Back to Archived Journals » Hypoxia » Volume 3
New perspectives on the molecular basis of the interaction between oxygen homeostasis and iron metabolism
Authors Recalcati S, Gammella E, Cairo G
Received 31 July 2015
Accepted for publication 5 November 2015
Published 11 December 2015 Volume 2015:3 Pages 93—103
DOI https://doi.org/10.2147/HP.S83537
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Prof. Dr. Dörthe Katschinski
Stefania Recalcati, Elena Gammella, Gaetano Cairo
Department of Biomedical Sciences for Health, University of Milan, Milan, Italy
Abstract: Oxygen and iron are two elements closely related from a (bio)chemical point of view. Moreover, they share the characteristic of being indispensable for life, while also being potentially toxic. Therefore, their level is strictly monitored, and sophisticated pathways have evolved to face variations in either element. In addition, the expression of proteins involved in iron and oxygen metabolism is mainly controlled by a complex interplay of proteins that sense both iron levels and oxygen availability (ie, prolyl hydroxylases, hypoxia inducible factors, and iron regulatory proteins), and in turn activate feedback mechanisms to re-establish homeostasis. In this review, we describe how cells and organisms utilize these intricate networks to regulate responses to changes in oxygen and iron levels. We also explore the role of these pathways in some pathophysiological settings.
Keywords: hypoxia, prolyl hydroxylases, hypoxia inducible factors, iron regulatory proteins, proteasome
Introduction
Iron and oxygen are two closely related elements: the large amount of iron that is present in the earth originated approximately 4.6 billion years ago from an oxygen-dependent process occurring during supernova explosions, the thermonuclear disruptions of a white dwarf star in which carbon and oxygen fused into radioactive nickel that decayed quickly into radioactive cobalt, which subsequently decayed into stable iron (56Fe). Recently, proof of this process was provided by the INTEGRAL satellite that detected gamma ray lines from the radioactive decay of nickel into iron.1 The interaction of iron and oxygen continued during the slow and long (1.5 billion years) accumulation in the atmosphere of the oxygen produced by photosynthesizing cyanobacteria that led to the present 20.9% atmospheric oxygen content. Over this period of time, living organisms adapted to exploit the higher metabolic efficiency of aerobic reactions and the chemical reactivity of iron for processes essential for life. In parallel, it has been necessary to evolve protection systems against the damage caused by the iron-catalyzed generation of highly reactive hydroxyl radicals from the less harmful superoxide and hydrogen peroxide (reactive oxygen species) continuously produced by every cell living in aerobic conditions.2 To this purpose, a complex regulatory pathway formed by a variety of proteins that bind, transport, and store iron has been developed, in order to maintain an appropriate iron balance in both the individual cells and the whole body.3
In addition, an adaptive response to hypoxia, both at the cellular and organismal levels and in populations living at high altitude, has evolved in order to ensure the delivery of appropriate levels of oxygen to tissues. Paradoxically, while activation of hypoxia inducible factors (HIF) confers cellular adaptation to hypoxia, in some highlanders (Tibetan and Ethiopians) variants in genes of the HIF pathway (eg, EGLN1 and EPAS1 coding for PHD2 and HIF-2α, respectively, see section: The HIF-hydroxylase system controls cellular oxygen homeostasis) that confer a hypo-responsiveness to hypoxia seem to have been crucial for adaptation to high altitude.4
HIF and IRPs are two interconnected sensors of both iron levels and oxygen availability
The interaction between oxygen and iron is not limited to the oxygen transport function performed by proteins containing heme iron, such as hemoglobin and myoglobin. In fact, the iron regulatory proteins (IRP1 and IRP2) that control cellular iron homeostasis and the hydroxylases that control the stability and activity of HIF sense both iron levels and oxygen availability, and in turn regulate both pathways in a coordinate and interconnected way.
The HIF-hydroxylase system controls cellular oxygen homeostasis
In this paragraph, we will offer only a brief summary of the role of HIF (HIF-1α and HIF-2α) as regulators of oxygen homeostasis, a topic comprehensively described in recent excellent reviews.5,6 The cells respond to conditions of insufficient oxygen availability by increasing the amount and activity of HIF transcription factors, which induce the expression of a large number of genes involved in the response to hypoxia.5,6 The oxygen-regulated HIF-1α and HIF-2α subunits form heterodimers with the constitutively expressed HIF-1β subunit and bind hypoxia responsive elements (HRE) in DNA regulatory regions that can be far from the transcription start site. A third HIF-α (HIF-3α), structurally and functionally distinct, is present in humans and mammals, but its role and mode of action are not completely clear. One reason for this is the presence of alternative splice variants coding for at least six isoforms with structural and functional differences that complicate the study of HIF-3α. Although several studies have led to the view that HIF-3α is a negative regulator of HIF-1α and HIF-2α, recent evidence suggests that HIF-3α activates a distinct transcriptional response to hypoxia, at least in zebrafish.7
The targets of HIF include genes with a direct role in the adaptation to hypoxia, as well as regulatory molecules with a secondary effect on gene expression, such as induction of transcriptional repressors that result in decreased expression of many genes under hypoxic conditions. Notably, genes coding for proteins directly or indirectly involved in iron metabolism are among the variety of HIF target genes (Table 1).8 Another connection between the hypoxic response and iron metabolism is represented by the iron requirement of the prolyl hydroxylases (PHDs) that regulate HIF-α stability. PHDs are members of a family of 2-oxoglutarate-dependent dioxygenases whose activity requires 2-oxoglutarate and oxygen as obligatory substrates and non-heme ferrous iron as cofactor.9,10 In addition to other important functions, such as collagen biosynthesis, these enzymes play an important role in gene expression. In fact, they are involved in the control of both epigenetic (eg, DNA demethylation and histone lysine demethylation) and transcriptional (eg, HIF) regulation. PHDs catalyze prolyl hydroxylation in the oxygen-dependent degradation domains of HIF-α. After this modification, the affinity of HIF-α for the E3 ubiquitin ligase von Hippel-Lindau (VHL) protein increases several hundred fold, thereby leading to HIF-α degradation by the proteasome (Figure 1).9,10 The mechanism by which PHDs act as hypoxia sensors is both efficient and sensitive to changes in oxygen availability, such that under normoxia the inducible HIF-α are extremely short lived, but as oxygen levels fall slightly, the decrease in PHDs activity allows HIF-α levels to rise and the hypoxic response to start. PHDs are also sensors of energy metabolism, as the Krebs cycle intermediates 2-oxoglutarate and succinate and fumarate are an activator or inhibitors, respectively, of 2-oxoglutarate-dependent dioxygenases enzymes. Moreover, PHDs activity is dependent on the availability of ferrous iron, and therefore, HIF-dependent transcription is also induced by iron deprivation, even under normoxia.9,10 Asparaginyl hydroxylation of HIF-1/2α by factor inhibiting HIF, which inhibits their transactivation activity, represents an additional mechanism preventing the hypoxic response when sufficient oxygen and cofactors are available.6
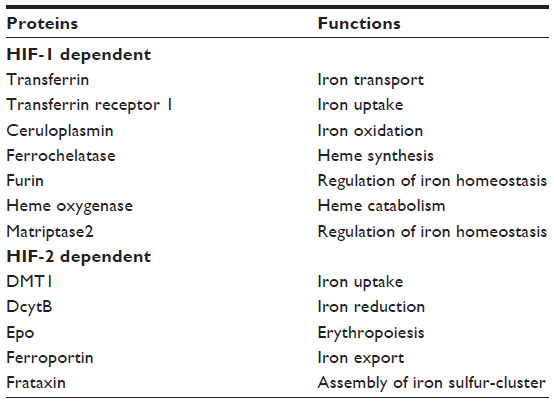 | Table 1 Proteins of iron metabolism regulated by HIF |
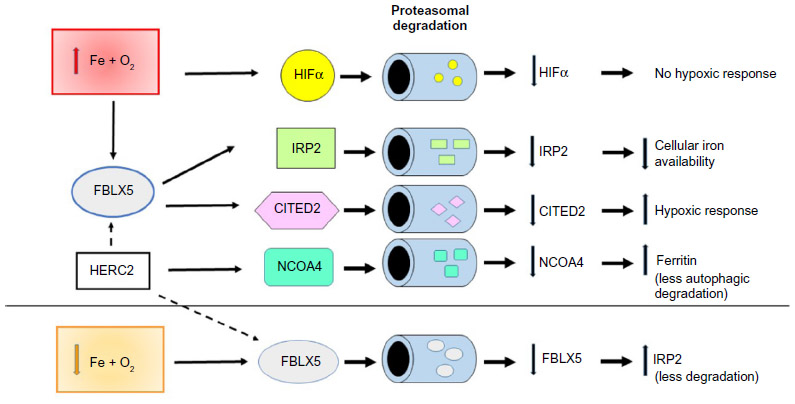 | Figure 1 Different levels of iron and oxygen promote the proteasomal degradation of key regulatory proteins of iron and oxygen metabolism. |
Despite extensive studies, the respective role of HIF-1α and HIF-2α has not been fully clarified. They bind the same HRE in vitro and share an overlapping set of target genes, but they also activate the transcription of distinct genes. For example, both HIF-α bind the HRE of erythropoietin (Epo), but selective inactivation in mice revealed that only HIF-2α is important for Epo production in vivo.11
In this context, the activity of the various components of the HIF pathway can be modulated by various factors, including tissue-specific expression and oxygen sensing. In relation to the interdependence of iron and oxygen homeostasis covered in this review, we would like to point out the direct regulation of HIF-2α mRNA stability by iron (see section: The IRE/IRP regulatory pathaway controls cellular iron homeostasis).
The IRE/IRP regulatory pathway controls cellular iron homeostasis
The challenging task of maintaining intracellular iron levels sufficient for essential cellular functions, but as low as possible to avoid reactive oxygen species formation, is mainly performed by IRP1 and IRP2, which tightly control intracellular iron metabolism by regulating at the post-transcriptional level the expression of genes coding for proteins involved in iron uptake, utilization, storage, and export.8,12–14
All these proteins are coded by mRNAs with conserved 25–30 nucleotide-long sequences in their untranslated regions that are able to form stem-loop structures called iron responsive elements (IREs).
IRP1 is a “moonlighting” protein that can perform two entirely different functions; when sufficient iron is available, IRP1 can assemble a [4Fe-4S] cluster becoming the cytosolic counterpart of mitochondrial aconitase and catalyzing the conversion of citrate and isocitrate, a function that may affect NADPH levels and lipid biosynthesis.15 Conversely, in conditions of iron deficiency, IRP1 cannot form the cluster and the apoform is able to bind IREs, and thus control the expression of a variety of mRNAs (Figure 2).
 | Figure 2 Differential iron- and oxygen-dependent regulation of IRP activity. |
IRP2, which is highly homologous to IRP1, binds IRE motifs with affinity and specificity similar to that of IRP1, but is controlled by a different mechanism. In fact, IRP2 is not able to assemble a [4Fe-4S] cluster, and hence is not regulated by the switch between an apoform and a cluster-containing form; conversely, the protein accumulated in iron-deficient cells and in iron-replete cells is rapidly targeted for proteasomal degradation by an E3 ubiquitin ligase complex that comprises FBXL5 (see section: The iron-oxygen axis: regulation at the cellular level).16
When iron inside the cell is scarce, active IRPs bind to IREs and stabilize the mRNA for transferrin receptor (TfR1) and divalent metal transporter DMT1, thus increasing the uptake of both transferrin-bound and unbound iron, while also decreasing translation of mRNAs for the iron storage protein ferritin and the iron exporter ferroportin. This coordinated regulation eventually enhances iron availability within the cell. Conversely, in iron-replete cells, the IRE-binding activity of both IRPs is decreased, resulting in efficient translation of ferritin and ferroportin mRNAs and lower stability of TfR1 and DMT1 mRNAs, eventually increasing iron sequestration and release over uptake (Figure 2).
Targeted deletion of either IRPs has extended our knowledge of their respective role and importance. Embryonic lethality in mice lacking both IRPs17 showed that the IRP/IRE regulatory system is vital. However, the two IRPs can compensate for each other, as expression of either IRP1 or IRP2 is sufficient for life. Early studies found mild hematopoietic defects and late-onset neurodegeneration in Irp2−/− mice, whereas mice lacking IRP1 were apparently normal.8,12–14 In line with these findings showing a dominant role for IRP2 in the regulation of iron homeostasis in mice, analysis of the comparative expression of the two IRPs indicated that IRP2 is the major regulator of intracellular iron metabolism in humans.18 However, more recent results showed an important role for IRP1 in the control of the IRE-containing mRNA for erythroid aminolevulinate synthase in anemic zebrafish.19 Moreover, it appears that the IRE in HIF-2α mRNA is predominantly bound by IRP1, as mice with deletion of IRP1 maintained on a low iron diet have derepressed HIF-2α expression that leads to pulmonary hypertension and polycythemia.20
The iron–oxygen axis: regulation at the cellular level
Both IRPs are regulated by multiple factors in addition to iron, including oxygen tension, which differentially controls their binding activity (reviewed and discussed in Recalcati et al,8 Pantopoulos et al,12 Zhang et al,13 Kühn,14 and Cairo and Recalcati21). Most studies suggest that hypoxia decreases IRP1 binding to IREs, and results in IRP1 acquiring aconitase activity whereas lack of oxygen increases IRP2 activity (Figure 2).8,21 In this context, a key role is played by FBXL5, a recently identified and characterized protein that is regulated in an iron- and oxygen-dependent manner thanks to an hemerythrin-like domain (Figure 1).16 When iron and oxygen are fully available, FBXL5 levels in the cell increase promoting IRP2 (and apo IRP1) proteasomal degradation. On the other hand, lack of iron, which impairs the assembly of the di-iron center in the hemerythrin-like domain, or hypoxia leads to FBXL5 polyubiquitination and degradation by the proteasome, thus stabilizing IRP2 and increasing iron availability. A recent study demonstrated that FBXL5 itself is constitutively targeted to ubiquitin-dependent proteasomal degradation through interaction with HERC2,22 a protein involved in the response to DNA damage. As iron is a cofactor of several DNA repair proteins, these findings reveal an interesting new link between iron and an essential function required for cell survival.
The effects of hypoxia on IRPs binding activity are not the only link between oxygen and iron homeostasis. As reported above, the lack of oxygen and/or iron inhibits the hydroxylation process that results in proteasomal degradation of HIF-1/2α, thereby increasing protein stability and transcriptional capacity.5,6
Moreover, the finding of a functional 5′ IRE in the mRNA for HIF-2α showed the existence of another physiologically relevant connection between oxygen and iron sensing.13,23 In normoxia, active IRP1 represses basal HIF-2α translation, whereas in hypoxic conditions the IRP1/IRE interaction is impaired and HIF-2α is efficiently translated, thus suggesting that IRP1 acts as a direct or indirect sensor of hypoxia. In hypoxia, the combination of increased HIF-2α mRNA translation due to impaired IRP1-binding activity and inhibited HIF-2α degradation leads to a strong increase in HIF-2α protein levels (Figure 3). Cellular iron deficiency hampers PHD activity as well, but also increases IRP1 activity, thus repressing HIF-2α translation. This suggests a negative feedback control of the HIF-mediated response when iron levels are low, in order to fine tune iron availability and utilization. This may be particularly relevant to modulate the expression of Epo, which is a major target of HIF-2α,11,24 thus potentially keeping in balance the rate of red blood cells production with iron availability. On the other hand, the demonstration that hypoxia and iron metabolism are coordinately regulated also in Caenorhabditis elegans,25 which does not have a circulatory system, suggests that this pathway has a role that extends beyond erythropoiesis.
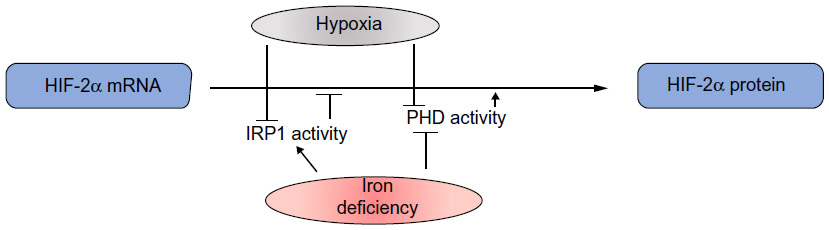 | Figure 3 Hypoxia activates two mechanisms to increase the amount of HIF-2α protein: derepression of HIF-2α translation through impaired binding of IRP1 to its IRE and prevention of HIF-2α degradation by inhibition of prolyl hydroxylase (PHD) activity. |
The role of IRP1 and IRP2 in the response to hypoxia has not been fully established; however, while not ruling out the possibility that endogenous IRP2 binds to HIF-2α IREs, the fact that IRP1 binds the IREs in HIF-2α mRNA challenges the concept (inferred by evidence that IRP2 is the major IRE-binding protein at physiological [3%–5%] oxygen tension)26 that IRP2 has a prominent role in the response to hypoxia. However, there is a possible interpretation for the differential response of the two IRPs to hypoxia. At low oxygen levels, in which the 4Fe-4S cluster of IRP1 is assembled promptly and IRP2 is stable, IRP2 is the main regulator of the translation of IRE-bearing mRNAs. Conversely, high oxygen concentrations result in stabilization of FBXL5 that binds IRP2 and induces its degradation, but the apoform of IRP1 can bind IREs and control iron balance. These mechanisms are expected to confer the cell the capacity to effectively regulate iron metabolism over a wide range of oxygen levels.
Notably, it has been recently shown that FBXL5 targets to proteasomal degradation CITED2, a nuclear protein that limits HIF transcriptional activity by competing for the coactivator CBP/p300.27 It seems therefore that when oxygen and iron are available, higher levels of FBXL5 lead to a decrease in CITED2, ultimately favoring HIF-dependent transcription (Figure 1). Although the meaning of this mechanism in the response to hypoxia remains to be established, this finding represents a novel additional link between iron and oxygen homeostasis.
Recent data point out an increasingly relevant role of mechanisms determining protein stability in intracellular iron metabolism (Figure 1). For example, the levels of NCOA4, which targets ferritin to autophagic turnover and thus affects iron availability at both the cellular and systemic levels, are regulated by HERC2-mediated proteasomal degradation.28 Clearly, in this context, the proteasome is a key player; therefore, one possible question is how iron homeostasis could be affected by changes in proteasomal activity, which can occur in a variety of pathophysiologic conditions including the immune response, neurodegenerative diseases, and cancer.29 For instance, will proteasomal inhibition lead (directly) to a higher amount of IRP2 or (indirectly) to a decrease in IRP2 levels because of FBXL5 stabilization? Although this problem has not been experimentally addressed in a specific way, a couple of recent papers provide some information. Multiple myeloma cell lines exposed to bortezomib, a proteasome inhibitor in use for treatment of this type of tumor, showed a 50% reduction in IRP-binding activity,30 an effect that the authors attributed to oxidative stress-mediated IRP2 degradation, but could well be the result of lower proteasomal degradation of FBXL5. Conversely, higher IRP2 levels were found in neuronal cells exposed to a different proteasomal inhibitor (lactacystin).31 These contrasting results may depend on the remarkably different experimental models, but also suggest that further studies are needed to investigate the role of the proteasome in cellular iron metabolism.
The rapid emergence of microRNAs (miRNAs) as key regulators of gene expression in a variety of biological processes expanded recently to the connection between iron and oxygen homeostasis. miRNA-210 was identified as a HIF target, induced by both hypoxia and iron deprivation,32 which regulates iron homeostasis through a complex mechanism. In fact, miRNA-210 directly inhibits TfR1 expression by direct binding to TfR1 mRNA. However, miRNA-210 concurrently suppresses ISCU, a protein involved in iron–sulfur cluster formation, thus switching IRP1 to the RNA-binding apoform, and leading to TfR1 mRNA stabilization. The net result of these contrasting mechanisms was decreased transferrin internalization (and possibly iron uptake) in miRNA-210-transfected breast cancer cells. On the other hand, previous studies have shown that TfR1 transcription is induced by hypoxia and iron deficiency.33–35 Therefore, it seems that numerous mechanisms of regulation of gene expression cooperate to amplify the range of control and response to stimuli. These recent advances support previous evidence indicating that precise regulation of TfR1 expression, and more generally of iron levels, is a key for cell activity, and probably depends on individual cell needs and specificities. HIF-1-dependent transcriptional upregulation may cooperate with IRP-dependent post-transcriptional control to expand the extent of response of iron homeostasis genes to oxygen or iron shortage. In fact, iron chelation induced TfR1 expression less efficiently in HIF-1-deficient hepatoma cells, which only depend on IRP-mediated upregulation, than in their wild-type counterpart,34 and is strongly induced in VHL-deficient renal carcinoma cells that overexpress HIF-1.36
The iron–oxygen axis: regulation at the systemic level
The two hormones most important to regulate iron and oxygen homeostasis at the systemic level are hepcidin and Epo, respectively. Hepcidin controls body iron balance by downregulating ferroportin, thus stopping iron efflux from duodenal and reticuloendothelial cells.37 Given their related physiological function, the idea that Epo could directly regulate hepcidin was attractive and initially supported by experimental data. For example, under conditions of elevated erythropoiesis, such as hypoxia or anemia, the enhanced iron absorption to face the higher demand of erythropoietic cells is accompanied by a strong inhibition of hepcidin expression.38,39 Therefore, the possibility of a direct effect of Epo on hepcidin has been explored. Indeed, Epo administration in mice (reviewed by Kautz and Nemeth39) and humans40,41 resulted in strong repression of hepcidin synthesis. Moreover, early studies found a direct repression of hepcidin transcription in Epo-treated cell lines.42,43 However, the idea of a direct effect of Epo was challenged by studies showing that administration of Epo to mice with damaged bone marrow due to exposure to cytotoxic agents or irradiation did not result in hepcidin suppression.44 Furthermore, it has been demonstrated that increasing liver iron content can stimulate hepcidin expression in conditions of permanently high Epo serum levels.45 More recently, the inhibitory effect of Epo on hepcidin synthesis was observed in mice lacking Epo receptor expression in hepatic cells, allowing us to show that the direct binding of Epo to liver receptors is not necessary to suppress hepcidin.46 In line with this conclusion, the erythroid factor erythroferrone (ERFE), which hinders hepcidin expression under conditions of high erythropoietic activity, was recently identified.47 Altogether, the available data indicate that the role of Epo is to stimulate the synthesis of ERFE in erythroblasts, which eventually downregulates hepcidin production in the liver (Figure 4).
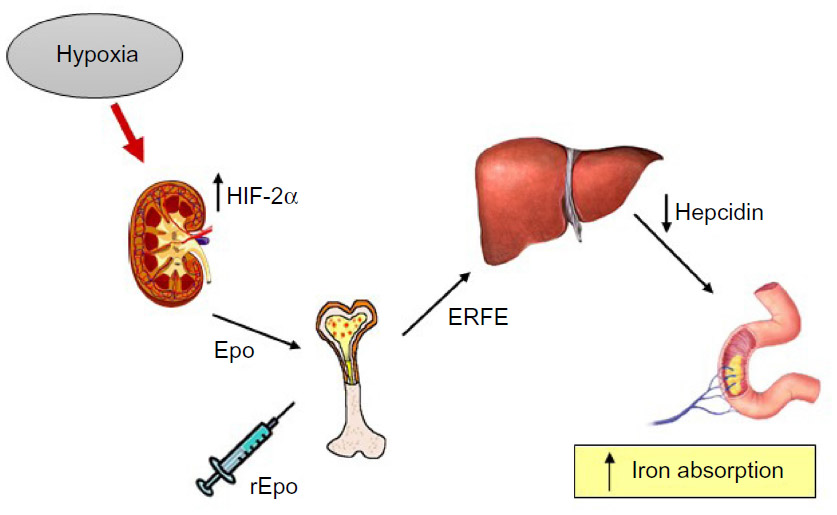 | Figure 4 Mechanisms of hepcidin downregulation by hypoxia and Epo. |
Similarly, it was initially proposed that the increased levels of HIF-1α present in livers of iron-deficient mice might repress hepcidin transcription.48 However, subsequent studies using liver-restricted deletion of various components of the HIF complex49,50 did not confirm a direct transcriptional suppression of hepcidin, and showed that the real role of HIF in this context is to induce Epo. The consequent stimulation of erythropoiesis then leads to hepcidin inhibition (Figure 4). In line with these findings, hepcidin was not suppressed in Ethiopians living at high altitude, who do not have a higher erythropoietic drive,51 as these populations chronically adapted to hypoxia by a pattern not involving increased hemoglobin concentration.4 Although in vivo the effect of hypoxia on hepcidin is probably indirect, several studies reported that activation of HIF by hypoxia, iron chelation, or PHDs inhibitors resulted in hepcidin downregulation in cultured cells. Also in this case, the effect appears to be indirect, because proteins that negatively control hepcidin expression are induced by HIF (Table 1). Iron-mediated hepcidin induction depends on the assembly on the cell surface of a complex consisting of bone morphogenetic proteins (BMPs), in particular BMP6, their receptors and hemojuvelin, a membrane protein that functions as a co-receptor for BMPs.37 Hemojuvelin is cleaved by two proteases, Matriptase2 and furin, resulting in the interruption of the hepcidin activation pathway and the release of a soluble form that acts as a decoy molecule for BMPs, respectively. The demonstration that expression of both Matriptase252 and furin53,54 is increased in hypoxia via HIF-1/2α may thus explain the hepcidin downregulation found in cell culture experiments. Recently, it has been shown that hypoxia inhibits hepcidin expression by a novel pathway involving platelet derived growth factor-BB.55 This growth factor is increased by hypoxia and regulates hepcidin by interfering with the CREB and CREB-H signaling pathways. As platelet derived growth factor-BB is stored in several cell types (eg, platelets) and promptly released in response to Epo,56 this finding may explain that even low-dose Epo injections in humans lead to early and considerable decrease in hepcidin levels that preceded any change in potentials mediators.40,41,57
Implications in pathophysiology
The IRP–HIF axis is important for various functions of specific tissues and organs, and its disruption is involved in several pathophysiological settings, for example erythropoiesis, which has been recently reviewed elsewhere.13,58 Here, we will focus on the intestine, the adipose tissue, and the lungs.
Iron/oxygen sensing and duodenal iron absorption
A number of recent studies provided convincing evidence that, in the context of duodenum, which is a key for iron absorption, the IRP 1/HIF-2α axis plays a predominant role. Iron deficiency specifically induced intestinal HIF-2α,59 and intestine-restricted inactivation indicated that HIF-2α is required to activate the reductase DcytB and the iron transporters DMT1 and ferroportin, and increase iron absorption in the gut of mice kept on an iron-deficient diet.59–61 Notably, it has been shown that dysregulation of the HIF-2α–iron axis in absorbing enterocytes plays a role in animal models of both primary50 and secondary62 iron overload. Moreover, experiments in mice with IRP1 inactivation and increased duodenal HIF-2α expression confirmed that the IRP 1/HIF-2α axis coordinates duodenal iron absorption according to systemic oxygen and iron availability.63
However, recent studies showing that FBXL5 also plays a relevant role at the systemic level, including the gut (see review Ruiz and Bruick16), led to reappraisal of the role of IRP2 in this context. Regulation of IRP-mediated iron homeostasis is disrupted in mice lacking FBXL5, and the unregulated IRP2 expression results in embryonic lethality due to iron overload in the early placenta. Mice with conditional deletion in the liver showed hepatic iron accumulation and died with acute liver damage when fed an iron-enriched diet.64 Intriguingly, despite liver overload, these mice express inappropriately low hepcidin levels, possibly because of impaired BMP6 signaling, and thus present systemic iron overload. Moreover, FBXL5 seems to have a special role in the intestine, as mice heterozygous for FBXL5 deletion fed an iron-deficient diet are able to maintain normal hematological values and avoid iron deficiency anemia by increasing DMT1-mediated iron absorption.
Role of iron and hypoxia in obesity
Both iron and oxygen homeostasis are modified in obesity. Animal studies have demonstrated that adipose tissues become hypoxic in obesity and, despite HIF-2α protective role, the consequent HIF signaling (mainly mediated by HIF-1α) contributes to the development of obesity-associated inflammation, insulin resistance, and other metabolic disorders that are strongly associated with obesity.65,66 The modifications of iron metabolism in obesity are multifarious and more difficult to put in a coherent framework, with strong differences between cell autonomous and systemic regulation.67 On the one hand, obese subjects often present body iron deficiency, and possibly anemia, probably because the chronic and low-grade inflammation that characterizes obesity68 leads to hepcidin upregulation and decreased circulating iron availability.69 On the other hand, in adipose tissue, the picture is more complex, as iron overload may play an important role in at least two cell types of obese adipose tissue: adipocytes and macrophages. Iron is required for adipogenesis (and adipocyte tissue expansion), probably because these cells are rich in mitochondria, which are the major iron sink of the cell.70,71 However, excess iron in adipocytes leads to decreased adiponectin release and impaired adipocyte insulin sensitivity, resulting in compromised systemic glucose tolerance.70,72 Therefore, in adipocytes, like in every other body cell, an optimal level of iron is required for homeostasis. Adipose tissue also contains macrophages that are mainly polarized toward a M2 anti-inflammatory phenotype in lean subjects, whereas in obesity the number of macrophages is greatly increased, and their polarization is shifted to the M1 classically activated phenotype.73 Interestingly, macrophage subtypes not only differ for the expression of classical inflammatory markers, but also have profound differences in iron handling.74 M1 macrophages are characterized by increased iron levels, whereas M2 macrophages have a pattern of gene expressions that favors uptake of heme iron and ferroportin-mediated iron release.75 As HIF-1α and HIF-2α have been mainly related to M1 and M2 phenotypes, respectively,76 the lower iron content that characterizes M2 macrophages is expected to favor HIF-2α stabilization and activity, with a favorable outcome in obesity. In fact, activated macrophage HIF-2α will relieve adipose tissue inflammation and insulin resistance.77 These data reveal another aspect of the complex interplay between iron and oxygen homeostasis and highlight its importance in relevant pathologies. However, the situation may be more complicated because the distinction of macrophages in the M1 and M2 subclasses is probably an oversimplification,78 and indeed M2-like macrophages with either low or high iron content have been isolated from adipose tissue.79
Role of iron in hypoxia-dependent pulmonary hypertension
Both hypoxia and iron have been demonstrated to play a key role in pulmonary hypertension, a common clinical problem characterized by progressive pulmonary vasculature remodeling leading to increased pulmonary arterial pressures.80
Both animal models, in which heterozygous deletion or activating mutation of HIF-α genes leads to protection from pulmonary hypertension and higher sensitivity to hypoxia, respectively, and a human genetic condition, Chuvash polycythemia, in which hypoxia sensing is increased because the VHL protein is inactive, provided a strong demonstration of the role of the HIF pathway in pulmonary hypertension (discussed in Zhang et al13).
Several studies have reported that iron deficiency may cooperate in the activation of HIF triggered by the conditions of persistent or intermittent hypoxia that are associated with pulmonary hypertension. Indeed, increasing iron bioavailability by intravenous iron loading effectively reversed the hypertensive response of the pulmonary vasculature to hypoxia.81 The effect of intravenous iron on the pulmonary circulation in human studies was observed both under laboratory conditions (normobaric hypoxia) and in subjects exposed to high altitude hypobaric hypoxia. Given the high prevalence of both nutritional and functional iron deficiency (in chronic diseases characterized by cytokine-induced iron sequestration in the reticuloendothelial compartment), the effect of iron status on oxygen sensing and hypoxia-induced pulmonary vasoconstriction would be potentially significant for human health.
Notably, mice with IRP 1 inactivation and consequent unrestricted HIF-2 activity develop spontaneous pulmonary hypertension, thus underscoring the physiological importance of the overlap between oxygen and iron sensing.20
Candidate HIF-dependent genes that may contribute to the development of hypoxic pulmonary hypertension in humans include endothelin and proteins involved in sodium and potassium transport. Interestingly, a recent study showed that hypoxia elevates miR-210, which in turn represses its target ISCU1/2 and downregulates Fe-S cluster formation, thus leading to mitochondrial dysfunction and promoting pulmonary hypertension.82 Therefore, iron deficiency can act both by impairing PHDs activity and by preventing Fe-S cluster assembly.
Conclusion
Recent results have advanced the identification of strong associations between iron metabolism and oxygen homeostasis. However, our understanding of this complex interplay remains incomplete. Further studies will clarify the mechanistic relationships between these pathways and their relevance in pathophysiological conditions and disease, possibly paving the way for more advanced therapeutic approaches aimed at re-establishing iron and/or oxygen homeostasis in clinical settings.
Acknowledgment
The authors gratefully acknowledge the grant support of MIUR (Project COFIN).
Disclosure
The authors report no conflicts of interest in this work.
References
Churazov E, Sunyaev R, Isern J, et al. Cobalt-56 γ-ray emission lines from the type Ia supernova 2014J. Nature. 2014;512(7515):406–408. | |
Sheftel AD, Mason AB, Ponka P. The long history of iron in the Universe and in health and disease. Biochim Biophys Acta. 2012;1820(3):161–187. | |
Andrews NC. Forging a field: the golden age of iron biology. Blood. 2008;112(2):219–230. | |
Beall CM. Human adaptability studies at high altitude: research designs and major concepts during fifty years of discovery. Am J Hum Biol. 2013;25(2):141–147. | |
Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408. | |
Bishop T, Ratcliffe PJ. Signaling hypoxia by hypoxia-inducible factor protein hydroxylases: a historical overview and fututre perspective. Hypoxia. 2014;2:197–213. | |
Zhang P, Yao Q, Lu L, Li Y, Chen PJ, Duan C. Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep. 2014;6(6):1110–1121. | |
Recalcati S, Minotti G, Cairo G. Iron regulatory proteins: from molecular mechanisms to drug development. Antioxid Redox Signal. 2010;13(10):1593–1616. | |
Markolovic S, Wilkins SE, Schofield CJ. Protein hydroxylation catalyzed by 2-oxoglutarate-dependent oxygenases. J Biol Chem. 2015;290(34):20712–20722. | |
Salminen A, Kauppinen A, Kaarniranta K. 2-Oxoglutarate-dependent dioxygenases are sensors of energy metabolism, oxygen availability, and iron homeostasis: potential role in the regulation of aging process. Cell Mol Life Sci. 2015;72(20):3897–3914. | |
Gale DP, Harten SK, Reid CD, Tuddenham EG, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2 alpha mutation. Blood. 2008;112(3):919–921. | |
Pantopoulos K, Porwal SK, Tartakoff A, Devireddy L. Mechanisms of mammalian iron homeostasis. Biochemistry. 2012;51(29):5705–5724. | |
Zhang DL, Ghosh MC, Rouault TA. The physiological functions of iron regulatory proteins in iron homeostasis – an update. Front Pharmacol. 2014;5:124. | |
Kühn LC. Iron regulatory proteins and their role in controlling iron metabolism. Metallomics. 2015;7(2):232–243. | |
Moreno M, Ortega F, Xifra G, Ricart W, Fernández-Real JM, Moreno-Navarrete JM. Cytosolic aconitase activity sustains adipogenic capacity of adipose tissue connecting iron metabolism and adipogenesis. FASEB J. 2015;29(4):1529–1539. | |
Ruiz JC, Bruick RK. F-box and leucine-rich repeat protein 5 (FBXL5): sensing intracellular iron and oxygen. J Inorg Biochem. 2014;133:73–77. | |
Smith SR, Cooperman S, Lavaute T, et al. Severity of neurodegeneration correlates with compromise of iron metabolism in mice with iron regulatory protein deficiencies. Ann NY Acad Sci. 2004;1012:65–83. | |
Recalcati S, Alberghini A, Campanella A, et al. Iron regulatory proteins 1 and 2 in human monocytes, macrophages and duodenum: expression and regulation in hereditary hemochromatosis and iron deficiency. Haematologica. 2006;91(3):303–310. | |
Wingert RA, Galloway JL, Barut B, et al. Deficiency of glutaredoxin 5 reveals Fe-S clusters are required for vertebrate haem synthesis. Nature. 2005;436(7053):1035–1039. | |
Ghosh MC, Zhang DL, Jeong SY, et al. Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of HIF2α. Cell Metab. 2013;17(2):271–281. | |
Cairo G, Recalcati S. Iron-regulatory proteins: molecular biology and pathophysiological implications. Expert Rev Mol Med. 2007;9(33):1–13. | |
Moroishi T, Yamauchi T, Nishiyama M, Nakayama KI. HERC2 targets the iron regulator FBXL5 for degradation and modulates iron metabolism. J Biol Chem. 2014;289(23):16430–16441. | |
Simpson RJ, McKie AT. Iron and oxygen sensing: a tale of 2 interacting elements? Metallomics. 2015;7(2):223–231. | |
Warnecke C, Zaborowska Z, Kurreck J, et al. Differentiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2alpha target gene in Hep3B and Kelly cells. FASEB J. 2004;18(12):1462–1464. | |
Ackerman D, Gems D. Insulin/IGF-1 and hypoxia signaling act in concert to regulate iron homeostasis in Caenorhabditis elegans. PLoS Genet. 2012;8(3):e1002498. | |
Meyron-Holtz EG, Ghosh MC, Rouault TA. Mammalian tissue oxygen levels modulate iron-regulatory protein activities in vivo. Science. 2004; 306(5704):2087–2090. | |
Machado-Oliveira G, Guerreiro E, Matias AC, Facucho-Oliveira J, Pacheco-Leyva I, Bragança J. FBXL5 modulates HIF-1α transcriptional activity by degradation of CITED2. Arch Biochem Biophys. 2015;576:61–72. | |
Mancias JD, Pontano Vaites L, Nissim S, et al. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife. 2015;4. | |
Grigoreva TA, Tribulovich VG, Garabadzhiu AV, Melino G, Barlev NA. The 26S proteasome is a multifaceted target for anti-cancer therapies. Oncotarget. 2015;6(28):24733–24749. | |
Campanella A, Santambrogio P, Fontana F, et al. Iron increases the susceptibility of multiple myeloma cells to bortezomib. Haematologica. 2013;98(6):971–979. | |
Li XP, Xie WJ, Zhang Z, Kansara S, Jankovic J, Le WD. A mechanistic study of proteasome inhibition-induced iron misregulation in dopamine neuron degeneration. Neurosignals. 2012;20(4):223–236. | |
Yoshioka Y, Kosaka N, Ochiya T, Kato T. Micromanaging Iron Homeostasis: hypoxia-inducible micro-RNA-210 suppresses iron homeostasis-related proteins. J Biol Chem. 2012;287(41):34110–34119. | |
Tacchini L, Bianchi L, Bernelli-Zazzera A, Cairo G. Transferrin receptor induction by hypoxia. HIF-1-mediated transcriptional activation and cell-specific post-transcriptional regulation. J Biol Chem. 1999;274(34):24142–24146. | |
Bianchi L, Tacchini L, Cairo G. HIF-1-mediated activation of transferrin receptor gene transcription by iron chelation. Nucleic Acids Res. 1999;27(21):4223–4227. | |
Lok CN, Ponka P. Identification of a hypoxia response element in the transferrin receptor gene. J Biol Chem. 1999;274(34):24147–24152. | |
Alberghini A, Recalcati S, Tacchini L, Santambrogio P, Campanella A, Cairo G. Loss of the von Hippel Lindau tumor suppressor disrupts iron homeostasis in renal carcinoma cells. J Biol Chem. 2005;280(34):30120–30128. | |
Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of Mammalian iron metabolism. Cell. 2010;142(1):24–38. | |
Camaschella C, Pagani A. Iron and erythropoiesis: a dual relationship. Int J Hematol. 2011;93(1):21–26. | |
Kautz L, Nemeth E. Molecular liaisons between erythropoiesis and iron metabolism. Blood. 2014;124(4):479–482. | |
Robach P, Recalcati S, Girelli D, et al. Alterations of systemic and muscle iron metabolism in human subjects treated with low-dose recombinant erythropoietin. Blood. 2009;113(26):6707–6715. | |
Ashby DR, Gale DP, Busbridge M, et al. Erythropoietin administration in humans causes a marked and prolonged reduction in circulating hepcidin. Haematologica. 2010;95(3):505–508. | |
Fein E, Merle U, Ehehalt R, Herrmann T, Kulaksiz H. Regulation of hepcidin in HepG2 and RINm5F cells. Peptides. 2007;28(5):951–957. | |
Pinto JP, Ribeiro S, Pontes H, et al. Erythropoietin mediates hepcidin expression in hepatocytes through EPOR signaling and regulation of C/EBPalpha. Blood. 2008;111(12):5727–5733. | |
Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006; 108(12):3730–3735. | |
Díaz V, Gammella E, Recalcati S, et al. Liver iron modulates hepcidin expression during chronically elevated erythropoiesis in mice. Hepatology. 2013;58(6):2122–2132. | |
Gammella E, Diaz V, Recalcati S, et al. Erythropoietin’s inhibiting impact on hepcidin expression occurs indirectly. Am J Physiol Regul Integr Comp Physiol. 2015;308(4):R330–R335. | |
Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46(7):678–684. | |
Peyssonnaux C, Zinkernagel AS, Schuepbach RA, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs). J Clin Invest. 2007;117(7):1926–1932. | |
Liu Q, Davidoff O, Niss K, Haase VH. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest. 2012;122(12):4635–4644. | |
Mastrogiannaki M, Matak P, Mathieu JR, et al. Hepatic hypoxia-inducible factor-2 down-regulates hepcidin expression in mice through an erythropoietin-mediated increase in erythropoiesis. Haematologica. 2012;97(6):827–834. | |
Lundgrin EL, Janocha AJ, Koch CD, et al. Plasma hepcidin of Ethiopian highlanders with steady-state hypoxia. Blood. 2013;122(11):1989–1991. | |
Lakhal S, Schödel J, Townsend AR, Pugh CW, Ratcliffe PJ, Mole DR. Regulation of type II transmembrane serine proteinase TMPRSS6 by hypoxia-inducible factors: new link between hypoxia signaling and iron homeostasis. J Biol Chem. 2011;286(6):4090–4097. | |
McMahon S, Grondin F, McDonald PP, Richard DE, Dubois CM. Hypoxia-enhanced expression of the proprotein convertase furin is mediated by hypoxia-inducible factor-1: impact on the bioactivation of proproteins. J Biol Chem. 2005;280(8):6561–6569. | |
Silvestri L, Pagani A, Camaschella C. Furin-mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis. Blood. 2008;111(2):924–931. | |
Sonnweber T, Nachbaur D, Schroll A, et al. Hypoxia induced downregulation of hepcidin is mediated by platelet derived growth factor BB. Gut. 2014;63(12):1951–1959. | |
Janmaat ML, Heerkens JL, de Bruin AM, Klous A, de Waard V, de Vries CJ. Erythropoietin accelerates smooth muscle cell-rich vascular lesion formation in mice through endothelial cell activation involving enhanced PDGF-BB release. Blood. 2010;115(7):1453–1460. | |
Robach P, Recalcati S, Girelli D, et al. Serum hepcidin levels and muscle iron proteins in humans injected with low- or high-dose erythropoietin. Eur J Haematol. 2013;91(1):74–84. | |
Shah YM, Xie L. Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterology. 2014;146(3):630–642. | |
Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Intestinal hypoxia-inducible transcription factors are essential for iron absorption following iron deficiency. Cell Metab. 2009;9(2):152–164. | |
Mastrogiannaki M, Matak P, Keith B, Simon MC, Vaulont S, Peyssonnaux C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest. 2009;119(5):1159–1166. | |
Taylor M, Qu A, Anderson ER, et al. Hypoxia-inducible factor-2α mediates the adaptive increase of intestinal ferroportin during iron deficiency in mice. Gastroenterology. 2011;140(7):2044–2055. | |
Anderson ER, Taylor M, Xue X, et al. Intestinal HIF2α promotes tissue-iron accumulation in disorders of iron overload with anemia. Proc Natl Acad Sci U S A. 2013;110(50):E4922–E4930. | |
Anderson SA, Nizzi CP, Chang YI, et al. The IRP1-HIF-2α axis coordinates iron and oxygen sensing with erythropoiesis and iron absorption. Cell Metab. 2013;17(2):282–290. | |
Moroishi T, Nishiyama M, Takeda Y, Iwai K, Nakayama KI. The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo. Cell Metab. 2011;14(3):339–351. | |
Trayhurn P. Hypoxia and adipocyte physiology: implications for adipose tissue dysfunction in obesity. Annu Rev Nutr. 2014;34:207–236. | |
Goossens GH, Blaak EE. Adipose tissue dysfunction and impaired metabolic health in human obesity: a matter of oxygen? Front Endocrinol (Lausanne). 2015;6:55. | |
Hubler MJ, Peterson KR, Hasty AH. Iron homeostasis: a new job for macrophages in adipose tissue? Trends Endocrinol Metab. 2015;26(2):101–109. | |
Shoelson SE, Herrero L, Naaz A. Obesity, inflammation, and insulin resistance. Gastroenterology. 2007;132(6):2169–2180. | |
Simcox JA, McClain DA. Iron and diabetes risk. Cell Metab. 2013;17(3):329–341. | |
Gabrielsen JS, Gao Y, Simcox JA, et al. Adipocyte iron regulates adiponectin and insulin sensitivity. J Clin Invest. 2012;122(10):3529–3540. | |
Moreno-Navarrete JM, Ortega F, Moreno M, Ricart W, Fernández-Real JM. Fine-tuned iron availability is essential to achieve optimal adipocyte differentiation and mitochondrial biogenesis. Diabetologia. 2014;57(9):1957–1967. | |
Dongiovanni P, Ruscica M, Rametta R, et al. Dietary iron overload induces visceral adipose tissue insulin resistance. Am J Pathol. 2013;182(6):2254–2263. | |
Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science. 2013;339(6116):172–177. | |
Cairo G, Recalcati S, Mantovani A, Locati M. Iron trafficking and metabolism in macrophages: contribution to the polarized phenotype. Trends Immunol. 2011;32(6):241–247. | |
Recalcati S, Locati M, Marini A, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol. 2010;40(3):824–835. | |
Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. | |
Choe SS, Shin KC, Ka S, Lee YK, Chun JS, Kim JB. Macrophage HIF-2α ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes. 2014;63(10):3359–3371. | |
Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. | |
Orr JS, Kennedy A, Anderson-Baucum EK, et al. Obesity alters adipose tissue macrophage iron content and tissue iron distribution. Diabetes. 2014;63(2):421–432. | |
Robinson JC, Graham BB, Rouault TC, Tuder RM. The crossroads of iron with hypoxia and cellular metabolism. Implications in the pathobiology of pulmonary hypertension. Am J Respir Cell Mol Biol. 2014;51(6):721–729. | |
Smith TG, Talbot NP, Privat C, et al. Effects of iron supplementation and depletion on hypoxic pulmonary hypertension: two randomized controlled trials. JAMA. 2009;302(13):1444–1450. | |
White K, Lu Y, Annis S, et al. Genetic and hypoxic alterations of the microRNA-210-ISCU1/2 axis promote iron-sulfur deficiency and pulmonary hypertension. EMBO Mol Med. 2015;7(6):695–713. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.