Back to Journals » Journal of Experimental Pharmacology » Volume 13
New Pharmacologic Approaches to Bronchopulmonary Dysplasia
Authors Roberts K, Stepanovich G, Bhatt-Mehta V, Donn SM ![]()
Received 7 December 2020
Accepted for publication 26 February 2021
Published 25 March 2021 Volume 2021:13 Pages 377—396
DOI https://doi.org/10.2147/JEP.S262350
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Bal Lokeshwar
Katelyn Roberts,1 Gretchen Stepanovich,1 Varsha Bhatt-Mehta,1,2 Steven M Donn1
1Division of Neonatal-Perinatal Medicine, Department of Pediatrics, Michigan Medicine, University of Michigan, Ann Arbor, MI, USA; 2College of Pharmacy, Michigan Medicine, University of Michigan, Ann Arbor, MI, USA
Correspondence: Steven M Donn
University of Michigan, 8-621 C.S. Mott Children’s Hospital, 1540 E. Medical Center Drive, Ann Arbor, MI, 48109-4254, USA
Tel +1 734 763-4109
Fax +1 734 763-7728
Email [email protected]
Abstract: Bronchopulmonary Dysplasia is the most common long-term respiratory morbidity of preterm infants, with the risk of development proportional to the degree of prematurity. While its pathophysiologic and histologic features have changed over time as neonatal demographics and respiratory therapies have evolved, it is now thought to be characterized by impaired distal lung growth and abnormal pulmonary microvascular development. Though the exact sequence of events leading to the development of BPD has not been fully elucidated and likely varies among patients, it is thought to result from inflammatory and mechanical/oxidative injury from chronic ventilatory support in fragile, premature lungs susceptible to injury from surfactant deficiency, structural abnormalities, inadequate antioxidant defenses, and a chest wall that is more compliant than the lung. In addition, non-pulmonary issues may adversely affect lung development, including systemic infections and insufficient nutrition. Once BPD has developed, its management focuses on providing adequate gas exchange while promoting optimal lung growth. Pharmacologic strategies to ameliorate or prevent BPD continue to be investigated. A variety of agents, to be reviewed henceforth, have been developed or re-purposed to target different points in the pathways that lead to BPD, including anti-inflammatories, diuretics, steroids, pulmonary vasodilators, antioxidants, and a number of molecules involved in the cell signaling cascade thought to be involved in the pathogenesis of BPD.
Keywords: prematurity, respiratory distress, bronchopulmonary dysplasia, chronic lung disease, pulmonary hypertension, pharmacologic management
Introduction
Bronchopulmonary dysplasia (BPD) was first described by Northway and colleagues in 1967, reporting the radiographic findings in a small group of infants who had been mechanically ventilated for respiratory distress syndrome (RDS). These infants were modestly premature compared to babies found in modern neonatal intensive care units (NICU). BPD was originally believed to result from exposure to high mechanical ventilator pressures and high concentrations of oxygen;1 this form of lung damage and maldevelopment is now referred to as “old BPD” or “classic BPD.” However, as neonatal practices evolved and smaller and more immature babies survived, the pathophysiology of BPD is now believed to be the consequence of decreased alveolarization and a developmental arrest of the lung; to distinguish between the two pathophysiologic processes, the latter was assigned the updated moniker, “new BPD.”2
The Pulmonary Injury Sequence
In the five decades since the Northway report, much has been learned about the pulmonary injury sequence. Affected infants are born at a time when the lung is still in embryologic stages where alveolarization has not begun; some are even in the saccular and canalicular phases (Figure 1). Respiratory insufficiency results in the initiation of mechanical ventilation, subjecting many infants to pressures and oxygen concentrations that are far greater than they would have experienced in utero. Additionally, there are characteristics of the preterm lung that increase susceptibility to injury, including surfactant deficiency, structural abnormalities, inadequate antioxidant defenses, and a chest wall that is more compliant than the lung. Even infants requiring minimal treatment remain at risk, but those at greatest risk are born before 30 weeks of gestation.3 Factors such as susceptibility genes and perinatal exposures may contribute in the early weeks of pregnancy. Late perinatal factors, including maternal illness, inadequate placental function, and exposure to antenatal steroids may also play a role. Glucocorticoid exposure (both prenatally and postnatally) may stimulate surfactant production and decrease inflammation, but as a potent inhibitor of DNA synthesis, it can also diminish lung repair and growth. Insufficient postnatal nutrition may prevent adequate growth of new lung tissue. Lung and systemic infections are commonplace in the NICU and may further contribute to lung injury. Exposure to high concentrations of inspired oxygen may generate oxygen free radicals and lead to further lung injury.3–5 Thus, BPD is a multi-factorial disorder, which explains why no single intervention has proved successful and why novel therapeutic interventions continue to be sought. While the original pathophysiology of BPD described by Northway et al included inflammation, alveolar fibroproliferation, and cystic changes, the pathophysiology of the new BPD involves impaired distal lung growth and dysregulated microvascular development with less fibrosis.5,6
 |
Figure 1 The pulmonary injury sequence representing the stages of lung development and factors contributing to BPD which should be the focus of prevention of the disease. Reprinted with permission of the American Thoracic Society. Copyright © 2014 American Thoracic Society. All rights reserved. McEvoy CT, Jain L, Schmidt B, Abman S, Bancalari E, Aschner JL. NHLBI Workshop on the primary prevention of chronic lung disease: Bronchopulmonary dysplasia. Ann Am Thorac Soc. 2014;11: S146–S153. Annals of the American Thoracic Society is an official journal of the American Thoracic Society.6 |
Epidemiology of BPD
The definition of BPD has changed over time as available ventilatory support and strategies to minimize lung injury in preterm infants have evolved. The most common contemporary definition is the need for supplemental oxygen for at least 28 days after birth, with the disease being classified as mild, moderate, and severe based upon the necessity for positive pressure and the need for supplemental oxygen at 36 weeks’ corrected gestational age. Several other definitions of BPD exist.3 Regardless of the criteria or timing to definitively make the diagnosis, the underlying pathophysiology remains the same.
Characteristics that modify the development of BPD include gestational age, birthweight, gender and pre- and post-natal risk factors. The most significant contributor to the development of BPD is prematurity; the incidence and severity of BPD are inversely proportional to gestational age, with the infants born at the earliest gestational ages more likely to develop BPD and the most severe lung disease. NICHD Neonatal Research Network (NRN) data published in 2010 indicate an incidence of BPD of 73% at 23 weeks compared to 23% at 28 weeks.7 Furthermore, the increased risk is seen with extremely low birth weight, male gender, exposure to chorioamnionitis, intrauterine growth restriction, prolonged use of mechanical ventilation/positive pressure, and exposure to high concentrations of supplemental oxygen.3,4 In infants with fetal growth restriction, the mechanisms leading to growth restriction are thought to contribute to the vulnerability of the developing lung leading to a higher risk of development of BPD in this group.8 Additional risk factors that may contribute to developing BPD include perinatal asphyxia, a patent ductus arteriosus, sepsis, the relative dysbiosis of the preterm airway, and genetic factors that have yet to be fully elucidated. Data from the NICHD NRN have been used to create a model to predict BPD at several time points based on gestational age, birthweight, gender, race/ethnicity, postnatal day, type of ventilatory support, and FiO2. This can be used to assist with BPD-associated therapeutic decision-making.9,10
Despite efforts to reduce the rate of preterm birth, in the United States, in 2018 (the most recent year for which the National Vital Statistics Reports are available) more than 100,000 infants were born <34 weeks’ gestation and more than 50,000 were <1500 g. While the rates of BPD at different institutions vary based upon the population served and differing clinical practices, BPD is the most common morbidity associated with preterm birth, affecting 10,000–15,000 infants annually in the United States.10,11 The reported rate of BPD by the Vermont Oxford Network is 22% to 36%,12 with the wide variability likely reflective of practice variations among the >750 NICU member institutions. Infants with BPD are at increased risk for mortality, prolonged hospitalization, chronic respiratory and cardiovascular issues, poor growth, and neurodevelopmental delay.
The primary goal of treatment of BPD is to provide adequate respiratory support and nutrition to promote optimal lung growth.13 Respiratory therapies range widely from noninvasive ventilatory support to mechanical ventilation with a tracheostomy. Several pharmacologic therapies may also be beneficial in the treatment of BPD. The following will review established and novel treatments to prevent or ameliorate BPD, presented in an order that parallels the pulmonary injury sequence.
New Pharmacologic Approaches
Surfactant
Physical trauma to the lung parenchyma, both “volutrauma,” from excess tidal volume delivery, and “atelectotrauma,” or damage as a result of shearing forces from expansion and collapse of smaller airways are contributory factors. In addition to the use of low tidal volumes and maintenance of alveolar expansion at end-expiration, surfactant, a phospholipoprotein generated by Type II pneumocytes within the pulmonary epithelium, improves lung compliance and may help reduce volutrauma and atelectotrauma. While the cells that contain and later release surfactant components may start to be evident as early as 24 weeks’ gestation, most preterm infants have some degree of surfactant deficiency. Surfactant reduces alveolar surface tension at the air-liquid interface and improves lung compliance, and different surfactant components play roles in cell signaling and defense.14
Surfactant Alone
Exogenous surfactant therapy, derived from bovine or porcine lungs or synthetic preparations, was introduced for commercial use in 1990 and has since been a mainstay of therapy for RDS. Early surfactant therapy often eliminates or reduces the need for aggressive mechanical ventilatory support and may reduce resultant lung injury. It is typically given soon after birth and can be re-administered to infants every 12 hours for persistently poor compliance, ventilation, and/or oxygenation.
Although Surfactant therapy has shown important improvements in survival, the rate of BPD has not changed, perhaps at least in part from the improved survival of the most preterm infants.10,15 Surfactant has traditionally been administered through an endotracheal tube, though newer techniques include administration directly into the trachea by a feeding tube or vascular catheter, obviating intubation. More recently, surfactant has been aerosolized and administered by continuous positive airway pressure (CPAP) utilizing a special delivery device. Clinical trials are underway to determine if this approach can eliminate the need for any airway manipulation in selected populations. Reduction in BPD is a study objective.16
Late Surfactant Therapy
Administration of surfactant beyond the first few days of life has been proposed to provide benefit to preterm infants, as endogenous surfactant may become physiologically dysfunctional or deficient over time in chronically ventilated preterm infants. A multi-center randomized controlled trial published in 2009 evaluated the administration of a synthetic surfactant to premature infants requiring >0.3 FiO2 between three and 10 days of age. Infants were randomized to placebo (sham air), 90, or 175 mg/kg surfactant phospholipid/dose, with up to five doses, given every 48 hours, as long as the infant continued to need mechanical ventilation. There was a trend toward decreased oxygen need and a lower incidence of BPD and mortality in the group receiving the higher surfactant dose, though the findings were not statistically significant.17 A multi-center randomized controlled trial published in 2016 investigated whether one dose of porcine-derived surfactant administered at 14 days of age in infants requiring significant respiratory support modified respiratory outcomes. While the time to extubation and the combined outcome of death or BPD at 36 weeks’ corrected age were not statistically different, the group that received surfactant did have less re-hospitalization for pulmonary issues at 1 year of age.18
A subsequent multi-center randomized controlled trial involving more than 1000 neonates born at ≤28 weeks, who continued to require mechanical ventilation between 7 and 14 days of life did not show benefit to “late” surfactant administration. Enrolled infants were randomized to receive up to five doses of bovine-derived surfactant or air; survival without BPD at 36 and 40 weeks was not improved by surfactant. Of note, all infants in this trial also received inhaled nitric oxide.19
Surfactant Plus Budesonide
Multiple observational and randomized controlled trials involving preterm and/or very low birthweight infants, in which a standard dose of surfactant with added budesonide (0.2–0.25 mg/kg) was administered into the trachea within the first several hours of life (2–8 hours), demonstrated a statistically significant reduction in the incidence of BPD as well as the need for additional surfactant. A meta-analysis performed in 2019 confirmed these findings and also concluded that in babies who received the early inhaled surfactant/steroid combination, there was no increase in retinopathy of prematurity, infection, or adverse neuromotor impairment up to 3 years of age.20–22
Anti-Inflammatories
The development of BPD is a complex and multi-factorial process. One common pathway is injurious inflammation. Pro-inflammatory and pro-fibrotic growth factors adversely affect the preterm lung’s microvascular and alveolar development, resulting in impaired gas exchange. While the inflammatory cascade involves many different cell types and signaling molecules, agents that block specific receptors or alter individual pathways have not yet been found to be beneficial in reducing the incidence of BPD. Some of these, however, bear discussion.
Inhaled Nitric Oxide (iNO): Anti-Inflammatory Role
iNO is a potent pulmonary vasodilator with minimal systemic effects. It acts via cGMP to reduce calcium within the pulmonary arterial smooth muscle cells, thereby relaxing the pulmonary arterial vasculature. Multiple studies have proposed anti-inflammatory mechanisms through which iNO reduces pulmonary inflammation in settings of potential pulmonary injury, such as mechanical ventilation or sepsis (reduced leukocyte migration, reduced pro-inflammatory cytokine levels and induction of apoptosis in injured cells).23,24 It is an established treatment in term and late preterm infants with hypoxemic respiratory failure accompanying persistent pulmonary hypertension of the newborn (PPHN) and is the only FDA-approved agent for such.10 However, it has been proposed to have a role in BPD reduction by positively influencing lung development. In multiple murine studies, term mice and rats exposed to either hyperoxia or inhibitors of Vascular Endothelial Growth Factor, VEGF (both insults result in impaired alveolarization, decreased vascular growth and pulmonary hypertension), exposure to iNO resulted in attenuation of these adverse effects. In rodents exposed to iNO, lung histologic studies and barium arteriograms demonstrated smaller and more alveoli, as well as enhanced pulmonary vascular density, respectively.25,26 These results were further supported by a study involving mechanically ventilated preterm lambs; lambs exposed to iNO for the first 3 weeks of life developed a greater number of alveoli, had reduced airway resistance, and had less pulmonary hypertension than unexposed lambs.27 A similar study in mechanically ventilated preterm baboons found comparable improvements in lung structure and function after exposure to iNO, as well as a reduction in abnormal elastin deposition typically seen in BPD.28
Despite promising animal studies, human studies have not consistently supported the role of iNO in reducing BPD. One randomized controlled trial (2006) concluded that iNO administered for at least 24 days to preterm infants who continued to require ventilator support between 7 and 21 days of age reduced the risk of BPD.29 However, a subsequent trial published in 2017 that investigated the routine use of iNO in preterm infants had contradictory findings. There was no difference in survival without BPD at 36 weeks’ corrected gestational age following a 24-day course of iNO given to infants born at <30 weeks’ gestation and <1250 g.30 Furthermore, a Cochrane review of relevant studies found no statistically significant benefit of iNO in BPD reduction with either “early” or “late” administration.31
Corticosteroids
Corticosteroids act in several ways to reduce inflammation. By binding to glucocorticoid receptors, they both inhibit pro-inflammatory signals and promote anti-inflammatory signals. Acutely, they decrease vasodilation and capillary permeability while decreasing leukocyte migration. Long-term action at glucocorticoid receptors mediates changes in gene expression that affect many inflammatory cell-signaling molecules, resulting in a broad reduction in inflammation. As inflammation is central to the development of BPD, administration of steroids may dampen injurious inflammation and decrease the likelihood of BPD in at-risk infants.
The use of antenatal steroids, typically given to a pregnant woman as a 48-hour course prior to preterm delivery, has been shown to reduce neonatal mortality, and the incidence of RDS and intraventricular hemorrhage. There are multiple mechanisms, including an increase in the surfactant pool and changes in the airspace epithelium that result in improved lung function. Despite decreasing the rate of RDS, however, a reduction in BPD has not been observed.32
Administration of systemic steroids after birth has shown benefit in facilitating extubation and reducing BPD, though systemic steroid administration is not without risk and includes hyperglycemia, hypertension, and spontaneous intestinal perforation (often with concomitant use of nonsteroidal anti-inflammatory agents). Dexamethasone, a potent steroidal agent, has historically been used in relatively high doses early in a premature infant’s postnatal course to provide pulmonary benefit (up to 8 mg/kg over 42 days);33 while acute improvement in gas exchange and lung mechanics has been observed, follow-up studies revealed an increase in adverse neurodevelopmental outcomes, especially cerebral palsy.34,35 However, BPD itself carries risks for adverse long-term neurodevelopmental outcomes, so steroid use has been considered beyond the first week of life for infants at high risk. The NICHD BPD calculator was generated to provide guidance for such situations.9
In a retrospective cohort study (2019) involving a small group of preterm infants at high risk for BPD, infants treated “moderately early” with dexamethasone at 14–28 days compared to those who received “delayed” treatment at 29–42 days, were discharged at an earlier corrected age, spent fewer days on mechanical ventilation and on supplemental oxygen.36 Dosing recommendations for systemic steroid administration have been in flux. In the “Dexamethasone: A Randomized Trial (DART),” published in 2006, much lower doses were administered; a 10-day tapering course was provided with a total dose of less than 0.9 mg/kg. The benefit was shown with improvements in respiratory status and a shorter duration of intubation without an increased risk of adverse events at age 2.37,38 However, the trial was prematurely terminated and did not enroll sufficient patients to be conclusive. Early low-dose hydrocortisone has also been studied. In the PREMILOC trial (2016), a 10-day tapering course of low-dose hydrocortisone that commenced on the first day of life resulted in an increase in survival without BPD. Follow-up studies revealed no increase in long-term adverse neurodevelopmental outcomes.39,40 Given the known risks and only potential benefits of postnatal steroids, the decision to administer steroids, as well as the optimal dosage and timing, remains challenging and unclear. Identifying biomarkers or genes that confer a greater likelihood of improved respiratory status with postnatal steroid administration would be a worthwhile contribution.
To this end, “precision therapeutics” aim to investigate whether certain genes or biomarkers predict the likelihood of a desired therapeutic response. In a secondary analysis of data from the TOLSURF trial (2019), variance in one single nucleotide polymorphism (SNP) within the Corticotriphin Releasing Hormone Receptor 1 (CRHR1) intron was associated with a significant difference in response to a course of exogenous postnatal steroid, thought to result from a difference in baseline endogenous corticosteroid concentrations as well as baseline airway inflammation.41 Furthermore, pharmacometabolomic research investigates whether levels of a variety of metabolites found in different body fluids change in parallel with certain clinical outcomes following a prescribed therapy. In a study from 2019, the decline in serum and urinary gluconic acid levels following steroid administration (presumably an indicator of reduced inflammation) correlated well with improved respiratory status.42 Continued evolution and advancements in these fields may help provide more targeted steroid therapy to infants who are most likely to benefit.
PF543: A Sphingosine Kinase 1 Inhibitor
Sphingolipids are cell signaling molecules involved in cell proliferation, differentiation, and apoptosis.43 Sphingosine can be generated by the hydrolysis of sphingolipids. Sphingosine Kinase 1 (Sphk1) is an enzyme that catalyzes the formation of sphingosine-1-phosphate (S1P) from Sphingosine. S1P promotes cell proliferation, survival, mobility, and cell-to-cell adhesion. Murine studies have shown it to recruit monocytes involved in the development of pulmonary fibrosis, as well as the generation of reactive oxygen species and Lysyl oxidase-associated excess collagen cross-linking. PF543 is a small molecule that inhibits Sphk1 and reduces S1P generation. Mice treated with PF543 have reduced BPD and airway remodeling when exposed to hyperoxia. Studies are currently underway to investigate the therapeutic efficacy of PF 543 provided at different stages of lung development.44,45
Clara Cell 10-kD Protein
Clara cell 10-kD protein (CC10), an anti-inflammatory and immunomodulatory protein generated by cells within the respiratory epithelium, has been theorized to alter the development of BPD, as concentrations are low in premature infants. Multiple animal studies demonstrated reduced lung inflammation, improved lung development, cytoarchitecture, and pulmonary function with administration of CC10, and led to a randomized trial involving 22 premature infants. Intratracheal administration of recombinant CC10 was found to be safe and resulted in decreased inflammation, as evidenced by decreased total cells, neutrophils, total protein and IL-6 in bronchoalveolar lavage samples from treated infants compared to placebo.46 A subsequent double-masked, placebo-controlled RCT published in 2019 involved 88 preterm infants who received a single dose of intratracheal recombinant CC10 to investigate whether CC10 administration resulted in clinically meaningful differences in lung development. Two different CC10 doses were trialed; neither was found to be effective in changing long-term respiratory outcomes at 12 months of age with no significant differences in growth parameters, adverse events, or mortality.47,48
Docosahexaenoic Acid (DHA)
DHA is an omega-3 fatty acid proposed as a therapy for BPD because of its anti-inflammatory properties. In a retrospective cohort study of 88 infants born at less than 30 weeks’ gestation, DHA concentrations were observed to decrease within the first few weeks of life, and infants with lower levels had a greater incidence of BPD, suggesting an association between DHA concentrations and pulmonary inflammation and development.49 DHA supplementation was also found to lower the incidence of BPD as a secondary outcome in a large study looking at neurodevelopmental outcomes.50 However, an RCT published in 2017 involving infants born at <29 weeks’ gestation found no significant difference in the frequency of BPD development between infants given enteral DHA supplementation compared to controls (soy emulsion).51 Furthermore, a meta-analysis evaluating four RCTs and nearly 2000 neonates (including the two aforementioned studies from 2009 to 2017) found no benefit to DHA supplementation in the reduction of BPD at 28 days, 36 weeks’ postmenstrual age or other neonatal morbidities, such as NEC, IVH, severe ROP, or sepsis.52
Antioxidants
Superoxide Dismutase
Superoxide dismutase is an intracellular enzyme that converts toxic superoxide radicals into less harmful hydrogen peroxide.53 Superoxide dismutase expression is decreased in the pulmonary vasculature of neonates, leading to endothelial dysfunction and impaired nitric oxide-induced vasodilation. Its use as an antioxidant might reduce BPD via blockade of the effects of free radicals on pulmonary tissue. Superoxide dismutase has been used as an adjunctive therapy to iNO in lamb models to decrease oxidative stress and restore eNOS coupling and shows promise as a future treatment in the neonatal population.54 In a randomized study of premature infants, 600–1200 g requiring surfactant for respiratory distress syndrome, infants randomized to receive intratracheal recombinant human CuZn superoxide dismutase every 48 hours while intubated had no difference in the incidence of death or BPD at 36 weeks postmenstrual age compared to placebo but did require less asthma medication, emergency department visits, and subsequent hospitalizations at 1 year of corrected age possibly indicating an improved pulmonary status.55
Antimicrobials
Bacterial colonization of the airway may begin while an infant is in utero, during the labor and delivery process, or soon after delivery.56 Multiple studies have shown that the bacterial diversity and most abundant organisms in the airway are different in preterm compared to term infants.56,57 Among preterm infants, diminished diversity and predominance of Ureaplasma at birth or Proteobacteria later in a neonate’s course may be associated with an increased risk of BPD.57 A neonate’s bacterial blueprint can be altered by a variety of causes, including intrapartum maternal antibiotics, antibiotics received during a neonate’s NICU course, presence of maternal chorioamnionitis, immaturity of the preterm neonate’s immune system, cesarean delivery, and altered colonization of the mother’s amniotic cavity or vaginal canal.56 The altered microbiota established in a preterm infant’s airway may predispose to BPD, resulting in a potential prophylactic role for antibiotics.
Azithromycin
Azithromycin is a macrolide antibiotic considered to be bacteriostatic by inhibiting protein synthesis. It also has non-specific anti-inflammatory properties. Azithromycin binds to the 50S subunit of the bacterial ribosome to inhibit translation of mRNA; this is particularly important as it confers activity against Ureaplasma. Intrauterine Ureaplasma infection has been reported to increase the risk of preterm labor and delivery, and infection in neonates is associated with the development of BPD. In a randomized controlled trial in preterm infants given IV Azithromycin 20mg/kg for 3 days, Ureaplasma was effectively eradicated in all infants receiving treatment compared to 84% of control infants who remained Ureaplasma-positive in the placebo group. However, overall mortality was similar between the two groups.58 A meta-analysis of clinical trials that administered 10 mg/kg/d of Azithromycin for 1 week followed by 5 mg/kg/d for 1–5 weeks showed a significant reduction in BPD in extremely premature infants compared to placebo regardless of infection or colonization with Ureaplasma, but the overall quality of evidence was low.59 Azithromycin is often administered once daily because of its long half-life (28–83 hours) in neonates.60 Concerns with Azithromycin use in the neonatal population include feeding intolerance, adverse neurologic and respiratory events, vomiting, diarrhea, and abdominal tenderness.
Clarithromycin
Clarithromycin is also a macrolide antibiotic. It binds to the 50S subunit of the bacterial ribosome and interferes with amino acid translocation during translation. Because of the continued concern for an association between Ureaplasma colonization or infection with the development of BPD, clarithromycin was evaluated in preterm infants. Infants with a birthweight of 750–1250 g who had a positive nasopharyngeal swab for Ureaplasma were randomized to IV Clarithromycin at a dose of 10 mg/kg twice per day for 10 days or placebo. With Clarithromycin treatment, 69% of patients had eradication of Ureaplasma and had a significantly lower incidence of BPD.61 The use of macrolides for the eradication of Ureaplasma in the neonatal population remains controversial; at present, the routine prophylactic use of macrolides for the prevention of BPD is not recommended.
Diuretics
While studies have yet to show the long-term benefit of diuretics to reduce BPD, they might provide benefit in mitigating pulmonary over-circulation in pulmonary hypertension.
Loop Diuretics: Furosemide and Bumetanide
Loop diuretics act by inhibiting the reabsorption of NaCl in the renal thick ascending loop of Henle. They are the most frequently used diuretics in the NICU and typically result in a rapid and vigorous diuresis. Loop diuretics also increase the concentration of prostaglandin E2 within the renal parenchyma. Between the PGE2 effects on the pulmonary vasculature and the reduction in total body water, loop diuretics decrease pulmonary interstitial fluid and improve lung compliance.10 They may provide benefit in pulmonary hypertension by reducing right ventricular distension and avoidance of impeding left ventricle filling.62 Commonly observed adverse side effects include electrolyte disturbances (especially hyponatremia, hypokalemia, hypocalcemia, and hypochloremic metabolic alkalosis), nephrocalcinosis, and ototoxicity. Bumetanide is much more potent than furosemide and may result in decreased electrolyte disturbances and ototoxicity compared to furosemide.10
Potassium-Sparing Diuretics: Spironolactone
Spironolactone is an aldosterone antagonist that acts at the cortical tubule to reduce the expression of the sodium-potassium exchange site in the apical membrane of the distal convoluted renal tubule. Inhibition of reabsorption results in diuresis with sparing of potassium. It is neither fast-acting nor potent in its diuretic effect and is commonly used in combination with other diuretics.
Aldosterone antagonists such as spironolactone may also have a role in preventing BPD as it relates to the development of pulmonary hypertension (PAH). A study performed on adults with pulmonary hypertension revealed that such patients had elevated serum aldosterone concentrations. Through multiple mechanisms, elevated aldosterone reduces endothelial NO production and promotes abnormal pulmonary extracellular remodeling.63,64 A comprehensive study investigated aldosterone’s effects on the development of pulmonary hypertension. In vitro, pulmonary arterial smooth muscle cells demonstrated reduced abnormal extracellular matrix remodeling when treated with spironolactone, and two experimental animal models of PAH revealed decreased arteriole muscularization with treatment that included spironolactone.65 There are currently two clinical trials underway to investigate whether spironolactone affects clinical and biochemical markers of pulmonary hypertension in adults.64
Carbonic Anhydrase Inhibitors
Carbonic anhydrase inhibitors (CAIs), such as acetazolamide, which act by catalyzing the forward and reverse reactions between carbon dioxide and water to form carbonic acid, are not typically used in the NICU. However, early studies have proposed a role in the amelioration of pulmonary hypertension via induced metabolic acidosis, which has been shown to reduce inflammation and pulmonary arterial smooth muscle differentiation. In addition to reducing pulmonary cytokine and chemokine concentrations, CAIs have also been shown to alter alveolar macrophage activation and function, blunting both pro- and anti-inflammatory signaling. Therefore, CAIs may have a role in reducing the development of PAH, and through this decrease in inflammation and improvement in pulmonary vascular homeostasis may reduce the development of BPD.66
Thiazide Diuretics: Hydrochlorothiazide
Thiazide diuretics result in diuresis by inhibiting the reabsorption of sodium and chloride at the sodium-chloride co-transporter within the distal convoluted tubule of the nephron. As the majority of sodium is reabsorbed proximally to this site of action, the degree of diuresis is typically much less than those of loop diuretics and the adverse electrolyte effects are also typically diminished in comparison, though hyponatremia, hypokalemia, hypomagnesemia, and hypophosphatemia can be seen. As resistance to loop diuretics may develop over time, the addition of a thiazide diuretic may provide benefit. If used in combination, the thiazide diuretic should be given roughly 30–60 minutes before the loop diuretic to optimize diuresis.10
Agents to Treat Pulmonary Hypertension
BPD is associated with pulmonary vascular disease, right ventricular hypertrophy, left ventricular hypertrophy, systemic hypertension, and the development of systemic to pulmonary collateral vessels. Approximately 25% of infants with significant BPD develop PAH with significant mortality if left untreated.67–69 The mechanisms for PAH include increased pulmonary arteriolar vasoconstriction and vascular structural remodeling, which then result in increased pulmonary vascular resistance. Underdevelopment of the pulmonary vascular bed may or may not be involved in this mechanism. Structural remodeling likely occurs following early lung injury, including oxygen free radical injury, ventilator-induced injury, and/or exposure to inflammatory mediators.70 These injuries then lead to smooth muscle cell proliferation and fibroblast incorporation into the vessel wall, which results in decreased vessel diameter (and increased vascular resistance) and decreased vascular compliance. There is also a disordered expression of angiogenic growth factors and their receptors, leading to dysmorphic capillaries seen in the thickened alveolar septae.54 PAH in a neonate with BPD can be manifest as respiratory insufficiency leading to chronic hypoxemia, hypercapnia, and acidosis, which then contributes to increased vascular tone. In infants with PAH and severe BPD, pulmonary vasodilators can improve oxygenation and pulmonary function acting by several mechanisms (Figure 2).
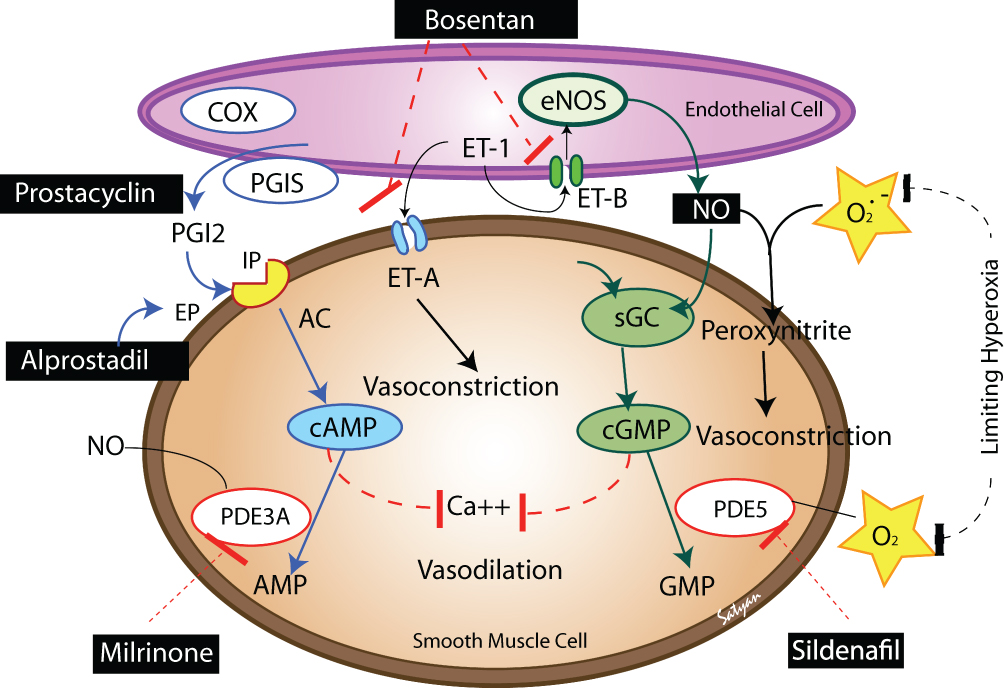 |
Figure 2 The various pathways to focus therapeutics for treatment of pulmonary hypertension which is a common complication in infants with BPD. Adapted from Semin Perinatol, 40(3), Lakshminrusimha S, Mathew B, Leach CL. Pharmacologic strategies in neonatal pulmonary hypertension other than nitric oxide, 160–73, Copyright (2016), with permission from Elsevier.80 |
Inhaled Nitric Oxide (iNO): Vasodilatory Role
Endogenous production of NO is needed for alveolar and vascular development. A disruption in NO signaling can lead to the characteristic lung and vascular injury findings of BPD. Identification of those infants at high risk for early pulmonary vascular disease allows utilization of strategies to prevent poor respiratory outcomes.71 Infants with BPD have pulmonary parenchymal changes that lead to increased dead space and thus ventilation-perfusion mismatch. This mismatch can be decreased by inhaled nitric oxide through increases in cGMP levels.72 Increased cGMP levels lead to the relaxation of the pulmonary arterial vasculature. iNO causes vasodilation and an improvement in blood flow to well-ventilated areas. It may also be involved in vascular remodeling, bronchodilation, and decreased inflammation via reduced endothelin and platelet-derived growth factor B, and decreased neutrophil adhesion to endothelial cells. iNO has been shown to be safe for use in infants with PPHN73 but is a treatment that is expensive and cumbersome for long-term administration. Its use in BPD is off-label.
Phosphodiesterase Type 5 (PDE5) Inhibitors
PDE5 inhibitors, which include sildenafil and tadalafil, are in another class of drugs that may be utilized to affect the NO signaling pathway involved in PAH. By inhibiting PDE5, they act on vascular, cardiac, and bronchial smooth muscle cells to increase cGMP concentrations by preventing its degradation in the lungs, thus increasing endogenous NO and promoting vasodilation. In addition, Sildenafil has been shown to promote angiogenesis and alveolar development via the vascular endothelial growth factor-NO-cGMP pathway, decrease lung inflammation, and reduce hyperoxia-induced lung injury in animal models.74 In a retrospective review of neonatal patients with BPD treated with Sildenafil, doses ranged from 0.2 to 8 mg/kg/day, with 88% of infants experiencing improved hemodynamics after a median duration of 40 days. It was concluded that Sildenafil is safe and effective for this patient population.75 PDE5 inhibitors may worsen gastroesophageal reflux, which can be a common issue in the preterm population.75 Increased mortality was reported in a clinical trial of patients with PAH randomized to Sildenafil monotherapy, but potential benefits may outweigh risks for certain patients, as Sildenafil treated patients appear to have favorable survival compared to historical controls.76 Of note, the STARTS trial (in patients with congenital heart disease) found increased mortality with high dose Sildenafil but only performed in children 1–17 years old and thus did not adequately evaluate younger infants with BPD/PAH.77 Tadalafil acts in the same manner as Sildenafil but may be a more favorable option because of less frequent dosing requirements (once daily as opposed to every 6–12 hours).67
L-Citrulline
L-Citrulline is an amino acid that has the potential to affect the NO and cyclic guanoside monophosphate (cGMP) cascade. Its use leads to conversion to L-arginine, which is the substrate of eNOS that produces nitric oxide. It is speculated that L-Citrulline can promote eNOS recoupling in PAH to improve nitric oxide production and prevent hypoxemia-induced injury in newborn piglets.78 Although L-Citrulline has been shown to improve pulmonary hypertension in animal models, it has not been adequately tested in the human neonatal population.
Milrinone
Milrinone is a Phosphodiesterase 3 (PDE3) inhibitor, whose use leads to increased cAMP and increased vasodilation. Milrinone also has inotropic and lusitropic effects, which can assist with the cardiovascular dysfunction often associated with BPD and PAH.13 Retrospective studies looking at milrinone use as an effective therapy for PAH showed consistent improvement in oxygenation.79 There are no randomized controlled studies in neonates, but milrinone has been shown to result in relaxation of pulmonary arteries in ovine models of PAH in a dose-dependent manner.80 There were a few cases of intracerebral hemorrhage raising concerns for safety in the preterm infant population, but subsequent case reports have shown improvement in oxygenation and cardiac output with Milrinone without the development of hemorrhage in a small group of late preterm infants.81
Bosentan
Bosentan is an Endothelin-1 (ET-1) receptor antagonist, whose use leads to the prevention of the effects of the neurohormone ET-1, which is released from vascular endothelium and causes vasoconstriction.82 In addition, Bosentan is speculated to have a direct anti-fibrotic effect, which is helpful in BPD.83 It is used most frequently in doses of 1–2 mg/kg/dose twice daily. Infants given Bosentan had often failed to respond optimally to Sildenafil. There have been concerns regarding the development of elevated liver transaminases with prolonged use, so levels need to be monitored frequently.84 In a double-masked clinical trial evaluating the safety and efficacy of Bosentan and Sildenafil, Bosentan was found to be as effective as Sildenafil in reducing pulmonary arterial pressure and cardiac dysfunction, and the length of required treatment was significantly shorter with Bosentan without additional adverse effects.85
Iloprost/Epoprostenol/Treprostinil
Iloprost, Epoprostenol, and Treprostinil are prostacyclin (PGI2) analogues, whose use leads to vasodilation. PGI2 is an arachidonic metabolite formed in the vascular endothelium; increased levels lead to greater cyclic adenosine monophosphate (cAMP) formation and therefore, calcium-induced smooth muscle relaxation (Figure 2).54 Iloprost is given as a continuous intravenous infusion at a dose of 0.2–10 ng/kg/min, or by inhalation at a dose of 2.5–5 mcg six to nine times per day. Adequately powered, multicenter randomized controlled trials are still needed to address efficacy, safety, mortality, and long-term outcomes in infants with BPD/PAH. Epoprostenol is given as a continuous IV infusion through a dedicated central venous catheter at a dose of 20 ng/kg/min. There are reported cases of improved pulmonary artery pressures and normalization of right ventricular dilation and function in the setting of BPD.86 Treprostinil can be given by continuous intravenous or subcutaneous (SQ) infusions. It is dosed up to 20 ng/kg/min SQ via slow titration. A transition from epoprostenol to treprostinil typically requires a 1.5–2.0 fold increase in dose. A major complication of subcutaneous administration of prostaglandin analogues is pain at the infusion site. Treprostinil has a longer half-life than Epoprostenol, so there is less of a concern for a severe pulmonary hypertensive episode if the infusion is abruptly stopped or interrupted.87 Recent studies have shown it to be safe in infants, with improvement in PAH and decreased respiratory support with no instances of severe site erythema, bleeding, bruising, or infection.88 Abrupt withdrawal and interruptions in the delivery of any prostaglandin analogue may precipitate pulmonary crises. Controlled weaning is recommended. Overall, there are limited data on the efficacy or safety of medications for the treatment of PAH in infants with BPD. In adults with connective tissue disorders and PAH, improved exercise capacity, less dyspnea, and fewer symptoms of PAH have been reported.89,90
Inhaled Pulmonary Vasodilators
Aerosolized prostacyclin mimics endogenous prostacyclin generated by pulmonary endothelium and is a potent vasodilator. Interest has grown in the use of aerosolized prostacyclin and prostacyclin analogues for the treatment of PAH during mechanical ventilation given its non-invasive administration and reduced systemic side effects compared to intravenous administration. Airway administration of these drugs can improve oxygenation, lower pulmonary vascular resistance, increase pulmonary blood flow, and reduce right-to-left shunting. There is a lack of clinical trials to address safety and efficacy. Clearance of prostacyclin occurs rapidly in the lung with a half-life of 6.5–10 minutes, but newer generation analogues have longer half-lives, up to 4.4–4.6 hours. Disadvantages include rebound PAH with severe hypoxemia if discontinuation is abrupt. There are no FDA-approved devices for aerosolizing prostacyclin in conjunction with mechanical ventilation and no administration device is currently available to accurately measure the delivered dosage of the continuous aerosolized drug. Another concern is the possible disturbance of the pH of the lung surface with an administration which can result in inflammation, bronchial hyper-reactivity, mucociliary lung clearance and increased epithelial damage, as prostacyclin must be dissolved in a buffer solution designed for intravenous administration rather than inhalation.91
Stem Cell Therapy
Mesenchymal Stem Cells (MSCs)
Mesenchymal stem cells (MSCs) are involved in lung development. Stem cell dysfunction is thought to prevent the self-repair of immature lung tissue leading to the development of BPD. Studies have also shown that depletion or dysfunction of endogenous stem cells increases the risk of BPD.71 In animal models, the use of exogenous stem cells as a supplement to impaired endogenous mechanisms can repair lung injury. The thought behind the use of MSCs to prevent BPD is through the prevention of lung inflammation via a paracrine effect rather than engraftment and differentiation into the target cell type. MSCs have also been shown to increase bronchoalveolar stem cell growth efficiency, improve secretion of vascular endothelial growth factor to improve angiogenesis, and have bactericidal effects.92 Transplanted stem cells can react to the microenvironment of the injured lung signals to enhance regeneration.93 MSCs are relatively heterogeneous and the source may affect the therapeutic efficacy.93 MSCs were discovered in umbilical cord tissue and cord blood to serve as a feasible source for the treatment of the neonatal population.71 MSCs obtained from gestational tissue have been shown to have lower immunogenicity, greater proliferation ability, and higher paracrine potency than MSCs from adult sources, but they have also raised ethical issues.
MSCs are able to inhibit the proliferation and function of immune cells, do not engraft, and then disappear, suggesting long-term survival is not required for beneficial gain.93 Since therapeutic efficacy is based on paracrine potency, it is possible that measuring paracrine factors, such as VEGF, could serve as a measurable biomarker for therapeutic efficacy. The optimal pre-conditioning regimen to ensure maximal paracrine potency, genetic engineering for overexpression of specific paracrine regenerative factors, identification of infants at highest risk of developing BPD, determining the appropriate route, and defining the appropriate timing (as benefits from MSC transplantation seem to occur only during the narrow, early inflammatory phase) are all key factors affecting future use of MSCs.93 The first Phase I clinical trial was completed in 2014 in nine premature infants with no adverse effects noted in the short-term or at 2-year follow-up.71 Of note, long-term studies in rats have shown sustained protective effects after the use of MSCs including improved alveolarization and angiogenesis at time periods similar to that of a human adolescent and middle-aged adult.93
Extracellular Vesicles
Extracellular vesicles (EVs) are lipid-bilayer enclosed vesicles that are released by cells containing bioactive compounds that play a role in cell-to-cell communication. There are three subclasses, including small EVs (secreted by the endosomal pathway and contain bioactive cargo from the parental cell), large EVs (which bud from the plasma membrane and have biomarkers that can indicate tissue injury), and apoptotic bodies (released from apoptotic cells containing DNA and proteins). Because of a lack of standardized processes for purification and characterization of EVs, this treatment is still considered experimental and limited to animal studies. Reported beneficial effects of MSC-EVs in rodents with BPD include improvement in pulmonary parenchymal and vascular development.94 It is speculated that umbilical cord MSC-EVs contain TSG-6, an immunomodulatory molecule with anti-inflammatory properties.95,96 Before EVs can be explored as a true therapeutic option in humans, more information about isolation techniques, administration, and safety will need to be explored.93
Growth Factors
Growth factors, including VEGF, platelet-derived growth factor-A, hypoxia-inducible factor-1alpha, and insulin-like growth factor, promote alveolar structural development and are decreased in BPD. Pigment epithelium-derived factor and endothelial monocyte activating polypeptide II, which are anti-angiogenic factors, are also decreased. IGF-1 is important for pulmonary development. IGFBP-1 can prevent growth by decreasing the bioavailability of IGF-1. Preterm infants who develop BPD have lower serum concentrations of IGF-1 after birth.97
A therapy to increase serum IGF-1 at early postnatal ages is a possible option to ameliorate BPD. In antenatal rat studies, treatment with IGF-1 and its binding protein was able to protect lung structure.98 VEGF is an angiogenic agent that helps with vascular development and repair in the developing lung. Animal models have shown that a decrease in VEGF is an important factor contributing to the development of BPD.99,100 HIFs are important in fetal lung development, and fetal preterm lambs and rats have decreased HIF-1 alpha when mechanically ventilated or exposed to high levels of oxygen.101,102 Intra-tracheal administration for the restoration of HIF-1alpha improved vascular and alveolar growth in hyperoxia-exposed newborn rat models.103 There are several growth factors that when elevated lead to clinical characteristics consistent with BPD, including EMAP II, TGF-alpha, TGF-beta, and CTGF (connective tissue growth factor), for which antibodies/inhibitors to these proteins have potential roles as future therapies.93
Micro-RNAs (miRs)
miRs are small non-coding RNAs that act to control gene expression of protein-encoding genes, with a dysregulation in the expression of miRs contributing to the development of lung disorders. There are several miRs that have been found to be associated with early lung development, which contribute to branching morphogenesis of the lung, maturation of lung epithelium, type II pneumocyte maturation, bud morphogenesis, and proliferation of mesenchymal progenitors. miRs also have a role in late lung development, including reduction in oxidative stress and cellular apoptosis, morphogenesis of alveoli and lung capillaries, and mediation of cell adhesion and migration. Decreased levels of certain miRs are associated with an increased risk of severe BPD. Improved miR-29b expression to improve alveolarization has been noted as a possible future treatment of severe BPD.104 miR-34a involved in signaling with Type II alveolar epithelial cell apoptosis and its inhibition is also a possible future therapy to improve or prevent hyperoxia-induced lung injury.105 More information regarding changes in miR levels in the lungs, systemic effects, and feasible miR targets will need to be gathered before it can serve as a therapeutic option for BPD.93
Other Immune Modulators
Several cell signaling molecules and proteins in a variety of inflammatory pathways have been proposed as targets to ameliorate the BPD inflammatory cascade. The challenge in modifying these inflammatory processes is that many of these targets, which may result in BPD if they are abundant or deficient, are necessary at certain stages for appropriate lung development. Proposed signaling molecules that may be targeted and may influence the development of BPD include Macrophage Migration Inhibitory Factor and the cytokine IL-1β. Surfactant Protein D (SP-D), which has pulmonary immune functions, has also been proposed as an immunomodulatory agent that may affect the development of BPD.93
Macrophage Migration Inhibitory Factor (MIF) is a protein involved in innate immunity and appears to be involved in lung development, angiogenesis, and host defense.93 In studies involving mice with genes that resulted in both absence and over-expression of MIF, abnormal pulmonary development resulted. However, over-expression of MIF appeared to provide some benefit when such mice were exposed to conditions that resulted in BPD in wild-type mice.106,107 In a similar study published in 2020, premature mice with lungs at the saccular stage of development (which correlates roughly to 27–36 weeks’ gestational age in humans) were randomized to either room air or a hyperoxic environment to induce BPD; administration of an agent that promotes MIF production resulted in improved lung, cardiac, and vascular findings at autopsy, consistent with a decreased likelihood of BPD development.108 While this study was promising, the effect on non-pulmonary organ systems when targeting the MIF pathway must also be investigated, as MIF is ubiquitously expressed.93
IL-1β is a predominantly pro-inflammatory cytokine. In mice, over-expression of IL-1β during the later stages of lung development results in a lung disease similar to BPD. IL-1RA anakinra, an IL-1RA antagonist that opposes the generation of IL-1β, has been used in some settings to treat disorders associated with IL-1β expression;109 however, the benefit of these types of agents may be limited because of the lack of modifications of other mediators within the IL-1β inflammatory pathway. Nevertheless, some benefit has been shown in anakinra-treated mice in BPD models induced by exposure to LPS and hypoxia,110,111 though studies of administration of anakinra at the stages of lung development in which preterm human infants are at greatest risk (late canalicular, early saccular) have not yet been performed.93
Surfactant protein D (SP-D), a protein within the surfactant molecule, is involved in host defense and is not present in exogenously administered surfactant preparations. Its administration may reduce the incidence of BPD. However, its hydrophobic properties render it unable to be extracted from lung tissue, and to date, an effective recombinant SP-D that could be administered to patients is not available.93
Conclusion
Bronchopulmonary dysplasia continues to be the leading cause of long-term respiratory morbidity in preterm infants. The etiology and pathogenesis are complex and involve multiple pathways. Current treatments and areas under investigation include anti-inflammatories, diuretics, steroids, pulmonary vasodilators, antioxidants, mesenchymal stem cells, and a number of molecules involved in the cell signaling cascade. These are summarized in Table 1. No single agent is likely to have a major impact on reducing its incidence or severity, but the active investigation of multiple pharmaceuticals gives promise of future success.
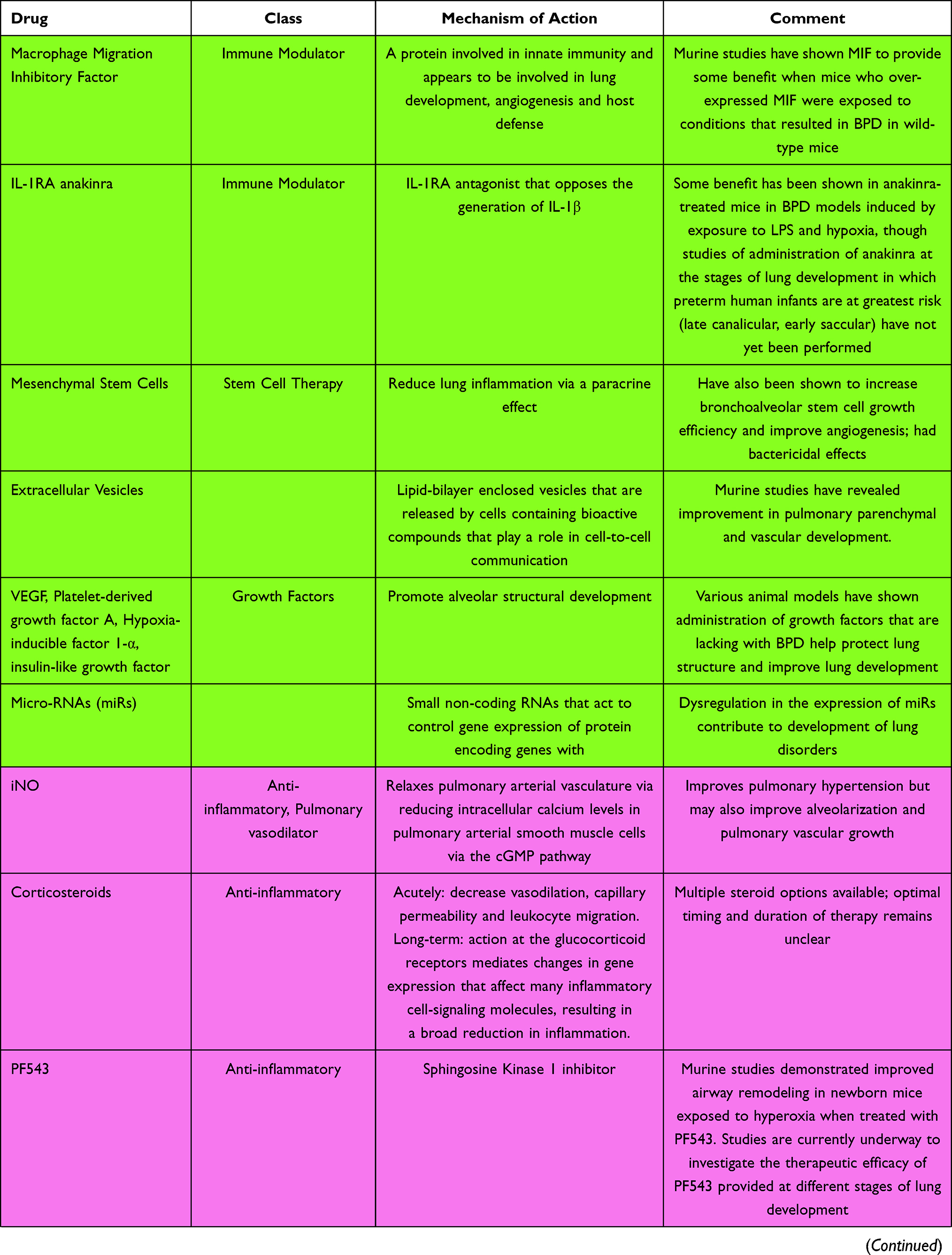 | 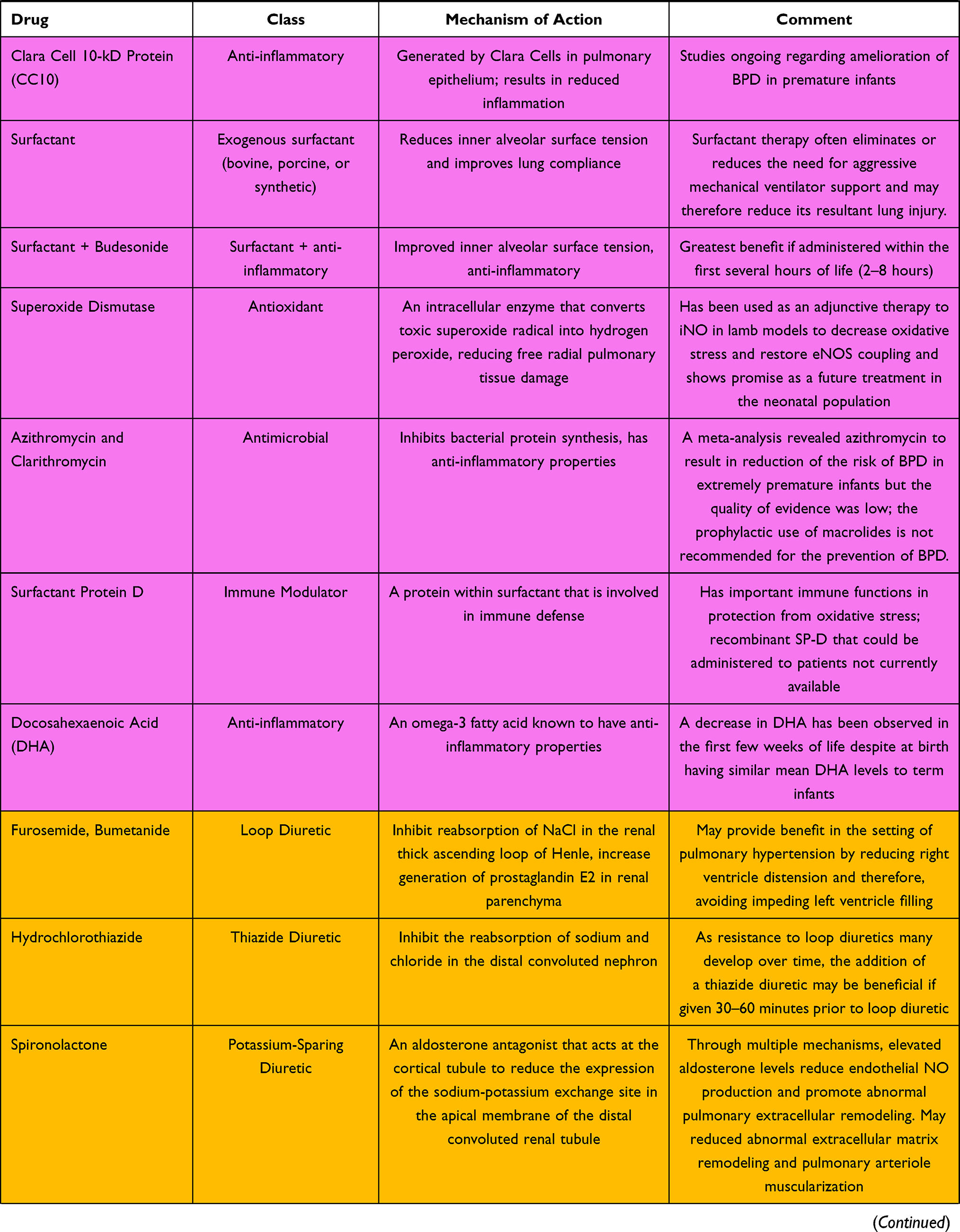 | 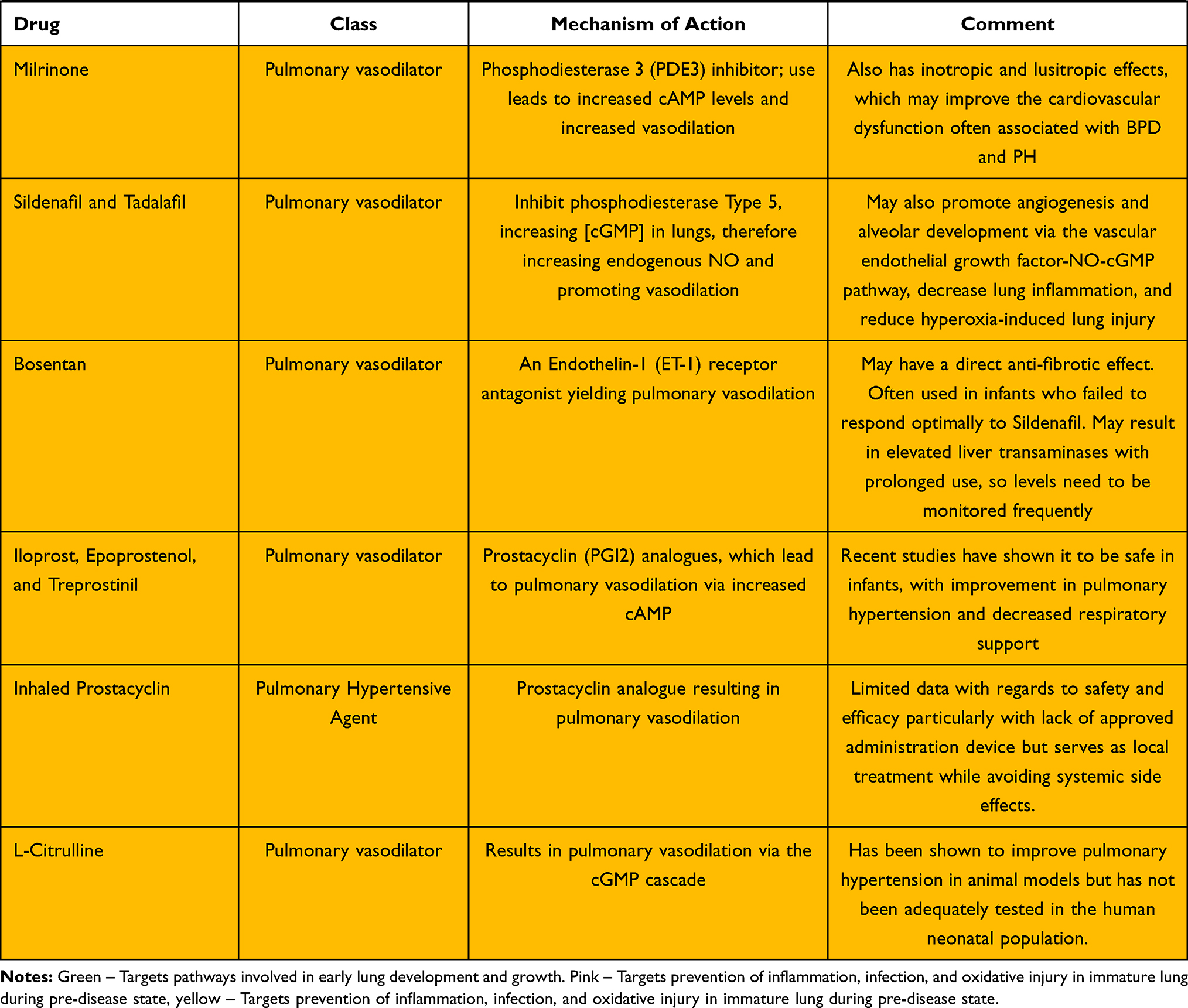 |
Table 1 A Summary of Drugs for the Treatment of BPD and Mechanisms of Action |
Disclosure
The authors report no conflicts of interest in this work.
References
1. Northway WH, Rosan RC, Porter DY. Pulmonary disease following respirator therapy of hyaline-membrane disease. Bronchopulmonary dysplasia. N Engl J Med. 1967;276(7):357–368. doi:10.1056/NEJM196702162760701
2. Day CL, Ryan RM. Bronchopulmonary dysplasia: new becomes old again! Pediatr Res. 2017;81(1–2):210–213. doi:10.1038/pr.2016.201
3. Jobe AH, Bancalari E. Bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2001;163(7):1723–1729. doi:10.1164/ajrccm.163.7.2011060
4. Thébaud B, Goss KN, Laughon M, et al. Bronchopulmonary dysplasia. Nat Rev Dis Primers. 2019;5(1):78.
5. Attar MA, Donn SM. Mechanisms of ventilator-induced lung injury in premature infants. Semin Neonatol. 2002;7(5):353–360. doi:10.1053/siny.2002.0129
6. McEvoy CT, Jain L, Schmidt B, Abman S, Bancalari E, Aschner JL. Bronchopulmonary dysplasia: NHLBI Workshop on the Primary Prevention of Chronic Lung Diseases. Ann Am Thorac Soc. 2014;11(Suppl3):S146–S153. doi:10.1513/AnnalsATS.201312-424LD
7. Stoll BJ, Hansen NI, Bell EF, et al. Neonatal outcomes of extremely preterm infants from the NICHD Neonatal Research Network. Pediatrics. 2010;126(3):443–456. doi:10.1542/peds.2009-2959
8. Trembath A, Laughon MM. Predictors of bronchopulmonary dysplasia. Clin Perinatol. 2012;39(3):585–601. doi:10.1016/j.clp.2012.06.014
9. Laughon MM, Langer JC, Bose CL, et al. Prediction of bronchopulmonary dysplasia by postnatal age in extremely premature infants. Am J Respir Crit Care Med. 2011;183(12):1715–1722. doi:10.1164/rccm.201101-0055OC
10. Goldsmith JP, Karotkin EH. Assisted Ventilation of the Neonate.
11. National Center for Health S. National Vital Statistics Reports: From the Centers for Disease Control and Prevention, National Center for Health Statistics, National Vital Statistics System. Monthly Vital Statistics Report (Hyattsville, Md). Hyattsville, Md: The Center; 1998.
12. Polin RA, Yoder MC. Workbook in Practical Neonatology. Elsevier Health Sciences; 2019.
13. Papoff P, Cerasaro C, Caresta E, Barbara CS, Midulla F, Moretti C. Current strategies for treating infants with severe bronchopulmonary dysplasia. J Matern Fetal Neonatal Med. 2012;25(Suppl 3):15–20. doi:10.3109/14767058.2012.712352
14. Creuwels LA, van Golde LM, Haagsman HP. The pulmonary surfactant system: biochemical and clinical aspects. Lung. 1997;175(1):1–39. doi:10.1007/PL00007554
15. St John EB, Carlo WA. Respiratory distress syndrome in VLBW infants: changes in management and outcomes observed by the NICHD Neonatal Research Network. Semin Perinatol. 2003;27(4):288–292. doi:10.1016/S0146-0005(03)00056-9
16. Herting E, Hartel C, Gopel W. Less invasive surfactant administration: best practices and unanswered questions. Curr Opin Pediatr. 2020;32(2):228–234. doi:10.1097/MOP.0000000000000878
17. Laughon M, Bose C, Moya F, et al. A pilot randomized, controlled trial of later treatment with a peptide-containing, synthetic surfactant for the prevention of bronchopulmonary dysplasia. Pediatrics. 2009;123(1):89–96. doi:10.1542/peds.2007-2680
18. Hascoët JM, Picaud JC, Ligi I, et al. Late surfactant administration in very preterm neonates with prolonged respiratory distress and pulmonary outcome at 1 year of age: a randomized clinical trial. JAMA Pediatr. 2016;170(4):365–372. doi:10.1001/jamapediatrics.2015.4617
19. Ballard RA, Keller RL, Black DM, et al. Randomized trial of late surfactant treatment in ventilated preterm infants receiving inhaled nitric oxide. J Pediatr. 2016;168:23–9.e4. doi:10.1016/j.jpeds.2015.09.031
20. Kothe TB, Sadiq FH, Burleyson N, Williams HL, Anderson C, Hillman NH. Surfactant and budesonide for respiratory distress syndrome: an observational study. Pediatr Res. 2020;87(5):940–945. doi:10.1038/s41390-019-0663-6
21. Zhong YY, Li JC, Liu YL, et al. Early intratracheal administration of corticosteroid and pulmonary surfactant for preventing bronchopulmonary dysplasia in preterm infants with neonatal respiratory distress syndrome: a meta-analysis. Curr Med Sci. 2019;39(3):493–499. doi:10.1007/s11596-019-2064-9
22. Yeh TF, Chen CM, Wu SY, et al. Intratracheal administration of budesonide/surfactant to prevent bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2016;193(1):86–95. doi:10.1164/rccm.201505-0861OC
23. Zhu Y, Guo C, Cao L, Gong X, Wang C, Sun B. Different effects of surfactant and inhaled nitric oxide in modulation of inflammatory injury in ventilated piglet lungs. Pulm Pharmacol Ther. 2005;18(4):303–313. doi:10.1016/j.pupt.2005.01.005
24. El Kebir D, Taha R, Hubert B, Gauvin D, Gangal M, Blaise G. The anti-inflammatory effect of inhaled nitric oxide on pulmonary inflammation in a swine model. Can J Physiol Pharmacol. 2005;83(3):252–258. doi:10.1139/y05-008
25. Tang JR, Markham NE, Lin YJ, et al. Inhaled nitric oxide attenuates pulmonary hypertension and improves lung growth in infant rats after neonatal treatment with a VEGF receptor inhibitor. Am J Physiol. 2004;287(2):L344–L351. doi:10.1152/ajplung.00291.2003
26. Lin YJ, Markham NE, Balasubramaniam V, et al. Inhaled nitric oxide enhances distal lung growth after exposure to hyperoxia in neonatal rats. Pediatr Res. 2005;58(1):22–29. doi:10.1203/01.PDR.0000163378.94837.3E
27. Bland RD, Albertine KH, Carlton DP, MacRitchie AJ. Inhaled nitric oxide effects on lung structure and function in chronically ventilated preterm lambs. Am J Respir Crit Care Med. 2005;172(7):899–906. doi:10.1164/rccm.200503-384OC
28. McCurnin DC, Pierce RA, Chang LY, et al. Inhaled NO improves early pulmonary function and modifies lung growth and elastin deposition in a baboon model of neonatal chronic lung disease. Am J Physiol2005;288(3):L450–L459. doi:10.1152/ajplung.00347.2004
29. Ballard RA, Truog WE, Cnaan A, et al. Inhaled nitric oxide in preterm infants undergoing mechanical ventilation. N Engl J Med. 2006;355(4):343–353. doi:10.1056/NEJMoa061088
30. Hasan SU, Potenziano J, Konduri GG, et al. Effect of inhaled nitric oxide on survival without bronchopulmonary dysplasia in preterm infants: a randomized clinical trial. JAMA Pediatr. 2017;171(11):1081–1089. doi:10.1001/jamapediatrics.2017.2618
31. Barrington KJ, Finer N, Pennaforte T. Inhaled nitric oxide for respiratory failure in preterm infants. Cochrane Database Syst Rev. 2017;1(1):Cd000509.
32. Jobe AH. Chapter 20: prenatal and postnatal steroids and pulmonary outcomes. In: Bancalari E, editor. The Newborn Lung.
33. Cummings JJ, D’Eugenio DB, Gross SJ. A controlled trial of dexamethasone in preterm infants at high risk for bronchopulmonary dysplasia. N Engl J Med. 1989;320(23):1505–1510. doi:10.1056/NEJM198906083202301
34. Doyle LW, Halliday HL, Ehrenkranz RA, Davis PG, Sinclair JC. Impact of postnatal systemic corticosteroids on mortality and cerebral palsy in preterm infants: effect modification by risk for chronic lung disease. Pediatrics. 2005;115(3):655–661. doi:10.1542/peds.2004-1238
35. Kothadia JM, O’Shea TM, Roberts D, Auringer ST, Weaver RG, Dillard RG. Randomized placebo-controlled trial of a 42-Day tapering course of dexamethasone to reduce the duration of ventilator dependency in very low birth weight infants. Pediatrics. 1999;104(1):22–27. doi:10.1542/peds.104.1.22
36. Cuna A, Lewis T, Dai H, Nyp M, Truog WE. Timing of postnatal corticosteroid treatment for bronchopulmonary dysplasia and its effect on outcomes. Pediatr Pulmonol. 2019;54(2):165–170. doi:10.1002/ppul.24202
37. Doyle LW, Davis PG, Morley CJ, McPhee A, Carlin JB, Investigators DS. Low-dose dexamethasone facilitates extubation among chronically ventilator-dependent infants: a multicenter, international, randomized, controlled trial. Pediatrics. 2006;117(1):75–83. doi:10.1542/peds.2004-2843
38. Doyle LW, Davis PG, Morley CJ, McPhee A, Carlin JB. Outcome at 2 years of age of infants from the DART study: a multicenter, international, randomized, controlled trial of low-dose dexamethasone. Pediatrics. 2007;119(4):716–721. doi:10.1542/peds.2006-2806
39. Baud O, Maury L, Lebail F, et al. Effect of early low-dose hydrocortisone on survival without bronchopulmonary dysplasia in extremely preterm infants (PREMILOC): a double-blind, placebo-controlled, multicentre, randomised trial. Lancet. 2016;387(10030):1827–1836. doi:10.1016/S0140-6736(16)00202-6
40. Ofman G, Perez M, Farrow KN. Early low-dose hydrocortisone: is the neurodevelopment affected? J Perinatol. 2018;38(6):636–638. doi:10.1038/s41372-018-0086-y
41. Lewis T, Truog W, Norberg M, Ballard PL, Torgerson D. Genetic variation in CRHR1 is associated with short-term respiratory response to corticosteroids in preterm infants at risk for bronchopulmonary dysplasia. Pediatr Res. 2019;85(5):625–633. doi:10.1038/s41390-018-0235-1
42. Lewis T, Chalise P, Gauldin C, Truog W. Pharmacometabolomics of respiratory phenotypic response to dexamethasone in preterm infants at risk for bronchopulmonary dysplasia. Clin Transl Sci. 2019;12(6):591–599. doi:10.1111/cts.12659
43. Lee J, Yeganeh B, Ermini L, Post M. Sphingolipids as cell fate regulators in lung development and disease. Apoptosis. 2015;20(5):740–757. doi:10.1007/s10495-015-1112-6
44. Cheresh P, Kim SJ, Huang LS, et al. The sphingosine kinase 1 inhibitor, PF543, mitigates pulmonary fibrosis by reducing lung epithelial cell mtDNA damage and recruitment of fibrogenic monocytes. Int J Mol Sci. 2020;21(16):5595. doi:10.3390/ijms21165595
45. Ha AW, Sudhadevi T, Ebenezer DL, et al. Neonatal therapy with PF543, a sphingosine kinase 1 inhibitor, ameliorates hyperoxia-induced airway remodeling in a murine model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2020;319(3):L497–L512. doi:10.1152/ajplung.00169.2020
46. Levine CR, Gewolb IH, Allen K, et al. The safety, pharmacokinetics, and anti-inflammatory effects of intratracheal recombinant human Clara cell protein in premature infants with respiratory distress syndrome. Pediatr Res. 2005;58(1):15–21. doi:10.1203/01.PDR.0000156371.89952.35
47. Davis JM, Pilon AL, Shenberger J, et al. The role of recombinant human CC10 in the prevention of chronic pulmonary insufficiency of prematurity. Pediatr Res. 2019;86(2):254–260. doi:10.1038/s41390-019-0419-3
48. Center TM Efficacy of recombinant human clara cell 10 protein (rhCC10) administered to premature neonates with respiratory distress syndrome; 2019. [(ClinicalTrials.gov Identifier: NCT01941745)]. Available from: https://clinicaltrials.gov/ct2/show/study/NCT01941745.
49. Martin CR, Dasilva DA, Cluette-Brown JE, et al. Decreased postnatal docosahexaenoic and arachidonic acid blood levels in premature infants are associated with neonatal morbidities. J Pediatr. 2011;159(5):743. doi:10.1016/j.jpeds.2011.04.039
50. Makrides M, Gibson RA, McPhee AJ, et al. Neurodevelopmental outcomes of preterm infants fed high-dose docosahexaenoic acid: a randomized controlled trial. JAMA. 2009;301(2):175–182. doi:10.1001/jama.2008.945
51. Collins CT, Makrides M, McPhee AJ, et al. Docosahexaenoic acid and bronchopulmonary dysplasia in preterm infants. N Engl J Med. 2017;376(13):1245–1255. doi:10.1056/NEJMoa1611942
52. Tanaka K, Tanaka S, Shah N, Ota E, Namba F. Docosahexaenoic acid and bronchopulmonary dysplasia in preterm infants: a systematic review and meta-analysis. J Matern Fetal Neonatal Med. 2020;1–9.
53. Gentyala RR, Ehret D, Suresh G, Soll R. Superoxide dismutase for preventing bronchopulmonary dysplasia (BPD) in preterm infants. Cochrane Database Syst Rev. 2019;2019(1):CD013232. doi:10.1002/14651858.CD013232
54. Martinho S, Adão R, Leite-Moreira AF, Brás-Silva C. Persistent pulmonary hypertension of the newborn: pathophysiological mechanisms and novel therapeutic approaches. Front Pediatr. 2020;8. doi:10.3389/fped.2020.00342
55. Davis JM, Parad RB, Michele T, Allred E, Price A, Rosenfeld W. Pulmonary outcome at 1 year corrected age in premature infants treated at birth with recombinant human CuZn superoxide dismutase. Pediatrics. 2003;111(3):469–476. doi:10.1542/peds.111.3.469
56. Gentle SJ, Lal CV. Predicting BPD: lessons learned from the airway microbiome of preterm infants. Front Pediatr. 2019;7:564. doi:10.3389/fped.2019.00564
57. Pammi M, Lal CV, Wagner BD, et al. Airway microbiome and development of bronchopulmonary dysplasia in preterm infants: a systematic review. J Pediatr. 2019;204:126–33.e2. doi:10.1016/j.jpeds.2018.08.042
58. Viscardi RM, Terrin ML, Magder LS, et al. Randomised trial of azithromycin to eradicate Ureaplasma in preterm infants. Arch Dis Child Fetal Neonatal Ed. 2020;105(6):615–622. doi:10.1136/archdischild-2019-318122
59. Smith C, Egunsola O, Choonara I, Kotecha S, Jacqz-Aigrain E, Sammons H. Use and safety of azithromycin in neonates: a systematic review. BMJ Open. 2015;5(12):e008194. doi:10.1136/bmjopen-2015-008194
60. Hammerschlag MR, Gelling M, Roblin PM, Kutlin A, Jule JE. Treatment of neonatal chlamydial conjunctivitis with azithromycin. Pediatr Infect Dis J. 1998;17(11):1049–1050. doi:10.1097/00006454-199811000-00020
61. Ozdemir R, Erdeve O, Dizdar EA, et al. Clarithromycin in preventing bronchopulmonary dysplasia in Ureaplasma urealyticum-positive preterm infants. Pediatrics. 2011;128(6):e1496–1501. doi:10.1542/peds.2011-1350
62. Fuso L, Baldi F, Di Perna A. Therapeutic strategies in pulmonary hypertension. Front Pharmacol. 2011;2:21. doi:10.3389/fphar.2011.00021
63. Maron BA, Opotowsky AR, Landzberg MJ, Loscalzo J, Waxman AB, Leopold JA. Plasma aldosterone levels are elevated in patients with pulmonary arterial hypertension in the absence of left ventricular heart failure: a pilot study. Eur J Heart Fail. 2013;15(3):277–283. doi:10.1093/eurjhf/hfs173
64. Prins KW, Thenappan T, Weir EK, Kalra R, Pritzker M, Archer SL. Repurposing medications for treatment of pulmonary arterial hypertension: what’s old is new again. J Am Heart Assoc. 2019;8(1):e011343. doi:10.1161/JAHA.118.011343
65. Aghamohammadzadeh R, Zhang YY, Stephens TE, et al. Up-regulation of the mammalian target of rapamycin complex 1 subunit Raptor by aldosterone induces abnormal pulmonary artery smooth muscle cell survival patterns to promote pulmonary arterial hypertension. FASEB J. 2016;30(7):2511–2527. doi:10.1096/fj.201500042
66. Hudalla H, Michael Z, Christodoulou N, et al. Carbonic anhydrase inhibition ameliorates inflammation and experimental pulmonary hypertension. Am J Respir Cell Mol Biol. 2019;61(4):512–524. doi:10.1165/rcmb.2018-0232OC
67. Hansmann G, Sallmon H, Roehr CC, et al. Pulmonary hypertension in bronchopulmonary dysplasia. Pediatr Res. 2020. doi:10.1038/s41390-020-0993-4
68. Weismann CG, Asnes JD, Bazzy-Asaad A, Tolomeo C, Ehrenkranz RA, Bizzarro MJ. Pulmonary hypertension in preterm infants: results of a prospective screening program. J Perinatol. 2017;37(5):572–577. doi:10.1038/jp.2016.255
69. Mourani PM, Sontag MK, Younoszai A, et al. Early pulmonary vascular disease in preterm infants at risk for bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2015;191(1):87–95. doi:10.1164/rccm.201409-1594OC
70. Mourani PM, Abman SH. Pulmonary vascular disease in bronchopulmonary dysplasia: pulmonary hypertension and beyond. Curr Opin Pediatr. 2013;25(3):329–337. doi:10.1097/MOP.0b013e328360a3f6
71. Mandell EW, Kratimenos P, Abman SH, Steinhorn RH. Drugs for the prevention and treatment of bronchopulmonary dysplasia. Clin Perinatol. 2019;46(2):291–310. doi:10.1016/j.clp.2019.02.011
72. Banks BA, Seri I, Ischiropoulos H, Merrill J, Rychik J, Ballard RA. Changes in oxygenation with inhaled nitric oxide in severe bronchopulmonary dysplasia. Pediatrics. 1999;103(3):610–618. doi:10.1542/peds.103.3.610
73. Barrington KJ, Finer N, Pennaforte T, Altit G. Nitric oxide for respiratory failure in infants born at or near term. Cochrane Database Syst Rev. 2017;1(1):Cd000399.
74. de Visser YP, Walther FJ, Laghmani El H, Boersma H, van der Laarse A, Wagenaar GT. Sildenafil attenuates pulmonary inflammation and fibrin deposition, mortality and right ventricular hypertrophy in neonatal hyperoxic lung injury. Respir Res. 2009;10(1):30. doi:10.1186/1465-9921-10-30
75. Mourani PM, Sontag MK, Ivy DD, Abman SH. Effects of long-term sildenafil treatment for pulmonary hypertension in infants with chronic lung disease. J Pediatr. 2009;154(3):379. doi:10.1016/j.jpeds.2008.09.021
76. Bhatt-Mehta V, Donn SM. Sildenafil for pulmonary hypertension complicating bronchopulmonary dysplasia. Expert Rev Clin Pharmacol. 2014;7(4):393–395. doi:10.1586/17512433.2014.922867
77. Barst RJ, Ivy DD, Gaitan G, et al. A randomized, double-blind, placebo-controlled, dose-ranging study of oral sildenafil citrate in treatment-naive children with pulmonary arterial hypertension. Circulation. 2012;125(2):324–334. doi:10.1161/CIRCULATIONAHA.110.016667
78. Fike CD, Dikalova A, Kaplowitz MR, Cunningham G, Summar M, Aschner JL. Rescue treatment with L-citrulline inhibits hypoxia-induced pulmonary hypertension in newborn pigs. Am J Respir Cell Mol Biol. 2015;53(2):255–264. doi:10.1165/rcmb.2014-0351OC
79. McNamara PJ, Laique F, Muang-In S, Whyte HE. Milrinone improves oxygenation in neonates with severe persistent pulmonary hypertension of the newborn. J Crit Care. 2006;21(2):217–222. doi:10.1016/j.jcrc.2006.01.001
80. Lakshminrusimha S, Mathew B, Leach CL. Pharmacologic strategies in neonatal pulmonary hypertension other than nitric oxide. Semin Perinatol. 2016;40(3):160–173. doi:10.1053/j.semperi.2015.12.004
81. McNamara PJ, Shivananda SP, Sahni M, Freeman D, Taddio A. Pharmacology of milrinone in neonates with persistent pulmonary hypertension of the newborn and suboptimal response to inhaled nitric oxide. Pediatr Crit Care Med. 2013;14(1):74–84. doi:10.1097/PCC.0b013e31824ea2cd
82. Rosenzweig EB, Ivy DD, Widlitz A, et al. Effects of long-term bosentan in children with pulmonary arterial hypertension. J Am Coll Cardiol. 2005;46(4):697–704. doi:10.1016/j.jacc.2005.01.066
83. Choudhary G, Troncales F, Martin D, Harrington EO, Klinger JR. Bosentan attenuates right ventricular hypertrophy and fibrosis in normobaric hypoxia model of pulmonary hypertension. J Heart Lung Transplant. 2011;30(7):827–833. doi:10.1016/j.healun.2011.03.010
84. Krishnan U, Krishnan S, Gewitz M. Treatment of pulmonary hypertension in children with chronic lung disease with newer oral therapies. Pediatr Cardiol. 2008;29(6):1082–1086. doi:10.1007/s00246-008-9260-x
85. Farhangdoust S, Mehralizadeh S, Bordbar A. Comparison of the effects of bosentan and sildenafil in the treatment of persistent pulmonary arterial hypertension in infants. J Clin Neonatol. 2020;9(4):249–254. doi:10.4103/jcn.JCN_5_20
86. Zaidi AN, Dettorre MD, Ceneviva GD, Thomas NJ. Epoprostenol and home mechanical ventilation for pulmonary hypertension associated with chronic lung disease. Pediatr Pulmonol. 2005;40(3):265–269. doi:10.1002/ppul.20238
87. Krishnan U, Feinstein JA, Adatia I, et al. Evaluation and management of pulmonary hypertension in children with bronchopulmonary dysplasia. J Pediatr. 2017;188(24):24–34.e1. doi:10.1016/j.jpeds.2017.05.029
88. Ferdman DJ, Rosenzweig EB, Zuckerman WA, Krishnan U. Subcutaneous treprostinil for pulmonary hypertension in chronic lung disease of infancy. Pediatrics. 2014;134(1):e274–e278. doi:10.1542/peds.2013-2330
89. Simonneau G, Barst RJ, Galie N, et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165(6):800–804. doi:10.1164/ajrccm.165.6.2106079
90. Oudiz RJ, Schilz RJ, Barst RJ, et al. Treprostinil, a prostacyclin analogue, in pulmonary arterial hypertension associated with connective tissue disease. Chest. 2004;126(2):420–427. doi:10.1378/chest.126.2.420
91. Davis MD, Donn SM, Ward RM. Administration of inhaled pulmonary vasodilators to the mechanically ventilated neonatal patient. Paediatr Drugs. 2017;19(3):183–192. doi:10.1007/s40272-017-0221-9
92. Ee MT, Thébaud B. The therapeutic potential of stem cells for bronchopulmonary dysplasia: “it’s about time” or “not so fast”? Curr Pediatr Rev. 2018;14(4):227–238. doi:10.2174/1573396314666180911100503
93. Bhandari V. Tantalizing therapeutics in bronchopulmonary dysplasia. San Diego: Elsevier Science & Technology; 2020. Available from: https://mgetit.lib.umich.edu/resolve?ctx_ver=Z39.88-2004&ctx_enc=info%3Aofi%2Fenc%3AUTF-8&rfr_id=info%3Asid%2Fsummon.serialssolutions.com&rft_val_fmt=info%3Aofi%2Ffmt%3Akev%3Amtx%3Abook&rft.genre=book&rft.title=Tantalizing+Therapeutics+in+Bronchopulmonary+Dysplasia&rft.au=Bhandari%2C+Vineet&rft.date=2020-01-01&rft.pub=Elsevier+Science+%26+Technology&rft.isbn=9780128189870&rft.externalDocID=EBC6216636.
94. Braun RK, Chetty C, Balasubramaniam V, et al. Intraperitoneal injection of MSC-derived exosomes prevent experimental bronchopulmonary dysplasia. Biochem Biophys Res Commun. 2018;503(4):2653–2658. doi:10.1016/j.bbrc.2018.08.019
95. Wisniewski HG, Vilcek J. TSG-6: an IL-1/TNF-inducible protein with anti-inflammatory activity. Cytokine Growth Factor Rev. 1997;8(2):143–156. doi:10.1016/S1359-6101(97)00008-7
96. Chaubey S, Thueson S, Ponnalagu D, et al. Early gestational mesenchymal stem cell secretome attenuates experimental bronchopulmonary dysplasia in part via exosome-associated factor TSG-6. Stem Cell Res Ther. 2018;9(1):173. doi:10.1186/s13287-018-0903-4
97. Löfqvist C, Hellgren G, Niklasson A, Engström E, Ley D, Hansen-Pupp I. Low postnatal serum IGF-I levels are associated with bronchopulmonary dysplasia (BPD). Acta Paediatr. 2012;101(12):1211–1216. doi:10.1111/j.1651-2227.2012.02826.x
98. Seedorf G, Kim C, Wallace B, et al. rhIGF-1/BP3 preserves lung growth and prevents pulmonary hypertension in experimental bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2020;201(9):1120–1134. doi:10.1164/rccm.201910-1975OC
99. Maniscalco WM, Watkins RH, Pryhuber GS, Bhatt A, Shea C, Huyck H. Angiogenic factors and alveolar vasculature: development and alterations by injury in very premature baboons. Am J Physiol Lung Cell Mol Physiol. 2002;282(4):L811–L823. doi:10.1152/ajplung.00325.2001
100. Tambunting F, Beharry KD, Waltzman J, Modanlou HD. Impaired lung vascular endothelial growth factor in extremely premature baboons developing bronchopulmonary dysplasia/chronic lung disease. J Investig Med. 2005;53(5):253–262. doi:10.2310/6650.2005.53508
101. Hosford GE, Olson DM. Effects of hyperoxia on VEGF, its receptors, and HIF-2alpha in the newborn rat lung. Am J Physiol Lung Cell Mol Physiol. 2003;285(1):L161–L168. doi:10.1152/ajplung.00285.2002
102. Grover TR, Asikainen TM, Kinsella JP, Abman SH, White CW. Hypoxia-inducible factors HIF-1alpha and HIF-2alpha are decreased in an experimental model of severe respiratory distress syndrome in preterm lambs. Am J Physiol Lung Cell Mol Physiol. 2007;292(6):L1345–L1351. doi:10.1152/ajplung.00372.2006
103. Vadivel A, Alphonse RS, Etches N, et al. Hypoxia-inducible factors promote alveolar development and regeneration. Am J Respir Cell Mol Biol. 2014;50(1):96–105. doi:10.1165/rcmb.2012-0250OC
104. Durrani-Kolarik S, Pool CA, Gray A, et al. miR-29b supplementation decreases expression of matrix proteins and improves alveolarization in mice exposed to maternal inflammation and neonatal hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2017;313(2):L339–l49. doi:10.1152/ajplung.00273.2016
105. Syed M, Das P, Pawar A, et al. Hyperoxia causes miR-34a-mediated injury via angiopoietin-1 in neonatal lungs. Nat Commun. 2017;8(1):1173. doi:10.1038/s41467-017-01349-y
106. Bry K, Whitsett JA, Lappalainen U. IL-1beta disrupts postnatal lung morphogenesis in the mouse. Am J Respir Cell Mol Biol. 2007;36(1):32–42. doi:10.1165/rcmb.2006-0116OC
107. Stouch AN, McCoy AM, Greer RM, et al. IL-1beta and inflammasome activity link inflammation to abnormal fetal airway development. J Immunol. 2016;196(8):3411–3420. doi:10.4049/jimmunol.1500906
108. Gilfillan M, Das P, Shah D, Alam MA, Bhandari V. Inhibition of microRNA-451 is associated with increased expression of macrophage migration inhibitory factor and mitigation of the cardio-pulmonary phenotype in a murine model of bronchopulmonary dysplasia. Respir Res. 2020;21(1):92. doi:10.1186/s12931-020-01353-9
109. Sibley CH, Plass N, Snow J, et al. Sustained response and prevention of damage progression in patients with neonatal-onset multisystem inflammatory disease treated with anakinra: a cohort study to determine three- and five-year outcomes. Arthritis Rheum. 2012;64(7):2375–2386. doi:10.1002/art.34409
110. Johnson BH, Yi M, Masood A, et al. A critical role for the IL-1 receptor in lung injury induced in neonatal rats by 60% O2. Pediatr Res. 2009;66(3):260–265. doi:10.1203/PDR.0b013e3181b1bcd2
111. Nold MF, Mangan NE, Rudloff I, et al. Interleukin-1 receptor antagonist prevents murine bronchopulmonary dysplasia induced by perinatal inflammation and hyperoxia. Proc Natl Acad Sci U S A. 2013;110(35):14384–14389. doi:10.1073/pnas.1306859110
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.