, rash, nausea, chest tightness, and elevated blood pressure) and resolved when the infusion was discontinued.52 Overall, KB001 was well tolerated at both doses. The pharmacokinetic analysis found a half-life of 11.9 days after a single infusion. There was a trend for inflammatory biomarkers to decrease after KB001 administration and increase in the placebo group, although the only statistically significant difference was a decrease in neutrophil count in the 10 mg/kg KB001 group compared to placebo (P<0.05).52 A repeat-dosing Phase II study in CF patients is ongoing to assess the durability of the anti-inflammatory effect of KB001 in CF patients with P. aeruginosa infections.53 These studies indicate that KB001 is a promising nonantibiotic treatment for P. aeruginosa in CF patients.
Arikace (liposomal amikacin)
Liposomal amikacin is the aminoglycoside antibiotic amikacin encapsulated in liposomal carriers (Table 1).54 The liposomal carriers are made of saturated lipid dipalmitoyl phosphatidylcholine and cholesterol.54 Importantly, these liposomal carriers are natural constituents of lung surfactant and are biocompatible.54 Liposomal amikacin is designed to be administered via inhalation and have a slow-release mechanism. Specifically, the release of the amikacin is triggered by rhamnolipids, which are surfactant molecules secreted by biofilm-localized P. aeruginosa.54 This mechanism allows liposomal amikacin to have superior in vivo efficacy compared to nonencased inhaled amikacin, and gives it the ability to penetrate biofilms.54
These factors indicate that liposomal amikacin has the characteristics necessary to treat chronic mucoid P. aeruginosa in CF patients. The pharmacokinetics and pharmacodynamics of liposomal amikacin 500 mg inhaled daily for 14 days were evaluated in 24 CF patients.55 This study was not able to correlate sputum concentration (the amount of drug in the sputum) to pulmonary function test changes, but did show a statistically significant increase in percentage predicted FEV1 (FEV1 % predicted) and forced expiratory flow of between 25% and 75% of forced vital capacity compared to baseline at both 7 and 14 days (7.49, P<0.001; 4.38, P=0.03; 0.49, P<0.001; 0.42, P=0.002; respectively).55
Another two Phase II studies of liposomal amikacin in CF patients with P. aeruginosa infections were conducted. One Phase II study evaluated the safety and efficacy of nebulized liposomal amikacin at doses of 70 mg, 140 mg, 280 mg, and 560 mg once daily for 28 days versus placebo.56 The adverse events were similar between all groups; the most common adverse effect in all groups was patients developing an acute pulmonary exacerbation. There was more dysphonia reported in the high-dose liposomal amikacin group, which was not unexpected given amikacin’s adverse-effect profile. The liposomal amikacin 560 mg dose versus placebo showed a greater relative change in FEV1 at day 28 (0.081 L ±0.161 versus 0.011 L ±0.101, P=0.033) and at day 56 (0.093 L ±0.203 versus –0.03 L ±0.119, P=0.003).56 P. aeruginosa density in the sputum decreased in the liposomal amikacin group compared to placebo. The open-label extension over six cycles of liposomal amikacin showed that pulmonary function-test improvement and sputum P. aeruginosa-density changes could be maintained over longer treatment.
Following the two Phase II studies, Phase III studies were conducted in both Europe and the US. The European study was conducted in both Europe and Canada, and 302 subjects were randomized to receive liposomal amikacin 560 mg once daily via eFlow® nebulizer or inhaled tobramycin solution (TOBI®) 300 mg nebulized twice a day for three treatment cycles. About 80% of patients had received inhaled antibiotics in the prior 12 months, and 75% of the patients were rolled over from the open-label study.57 The study showed improvements in lung function with liposomal amikacin and a decrease in P. aeruginosa sputum density.57 The Phase III study in the US was delayed, due to a Food and Drug Administration (FDA) hold on the study following some data suggesting inhalation carcinogenicity in rats. Further studies were conducted in dogs to assess this risk, and the FDA lifted the hold on clinical trials with liposomal amikacin.58 The US Phase III study of inhaled liposomal amikacin (NCT 01316276) is ongoing.
Liposomal encased antibiotics are a promising treatment strategy for CF patients with P. aeruginosa infections that produce biofilms. The ability to target therapy to the sputum and allow release and penetration into the biofilms may be particularly useful in patients with multidrug-resistant P. aeruginosa. Although not currently on the market, liposomal amikacin will likely play an important role in the treatment of P. aeruginosa infections in patients that have failed other treatment options.
Gallium
Gallium is a group IIIA transition metal that can disrupt bacterial iron metabolism and has been shown to have antipseudomonal properties (Table 1).59,60 Gallium competes with iron that bacteria utilize, since gallium cannot undergo the redox cycle necessary for activity to some iron-centered enzymes.61 Gallium was found to inhibit P. aeruginosa growth, prevent biofilm formation, and have bactericidal activity in vitro.59 Even in the presence of CF sputum, gallium was effective at killing P. aeruginosa in the laboratory.62
Gallium is traditionally administered by IV, however it has been given in animal studies aerosolized. The inhaled administration of gallium resulted in higher local concentrations in the lungs than the IV formulation in rats.63,64 It has also been hypothesized that the combination of gallium with other, traditional antibiotics may increase the antibiotic’s activity against multidrug-resistant organisms. For instance, one in vitro study examined the coencapsulation of gallium with gentamicin in liposomes. This combination was more effective in vitro at eradicating antibiotic-resistant P. aeruginosa than gentamicin alone.65
Despite promising in vitro and animal data, there has been only one Phase I pharmacokinetic study using IV gallium nitrate in patients with CF. This study was an open-label, unblended, nonrandomized study that assessed the safety and pharmacokinetics of two different doses of gallium nitrate:100 mg/m2/day and 200 mg/m2/day.66 The 20 adult CF patients that completed the study were infected with P. aeruginosa and had a baseline FEV1 > 30%. No serious adverse events were reported; however, there were nine adverse events that were deemed study-related. The pharmacokinetic analysis showed two-phase elimination, sputum concentrations reached biofilm inhibitory and bactericidal activity on day 14 in 100% of the 100 mg/m2/day group and 78% of the 200 mg/m2/day group. FEV1 increased in both groups after a single infusion (day 14 +118 mL [95% confidence interval {CI} 41–196]), but the change was greater in the higher-dosing group (FEV1 increase at day 14 +169 mL [95% CI 8–260]).66 P. aeruginosa sputum density had high variability, but the mean change at day 14 was −6×106 colony forming units per gram (95% CI -117.3 to 105.7). This Phase I study showed promising results of IV gallium nitrate for clinical application in the treatment of P. aeruginosa in CF patients. More clinical trials need to be conducted to delineate the benefit of gallium therapy in CF patients. It may also prove beneficial as an adjunctive therapy with other antibiotics.
Anti-Pseudomonas immunoglobulin Y
Anti-Pseudomonas immunoglobulin Y (IgY) (Table 1) is produced from the eggs of chickens that are immunized with P. aeruginosa.67 Anti-Pseudomonas IgY molecules bind to P. aeruginosa flagellin, which may decrease the ability of P. aeruginosa to attach to epithelial cells and infect the lungs.68 This treatment has been studied to prevent infection and prolong time to reinfection with P. aeruginosa. Patients gargle the IgY solution in the evenings after food intake and teeth brushing for 2 minutes and then swallow the solution.69
An initial IgY feasibility study was conducted in 17 Swedish CF patients using 50 mg IgY daily.67 Patients with a positive culture for P. aeruginosa were treated with antipseudomonal antibiotics for eradication. The study found that IgY treatment prolonged the time between the first and second P. aeruginosa cultures, compared to a control group (P=0.015).67
Following the initial feasibility study, the Swedish Medical Products Agency gave permission for CF patients with intermittent P. aeruginosa infection to be treated with IgY.69 Patients on this open-label treatment were studied for the safety and effectiveness of IgY. The study included 17 CF patients using IgY, and a small group of patients were used as the control group. In the treatment group, patients had 2.3 cultures positive for P. aeruginosa per 100 treatment months compared to seven per 100 months in the control group (P=0.028).69 However, P. aeruginosa cultures were not counted after the patient developed chronic P. aeruginosa infection. Overall, IgY was well tolerated and had no impact on pulmonary function or body mass index.69 A case report of a patient with CF that had received 10 years of IgY treatment reported no adverse effects.69
These initial67–69 reports led to two ongoing studies assessing the efficacy of IgY to prevent initial or recurrent P. aeruginosa infections. The first study is a postmarketing study of the IgY gargle, assessing its ability to prevent initial P. aeruginosa infections.70 The second study is a Phase III study comparing 50 mg of IgY gargled nightly compared to placebo in CF patients after a successfully treated acute or intermittent P. aeruginosa infection.71 This double-blind placebo-controlled study will help to delineate the efficacy of IgY and its ability to prevent the recurrence of P. aeruginosa in the CF airway.
Inhaled levofloxacin
Levofloxacin is a fluoroquinolone antibiotic with activity against P. aeruginosa, and this drug is commonly used orally in patients with CF. Levofloxacin is currently available as an oral and IV antibiotic, and has been reformulated to an inhaled product for use with CF patients with chronic P. aeruginosa infections (Table 1). A pharmacokinetic study of inhaled levofloxacin (MP-376, Aeroquin™) in CF patients found that it was well tolerated and produced high sputum concentrations and low serum concentrations.72 Inhaled levofloxacin is formulated to be nebulized using a customized eFlow nebulizer (PARI Pharma, Munich, Germany) with an average nebulization time of 4–6 minutes.73
A placebo-controlled study was conducted using three different dosing strategies of inhaled levofloxacin 120 mg daily, 240 mg daily, and 240 mg twice a day.73 The study randomized 151 CF patients >16 years old with chronic P. aeruginosa infections. The study population had a significant history of inhaled antibiotic use, and was required to have received at least three courses of inhaled antibiotics within the past year. The results showed a decrease in sputum P. aeruginosa density in all levofloxacin groups, and the 240 mg twice a day group had a 0.96 log difference compared to placebo (P=0.001). There was an increase in FEV1 of 8.7% in the 240 mg twice a day group compared to placebo (P=0.003), and there was a reduction in need for other antipseudomonal antibiotics in the treatment groups.73 Overall, inhaled levofloxacin was well tolerated; however, patients complained about the taste of the medication. The most common side effects were cough, diarrhea, and headache.73 This Phase II study showed promising results in a previously heavily antibiotic-treated CF patient population. These results led to two Phase III studies of inhaled levofloxacin.
Phase III studies have been completed, but results were not available at the time of this review. One of the Phase III studies assessed the time to the next CF exacerbation in patients ≥12 years of age treated with inhaled levofloxacin 240 mg twice a day for 28 days compared to placebo.74 Like the Phase II study, the patient population had chronic P. aeruginosa infections and had previously received inhaled antibiotics. The second Phase III study compared inhaled levofloxacin to tobramycin-inhalation solution in 28-day cycled regimens in CF patients ≥12 years of age.75 The results of these two Phase III studies will help delineate the efficacy of inhaled levofloxacin in CF patients and help determine its role in the treatment of chronic P. aeruginosa in CF patients. The fast delivery of this drug makes it an appealing option for patients, and twice-daily dosing would be more convenient then inhaled aztreonam with dosing three times a day.
Inhaled ciprofloxacin
Like levofloxacin, ciprofloxacin is a fluoroquinolone antibiotic with activity against P. aeruginosa. Ciprofloxacin is currently available as an oral tablet and an IV solution. Ciprofloxacin has been formulated for inhalation into a dry-powder inhaler (Table 1).76 Inhaled ciprofloxacin is not only being studied in CF patients but also in patients with non-CF bronchiectasis. The dry-powder inhaler for ciprofloxacin is a capsule-based system, and each capsule contains 32.5 mg of active ciprofloxacin.76 In healthy volunteers, inhaled ciprofloxacin was well tolerated with low systemic absorption.77
A Phase I pharmacokinetic dose-escalation study was conducted in adult CF patients with chronic P. aeruginosa. Patients received 32.5 mg daily, 65 mg daily, 32.5 mg twice a day, or placebo for 7 days.76 Twenty-five patients were enrolled in the study, and overall the inhaled ciprofloxacin was well tolerated. The inhaled ciprofloxacin produced high levels in the sputum, with low systemic absorption.76
A randomized Phase II study of inhaled ciprofloxacin was conducted comparing 32.5 mg twice a day, 48.75 mg twice a day, or placebo for 28 days. This study included CF patients ≥12 years of age with FEV1 35%–80% and chronic P. aeruginosa colonization.78 Published preliminary data reported that the inhaled ciprofloxacin was well tolerated. Final data had not been published at the time of this review. Inhaled ciprofloxacin is early in the development process for CF, but may potentially treat chronic P. aeruginosa.
Ceftolozane/tazobactam
CF patients are commonly treated with IV antipseudomonal antibiotics during acute pulmonary exacerbations. There are a limited number of antibiotics with activity against P. aeruginosa, and there has been a lack of new antibiotics produced to target those resistant to traditional options. Ceftolozane/tazobactam is a new broad-spectrum cephalosporin/β-lactamase-inhibitor combination with activity against P. aeruginosa (Table 1).79 Ceftolozane has activity against highly resistant P. aeruginosa, including strains resistant to aminoglycosides, fluoroquinolones, and other β-lactams. The addition of tazobactam extends its spectrum to include many extended-spectrum β-lactamase-producing bacteria and also provides coverage for some anaerobes.79 Phase III clinical trials have been completed with ceftolozane/tazobactam for the treatment of complicated urinary tract infections and intra-abdominal infections. There is also a study evaluating its efficacy in the treatment of ventilator-associated bacterial pneumonia.79 Ceftolozane/tazobactam was well tolerated in clinical studies; most adverse effects were mild, including abdominal pain, infusion-site reactions, nausea, headache, paresthesia, somnolence, flushing, diarrhea, and constipation.79 Ceftolozane/tazobactam has not been studied in CF patients, but it is an exciting new option for the treatment of multidrug-resistant P. aeruginosa.
Biofilm screening
Biofilm formation is one of the most difficult-to-overcome factors associated with the treatment of P. aeruginosa infection. Therapeutically important antibiotics like tetracycline, fluoroquinolones, and aminoglycosides typically become less effective as P. aeruginosa cells adapt themselves to form biofilms in the CF lung environment.80–82 Moreover, it has been reported recently that at subinhibitory concentrations, some antibiotics in fact stimulate biofilm formation.80 Discovery of novel drugs that specifically inhibit or disrupt biofilms holds the key to minimize or eradicate chronic P. aeruginosa infection in CF patients.
Naturally occurring antimicrobial peptides (AMPs) from animals and plants have been found to exert some antibiofilm effects (Table 2). AMPs contribute significantly to host defense.83,84 Low occurrence of inducing resistance, broad activity spectrum, and rapid mechanism of action make AMPs attractive candidates for the development of novel antimicrobial drugs.85–87 In search of potential AMPs with antibiofilm activity, initial studies reported that the C-terminal domain of human cathelicidin (LL-37), pexiganan, and modified oligo-N-substituted glycines (peptoids) showed significant antibiofilm activity against P. aeruginosa at MICs that were eight to 30 times lower than the MICs of antibiotics needed for comparable results.85 Later, the engineered derivatives LL7-37 and LL7-31 were found to overcome some of the limitations of LL-37, such as cytotoxicity against eukaryotic cells, cost of synthesis, and sensitivity toward cell-secreted proteases.88 Additionally, bovine myeloid antimicrobial peptide (BMAP)-27, BMAP-28, and synthetic P19(9/B) effectively inhibited bacterial biofilm formation and destroyed preformed biofilm.89 Moreover, a human parotid secretory protein with an additional lysine residue, GL13K, was reported to be effective in reducing biofilms of P. aeruginosa.90
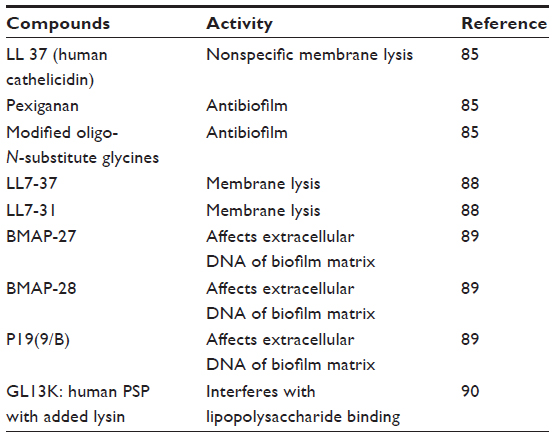 | Table 2 Antimicrobial peptides |
Quorum sensing is cell-to-cell signaling used by bacteria for communal behavior via global gene regulation. P. aeruginosa utilizes quorum-signaling systems to regulate various virulence factors, including biofilm formation.91 Therefore, anti-quorum-sensing molecules have been researched by many research groups as a means of inhibiting and disrupting P. aeruginosa-biofilm formation (Table 3). Brominated furanone derivatives have been reported to be inhibitory against P. aeruginosa quorum-signal activation, leading to biofilm reduction.92 Compounds found in various herb extracts (eg, methyl eugenol, andrographolide, diterpenoid lactone), synthesized 14-lipolyl andrographolide, and clove oil have also been found to cause damage to P. aeruginosa biofilms by affecting quorum-sensing signaling.93–96 Similarly, biofilm inhibition was also found with concurrent use of tobramycin and garlic extract.97 In addition, various synthetic inhibitors of quorum signaling, such as meta-bromo-thiolactone and ambroxol, have been proposed to be effective against P. aeruginosa biofilm.98,99
 | Table 3 Anti-quorum-sensing lead molecules |
Currently, a dry-powder formulation of alginate oligosaccharide (OligoG) is in clinical Phase II trials to assess the efficacy and safety in CF adults.100 Recently, OligoG has been found to inhibit bacterial biofilm and reduce sputum viscosity ex vivo.101,102
Administration of DNase I has also been shown to reduce P. aeruginosa-biofilm formation.103 This treatment appears to destroy the biofilm matrix, which contains extracellular DNA required for biofilm stability.
Additionally, photodynamic chemotherapy has also been promoted as a promising avenue of therapy for P. aeruginosa biofilms. In one study, toluidine blue and meso-tetra-N-methyl-4-pyridyl-prophine-tetratosylate used in combination with red light demonstrated P. aeruginosa-biofilm inhibition.104 Even though light and drug-molecule diffusion through mucus and the thick biofilm matrix may be a drawback, photodynamic chemotherapy could lead to unique solutions to the problem of biofilm antibiotic resistance.
Future directions
As we look at the present state of antimicrobial therapy development, with an eye toward the future, it is evident that there are many new promising avenues for the treatment of chronic P. aeruginosa-biofilm infections. For existing antibiotics, novel formulations and unique combinations with other antibiotics and with less traditional antimicrobial compounds may yield new life for these old drugs. Furthermore, the discovery and use of exciting new drugs will provide more and perhaps better options for treating biofilms. Therefore, the future of antimicrobial therapy for chronic P. aeruginosa infections in CF is bright.
Acknowledgment
This work was supported by funds from Indiana University–Purdue University Indianapolis to G Anderson.
Disclosure
The authors report no conflicts of interest in this work.
References
Lipuma JJ. The changing microbial epidemiology in cystic fibrosis. Clin Microbiol Rev. 2010;23(2):299–323. | |
Grosse SD, Boyle CA, Botkin JR, et al. Newborn screening for cystic fibrosis: evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recomm Rep. 2004;53(RR-13):1–36. | |
Southern KW, Munck A, Pollitt R, et al. A survey of newborn screening for cystic fibrosis in Europe. J Cyst Fibros. 2007;6(1):57–65. | |
Kumar V, Abbas AK, Fausto N, Mitchell RS. Robbins Basic Pathology. 8th ed. Philadelphia: Elsevier; 2007. | |
Bobadilla JL, Macek M Jr, Fine JP, Farrell PM. Cystic fibrosis: a worldwide analysis of CFTR mutations – correlation with incidence data and application to screening. Hum Mutat. 2002;19(6):575–606. | |
Roomans GM. Pharmacological treatment of the basic defect in cystic fibrosis. Cell Biol Int. Epub May 8, 2014. | |
Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiol Rev. 1999;79(Suppl 1):S23–S45. | |
Mehta A. CFTR: more than just a chloride channel. Pediatr Pulmonol. 2005;39(4):292–298. | |
Quinton PM. Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet. 2008;372(9636):415–417. | |
Reisin IL, Prat AG, Abraham EH, et al. The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. J Biol Chem. 1994;269(32):20584–20591. | |
Schwiebert EM, Egan ME, Hwang TH, et al. CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell. 1995;81(7):1063–1073. | |
Stutts MJ, Canessa CM, Olsen JC, et al. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269(5225):847–850. | |
Vankeerberghen A, Cuppens H, Cassiman JJ. The cystic fibrosis transmembrane conductance regulator: an intriguing protein with pleiotropic functions. J Cyst Fibros. 2002;1(1):13–29. | |
Matsui H, Grubb BR, Tarran R, et al. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95(7):1005–1015. | |
Boucher RC. Airway surface dehydration in cystic fibrosis: pathogenesis and therapy. Annu Rev Med. 2007;58:157–170. | |
Matsui H, Wagner VE, Hill DB, et al. A physical linkage between cystic fibrosis airway surface dehydration and Pseudomonas aeruginosa biofilms. Proc Natl Acad Sci U S A. 2006;103(48):18131–18136. | |
Lethem MI, James SL, Marriott C. The role of mucous glycoproteins in the rheologic properties of cystic fibrosis sputum. Am Rev Respir Dis. 1990;142(5):1053–1058. | |
Smith JJ, Travis SM, Greenberg EP, Welsh MJ. Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell. 1996;85(2):229–236. | |
Zabner J, Smith JJ, Karp PH, Widdicombe JH, Welsh MJ. Loss of CFTR chloride channels alters salt absorption by cystic fibrosis airway epithelia in vitro. Mol Cell. 1998;2(3):397–403. | |
Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM. Human β-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell. 1997;88(4):553–560. | |
Pezzulo AA, Tang XX, Hoegger MJ, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature. 2012;487(7405):109–113. | |
Ratner D, Mueller C. Immune responses in cystic fibrosis: are they intrinsically defective? Am J Respir Cell Mol Biol. 2012;46(6):715–722. | |
Bayes HK, Bicknell S, MacGregor G, Evans TJ. T helper cell subsets specific for Pseudomonas aeruginosa in healthy individuals and patients with cystic fibrosis. PLoS One. 2014;9(2):e90263. | |
Lyczak JB, Cannon CL, Pier GB. Lung infections associated with cystic fibrosis. Clin Microbiol Rev. 2002;15(2):194–222. | |
Lyczak JB, Cannon CL, Pier GB. Establishment of Pseudomonas aeruginosa infection: lessons from a versatile opportunist. Microbes Infect. 2000;2(9):1051–1060. | |
Harrison F. Microbial ecology of the cystic fibrosis lung. Microbiology. 2007;153(Pt 4):917–923. | |
Donlan RM, Costerton JW. Biofilms: survival mechanisms of clinically relevant microorganisms. Clin Microbiol Rev. 2002;15(2):167–193. | |
Costerton JW, Lewandowski Z, Caldwell DE, Korber DR, Lappin-Scott HM. Microbial biofilms. Annu Rev Microbiol. 1995;49:711–745. | |
Worlitzsch D, Tarran R, Ulrich M, et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest. 2002;109(3):317–325. | |
Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003;168(8):918–951. | |
Costerton JW, Stewart PS, Greenberg EP. Bacterial biofilms: a common cause of persistent infections. Science. 1999;284(5418):1318–1322. | |
Romling U, Fiedler B, Bosshammer J, et al. Epidemiology of chronic Pseudomonas aeruginosa infections in cystic fibrosis. J Infect Dis. 1994;170(6):1616–1621. | |
Høiby N, Bjarnsholt T, Givskov M, Molin S, Ciofu O. Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents. 2010;35(4):322–332. | |
Mogayzel PJ Jr, Naureckas ET, Robinson KA, et al. Cystic fibrosis pulmonary guidelines. Chronic medications for maintenance of lung health. Am J Respir Crit Care Med. 2013;187(7):680–689. | |
Høiby N. Recent advances in the treatment of Pseudomonas aeruginosa infections in cystic fibrosis. BMC Med. 2011;9:32. | |
Vakulenko SB, Mobashery S. Versatility of aminoglycosides and prospects for their future. Clin Microbiol Rev. 2003;16(3):430–450. | |
Bryan LE, Haraphongse R, Van den Elzen HM. Gentamicin resistance in clinical-isolates of Pseudomonas aeruginosa associated with diminished gentamicin accumulation and no detectable enzymatic modification. J Antibiot (Tokyo). 1976;29(7):743–753. | |
Hooper DC. Emerging mechanisms of fluoroquinolone resistance. Emerg Infect Dis. 2001;7(2):337–341. | |
Nordmann P, Guibert M. Extended-spectrum beta-lactamases in Pseudomonas aeruginosa. J Antimicrob Chemother. 1998;42(2):128–131. | |
Chen HY, Yuan M, Livermore DM. Mechanisms of resistance to beta-lactam antibiotics amongst Pseudomonas aeruginosa isolates collected in the UK in 1993. J Med Microbiol. 1995;43(4):300–309. | |
Strateva T, Yordanov D. Pseudomonas aeruginosa – a phenomenon of bacterial resistance. J Med Microbiol. 2009;58(Pt 9):1133–1148. | |
Starkey M, Hickman JH, Ma L, et al. Pseudomonas aeruginosa Rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol. 2009;191(11):3492–3503. | |
Yahav D, Farbman L, Leibovici L, Paul M. Colistin: new lessons on an old antibiotic. Clin Microbiol Infect. 2012;18(1):18–29. | |
Bosso JA, Liptak CA, Seilheimer DK, Harrison GM. Toxicity of colistin in cystic fibrosis patients. DICP. 1991;25(11):1168–1170. | |
Brown RB, Sands M, Morris AB. Seizure propensity with imipenem. Arch Intern Med. 1990;150(7):1551. | |
Linden P. Safety profile of meropenem: an updated review of over 6,000 patients treated with meropenem. Drug Saf. 2007;30(8):657–668. | |
Gilead Sciences. Phase 3 study of aztreonam for inhalation solution (AZLI) in a continuous alternating therapy regimen for the treatment of chronic Pseudomonas aeruginosa infection in patients with CF (AZLI CAT). Available from: http://clinicaltrials.gov/show/NCT01641822. NLM identifier:NCT01641822. Accessed August 21, 2014. | |
Novartis Pharmaceuticals. Tobramycin inhalation powder (TIP) administered once daily continuously versus TIP administered BID in 28 day on/28 day off cycles. Available from: http://clinicaltrials.gov/show/NCT02015663. NLM identifier:NCT02015663. Accessed August 21, 2014. | |
Döring G, Flume P, Heijerman H, Elborn JS Treatment of lung infection in patients with cystic fibrosis: current and future strategies. J Cyst Fibros. 2012;11(6):461–479. | |
Proesmans M, Vermeulen F, Boulanger L, Verhaegen J, De Boeck K. Comparison of two treatment regimens for eradication of Pseudomonas aeruginosa infection in children with cystic fibrosis. J Cyst Fibros. 2013;12(1):29–34. | |
Taccetti G, Bianchini E, Cariani L, et al. Early antibiotic treatment for Pseudomonas aeruginosa eradication in patients with cystic fibrosis: a randomised multicentre study comparing two different protocols. Thorax. 2012;67(10):853–859. | |
Milla CE, Chmiel JF, Accurso FJ, et al. Anti-PcrV antibody in cystic fibrosis: a novel approach targeting Pseudomonas aeruginosa airway infection. Pediatr Pulmonol. 2014;49(7):650–658. | |
KaloBios Pharmaceuticals. Study to evaluate the effect of KB001-A on time-to-need for antibiotic treatment. Available from: http://clinicaltrials.gov/show/NCT01695343. NLM identifier: NCT01695343. Accessed August 21, 2014. | |
Meers P, Neville M, Malinin V, et al. Biofilm penetration, triggered release and in vivo activity of inhaled liposomal amikacin in chronic Pseudomonas aeruginosa lung infections. J Antimicrob Chemother. 2008;61(4):859–868. | |
Okusanya OO, Bhavnani SM, Hammel J, et al. Pharmacokinetic and pharmacodynamic evaluation of liposomal amikacin for inhalation in cystic fibrosis patients with chronic pseudomonal infection. Antimicrob Agents Chemother. 2009;53(9):3847–3854. | |
Clancy JP, Dupont L, Konstan MW, et al. Phase II studies of nebulised Arikace in CF patients with Pseudomonas aeruginosa infection. Thorax. 2013;68(9):818–825. | |
Bilton D, Pressler T, Isabelle F, et al. Phase 3 efficacy and safety data from randomized, multicenter study of liposomal amikacin for inhalation (Arikace) compared with TOBI® in cystic fibrosis patients with chronic infection due to Pseudomonas aeruginosa [abstract]. Pediatr Pulmonol. 2013;48(S36):290. | |
CenterWatch. FDA lifts hold on Insmed’s Arikace. 2012. Available from: http://www.centerwatch.com/news-online/article/2785/fda-lifts-hold-on-insmeds-arikace#sthash.0ZMXjApN.dpbs. Accessed August 20, 2014. | |
Kaneko Y, Thoendel M, Olakanmi O, Britigan BE, Singh PK. The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J Clin Invest. 2007;117(4):877–888. | |
García-Contreras R, Peréz-Eretza B, Lira-Silva E, et al. Gallium induces the production of virulence factors in Pseudomonas aeruginosa. Pathog Dis. 2014;70(1):95–98. | |
Switzer B, Goss C, Abdalla M, Singh P, Britigan B. Gallium has potential as a new therapeutic agent for M. abscessus infections [abstract]. Pediatr Pulmonol. 2013;48(S36):334–335. | |
Kakiuchi T, Pham B, Lechuga-Ballesteros D, Patzer EJ. Bactericidal activity of anti-infective panaecin™ (gallium nitrate) on antibiotic resistant P. aeruginosa from clinical isolates and in sputum from CF patients [abstract]. Pediatr Pulmonol. 2008;43(S31):318. | |
Troung-Le V, Martin R, Ohtake S, Yee L, Patzer EJ, Lechuga-Ballesteros D. Intratracheal delivery of gallium results in higher local lung concentration and longer lung residence time compared to intravenous delivery in the rat [abstract]. Pediatr Pulmonol. 2009;44(S32):307–308. | |
Yee L, Adamcakova-Dodd A, Binuraj A, et al. Pharmacokinetics, acute inhalation toxicity, and bacterial activities of anti-infective Panaecin™ (formulated gallium nitrate) [abstract]. Pediatr Pulmonol. 2010;45(S33):351. | |
Halwani M, Yebio B, Suntres ZE, Alipour M, Azghani AO, Omri A. Co-encapsulation of gallium with gentamicin in liposomes enhances antimicrobial activity of gentamicin against Pseudomonas aeruginosa. J Antimicrob Chemother. 2008;62(6):1291–1297. | |
Goss CH, Hornick DB, Aitken ML, et al. Phase 1 pharmacokinetic and safety study of intravenous Ganite™ (gallium nitrate) in CF [abstract]. Pediatr Pulmonol. 2012;47(S35):303. | |
Kollberg H, Carlander D, Olesen H, Wejåker PE, Johannesson M, Larsson A. Oral administration of specific yolk antibodies (IgY) may prevent Pseudomonas aeruginosa infections in patients with cystic fibrosis: a phase I feasibility study. Pediatr Pulmonol. 2003;35(6):433–440. | |
Nilsson E, Amini A, Wretlind B, Larsson A. Pseudomonas aeruginosa infections are prevented in cystic fibrosis patients by avian antibodies binding Pseudomonas aeruginosa flagellin. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;856(1–2):75–80. | |
Nilsson E, Kollberg H, Johannesson M, Wejåker PE, Carlander D, Larsson A. More than 10 years’ continuous oral treatment with specific immunoglobulin Y for the prevention of Pseudomonas aeruginosa infections: a case report. J Med Food. 2007;10(2):375–378. | |
Immunsystem. Anti-Pseudomonas IgY to prevent infections in cystic fibrosis (PseudIgY). Available from: http://clinicaltrials.gov/show/NCT00633191. NLM identifier:NCT00633191. Accessed August 21, 2014. | |
Mukoviszidose Institut. Efficacy study of IgY (antibody against Pseudomonas) in cystic fibrosis patients (PsAer-IgY). Available from: http://clinicaltrials.gov/show/NCT01455675. NLM identifier:NCT01455675. Accessed August 21, 2014. | |
Geller DE, Flume PA, Griffith DC, et al. Pharmacokinetics and safety of MP-376 (levofloxacin inhalation solution) in cystic fibrosis subjects. Antimicrob Agents Chemother. 2011;55(6):2636–2640. | |
Geller DE, Flume PA, Staab D, Fischer R, Loutit JS, Conrad DJ. Levofloxacin inhalation solution (MP-376) in patients with cystic fibrosis with Pseudomonas aeruginosa. Am J Respir Crit Care Med. 2011;183(11):1510–1516. | |
Mpex Pharmaceuticals. MP-376 (Aeroquin™, levofloxacin for inhalation) in patients with cystic fibrosis. Available from: http://clinicaltrials.gov/show/NCT01180634. NLM identifier:NCT01180634. Accessed August 21, 2014. | |
Mpex Pharmaceuticals. Trial of aeroquin versus tobramycin inhalation solution (TIS) in cystic fibrosis (CF) patients. Available from: http://clinicaltrials.gov/show/NCT01270347. NLM identifier:NCT01270347. Accessed September 10, 2014. | |
Stass H, Weimann B, Nagelschmitz J, Rolinck-Werninghaus C, Staab D. Tolerability and pharmacokinetic properties of ciprofloxacin dry powder for inhalation in patients with cystic fibrosis: a phase I, randomized, dose-escalation study. Clin Ther. 2013;35(10):1571–1581. | |
Stass H, Baumann-Noss S, Delesen H, et al. Pharmacokinetics of ciprofloxacin PulmoSphere inhalational powder. J Cyst Fibros. 2008;7:S26. | |
Dorkin H, Criollo M, Reimnitz P, Alder J, Hampel B. Randomized, double-blind, placebo-controlled, multicenter study to evaluate the safety and efficacy of inhaled ciprofloxacin compared with placebo in patients with cystic fibrosis – a phase IIB study of ciprofloxacin dry powder for inhalation (DPI) [abstract]. Pediatr Pulmonol. 2011;46(S43):296. | |
Zhanel GG, Chung P, Adam H, et al. Ceftolozane/tazobactam: a novel cephalosporin/beta-lactamase inhibitor combination with activity against multidrug-resistant Gram-negative bacilli. Drugs. 2014;74(1):31–51. | |
Hoffman LR, D’Argenio DA, MacCoss MJ, Zhang Z, Jones RA, Miller SI. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature. 2005;436(7054):1171–1175. | |
Linares JF, Gustafsson I, Baquero F, Martinez JL. Antibiotics as intermicrobial signaling agents instead of weapons. Proc Natl Acad Sci U S A. 2006;103(51):19484–19489. | |
Singh PK, Schaefer AL, Parsek MR, Moninger TO, Welsh MJ, Greenberg EP. Quorum-sensing signals indicate that cystic fibrosis lungs are infected with bacterial biofilms. Nature. 2000;407(6805):762–764. | |
Lai Y, Gallo RL. AMPed up immunity: how antimicrobial peptides have multiple roles in immune defense. Trends Immunol. 2009;30(3):131–141. | |
Zanetti M. Cathelicidins, multifunctional peptides of the innate immunity. J Leukoc Biol. 2004;75(1):39–48. | |
Kapoor R, Wadman MW, Dohm MT, Czyzewski AM, Spormann AM, Barron AE. Antimicrobial peptoids are effective against Pseudomonas aeruginosa biofilms. Antimicrob Agents Chemother. 2011;55(6):3054–3057. | |
Pompilio A, Scocchi M, Pomponio S, et al. Antibacterial and anti-biofilm effects of cathelicidin peptides against pathogens isolated from cystic fibrosis patients. Peptides. 2011;32(9):1807–1814. | |
Hancock RE, Sahl HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol. 2006;24(12):1551–1557. | |
Nagant C, Pitts B, Nazmi K, et al. Identification of peptides derived from the human antimicrobial peptide LL-37 active against biofilms formed by Pseudomonas aeruginosa using a library of truncated fragments. Antimicrob Agents Chemother. 2012;56(11):5698–5708. | |
Pompilio A, Crocetta V, Scocchi M, et al. Potential novel therapeutic strategies in cystic fibrosis: antimicrobial and anti-biofilm activity of natural and designed alpha-helical peptides against Staphylococcus aureus, Pseudomonas aeruginosa, and Stenotrophomonas maltophilia. BMC Microbiol. 2012;12:145. | |
Hirt H, Gorr SU. Antimicrobial peptide GL13K is effective in reducing biofilms of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2013;57(10):4903–4910. | |
de Kievit TR. Quorum sensing in Pseudomonas aeruginosa biofilms. Environ Microbiol. 2009;11(2):279–288. | |
Liu HB, Lee JH, Kim JS, Park S. Inhibitors of the Pseudomonas aeruginosa quorum-sensing regulator, QscR. Biotechnol Bioeng. 2010;106(1):119–126. | |
Zeng X, Liu X, Bian J, et al. Synergistic effect of 14-alpha-lipoyl andrographolide and various antibiotics on the formation of biofilms and production of exopolysaccharide and pyocyanin by Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2011; 55(6):3015–3017. | |
Packiavathy IA, Agilandeswari P, Musthafa KS, Pandian SK, Ravi AV. Antibiofilm and quorum sensing inhibitory potential of Cuminum cyminum and its secondary metabolite methyl eugenol against Gram negative bacterial pathogens. Food Res Int. 2012;45(1):85–92. | |
Husain FM, Ahmad I, Asif M, Tahseen Q. Influence of clove oil on certain quorum-sensing-regulated functions and biofilm of Pseudomonas aeruginosa and Aeromonas hydrophila. J Biosci. 2013;38(5):835–844. | |
Murugan K, Sangeetha S, Kalyanasundaram VB, Al-Sohaibani S. In vitro and in silico screening for Andrographis paniculata quorum sensing mimics: new therapeutic leads for cystic fibrosis Pseudomonas aeruginosa biofilms. Plant Omics. 2013;6(5):340–346. | |
Bjarnsholt T, Jensen PØ, Rasmussen TB, et al. Garlic blocks quorum sensing and promotes rapid clearing of pulmonary Pseudomonas aeruginosa infections. Microbiology. 2005;151(Pt 12):3873–3880. | |
O’Loughlin CT, Miller LC, Siryaporn A, Drescher K, Semmelhack MF, Bassler BL. A quorum-sensing inhibitor blocks Pseudomonas aeruginosa virulence and biofilm formation. Proc Natl Acad Sci U S A. 2013;110(44):17981–17986. | |
Lu Q, Yu J, Yang X, et al. Ambroxol interferes with Pseudomonas aeruginosa quorum sensing. Int J Antimicrob Agents. 2010;36(3):211–215. | |
AlgiPharma A phase IIB study of OligoG in subjects with cystic fibrosis (SMR-2984). Available from: http://clinicaltrials.gov/ct2/show/NCT02157922. NLM identifier:NCT02157922. Accessed August 21, 2014. | |
Powell LC, Sowedan A, Khan S, et al. The effect of alginate oligosaccharides on the mechanical properties of Gram-negative biofilms. Biofouling. 2013;29(4):413–421. | |
Powell LC, Pritchard MF, Emanuel C, et al. A nanoscale characterization of the interaction of a novel alginate oligomer with the cell surface and motility of Pseudomonas aeruginosa. Am J Respir Cell Mol Biol. 2014;50(3):483–492. | |
Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. Extracellular DNA required for bacterial biofilm formation. Science. 2002;295(5559):1487. | |
Donnelly RF, McCarron PA, Cassidy CM, Elborn JS, Tunney MM. Delivery of photosensitisers and light through mucus: investigations into the potential use of photodynamic therapy for treatment of Pseudomonas aeruginosa cystic fibrosis pulmonary infection. J Control Release. 2007;117(2):217–226. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.