")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 15
New therapies for the treatment of heart failure: a summary of recent accomplishments
Authors Machaj F , Dembowska E , Rosik J , Szostak B , Mazurek-Mochol M , Pawlik A
Received 6 July 2018
Accepted for publication 19 November 2018
Published 22 January 2019 Volume 2019:15 Pages 147—155
DOI https://doi.org/10.2147/TCRM.S179302
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Garry Walsh
Filip Machaj,1 Elzbieta Dembowska,2 Jakub Rosik,1 Bartosz Szostak,1 Małgorzata Mazurek-Mochol,2 Andrzej Pawlik1
1Department of Physiology, Pomeranian Medical University, Szczecin, Poland; 2Department of Periodontology, Pomeranian Medical University, Szczecin, Poland
Abstract: Despite continuous efforts to prevent cardiovascular diseases (CVDs), heart failure prevails as the number one cause of death in developed countries. To properly treat CVDs, scientists had to take a closer look at the factors that contribute to their pathogenesis and either modernize current pharmaceuticals or develop brand new treatments. Enhancement of current drugs, such as tolvaptan and omecamtiv mecarbil, sheds new light on already-known therapies. Tolvaptan, a vasopressin antagonist, could be adopted in heart failure therapy as it reduces pre- and afterload by decreasing systolic blood pressure and blood volume. Omecamtiv mecarbil, which is a myosin binding peptide, could aid cardiac contractility. The next generation vasodilators, serelaxin and ularitide, are based on naturally occurring peptides and they reduce peripheral vascular resistance and increase the cardiac index. In combination with their anti-inflammatory properties, they could turn out to be extremely potent drugs for heart failure treatment. Cardiotrophin has exceeded many researchers’ expectations, as evidence suggests that it could cause sarcomere hypertrophy without excessive proliferation of connective tissue. Rapid progress in gene therapy has caused it to finally be considered as one of the viable options for the treatment of CVDs. This novel therapeutic approach could restore stable heart function either by restoring depleted membrane proteins or by balancing the intracellular calcium concentration. Although it has been set back by problems concerning its long-term effects, it is still highly likely to succeed.
Keywords: heart failure, therapy, cardiovascular diseases
Introduction
Cardiovascular disease (CVD) is currently one of the most common causes of mortality and morbidity in developed countries.1 In the last 20 years, strong emphasis was placed on improving survival and quality of life in patients with heart failure (HF); however, despite these great efforts, the HF 1-year mortality rate has only slightly declined and the 5-year mortality rate has not declined at all over the last 10 years.2
The current goals of HF research include the short-term improvement of clinical status and quality of life as well as the long-term targets of reducing all-cause readmission and, most importantly, reducing mortality. Thus, the emphasis is on identifying drugs with detrimental long-term effects.3
The standard therapy for HF involves the use of inotropic agents, vasodilators, and loop diuretics. These drugs, in combination with angiotensin-converting-enzyme inhibitors and beta-blockers, are effective forms of evidence-based therapies. The existing medical therapies for HF have brought about moderate success (52.6% of patients die within 5 years);2 however, there is still an unmet need for new pharmaceuticals that could be beneficial in HF treatment.4
This article aims to summarize the clinical progress of new therapeutic agents that could become standard HF therapies in the future (Table 1).
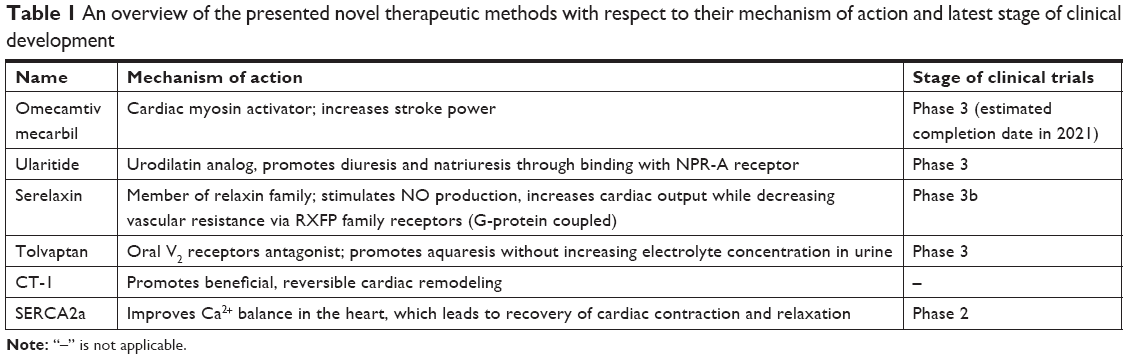 | Table 1 An overview of the presented novel therapeutic methods with respect to their mechanism of action and latest stage of clinical development |
Omecamtiv mecarbil
Treatment guidelines state that positive inotropic agents may be a therapeutic option in some specific types of HF, such as in patients with a low ejection volume, cardiogenic shock or low tissue perfusion. However, administering inotropic agents has no positive effects on mortality or hospitalization time, and these substances also have proarrhythmic effects.5 The use of standard inotropic agents increases cAMP levels in cardiomyocytes through various mechanisms. They produce a higher calcium concentration in the cytoplasm, which leads to better contractility; however, this is also accompanied by a higher oxygen use, which could have negative effects on patients’ health.6–8 These disadvantages of inotropic agents mean that there is a strong incentive to develop new and safer substances with similar pharmacological effects. One such substance could be the specific cardiac myosin activator, omecamtiv mecarbil.6–10
Omecamtiv mecarbil binds to the catalytic domain of myosin with high affinity and shifts the equilibrium of ATP hydrolysis toward ADP-P during the stroke. The change of equilibrium increases the number of myosin heads ready to bind to actin filaments. This results in a more powerful stroke without increasing the calcium level or oxygen use in cardiomyocytes (Figure 1).6,7,9
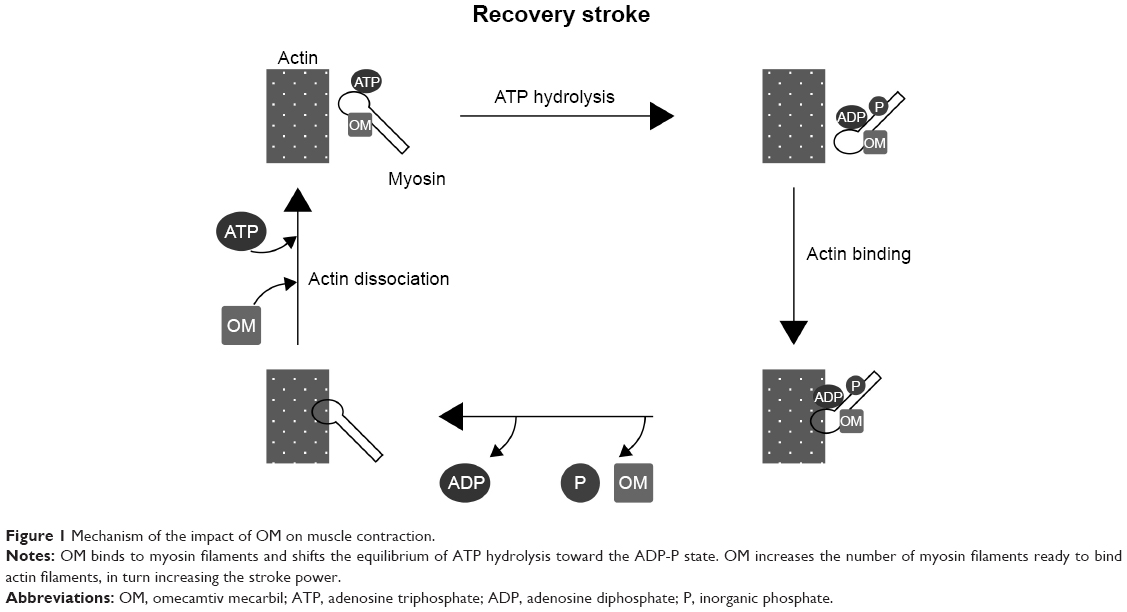 | Figure 1 Mechanism of the impact of OM on muscle contraction. |
The Acute Treatment with Omecamtiv Mecarbil to Increase Contractility in Acute Heart Failure (ATOMIC-HF) study was a phase 2 prospective trial that compared a placebo and omecamtiv mecarbil in patients with acute HF. The results showed that use of the cardiac myosin activator as a treatment did not match the predicted dyspnea relief, except in the higher administration group; however, omecamtiv mecarbil increased systolic ejection time (SET) and was well tolerated.7,8,11
The Chronic Oral Study of Myosin Activation to Increase Contractility in Heart Failure (COSMIC-HF) study showed better results for the treatment of chronic HF with omecamtiv mecarbil. This was a phase 2 trial that was designed to show the pharmacokinetics and optimal oral dose of omecamtiv mecarbil in the treatment of chronic HF. Changes in SET, stroke volume, end-diastolic diameter and N-terminal prohormone of brain natriuretic peptide (NT-proBNP) levels were also examined.7,11 Patients received a placebo or different dose of omecamtiv mecarbil. The results showed that the therapy was safe and well tolerated. It also led to an increased SET and stroke volume but decreased the heart rate, NT-proBNP level, and left ventricular volume.7 These results were very promising, therefore there are huge expectations for phase 3 trials in a larger group of patients.7,10,11
The Multicenter Study to Assess the Efficacy and Safety of Omecamtiv Mecarbil on Mortality and Morbidity in Subjects With Chronic Heart Failure With Reduced Ejection Fraction (GALACTIC-HF) study is a phase 3 clinical trial that is estimated to conclude in 2021. The study will include 8,000 patients with HF and its main outcome is to measure the time of the first HF event or CV death during omecamtiv mecarbil therapy.9
Ularitide
The natriuretic peptide hormone family includes ANP, BNP, CNP, DNP and urodilatin. There are three main natriuretic peptide receptors (NPRs): NPR-A, NPR-B, and NPR-C.12 Elevated levels of natriuretic peptides (BNP >35 pg/mL or NT-proBNP >125 pg/mL) are indicative of HF.3 Downregulation or desensitization of NPRs leads to beneficial effects in HF treatment, which makes them a target for pharmacological therapy.13
Ularitide is a synthetic analog of urodilatin, which is a kidney peptide hormone secreted by the distal tubule, collecting duct cells in response to increased pressure. It binds to NPR-A and results in increased diuresis and natriuresis (Figure 2).14 Other pharmacological effects of ularitide include vasodilation and inhibition of the renin-angiotensin-aldosterone system.15,16 Clinically, it demonstrates prolonged activity in comparison with atrial peptides.17
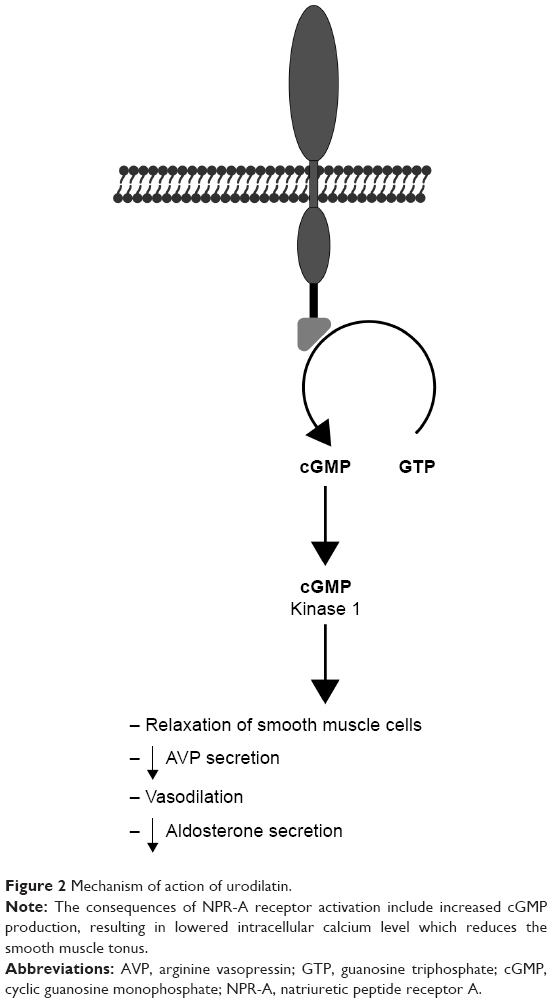 | Figure 2 Mechanism of action of urodilatin. |
Initial phase 1 studies found that ularitide decreases pulmonary capillary wedge pressure (PCWP) in healthy men18 and decreases systemic vascular resistance and PCWP and increases cardiac index and stroke volume in patients with acute HF (AHF).19 These preliminary studies were followed by the phase 2 SIRIUS I study, which explored the potential clinical value of ularitide. Patients subjected to ularitide treatment showed improved PCWP, reduced NT-proBNP, and improved dyspnea without renal function effects.20
SIRIUS II was a randomized controlled trial in 221 patients which further defined the hemodynamic effects of ularitide infusion. Its secondary endpoints included PCWP changes and patient-assessed dyspnea improvements. All ularitide groups showed significant reductions in PCWP and dyspnea, and the higher-dose groups also showed reductions in systemic vascular resistance and increased cardiac index at 6 hours. As in the SIRIUS I study, a dose-dependent reduction in systolic blood pressure (SBP) was noted in some patients.21
TRUE-AHF was a randomized double-blind parallel-group placebo-controlled trial in 2,157 patients. The goal of the study was to evaluate the effect of ularitide on CV mortality and 48-hour clinical status. Secondary endpoints included, among others, the length of stay, events of worsening HF, and proBNP changes.22 The drug produced satisfactory hemodynamic effects such as reduced SBP and decreased NT-proBNP levels; however, it did not significantly impact any of the primary or secondary endpoints. Death from CV causes occurred in 236 patients in the ularitide group and 225 patients in the placebo group.23 Therefore the findings of this study indicate that ularitide therapy can produce beneficial hemodynamic effects and reduce cardiac wall stress; however, it does not reduce myocardial injury or affect disease progression as indicated by the absence of change in the risk of CV death.24
Serelaxin
Relaxin is a member of the relaxin family, which includes peptides that are structurally similar to insulin. It was first extracted from the corpus luteum of pigs and has since been shown to play a role in modulating CV changes in pregnancy. Four relaxin receptors have been identified to date (RXFP1–4), all of which are G protein–coupled receptors. Their presence in the heart, lung, kidney, and blood cells is particularly important in CV therapy. Through stimulation of nitric oxide and VEGF production and inhibition of vasoconstrictors, relaxin can increase plasma volume and cardiac output and decrease blood pressure and vascular resistance (Figure 3). These results from animal models make relaxin a potential candidate for HF treatment.4,25
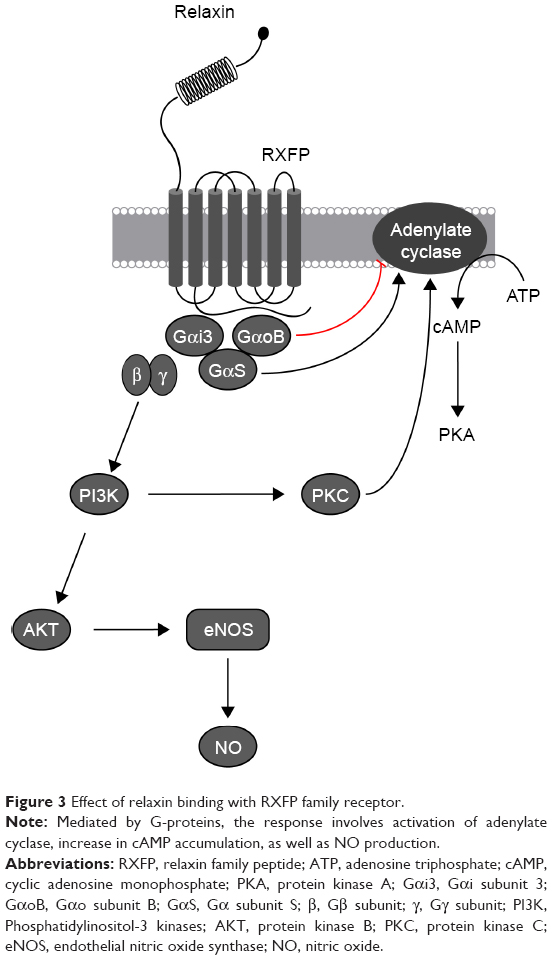 | Figure 3 Effect of relaxin binding with RXFP family receptor. |
Serelaxin is a synthetic recombinant of relaxin that displays identical hemodynamic properties. Initial investigation of relaxin in humans began with phase 1 trials, which examined the drug’s safety and tolerability, whereas the subsequent phase 2 trials focused on its hemodynamic effect and clinical efficacy. The primary phase 1 trials assessed if serelaxin was capable of increasing renal plasma flow and natriuresis,26 reducing PCWP, improving the cardiac index, and reducing vascular resistance.27 These preliminary studies were followed by a dose-finding pilot trial called pre-RELAX-AHF, which was a placebo-controlled, parallel-group phase 2 study that enrolled 234 patients with AHF. The results from this study revealed that a 30 μg/kg/day serelaxin infusion produced the best dyspnea improvement with a reduction in CV death or readmission due to heart or renal failure.28
These promising results initiated a phase 3 trial (RELAX-AHF) that was completed in 2012. The study enrolled 1,161 patients hospitalized for AHF, all of whom suffered from dyspnea, congestion, and elevated BNP levels. Each dose produced a clinical benefit; however, a 48-hour infusion of serelaxin at a dose of 30 μg/kg/day presented the greatest improvement in HF symptoms with sparse adverse effects. Investigators found that there was a great reduction in patient-assessed dyspnea over 5 days. The 60-day survival, death, or readmission rate did not significantly differ in serelaxin and placebo groups; however, it reduced HF worsening by 47% by day 5 and both all-cause and CV 180-day mortality by 37%. However, hypotensive episodes were noted in the serelaxin group.29 In light of the results of the RELAX-AHF trial, two global phase 3 trials were designed: RELAX-AHF-2 and RELAX-AHF-ASIA.
RELAX-AHF-ASIA was a randomized, double-blind, placebo-controlled phase 3 trial that aimed to evaluate the effects of serelaxin in Asian patients, unlike previous trials, which targeted primarily Caucasian patients. The study aimed to recruit 1,520 patients with AHF and assess the symptom relief and clinical outcomes with the endpoint being treatment success, treatment failure, or no change in status. The secondary endpoints included HF worsening by day 5 and deaths by day 180.30
RELAX-AHF-2 aimed to confirm the beneficial effects of serelaxin on 180-day CV death and worsening HF by day 5, with secondary endpoints including 180-day all-cause mortality, CV death or rehospitalization, and length of hospitalization. Patients were randomized to receive 48-hour serelaxin infusions (30 μg/kg/day) or a placebo in addition to their standard therapy.31 However, RELAX-AHF-2 did not meet any of its endpoints. There was no difference in CV mortality at 180 days and the reduction of worsening HF was not statistically significant. Serelaxin also failed to meet its secondary endpoints (ie, all-cause 180-day mortality, length of hospitalization, and rehospitalization due to heart or renal failure). The results of the study indicate that serelaxin is a safe but inefficacious drug (ie, it does not improve the outcomes compared with the placebo). These results were contrary to the findings of the RELAX-AHF trial, where a hemodynamic improvement was accompanied by a reduction in mortality.32
Tolvaptan
One consequence of HF is fluid retention, which leads to edemas and increased pulmonary congestion. The main causes of fluid retention are sodium retention and neurohumoral abnormalities.33,34 Arginine vasopressin (AVP) levels increase during HF, and higher levels of AVP are characteristic of the advanced stage of this disease. The main role is played by the nonosmotic regulatory pathway. Reduced cardiac output and decreased blood pressure lead to activation of baroreceptors and the renin-angiotensin-aldosterone system, which leads to increased AVP release from the pituitary gland.35
Increased vasopressin levels and the use of loop diuretics can cause hyponatremia.34 Loop diuretics are commonly used in HF treatment;33,36,37 however, up to 30% of patients are resistant to this type of therapy. Other side effects of loop diuretic therapy include electrolyte imbalance or hypotension.34,37,38
Tolvaptan is an oral vasopressin type 2 receptor antagonist that affects collecting ducts and increases aquaresis. Urine volume is increased but there is no change in the excretion of electrolytes.33–35,39 Studies show that tolvaptan increases serum sodium levels in patients with hyponatremia34,35,37,39 and has a lower tendency for worsening renal function than standard diuretics.36,38
Many studies assessed the beneficial effects of tolvaptan in the treatment of HF. For example, the Efficacy of Vasopressin Antagonism in Heart Failure: Outcome Study with Tolvaptan (EVEREST) trial checked the long- and short-term safety and efficacy of tolvaptan administration in addition to standard HF treatment. This was a double-blind, placebo-controlled test in 1,567 patients with HF. Patients were asked to complete the Kansas City Cardiomyopathy Questionnaire to assess their health-related quality of life (HRQOL) at 1 week after discharge. The results showed dyspnea relief in most patients; however, there was no improvement in the HRQOL score. There was also no association with mortality or dyspnea as assessed by a physician.40
The Qualification of Efficacy and Safety in the Study of Tolvaptan in Cardiac Oedema (QUEST) study was a prospective observational study. Patients were treated with tolvaptan (administered daily) for a standard observation time (eg, 14 days). The trial assessed HF symptoms, such as lower limb edema, dyspnea, pulmonary congestion, jugular venous distention, and hepatomegaly, during the treatment. Body weight and urine volume were also evaluated. All data were recorded from baseline to the end of treatment at day 14. The results showed a shrinking prevalence of these symptoms, a reduction in body weight, and an increase in diuresis; however, some patients developed hypernatremia with higher doses of tolvaptan.37
CT-1
CT-1 is a novel drug that is based on gp130, which forms part of the receptors for cytokines including IL-6, IL-11, and CT-1. These cytokines and their receptors are commonly associated with inflammation.
CT-1 is often connected with pathological hypertrophy of the heart,41 and elevated CT-1 plasma levels are associated with myocardial fibrosis.42 This substance is believed to play a key role in the stimulation of fibroblasts. Induction of cardiomyocyte hypertrophy and collagen synthesis is also connected with higher CT-1 levels. Furthermore, there is also a positive correlation between CT-1 plasma levels and heart valve disease.42 According to recent research, CT-1 is important for physiological hypertrophy of the heart, eg, research conducted on cardiomyocytes showed an instant modification in cell function and structure and a significant change in the length-to-width ratio. This change was different from those caused by pathological hypertrophy agents such as α-adrenergic agonists.43 Importantly, CT-1 also slows down the pathological hypertrophy induced by the aforementioned agents. The curbing of this unfavorable process is also observed in people who practice regular aerobic exercise. Therefore it is possible that hypertrophy is also curbed naturally (after exercise) by CT-1.43
CT-1 not only influences single cardiomyocytes but it also affects the whole cardiac muscle. These changes are reminiscent of those observed in sportsmen (eg, heart hypertrophy). Cardiac wall thickening occurred without changes in the internal diameter of the ventricle.43 The changes brought about by isoproterenol or phenylephrine are not as beneficial as those caused by CT-1, and isoproterenol and phenylephrine produce a negative change in the left-to-right ventricle internal diameter ratio.
Another difference between physiological and pathological hypertrophy is reversibility. Only changes caused by CT-1 therapy are fully reversible, which resembles physiological hypertrophy.
The molecular process responsible for the difference between CT-1 and phenylephrine activity is probably associated with the STAT1:STAT3 balance.44 STAT3 is anti-inflammatory (probably activated by CT-1), while STAT1 is proinflammatory (probably activated by α-adrenergic agonists). Caspases are also important for CT-1 activity, and knockout of CASP-3 or CASP-9 stops the positive effect of CT-1 stimulation.43 This suggests that beneficial hypertrophy depends on gp130, CASP-3, CASP-9, and STAT3 domination over STAT1. Another effect of CT-1 stimulation (probably via STAT3)45 is angiogenesis, which is vital for providing energy for sarcomeres (oxygen consumption after CT-1 therapy is increased).43 CK2, which is a caspase inhibitor, is activated by CT-1 and is the reason why CT-1-induced caspase activation is restricted and does not cause pathological changes.43
Another research group has shown a positive influence of CT-1 on human health.46 CT-1 depletion in a murine model can produce insulin resistance and lead to many subsequent negative consequences; however, CT-1 delivery can prevent insulin resistance. CT-1 also affects the regulation of food intake and leads to a noticeable reduction in consumption.
A separate study also showed the importance of CT-1 as a CV insufficiency therapy. A drug that could simultaneously promote beneficial heart remodeling, impede pathological changes, and fight against obesity (induced by increased food intake and insulin resistance) would solve the two main problems in those suffering from CV insufficiency.
Gene therapy
Gene therapy is believed to be a great hope for rare congenital diseases; however, many scientists think this is an unreal expectation due to its lack of spectacular success. Cardiology could also benefit from gene therapy. Supplementation of genes for molecules such as SERCA2a, SUMO1 (S100A1, a calcium sensor), and PP1 inhibitor-1 (an upstream regulator of SERCA2a via inhibition of PP 1) could be the future of modern cardiology. Other ways to treat CV insufficiency include upregulating the β-adrenergic receptor (by stopping GRK2 from using the genes of its inhibitors)47 and overexpressing microRNAs or other types of RNA to regulate gene expression and prevent hypertrophy and fibrosis.48,49 The regulation of calcium levels inside cardiomyocytes is often imbalanced in the heart of a person with CV insufficiency, therefore proteins that regulate calcium levels are the main targets of gene therapy. The use of SERCA2a, which transfers Ca2+ from the cytosol to the lumen of the sarcoplasmic reticulum, in gene therapy bodes very well. Supplying a cell with genes for SERCA2a restores the intracellular calcium concentration in different phases of myocardial activity (Figure 4). A 12-month observational study concluded that a high-dose treatment significantly decreases the risk of CV events (HR =0.12; P=0.003) and reduces the average time of hospitalization 11-fold vs a placebo (P=0.05).50 An additional 3-year experiment proved that this gene therapy is safe and reduces the risk of CV events.51
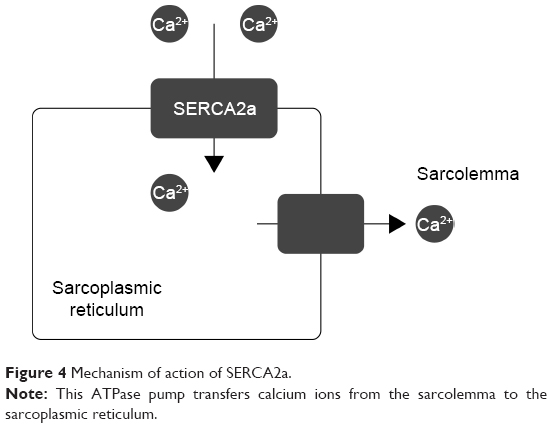 | Figure 4 Mechanism of action of SERCA2a. |
A percutaneous technique was used to insert genes into patients’ hearts; however, the main disadvantage of this method is that it is invasive. AAV1, AAV6, and AAV9 can be used as vectors in gene therapy;52–54 however, further trials must be conducted to determine the best way to transfer DNA to cardiomyocytes.
The two main problems of gene therapy for CVDs include: 1) the reaction of the patient’s immune system, which could prevent AAV penetration, and 2) worsening of the therapy’s effect over a prolonged period of time. These problems probably led to the results being far below the expectations of more optimistic researchers. Despite good results in the first stages of the study, CUPID 2 did not reach an endpoint with a substantial improvement in patient conditions.55
This last result should not discourage researchers, as vector modifications and technical improvements may lead to more refined therapies in the near future. Overcoming the present-day problems of gene therapy would be a great milestone, not only for cardiology but for medicine as a whole. Increasing numbers of gene therapy tests, not only in cardiology, is a good sign that high hopes are fully justified.
Conclusion
CVD is undoubtedly one of the biggest challenges for modern medicine. Increasing numbers of people suffering from CVD requires scientists to develop new therapeutic methods. Increasing knowledge of the pathological mechanisms of heart disease means that it is possible to employ therapies that take advantage of new pharmaceutical targets. The continuous progress of physiology and cardiology increases the chances of finding a highly successful therapy for the treatment of HF.
The outcomes of the aforementioned therapeutic methods are promising (the latest clinical trials summarized in Table 2); however, all require further research before they become efficacious therapeutic options. It should be mentioned that patients enrolled in clinical trials were treated not only with clinical trial medication, but also received standard HF treatment.
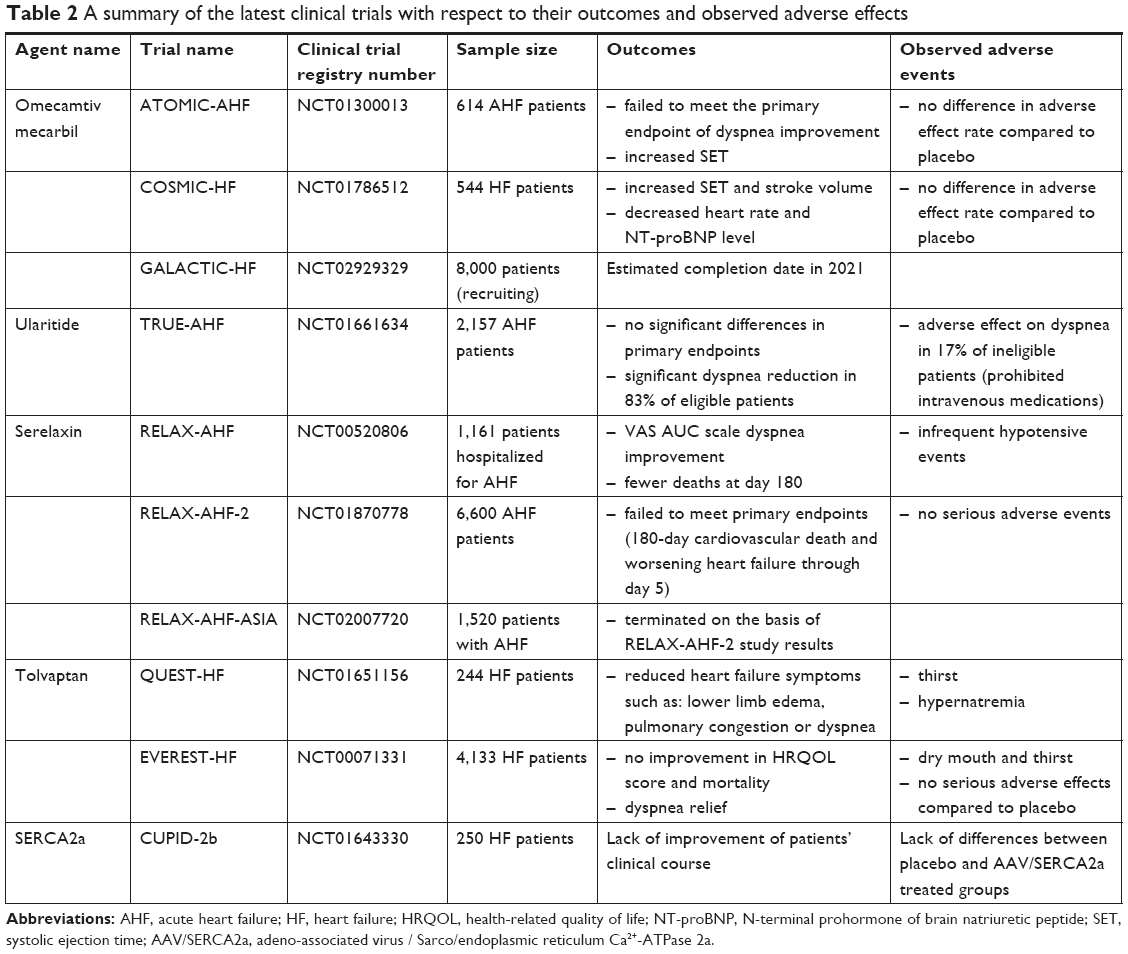 | Table 2 A summary of the latest clinical trials with respect to their outcomes and observed adverse effects |
Acknowledgment
The authors would like to thank Mikołaj Kozłowski for the figures, which helped greatly to improve the quality of the paper.
Disclosure
The authors report no conflicts of interest in this work.
References
Braunwald E. Research advances in heart failure: a compendium. Circ Res. 2013;113(6):633–645. | ||
Benjamin EJ, Blaha MJ, Chiuve SE. American Heart Association Statistics Committee and Stroke Statistics Subcommittee, 2017. Heart Disease and Stroke Statistics-2017 Update: a report from the American Heart Association. Circulation. 2017;135:e146–e603. | ||
Ponikowski P, Voors AA, Anker SD. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2016;2016(18):891–975. | ||
Ghosh RK, Banerjee K, Tummala R, Ball S, Ravakhah K, Gupta A. Serelaxin in acute heart failure: most recent update on clinical and preclinical evidence. Cardiovasc Ther. 2017;35(1):55–63. | ||
Aljundi AHS, Mohammed SFK, Patel A, et al. Inotropic agents use in patients hospitalized with acute decompensated heart failure: a retrospective analysis from a 22-year registry in a Middle-Eastern Country (1991–2013). BMC Cardiovasc Disord. 2016;16(1):47. | ||
Tariq S, Aronow W. Use of inotropic agents in treatment of systolic heart failure. Int J Mol Sci. 2015;16(12):29060–29068. | ||
Greenberg B. Novel therapies for heart failure-where do they stand? Circ J. 1891;2016(80):1882. | ||
Teerlink JR, Felker GM, Mcmurray JJV, et al. Acute treatment with omecamtiv mecarbil to increase contractility in acute heart failure: The ATOMIC-AHF Study. J Am Coll Cardiol. 2016;67(12):1444–1455. | ||
Planelles-Herrero VJ, Hartman JJ, Robert-Paganin J, Malik FI, Houdusse A. Mechanistic and structural basis for activation of cardiac myosin force production by omecamtiv mecarbil. Nat Commun. 2017;8(1):190. | ||
Hashem S, Tiberti M, Fornili A. Allosteric modulation of cardiac myosin dynamics by omecamtiv mecarbil. PLoS Comput Biol. 2017;13(11):e1005826. | ||
Starling RC. Cardiac myosin activators for the treatment of heart failure stop now or push ahead. J Am Coll Cardiol. 2016;67(12):1456–1458. | ||
Gassanov N, Biesenbach E, Caglayan E, et al. Natriuretic peptides in therapy for decompensated heart failure. Eur J Clin Pharmacol. 2012;68(3):223–230. | ||
Potter LR, Abbey-Hosch S, Dickey DM. Natriuretic peptides, their receptors, and cyclic guanosine monophosphate-dependent signaling functions. Endocr Rev. 2006;27(1):47–72. | ||
Anker SD, Ponikowski P, Mitrovic V, Peacock WF, Filippatos G. Ularitide for the treatment of acute decompensated heart failure: from preclinical to clinical studies. Eur Heart J. 2015;36(12):715–723. | ||
Bestle MH, Olsen NV, Christensen P, Jensen BV, Bie P. Cardiovascular, endocrine, and renal effects of urodilatin in normal humans. Am J Physiol. 1999;276(3 Pt 2):R684–R695. | ||
Schmitt M, Gunaruwan P, Payne N, et al. Effects of exogenous and endogenous natriuretic peptides on forearm vascular function in chronic heart failure. Arterioscler Thromb Vasc Biol. 2004;24(5):911–917. | ||
Saxenhofer H, Raselli A, Weidmann P, et al. Urodilatin, a natriuretic factor from kidneys, can modify renal and cardiovascular function in men. Am J Physiol. 1990;259(5 Pt 2):F832–F838. | ||
Kentsch M, Ludwig D, Drummer C, Gerzer R, Müller-Esch G. Haemodynamic and renal effects of urodilatin in healthy volunteers. Eur J Clin Invest. 1992;22(5):319–325. | ||
Kentsch M, Ludwig D, Drummer C, Gerzer R, Müller-Esch G. Haemodynamic and renal effects of urodilatin bolus injections in patients with congestive heart failure. Eur J Clin Invest. 1992;22(10):662–669. | ||
Mitrovic V, Lüss H, Nitsche K, et al. Effects of the renal natriuretic peptide urodilatin (ularitide) in patients with decompensated chronic heart failure: a double-blind, placebo-controlled, ascending-dose trial. Am Heart J. 2005;150(6):1239. | ||
Mitrovic V, Seferovic PM, Simeunovic D, et al. Haemodynamic and clinical effects of ularitide in decompensated heart failure. Eur Heart J. 2006;27(23):2823–2832. | ||
Packer M, Holcomb R, Abraham WT, et al. Rationale for and design of the TRUE-AHF trial: the effects of ularitide on the short-term clinical course and long-term mortality of patients with acute heart failure. Eur J Heart Fail. 2017;19(5):673–681. | ||
Packer M, O’Connor C, Mcmurray JJV, et al. Effect of ularitide on cardiovascular mortality in acute heart failure. N Engl J Med. 2017;376(20):1956–1964. | ||
Packer M. Short- and long-term effect of immediate vasodilator therapy in acute decompensated heart failure. Presented at American Heart Association Scientific Session. November 13, 2016. New Orleans, LA. | ||
Bathgate RA, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev. 2013;93(1):405–480. | ||
Smith MC, Danielson LA, Conrad KP, Davison JM. Influence of recombinant human relaxin on renal hemodynamics in healthy volunteers. J Am Soc Nephrol. 2006;17(11):3192–3197. | ||
Dschietzig T, Teichman S, Unemori E, et al. Intravenous recombinant human relaxin in compensated heart failure: a safety, tolerability, and pharmacodynamic trial. J Card Fail. 2009;15(3):182–190. | ||
Teerlink JR, Metra M, Felker GM, et al. Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study. The Lancet. 2009;373(9673):1429–1439. | ||
Teerlink JR, Cotter G, Davison BA. RELAXin in Acute Heart Failure (RELAX-AHF) Investigators. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. 2013;381:29–39. | ||
Sato N, Lam CS, Teerlink JR, et al. Evaluating the efficacy, safety, and tolerability of serelaxin when added to standard therapy in asian patients with acute heart failure: design and rationale of RELAX-AHF-ASIA trial. J Card Fail. 2017;23(1):63–71. | ||
Teerlink JR, Voors AA, Ponikowski P, et al. Serelaxin in addition to standard therapy in acute heart failure: rationale and design of the RELAX-AHF-2 study. Eur J Heart Fail. 2017;19(6):800–809. | ||
Teerlink J. Serelaxin in acute heart failure presented at Heart Failure 2017 and the 4th World Congress on Acute Heart Failure. Paris, France, 29 April, 2017. | ||
Inomata T, Ikeda Y, Kida K, et al. Effects of additive tolvaptan vs. Increased furosemide on heart failure with diuretic resistance and renal impairment – results from the K-STAR study. Circ J. 2018;82(1):159–167. | ||
Pose A, Almenar L, Gavira JJ, et al. Benefit of tolvaptan in the management of hyponatraemia in patients with diuretic-refractory congestive heart failure: the SEMI-SEC project. ESC Heart Failure. 2017;4(2):130–137. | ||
Vinod P, Krishnappa V, Chauvin AM, Khare A, Raina R. Cardiorenal syndrome: role of arginine vasopressin and vaptans in heart failure. Cardiol Res. 2017;8(3):87–95. | ||
Nomoto H, Satoh Y, Kamiyama M, et al. Mechanisms of diuresis for acute decompensated heart failure by tolvaptan. Int Heart J. 2017;58(4):593–600. | ||
Kinugawa K, Sato N, Inomata T. Efficacy and safety of tolvaptan in heart failure patients with volume overload – an Interim result of post-marketing surveillance in Japan. Circ J. 2014;78:844–852. | ||
Sağ S, Aydın Kaderli A, Yıldız A, et al. Use of tolvaptan in patients hospitalized for worsening chronic heart failure with severe hyponatremia: The initial experience at a single-center in Turkey. Turk Kardiyol Dern Ars. 2017;45(5):415–425. | ||
Uemura Y, Shibata R, Takemoto K, et al. Safety and efficacy of long-term use of tolvaptan in patients with heart failure and chronic kidney disease. Circ J. 2017;81(11):1736–1738. | ||
Ambrosy AP, Khan H, Udelson JE, et al. Changes in dyspnea status during hospitalization and postdischarge health-related quality of life in patients hospitalized for heart failure: findings from the EVEREST trial. Circ Heart Fail. 2016;9(5):e002458. | ||
López B, González A, Querejeta R, Larman M, Rábago G, Díez J. Association of cardiotrophin-1 with myocardial fibrosis in hypertensive patients with heart failure. Hypertension. 2014;63(3):483–489. | ||
Calabrò P, Limongelli G, Riegler L, et al. Novel insights into the role of cardiotrophin-1 in cardiovascular diseases. J Mol Cell Cardiol. 2009;46(2):142–148. | ||
Abdul-Ghani M, Suen C, Jiang B, et al. Cardiotrophin 1 stimulates beneficial myogenic and vascular remodeling of the heart. Cell Research. 2017;27(10):1195–1215. | ||
Regis G, Pensa S, Boselli D, Novelli F, Poli V. Ups and downs: the STAT1:STAT3 seesaw of Interferon and gp130 receptor signalling. Semin Cell Dev Biol. 2008;19(4):351–359. | ||
Obana M, Maeda M, Takeda K, et al. Therapeutic activation of signal transducer and activator of transcription 3 by interleukin-11 ameliorates cardiac fibrosis after myocardial infarction. Circulation. 2010;121(5):684–691. | ||
Moreno-Aliaga MJ, Pérez-Echarri N, Marcos-Gómez B, et al. Cardiotrophin-1 is a key regulator of glucose and lipid metabolism. Cell Metab. 2011;14(2):242–253. | ||
Williams ML, Hata JA, Schroder J, et al. Targeted beta-adrenergic receptor kinase (betaARK1) inhibition by gene transfer in failing human hearts. Circulation. 2004;109(13):1590–1593. | ||
Karakikes I, Chaanine AH, Kang S, et al. Therapeutic cardiac-targeted delivery of miR-1 reverses pressure overload-induced cardiac hypertrophy and attenuates pathological remodeling. J Am Heart Assoc. 2013;2(2):e000078. | ||
Ganesan J, Ramanujam D, Sassi Y, et al. MiR-378 controls cardiac hypertrophy by combined repression of mitogen-activated protein kinase pathway factors. Circulation. 2013;127(21):2097–2106. | ||
Jessup M, Greenberg B, Mancini D, et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID). Circulation. 2011;124(3):304–313. | ||
Zsebo K, Yaroshinsky A, Rudy JJ, et al. Long-Term Effects of AAV1/SERCA2a Gene Transfer in Patients With Severe Heart Failure. Circulation Research. 2014;114(1):101–108. | ||
Beeri R, Chaput M, Guerrero JL, et al. Gene delivery of sarcoplasmic reticulum calcium ATPase inhibits ventricular remodeling in ischemic mitral regurgitation. Circ Heart Fail. 2010;3(5):627–634. | ||
Fish KM, Ladage D, Kawase Y, et al. AAV9.I-1c Delivered via direct coronary infusion in a porcine model of heart failure improves contractility and mitigates adverse remodeling. Circulation: Heart Failure. 2013;6(2):310–317. | ||
Hammoudi N, Ishikawa K, Hajjar RJ. Adeno-associated virus-mediated gene therapy in cardiovascular disease. Current Opinion in Cardiology. 2015;30(3):228–234. | ||
Greenberg B, Butler J, Felker GM, et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. The Lancet. 2016;387(10024):1178–1186. |
 © 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2019 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.