Back to Journals » International Journal of Women's Health » Volume 7
New perspectives on targeted therapy in ovarian cancer
Authors Coward JI, Middleton K, Murphy F
Received 5 October 2014
Accepted for publication 11 November 2014
Published 4 February 2015 Volume 2015:7 Pages 189—203
DOI https://doi.org/10.2147/IJWH.S52379
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Elie Al-Chaer
Jermaine IG Coward,1–3 Kathryn Middleton,1 Felicity Murphy1
1Mater Health Services, Raymond Terrace, South Brisbane, QLD, Australia; 2Inflammtion and Cancer Therapeutics Group, Mater Research, University of Queensland, Translational Research Institute, Woolloongabba, Brisbane, QLD, Australia; 3School of Medicine, University of Queensland, Brisbane, QLD, Australia
Abstract: Epithelial ovarian cancer remains the most lethal gynecologic malignancy. During the last 15 years, there has been only marginal improvement in 5 year overall survival. These daunting statistics are compounded by the fact that despite all subtypes exhibiting striking heterogeneity, their systemic management remains identical. Although changes to the scheduling and administration of chemotherapy have improved outcomes to a degree, a therapeutic ceiling is being reached with this approach, resulting in a number of trials investigating the efficacy of targeted therapies alongside standard treatment algorithms. Furthermore, there is an urge to develop subtype-specific studies in an attempt to improve outcomes, which currently remain poor. This review summarizes the key studies with antiangiogenic agents, poly(adenosine diphosphate [ADP]-ribose) inhibitors, and epidermal growth factor receptor/human epidermal growth factor receptor family targeting, in addition to folate receptor antagonists and insulin growth factor receptor inhibitors. The efficacy of treatment paradigms used in non-ovarian malignancies for type I tumors is also highlighted, in addition to recent advances in appropriate patient stratification for targeted therapies in epithelial ovarian cancer.
Keywords: antiangiogenic therapy, high-grade serous, low grade ovarian cancer, PARP inhibition, cancer-related inflammation
Introduction
Epithelial ovarian cancer (EOC) is the most lethal gynecologic malignancy and the fifth most common cause of cancer-related death in women. The estimated annual incidence of this disease worldwide is just over 200,000 individuals, with approximately 125,000 deaths.1 Significant advances in the understanding of the natural history of the disease and thorough initial staging, along with surgical and chemotherapeutic management, have improved the short-term course of ovarian carcinoma. However, despite such improvements, most patients relapse after primary treatment and succumb to disease progression. The risk for ovarian cancer increases with age. The majority of patients are postmenopausal, with 80% of cases diagnosed being older than 50 years, and a peak incidence of 61.8 per 100,000 women is observed in the 60–64 year old age group (Cancer Research UK data). Racial and geographical variations are also evident for this disease. For example, lower incidences are seen in African-American and Afro-Caribbean women compared with their Caucasian counterparts.2,3 In addition, the rate of EOC is significantly higher in Europe and the United States compared with in Sub-Saharan Africa and Japan, respectively.4,5
Surgical management (consisting of total abdominal hysterectomy and bilateral salpingo-oophorectomy, together with omentectomy, peritoneal washings, and pelvic lymph node sampling) is essential to EOC diagnosis, staging, and treatment. The ultimate aim is to resect all macroscopic tumor (ie, optimal debulking) within the pelvis and to perform careful surveillance of the abdomen to detect any subtle sites of metastatic disease. The extent of debulking profoundly influences prognosis in EOC. With respect to optimally debulked (<1 cm residual disease) and suboptimally debulked (>1 cm residual disease) patients, Chi et al reported significant differences in both 5 year progression-free survival (PFS) rates (31% versus 14%, respectively [hazard ratio {HR}, 0.757; 95% confidence interval {CI}, 0.601–0.953; P=0.01]) and 5 year overall survival (OS) rates (47% versus 35%, respectively [HR, 0.764; 95% CI, 0.592–0.987; P=0.03]).6
With the exception of good-prognosis early-stage disease (ie, stage Ia and Ib, grade 1), in which standard care involves cytoreductive surgery followed by observation,7,8 chemotherapy remains the principal form of adjuvant and neoadjuvant treatment for EOC. During the last 40 years, a myriad trials have been conducted to establish a “gold standard” of therapy and to validate the optimal timing and mode of cytotoxic delivery. Arguably, the most significant initial advance in chemotherapeutic management was the introduction of paclitaxel to the treatment armamentarium. This was reported in the pivotal Gynecologic Oncology Group (GOG)-111 study in 1996 (n=386), in which the combination of cisplatin and paclitaxel conferred a substantial survival advantage over cisplatin and cyclophosphamide (PFS, 18 versus 13 months [P<0.001]; OS, 38 versus 24 months [P<0.001]).9 After this, the Arbeitsgemeinschaft Gynaekologische Onkologie (AGO) study in 2000 (n=208) established equivalent PFS (16 months) and OS (31 months) for cisplatin/paclitaxel and carboplatin/paclitaxel doublets.10 The toxicity profile favoring the carboplatin/paclitaxel regimen has now established this as standard of care in the first-line setting. Although numerous single agents (eg, gemcitabine, doxorubicin, topotecan) have known activity in recurrent disease, their addition to standard primary treatment failed to prolong survival.11 Only relatively recently has OS improved as a consequence of chemotherapy, with the Japanese Gynecologic Oncology Group (JGOG) 3016 (n=637) study confirming superior efficacy by altering treatment schedules.12 This trial evaluated dose-dense treatment with weekly paclitaxel alongside carboplatin three times weekly against traditional administration three times weekly for both drugs. Median PFS was significantly extended in the dose-dense treatment group compared with in the conventional treatment group (28.2 months [95% CI, 22.3–33.8 months] versus 17.5 months [95% CI, 15.7–21.7 months]; HR, 0.76; 95% CI, 0.62–0.91; P=0.0037). Importantly, median OS was 100.5 months (95% CI, 65.2–∞ months) in the dose-dense treatment group and 62.2 months (95% CI, 52.1–82.6 months) in the conventional treatment group (HR, 0.79; 95% CI, 0.63–0.99; P=0.039).12
As EOC has a predilection for peritoneal dissemination, there has been increasing interest in the role of intraperitoneal (ip) chemotherapy. One of the advantages of this approach relates to the higher concentrations of cisplatin and paclitaxel that can be achieved in the peritoneal cavity compared with plasma.13,14 One of the largest definitive studies testing this hypothesis in the first-line setting was the GOG 172 study.15 This study evaluated 417 patients with optimally debulked (<1 cm) stage III disease who were randomized to receive six courses every 3 weeks of either standard intravenous (iv) cisplatin/paclitaxel or iv paclitaxel (day 1) with ip cisplatin (day 2), followed by ip paclitaxel (day 8). Despite only 42% of the ip group completing all six cycles, this cohort had superior median PFS (23.8 versus 18.3 months; P=0.05) and OS (65.6 versus 49.7 months; P=0.03).15 However, ip therapy had a less favorable toxicity profile, with significant myelosuppression, neuropathy, and abdominal pain and poor quality of life and catheter complications.15 A recent update of this study at the 2013 Society of Gynecologic Oncology (SGO) Annual Meeting on Women’s Cancer confirmed that the 5 year OS rate among patients treated with ip therapy increased from 18% with the completion of one or two cycles to 33% with three or four cycles, to 59% for patients who completed five or six cycles of treatment.16
These results are further substantiated by a subsequent systematic Cochrane review of nine randomized studies (n=2,119), which confirms that in comparison with iv chemotherapy, ip administration confers a definite PFS advantage (five studies, 1,311 women; HR, 0.78; 95% CI, 0.70–0.86) and OS benefit (eight studies, 2,026 women; HR, 0.81; 95% CI, 0.72–0.90).17 Despite this, ip administration has yet to become an international standard of care and currently is usually reserved for patients who have optimally debulked or minimal residual disease. Furthermore, skepticism still exists about this approach in view of the significant toxicity and the possibility that any OS advantage may be a result of the weekly administration of paclitaxel as opposed to the ip delivery.
Although changes to both chemotherapy schedules and routes of administration are associated with improved survival, it appears that a therapeutic ceiling with these drugs has been reached. EOC still lags behind a number of common solid malignancies in terms of the sluggish incremental extension in OS during the last 20 years.18 Within this time, the advent of targeted therapy has undoubtedly revolutionized the therapeutic landscape in oncology. This review synthesizes the key studies using novel therapies either as single agents or alongside standard chemotherapy to improve the current dismal statistics for EOC.
Antiangiogenic therapy
Bevacizumab
EOC is unique among solid organ malignancies in its natural history, partly because of its pattern of spread within the peritoneum. Angiogenesis is thought to be particularly crucial in the persistence of EOC and so has been a key target in clinical research. Antiangiogenic agents have been evaluated extensively in the treatment of EOC and found to have some clinical efficacy, both as single agents as well as in combination with chemotherapy (Table 1). Bevacizumab, a monoclonal antibody that binds to the vascular endothelial growth factor (VEGF)-receptor ligand VEGF-A, has been most extensively investigated in clinical research. Phase II trials of bevacizumab alone in heavily pretreated patients demonstrated mixed results. In a Phase II trial of 62 patients, 13 patients (21.0%) experienced clinical responses (two complete responses [CR] and eleven partial responses [PR]; median duration, 10 months), and 25 patients (40.3%) survived progression-free for at least 6 months, with no gastrointestinal (GI) perforations.19 Another Phase II trial, which demonstrated a 15.9% PR rate, also found an 11% risk for GI perforation and was stopped early because of safety concerns. In this trial, the risk for GI perforation was highest in patients who had received three prior lines of treatment.20 This is the only trial to report such a higher rate of GI perforation with the use of bevacizumab.
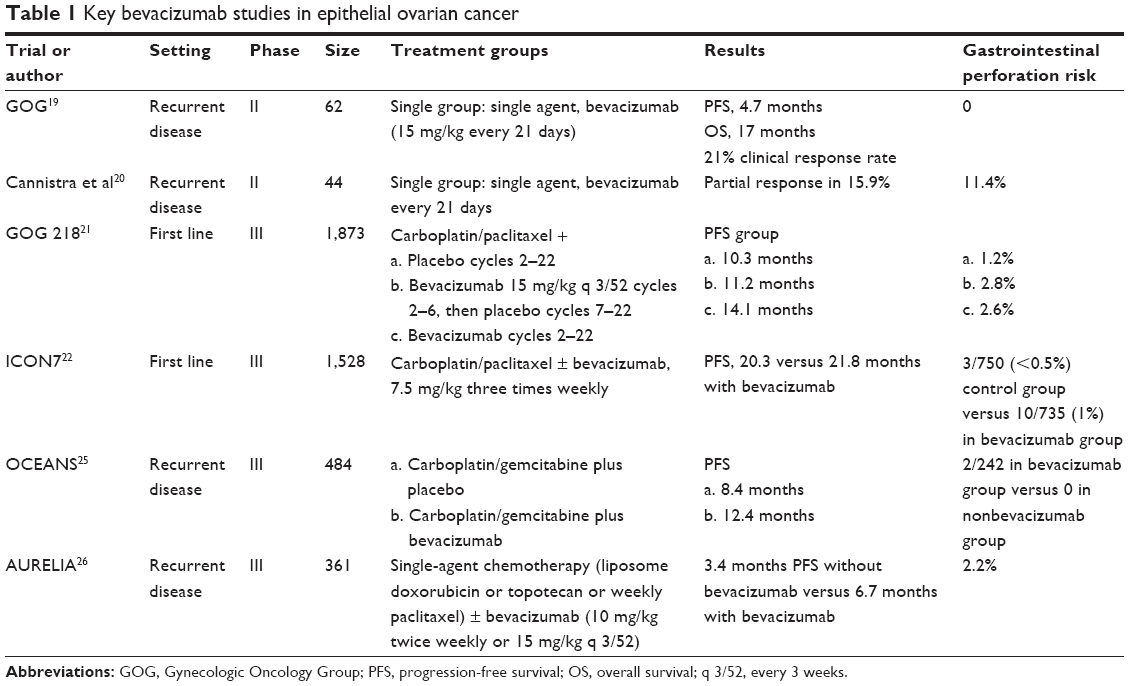 | Table 1 Key bevacizumab studies in epithelial ovarian cancer |
The landmark GOG-218 and International Collaborative Ovarian Neoplasm group (ICON) 7 trials evaluated the addition of bevacizumab to carboplatin/paclitaxel in first-line therapy.21,22 ICON7 found that the greatest benefit of adding bevacizumab was seen in a higher-risk population with stage III–IV disease and residual disease greater than 1 cm. In this population, PFS at 42 months was 14.5 months with standard therapy alone and 18.1 months with bevacizumab added, with respective median OS of 28.8 and 36.6 months.22 On the basis of these trials, the European Society for Medical Oncology clinical practice guidelines suggest bevacizumab in addition to chemotherapy in patients with poor prognostic features, such as stage IV or suboptimally debulked disease.23
Interestingly, it appears that predictive biomarkers for this treatment are on the horizon. At the American Society of Clinical Oncology (ASCO) 2014 annual meeting, Gourley et al presented data that highlighted differential responses between patients in the ICON7 study exhibiting particular genomic signatures.24 Using unsupervised hierarchical clustering, Gourley et al identified three subgroups: one with angiogenic repression/immune gene upregulation (immune molecular subgroup) and two with angiogenic gene upregulation (proangiogenic subgroup). Using a 63-gene signature to separate these subgroups, in the chemotherapy control group, the immunogenic subgroup had a significantly superior PFS (HR, 0.47; 95% CI, 0.32–0.71; P<0.001) and OS (HR, 0.45; 95% CI, 0.26–0.79; P=0.005) compared with the proangiogenic groups.24 The authors hypothesized this could be a result of either improved antitumor immunity or less-developed tumor vascularity in the immunogenic group.24 However, patients with the proangiogenic gene signature had significantly improved PFS when treated with carboplatin/paclitaxel and bevacizumab (17.4 months) compared with chemotherapy alone (12.3 months). This contrasted with patients harboring an immunogenic/angiogenic repression gene signature where PFS was significantly impeded by the addition of bevacizumab (P=0.015), with a median PFS of 18.5 months when treated with bevacizumab compared with 35.8 months when receiving carboplatin/paclitaxel alone.24
In relapsed disease, both the OCEANS (Ovarian Cancer Study Comparing Efficacy and Safety of Chemotherapy and Anti-Angiogenic Therapy in Platinum-Sensitive Recurrent Disease) and AURELIA (Avastin Use in Platinum-Resistant Epithelial Ovarian Cancer) trials have evaluated the addition of bevacizumab to chemotherapy and demonstrated an improvement in PFS.25,26 In AURELIA, in patients with relapsed platinum-resistant ovarian cancer, median PFS was 3.4 months with chemotherapy alone versus 6.7 months with the addition of bevacizumab. Median OS was 13.3 months (95% CI, 11.9–16.4 months) with chemotherapy alone versus 16.6 months (95% CI, 13.7–19.0 months) with bevacizumab plus chemotherapy, which did not reach statistical significance, but 40% of patients receiving chemotherapy crossed over to single-agent bevacizumab on progression, compromising the OS data.26
In the OCEANS trial, the addition of bevacizumab to carboplatin/gemcitabine in patients with relapsed platinum-sensitive ovarian cancer also improved PFS, with PFS of 12.4 months in the chemotherapy plus bevacizumab group versus 8.4 months in the chemotherapy plus placebo group.25 OS data were not mature when this study was published, but the preliminary analysis did not demonstrate an OS difference, at 33.3 and 35.2 months for the bevacizumab and placebo groups, respectively.25 The large crossover in these trials also significantly affected the interpretation of OS data.
The benefit of the antiangiogenic agents does come at the cost of additional toxicity. In ICON7, the risks included increased risk from perforation, in addition to well-recognized toxicities including bleeding, thromboembolic events, and hypertension.27 The AURELIA study revealed a 2.2% risk for perforation with the addition of bevacizumab;26 however, the risk for perforation was lower than might have been expected, given that patients with ovarian cancer are at a higher risk for perforation than patients with other solid organ malignancies. In general, in these trials, it seems the increased risk for perforation with the addition of bevacizumab is small and does not outweigh its possible clinical benefit. Antiangiogenics are also being trialed in combination with agents other than cytotoxic chemotherapy. Both bevacizumab and cediranib (a VEGF2 inhibitor) have been combined with mammalian target of rapamycin (mTOR) inhibitors in clinical trials that are underway.28 Undeniably, the success witnessed with bevacizumab in various settings for EOC has provided a useful platform for the introduction of other antiangiogenic agents.
Aflibercept (VEGF trap)
Aflibercept is a heterodimeric molecule consisting of domains of vascular endothelial growth factor 1 (VEGFR1) and VEGFR2 with immunoglobulin G Fc. Although it has a lower molecular weight than bevacizumab, it possesses a higher affinity for VEGF isoforms including VEGF-A, VEGF-B, and placental growth factor.29 Inevitably, this broader spectrum of antiangiogenic activity led to a number of Phase II studies in recurrent EOC. Coleman et al treated 46 patients in this setting with a combination of aflibercept (6 mg/kg iv) and docetaxel (75 mg/m2 iv) every 3 weeks until evidence of progressive disease.30 They reported an impressive 54% objective response rate (ORR), which included ten patients with CR and a median PFS and OS of 6.2 months and 24.3 months, respectively.30 In view of the intricate links between VEGF signaling and development of ascites, subsequent studies with aflibercept have focused on its potential in decreasing the frequency of paracentesis. In a double-blind randomized trial in 55 patients with EOC, Gotlieb et al reported that although aflibercept (4 mg/kg iv every 2 weeks) significantly extended time to ascitic drainage compared with placebo (55.1 versus 23.3 days, respectively; P=0.0019), there was no effect on OS.31 Furthermore, it was associated with more grade 3/4 toxicities, including fatal GI adverse effects.31 The current Australia and New Zealand Gynaecological Oncology Group (ANZGOG) REZOLVE II study aims to address whether the intraperitoneal administration of aflibercept could result in OS advantages not witnessed in the Gotlieb trial.
Nintedanib
Nintedanib (BIBF 1120) was the first triple angiokinase inhibitor developed with the principle of impeding tumor angiogenesis at multiple levels through targeting VEGF, fibroblast growth factor (FGF), and platelet-derived growth factor (PDGF). The first placebo-controlled Phase II study investigating nintedanib (250 mg twice daily) in 83 patients with recurrent EOC revealed some PFS advantages that were offset by significantly more GI toxicities.32 A subsequent double-blind Phase III study in the first-line setting comparing nintedanib combined with either carboplatin/paclitaxel or placebo confirmed significantly prolonged PFS with the nintedanib/chemotherapy group (18.3 versus 16.6 months; HR, 0.84; 95% CI, 0.72–0.98; P=0.0239).33
Trebananib (AMG 386)
The interaction between angiopoietin (Ang) 1 and Ang-2 ligands and their respective Tie-2 receptors within the angiopoietin signaling axis plays an integral role in the stabilization of tumor vascular integrity.34 Trebananib (AMG 386), a neutralizing peptide developed to block Ang-1/Ang-2 and Tie-2 binding, has garnered some interesting results in a series of consecutive TRINOVA Phase III studies for patients with primary or relapsed EOC. The first of these trials (TRINOVA-1; n=919) focused on the combination of paclitaxel with either trebananib or placebo with the trebananib-containing group associated with significant extension in PFS (7.2 versus 5.4 months; HR, 0.66; 95% CI, 0.57–0.77; P<0.001).35 The subsequent TRINOVA-2 (n=223; pegylated liposomal doxorubicin with trebananib or placebo in recurrent EOC; Clinicaltrials.gov identifier NCT01281254) and TRINOVA-3 (n=2,000; EOC first-line therapy with carboplatin/paclitaxel with trebananib or placebo; NCT01493505) interim analyses are awaited.
Pazopanib
Pazopanib is a multifunctional tyrosine kinase inhibitor that principally targets VEGFR (VEGFR-1, VEGFR-2, VEGFR-3), platelet derived growth factor receptor (PDGFR) (PDGFRα and PDGFRβ), fibroblast growth factor receptor (FGFR), and c-kit. Additional targets include colony-stimulating factor 1, lymphocyte-specific tyrosine kinase, and interleukin (IL)-2-inducible T-cell kinase.36 Its broad spectrum of antiangiogenic activity has forged some success in treating renal clear cell carcinoma and soft tissue sarcoma. However, after a small selection of Phase II and III studies, pazopanib is now emerging as an effective novel therapeutic agent in EOC. Friedlander et al published one of the first reports of its efficacy in a Phase II open-label study conducted in 36 patients with recurrent disease taking 800 mg pazopanib daily.37 Eleven patients (31%) obtained CA-125 response (95% CI, 16%–48%). Furthermore, the median response duration was 113 days, with 18% ORR in patients with measurable disease.37 Numerous additional Phase I/II studies are currently investigating the use of pazopanib in combination with various cytotoxic agents as a salvage regimen in the platinum-resistant setting.36 One such study is the Phase I/II Study of Pazopanib (GW786034) and Cyclophosphamide in Patients With Platinum-resistant Recurrent, Pre-treated Ovarian Cancer (PACOVAR), which aims to investigate the feasibility and efficacy of pazopanib in combination with metronomic cyclophosphamide. Conceptually, this is particularly intriguing in view of the potential additive antiangiogenic effects that could be realized with this strategy. Although a plethora of studies are ongoing for recurrent/resistant EOC, there has been some recent excitement generated from a Phase III study investigating pazopanib as a maintenance therapy in the first-line setting.38 Du Bois et al studied 940 patients with histologically proven EOC, primary peritoneal (PPC), or fallopian tube cancer (stage II–IV) and no evidence of progressive disease after primary platinum-paclitaxel-based chemotherapy. Subsequently, evaluable subjects were randomized to receive either 800 mg pazopanib daily or placebo for up to 2 years. Interestingly, maintenance pazopanib significantly prolonged median PFS compared with placebo by 5.6 months (17.9 versus 12.3 months, respectively; HR, 0.77; 95% CI, 0.64–0.91; P=0.0021).38 Inevitably, this was slightly offset by grade 3/4 toxicities, with hypertension (30.8%), neutropenia (9.9%), hepatotoxicity (9.4%), and diarrhea (8.2%) being the most frequently encountered.38 Although OS advantages have yet to be seen, the authors speculate that this may eventuate with future subgroup analyses.38
Sunitinib and sorafenib
Sunitinib also displays potent antiangiogenic properties through inhibition of multiple kinases, including VEGF, PDGF, c-Kit, and FMS-like tyrosine kinase-3 (Flt-3).39 To date, data confirming the potential efficacy of this agent emanate from three monotherapeutic Phase II studies in patients with recurrent EOC. Initial reports suggested that sunitinib (50 mg/day orally 4 of 6 weeks) yielded only modest benefits. In 30 evaluable patients with recurrent EOC/PPC, Biagi et al noted only a 3.3% PR rate and 10% CA-125 response, which was only evident in platinum-sensitive disease.40 A subsequent larger study (n=73) randomized patients with recurrent platinum-resistant ovarian cancer to either intermittent (50 mg/day, 4 of 6 weeks) or continuous (37.5 mg/day) dosing of sunitinib.41 The authors reported an ORR of 16.7% versus 5.4% (median PFS, 4.8 versus 2.9 months; median OS, 13.6 versus 13.7 months for the intermittent and continuous groups, respectively).41 The most recent trial in this same setting by Campos et al similarly showed a poor ORR of 8.3% and median PFS of 9.9 weeks.42 Of note, in all aforementioned trials, common adverse effects included hypertension, GI toxicity, hand–foot syndrome, and fatigue.
Sorafenib represents another multi-tyrosine kinase inhibitor (targeting VEGFR, PDGFR, and raf kinases), which exhibits a modicum of activity in EOC. A Phase II study by Matei et al (n=71) observed the effects of sorafenib monotherapy (400 mg orally twice daily) in recurrent ovarian cancer and revealed a 3.4% PR rate alongside a significant incidence of grade 3/4 toxicities mainly involving skin rash and GI and metabolic effects.43 In view of this, later studies have focused on combinatorial trials with chemotherapy or as maintenance therapy in the first-line setting, but they have equally shown poor tolerability and no PFS or OS advantage over chemotherapy alone.44,45
Cediranib
In parallel with the aforementioned multiangiogenic targeted therapies, cediranib (oral anti-VEGFR-1, VEGFR-2, VEGFR-3, and c-kit) also serves as an effective therapeutic strategy with some favorable results generated from both Phase II and III studies. Matulonis et al treated 46 patients with recurrent EOC, PPC, or fallopian tube cancer with oral cediranib originally dosed at 45 mg daily.46 This was eventually reduced to 30 mg because of significant toxicities in the initial stages of the study. Encouragingly, 17% of the patients achieved PR, and 13% SD, resulting in a 30% clinical benefit rate. Median PFS was 5.4 months, and the OS has not been reached to date. Grade 3 toxicities were evident in more than 20% of cases, and most included hypertension, diarrhea, and fatigue.46 This trial laid the foundations for the two-stage three-arm Phase III ICON6 trial, which observed the effects of cediranib (20 mg daily) in patients with recurrent platinum-sensitive disease (ie, platinum-free interval >12 months).47 Patients were equally randomized to standard chemotherapy (ie, platinum doublet), concurrent chemotherapy and cediranib, or concurrent therapy followed by maintenance cediranib. With restricted mean survival times the concurrent maintenance group conferred significant superiority over standard treatment in terms of both PFS (12.6 versus 9.4 months; HR 0.68; 95% CI, 0.54–0.87; log rank test P=0.0022) and OS (20.3 versus 17.6 months; HR, 0.70; 95% CI, 0.51-0.99; P=0.0419).47
Targeting type I ovarian cancer
In contrast to high-grade serous ovarian cancer (HGSC), type I ovarian cancers behave as biologically distinct entities and are commonly resistant to chemotherapy. Therefore, the advent of targeted therapies is particularly promising for the management of this challenging patient cohort. In contrast to type II ovarian tumors, which have a high frequency of p53 mutations and genetic instability, type I tumors (which include low-grade serous, endometrioid, mucinous, and clear cell carcinomas) more often have mutations in genes encoding protein kinases and other signaling molecules, including Kras, BRAF, PI3KCA and Erb2, CTNNB1, and PTEN.48 As a consequence, this provides a range of novel therapeutic targets (Table 2).
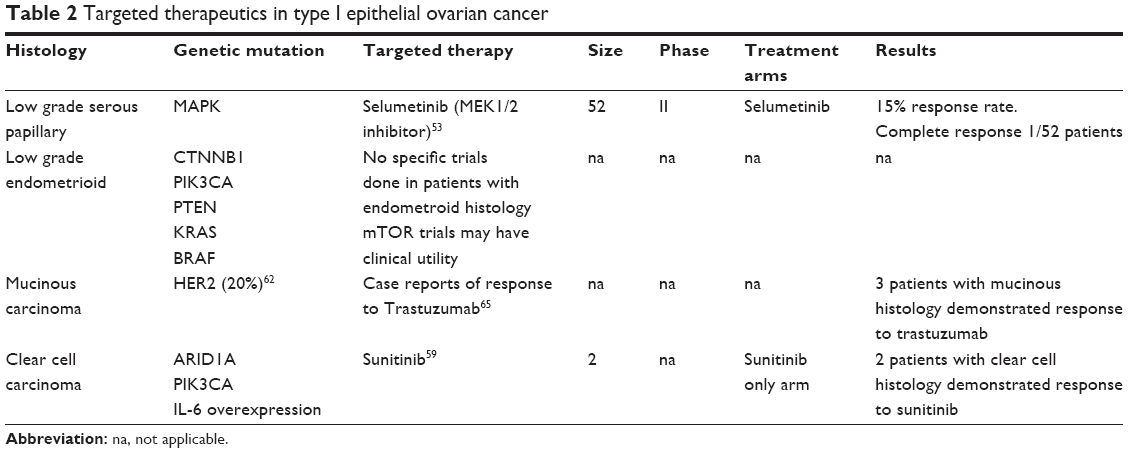 | Table 2 Targeted therapeutics in type I epithelial ovarian cancer |
Low-grade serous ovarian carcinoma
Low-grade serous carcinoma (LGSC) of the ovary, although generally having a more indolent course, has notably extremely poor response rates to chemotherapy.49,50 LGSC is purported to develop in a stepwise process of tumorigenesis, progressing from serous cystadenomas to serous borderline tumors and, eventually, to invasive LGSC. This is notably distinct from the pathway of development of HGSC, where p53 is commonly mutated.51,52 Furthermore, the LGSC group is frequently characterized by driver mutations in the MAPK pathway, which has created a surge of interest in small molecules targeting this pathway.51
The MEK inhibitors have been trialed in this population with some success. The GOG Phase II trial of selumetinib (a MEK 1/2 inhibitor) in 52 patients with recurrent LGSC reported a response rate of 15% and 65% of patients with stable disease. One patient had a complete response, two (6%) patients had BRAF mutations, and 14 (41%) had KRAS mutations, but there was no significant correlation between mutational status and response to treatment.53 Another MEK inhibitor trial, MILO (MEK inhibitor in Low-Grade Serous Ovarian Cancer), a Phase III trial of MEK162 versus physician’s choice of chemotherapy in patients with recurrent or persistent low-grade serous carcinomas of ovary, fallopian tube, or PPC, is currently open and aiming to recruit 300 patients internationally, including assessments of their mutational status (NCT01849874). An international randomized Phase II/III study of another MEK inhibitor, trametinib, compared with standard therapy is also under development for women with recurrent LGSC, again with a more extensive plan to characterize mutational status and correlate it to treatment response (NCT02101788). In view of potential resistance to MEK being mediated via the PI3K/AKT pathway, preclinical studies suggest that dual blockade of these pathways may result in greater efficacy, and a trial is currently being developed to investigate this further.54
Another potential treatment avenue is using the wild-type p53 gene in patients with LGSC. Nutlin-2 is an inhibitor of p53 breakdown, which binds to the pocket of murine double minute, where it binds TP53 and mediates rapid degradation of TP53 through the ubiquitin-proteasome pathway. Inhibiting this pathway can increase expression of wild-type TP53 and induce apoptosis and has been proposed as a possible therapeutic target not yet addressed by any investigational agent.55,56
Ovarian clear cell carcinoma
Ovarian clear cell cancer (OCCC) patients also experience disappointing responses to chemotherapy, whether it is standard carboplatin/paclitaxel or alternative regimens such as irinotecan/cisplatin.57 From a genomic perspective, up to 50% of OCCCs are characterized by somatic inactivating mutations in AT-rich interactive domain-containing protein 1A (ARID1A), and more than 30% of cases consist of activating mutations in the p110α catalytic subunit of PI3K (PIK3CA). Although a small Phase II study is currently investigating the addition of temsirolimus (mTOR inhibitor) to standard first-line chemotherapy (NCT01196429), to date, there are a paucity of trials confirming the efficacy of targeting the PI3K signaling pathway in OCCC.58 Another omnipresent feature in this subtype is the overexpression of the proinflammatory cytokine IL-6, which could also represent a potential therapeutic target.59 Furthermore, emerging in vivo and clinical evidence suggests that sunitinib may be an effective therapy for OCCC.60 Sunitinib has been reported to have some clinical efficacy in the setting of platinum-resistant clear cell ovarian cancer. Moreover, Anglesio et al highlighted two cases of patients with OCCC that had progressed on platinum chemotherapy who had a response to sunitnib.59 Clearly, more extensive clinical investigation is warranted for these approaches before they are incorporated into standard therapeutic management.
Mucinous ovarian carcinoma
In parallel with LGSC and clear cell subtypes, mucinous ovarian carcinomas are also more resistant to carboplatin/paclitaxel than HGSC.61 As these tumors have more biological similarities to colorectal tumors, the GOG0241 study was conceived to assess the appropriateness of applying standard regimens in this disease to mucinous ovarian carcinomas. This Phase III study recruited patients in the United States and the United Kingdom and compared carboplatin and paclitaxel +/− bevacizumab versus oxaliplatin and capecitabine +/− bevacizumab as first-line therapy in newly diagnosed or recurrent mucinous ovarian carcinomas (NCT01081262). Unfortunately, this study has recently been suspended because of poor patient accrual.
Mucinous carcinomas have also been found to overexpress human epidermal growth factor receptor 2 (HER2) at a rate of about 20%.62 Trastuzumab has only been investigated in a single Phase II trial of unselected ovarian cancer patients, but further trials in the HER2 overexpressing subpopulation would be of interest.63 In this Phase II trial of trastuzumab in patients with all types of advanced ovarian cancer and HER2 over-expression exhibited an ORR of 7.3% (of 45 patients), with one CR and two PRs.64 There are isolated reports in the literature of patients with HER2 amplified mucinous ovarian cancers responding to trastuzumab therapy. McAlpine et al reported a series of three patients with mucinous ovarian cancers who received trastuzumab with chemotherapy with some apparent clinical utility.65
Endometrioid ovarian carcinoma
Endometriosis is associated with endometrioid ovarian carcinomas, which also exhibit distinct pathways of tumorigenesis.48 Endometrioid ovarian carcinomas have been found to harbor a range of mutations in cell signaling pathways, including activating mutations in CTNNB1, which encodes β-catenin, and PIK3CA (approximately 20% mutation rate), in addition to PTEN (~20%) and less commonly, in KRAS and BRAF.28,48 Given this, it is possible that the mTOR inhibitors may have efficacy in this population. mTOR inhibitors have been used in a number of Phase I and Phase II trials in the treatment of EOC patients. Temsirolimus was studied as a single agent in an unselected population of patients with recurrent EOC and was found to have ORR of 9.3%.66 In the unselected population, as a single agent, in combination with chemotherapy, or in combination with bevacizumab, early results have been fairly disappointing. However, in a more select population, possibly with endometrioid histology, a higher level of efficacy may be seen.28
Poly(adenosine diphosphate [ADP]-ribose) polymerase inhibitors
Nearly a decade ago, preclinical data emerged suggesting a role for inhibitors of the poly(adenosine diphosphate [ADP]-ribose) polymerases (PARPs) in treating tumors with deficient DNA repair by homologous recombination (Table 3).67,68 The PARP enzymes play a vital role in cellular DNA repair, coordinating base-excision repair pathways.69 Inhibition of PARP results in an excess of single-strand breaks, which causes double-strand breaks during replication. Such defects are usually repaired by homologous recombination, a highly efficient process requiring intact BRCA proteins. Tumors with defective homologous recombination, including BRCA1/2 mutation-associated cancers, display particular sensitivity to PARP inhibition. In this situation, double-stranded DNA breaks are repaired by homologous end joining, which is error prone and causes genomic instability and cell death. This approach exploits the concept of synthetic lethality, in which significant lethal synergy occurs between two otherwise nonlethal events or, in this scenario, where PARP inhibition in the setting of defective BRCA protein leads to tumor cell death.70
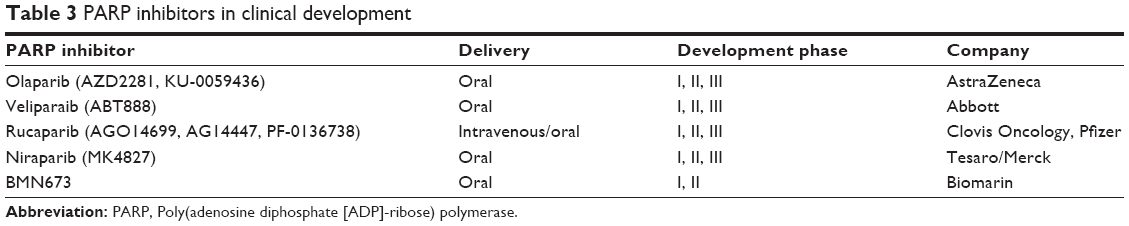 | Table 3 PARP inhibitors in clinical development |
Female carriers of germline mutations in BRCA1 and BRCA2 are at high risk of developing ovarian cancer, with lifetime risks of nearly 40% and 11%, respectively.71 Mutation in BRCA1 or BRCA2 is seen in 10%–20% of ovarian cancers, and defects in other homologous recombination pathway genes in a further 6%.72 However, in the most common form of malignant epithelial ovarian cancer, HGSC, defects in homologous recombination occur in up to 50% of cases, including germline or somatic loss-of-function mutations of BRCA1 or BRCA2, epigenetic silencing of BRCA1, and defects in other genes in this class including RAD51D, ATM, PALB2, RAD51C, and BRIP1.73 Patients with homologous recombination-deficient ovarian cancer typically demonstrate a “BRCAness” phenotype that is similar to that of patients with BRCA-mutated tumors, who generally exhibit better outcomes compared with patients with sporadic ovarian cancer, including improved platinum sensitivity and overall survival.74,75
The mechanism of action of small molecule inhibitors of PARP are, first, catalytic inhibition of repair of single-strand breaks and second, trapping of PARP on DNA forming cytotoxic PARP-DNA complexes.76 The first in-human trial of an oral PARP inhibitor, olaparib, was undertaken in patients with refractory solid tumors and was noteworthy for evidence of antitumor response in the subset of patients with known BRCA mutations and the absence of significant toxicity.77 The maximum tolerated dose was 400 mg twice daily, and the most common toxicities were mild fatigue, nausea, and vomiting. In an expansion cohort of this study in BRCA carriers with ovarian cancer, durable responses were seen correlating with platinum sensitivity. The response rate in platinum-sensitive patients was 69% compared with 45% in platinum-resistant patients and 23% in those with platinum refractory disease.78
In a Phase II trial of olaparib in patients with BRCA-mutated recurrent ovarian cancer, there was evidence of a dose-response relationship, with a response rate of 13% at 100 mg twice daily compared with 33% at 400 mg twice daily.79 Common adverse events included mild to moderate fatigue, nausea, diarrhea, and anemia. In a second Phase II study of olaparib in patients with recurrent high-grade serous or undifferentiated ovarian cancer with or without BRCA1/2 mutation, there was objective response in 41% of mutation-positive patients compared with 24% patients with sporadic disease.80 In both groups of patients, response rates were higher in those with platinum sensitivity, as seen previously.
This was followed by a randomized open-label Phase II trial of olaparib compared with standard therapy with pegylated liposomal doxorubicin (PLD) in patients with germline BRCA mutations and recurrent ovarian cancer after platinum failure. Olaparib 200 mg and 400 mg twice daily demonstrated response rates of 25% and 31%, respectively, compared with 18% with PLD, but there was no significant difference in PFS.81 There was improved tolerability of olaparib compared with PLD, suggesting PARP inhibition would be a reasonable option for this patient group.
Olaparib has also been investigated as maintenance monotherapy for platinum-sensitive relapsed HGSC. In a randomized placebo-controlled Phase II trial, olaparib demonstrated a PFS of 8.4 months compared with 4.8 months with placebo (HR for progression or death, 0.35; 95% CI, 0.25–0.49; P<0.001).82 A preplanned subgroup analysis after retrospective determination of BRCA status highlighted significant prolongation of PFS in BRCA-mutated patients with olaparib (11.2 months, versus 4.3 months with placebo).83 In patients without BRCA mutation, there was a less pronounced increase in PFS with olaparib (7.4 months compared with 5.5 months with placebo). There was no benefit in terms of OS in an interim analysis; however, 23% of BRCA-mutated patients who received placebo were eligible for PARP inhibitor therapy at progression.83 In view of this, as of June 2014, the US Food and Drug Administration’s Oncological Drugs Advisory Committee has voted against the accelerated use of olaparib in this setting. However, Phase III trials are under way to further evaluate the role of olaparib in patients with BRCA-mutated ovarian cancer after first-line platinum-based chemotherapy (Olaparib Monotherapy in Patients With BRCA Mutated Ovarian Cancer Following First Line Platinum Based Chemotherapy [NCT01844986]) and in BRCA-mutated patients with recurrent platinum-sensitive, high-grade serous cancer (Olaparib Treatment in BRCA Mutated Ovarian Cancer Patients After Complete or Partial Response to Platinum Chemotherapy [NCT01874353]). Hence, it appears that future Oncological Drugs Advisory Committee approval for olaparib in ovarian cancer hinges on the outcome of these studies.
PARP inhibitors have also been investigated in combination with chemotherapy as a result of potential synergy with platinum agents suggested in preclinical models.84,85 Initial Phase I studies of cytotoxics and olaparib have demonstrated dose-limiting myelosuppression requiring intermittent dosing of olaparib.86–88 However, promising antitumor activity was demonstrated in a Phase I trial of olaparib (50 mg twice a day, days 1–5) and cisplatin (60 mg/m2), with an ORR of 43% in patients with relapsed BRCA1/2 mutated ovarian cancer. Carboplatin (AUC 5) has also been assessed in combination with olaparib (400 mg twice a day, days 1–7), with a response rate of 44% in patients with recurrent ovarian cancer.89 In both of these studies, nearly half of the patients required growth factor support, and it is likely that hematologic toxicity will impose challenges in the further investigation of these combinations. Olaparib has also been investigated in combination with the antiangiogenic agent cediranib, with promising results presented at ASCO 2014.90 In a randomized Phase II trial in patients with recurrent platinum-sensitive high-grade serous or BRCA-mutated ovarian cancer, interim analysis revealed a response rate of 84% in patients receiving cediranib plus olaparib.90 The median PFS was 17.7 months in patients receiving both agents compared with 9.0 months in patients receiving olaparib monotherapy. However, the interim analysis also noted an unexpected PFS advantage with the combination group for BRCA wild-type/unknown (16.5 months in the combination group versus 5.7 months for olaparib; P=0.008) compared with BRCA-mutated patients (PFS of 19.4 months in the combination group versus 16.5 months with olaparib; P=0.16).90 It has to be noted that stratification of BRCA status for PFS was not a prespecified study endpoint. In addition, this study did not contain a comparative cedirinib monotherapy group. Nevertheless, this interesting finding clearly requires further investigation.
There are a number of other PARP inhibitors in early clinical development. Niraparib, a novel inhibitor of PARP1 and PARP2, demonstrated a 40% response rate in BRCA-mutated ovarian cancer in a Phase I trial, including a median duration of response of 431 days.91 Niraparib is being further explored in a randomized placebo-controlled Phase III trial as maintenance therapy in patients with platinum-sensitive BRCA-mutated ovarian cancer or HGSC (Maintenance Study With Niraparib Versus Placebo in Patients With Platinum Sensitive Ovarian Cancer [NCT01847274]).
As PARP inhibitors are still relatively new agents in the clinical arena, mechanisms of therapeutic resistance are largely based on preclinical models. To date, mechanisms of resistance include secondary mutations that restore functional BRCA1 or BRCA2 genes, reduced expression of nonhomologous end-joining factor 53BP1, and increased cellular drug efflux via increased expression of the P-glycoprotein pump.92 Elucidating the clinical effect of these putative resistance pathways requires clinical trial designs that incorporate repeat blood and tumor sampling at progression to allow comprehensive biomarker and genomic analyses.
The HER family
The ERBB/HER family of receptor tyrosine kinases plays a key role in cell growth and survival, and dysregulation is implicated in the pathogenesis of numerous malignancies.93 The epidermal growth factor receptor (EGFR or ERBB1) is expressed in 25%–50% of ovarian cancers, with conflicting data in terms of the prognostic implications.94 Clinical trials of EGFR inhibition in ovarian cancer have been disappointing to date. Phase II trials with cetuximab (EGFR chimeric monoclonal antibody) and gefitinib (EGFR tyrosine kinase inhibitor) both demonstrated limited activity in unselected patients with recurrent ovarian cancer.95,96 EGFR-activating mutations were present in 3.5% of tumors analyzed, including the only patient on study with an objective response. Similarly, erlotinib showed no benefit compared with observation alone in a randomized Phase III trial of patients with high-risk ovarian cancer after first-line chemotherapy.97
With respect to HER2 blockade, Bookman et al conducted a Phase II study using trastuzumab in 837 patients with recurrent or persistent EOC. The authors also reported a low frequency of mandated HER2 overexpression (ie, 2+/3+) amounting to 11.4% and found no relationship between HER2 expression and either survival or response to trastuzumab. Furthermore, the 7.3% ORR and 2.0 month PFS seen in this study was far inferior to analogous studies with single agent trastuzumab in breast cancer.98 In a subsequent GOG study, the rate of HER2 gene amplification by fluorescence in situ hybridization was only 7% and yielded no predictive or prognostic value.99 Pertuzumab, a recombinant humanized monoclonal antibody and HER dimerization inhibitor, has also demonstrated limited benefit in ovarian cancer, both as a single agent and in combination with platinum chemotherapy.100,101
A more recent report analyzing tissue arrays from 202 ovarian carcinoma patient samples demonstrated high levels of expression of HER3, HER4, and the hepatocyte growth factor, MET.102 HER3 activation and MET activation were present in 79% and 56% of cases, respectively. HER3 is thought to activate PI3K signaling in EGFR, HER2, and MET oncogene-addicted cancers; it has been associated with a poorer median survival in ovarian cancer.103,104 Preclinical work on MM-121, a HER3-monoclonal antibody, demonstrated significant inhibition of tumor growth in ovarian cancer cell lines and in xenograft models.104,105 A randomized Phase II trial of MM-121 with paclitaxel in platinum-resistant/refractory advanced ovarian cancer is ongoing (NCT01447706).
Targeting cancer-related inflammation
The intricate links between chronic inflammation and tumorigenesis postulated by Virchow in the 19th century106 have since been established in a host of malignancies. This particular paradigm is lucidly exemplified in EOC, with numerous cytokines and inflammatory cells playing key roles in processes such as tumor cell proliferation, migration, apoptotic evasion, and angiogenesis, which together promote a fertile microenvironment facilitating tumorigenesis. Recent translational studies have witnessed a modicum of success in targeting proinflammatory cytokines such as tumor necrosis factor α (TNF-α) and IL-6 (itself induced by TNF-α), which both act as principal orchestrators for these aforementioned tumorigenic processes. Madhusudan et al conducted a Phase II study with the TNF-α antagonist etanercept.107 In this trial, six of 30 patients with advanced ovarian cancer showed prolonged disease stabilization, and whole-blood cytokine assay showed a significant decrease in IL-6 levels (11 of 17 patients).107 In a subsequent Phase I clinical trial in patients with advanced cancer, Brown et al investigated the clinical efficacy and biological activity of infliximab (anti-TNF antibody).108 In this study, 41 patients with advanced cancer (eight of whom had EOC) received infliximab at 5 mg/kg (n=21) or 10 mg/kg (n=20) iv at 0 and 2 weeks and then every 4 weeks. Before iv infusion of 5 or 10 mg/kg infliximab, IL-6 levels were measured in plasma with levels ranging from undetectable to 35 pg/mL (median, 20 pg/mL). During the antibody infusion, serial samples of plasma were obtained over a 24–96 hour period. Plasma IL-6 levels decreased significantly in all patients at 24 and 48 hours after the first treatment with both doses. However, at the end of the study, IL-6 levels had started to rise, although not significantly. Furthermore, one of the eight patients with EOC attained stable disease.108 These observations influenced a subsequent Phase II study reported by Coward et al which investigated the efficacy of anti-IL-6 therapy with siltuximab in 18 patients with platinum-resistant EOC.109 The primary endpoint was response rate as assessed by combined Response Evaluation Criteria in Solid Tumors and CA125 criteria. One patient of 18 who were evaluable had a partial response, and seven others had periods of disease stabilization, four of which were treated for 6 months. None of the serious adverse events reported in the study were attributable to siltuximab, and it was generally well tolerated.109
In light of the resounding success of immunotherapy within malignant melanoma and non-small-cell lung cancer, there have been attempts to recapitulate these findings in EOC. Programmed death 1 (PD-1) is a coinhibitory receptor expressed on activated T cells, and ligand binding (PD-L1 and PD-L2) acts to regulate antitumor immunity. Nivolumab is a humanized monoclonal antibody that blocks this interaction and has recently been shown to confer some efficacy in a Phase I study in 15 patients (1 mg/kg, n=10; 3 mg/kg, n=5) with platinum-resistant EOC.110 In 13 evaluable patients, the interim analysis reported three PRs with a duration of response at 4, 5, and 10 months; the low-dose cohort was not associated with any significant adverse effects. With respect to PDL-1 inhibition, Brahmer et al conducted a multicenter Phase I study treating in 207 patients, including 17 with ovarian cancer. Of these, only one obtained an objective response.111 Although there is a current Phase II study with ipilimumab (anti CTLA-4 monoclonal antibody), in patients with platinum-sensitive EOC, recruitment has been halted (NCT01611558).
Folate antagonists
Folate receptors (FRs), specifically FR-α, are highly expressed in up to 80% of nonmucinous ovarian cancer.112–114 FR-α is integral to tumor folate transport and also facilitates chemoresistance and poor survival outcomes.114,115 Hence, it intuitively appears to represent a suitable therapeutic target. An initial study with the FR-α monoclonal antibody, farletuzumab, investigated its efficacy given alone and then subsequently in combination with carboplatin/paclitaxel at time of progression in patients with platinum-sensitive recurrence. Objective response was seen in 75% and CA125 declined in 80% of participants.116 Unfortunately, a follow-up study with farletuzumab in combination with weekly paclitaxel in platinum-resistant/refractory patients failed to reach its end point.117 However, a current Phase III study is investigating this agent in combination with carboplatin/paclitaxel in the first-line setting. In addition, intriguing results have been reported using an FR antibody conjugate of folic acid and desacetylvinblastine (EC145). In the Phase II PRECEDENT (Platinum Resistant Ovarian Cancer Evaluation of Doxil and Vintafolide [MK-8109, EC145] Combination Therapy [8109-009, EC-FV-04]) study, patients with recurrent platinum-resistant EOC were randomized to either PLD or PLD and EC145, with superior PFS seen in the combination group (24.0 versus 11.7 weeks; P=0.014).118 Interestingly, using whole-body single-photon emission computerized tomography scanning with Tc-labeled folate, increased response was evident in patients with higher folate receptor expression.118
Insulin signaling inhibition
The insulin-like growth factor (IGF) system has integral roles in numerous physiologic and pathologic processes. It consists of three ligands (IGF-1, IGF-2, and insulin) and their respective cell surface receptors (IGF-1R, IGF-2R, IR), all of which share a structural and functional homology.119–121 Their interaction initiates a surge of downstream signaling events predominantly via Ras-Raf-MAPK and PI3K-Akt transduction pathways, which facilitate tumorigenesis by promoting cellular proliferation, angiogenesis, invasion, and metastatic potential and inhibiting apoptosis. Manipulation of this pathway is currently being explored in various EOC settings. The dual IR/IGFR inhibitor, OSI906, has been investigated as part of a three-armed study in patients with platinum-resistant/refractory disease. The trial has randomized 199 patients to receive weekly paclitaxel either alone or in combination with OSI906, dosed either continuously or intermittently (NCT00889382). The interim analysis is pending. A single-agent IGFR monoclonal antibody study in platinum-sensitive disease is currently active (NCT00719212), but from a biologic perspective, the lack of dual IR blockade is likely to potentiate resistance to IGFR inhibition.122 Combinatorial studies with anti-IGFR/PI3K/mTOR inhibitors are also currently under way (NCT01243762).
Conclusion
In comparison to the significant advances in survival witnessed in other malignancies, undoubtedly, progress in this respect with EOC has been relatively sluggish. Taking into consideration the recent progress in our understanding of the biology of this disease, particularly with the type I and II dichotomy, it is appears counterintuitive to treat such a disparate collection of subtypes identically. Although the gold standard of the carboplatin and paclitaxel doublet regimen has remained the “soup de jour” for more than a decade, small increments in extending OS have only been realized with the addition of targeted therapies described in this review within the last few years. However, despite some encouraging results, such novel regimens have yet to become established treatments approved by the US Food and Drug Administration in EOC. Again, this highlights a significant gulf in the evolution of targeted agents that have now become embedded as standard of care in tumor types including breast, colorectal, and lung cancers. Specifically, the success witnessed in these tumor types was ultimately born from appropriate patient stratification. Unquestionably, the effect of trastuzumab, cetuximab, and erlotinib for these respective diseases would not have emerged without this stratification.
There is certainly evolving evidence of this practice bearing fruit in EOC. This has clearly been exemplified by the ICON7 subgroup analysis confirming that the OS advantage for first-line bevacizumab is restricted to patients with suboptimally debulked disease.22 It appears that such stratification could be enriched even further by the data presented by Gourley et al at ASCO 2014, whereby patients with a proangiogenic gene signature had significantly improved PFS when treated with carboplatin/paclitaxel and bevacizumab compared with chemotherapy alone. However, it appears that the addition of bevacizumab was detrimental to PFS in the immunogenic subgroup.24
With respect to PARP inhibition with olaparib in maintenance treatment for recurrent platinum-sensitive HGSC, PFS advantages appear to be enhanced in BRCA-mutated patients compared with BRCA wild-type.83 However, intriguing observations have been noted with combinations of olaparib and cediranib, in which PFS was significantly extended with this doublet over olaparib alone for BRCA wild-type/unknown status but not for BRCA-mutated patients.90 Hence this adds a new level of complexity to the stratification of patients for PARP inhibition when combined with other agents.
Another area of burgeoning excitement surrounds targeted therapy for type I EOC. By definition, low-grade tumors are inherently chemoresistant, yet chemotherapy still represents the standard of care for these diseases, which generally exhibit 4% ORR with this approach.49,50 For this reason alone, there is a desperate urge to change this outmoded paradigm. Moreover, these subtypes are signified by particular aberrant signaling pathways, which can be targeted by novel small molecule inhibitors. Indeed, this has resulted in numerous studies investigating the efficacy of MEK blockade in LGSC.
Furthermore, there is evidence confirming that lessons are being learned with the use of agents established in non-ovarian malignancies for EOC subtypes that share biological similarities to these diseases. For example, sunitinib, which is standard first-line therapy for metastatic renal clear cell carcinoma, has shown efficacy in OCCC59 and is now being further investigated in Phase II studies (NCT01824615 and NCT00979992). Similarly, the benefits of PI3K/mTOR inhibitors with endometrioid endometrial cancer certainly serve as a platform for developing studies with these drugs in endometrioid EOC.
Undoubtedly, future challenges will revolve around evading resistance to these new therapies, and various combinatorial studies are being designed to address these issues. Nevertheless, by expanding on these aforementioned trials with a deeper appreciation for the heterogeneity of EOC, it is certainly feasible that significant prolongation of survival could be achieved by adopting this philosophy.
Disclosure
The authors report no conflicts of interest in this work.
References
Sankaranarayanan R, Ferlay J. Worldwide burden of gynaecological cancer: the size of the problem. Best Pract Res Clin Obstet Gynaecol. 2006;20(2):207–225. | ||
Grulich AE, Swerdlow AJ, Head J, Marmot MG. Cancer mortality in African and Caribbean migrants to England and Wales. Br J Cancer. 1992;66(5):905–911. | ||
Whittemore AS, Harris R, Itnyre J; Collaborative Ovarian Cancer Group. Characteristics relating to ovarian cancer risk: collaborative analysis of 12 US case-control studies. IV. The pathogenesis of epithelial ovarian cancer. Am J Epidemiol. 1992;136(10):1212–1220. | ||
Daly M, Obrams GI. Epidemiology and risk assessment for ovarian cancer. Semin Oncol. 1998;25(3):255–264. | ||
Parkin DM, Muir CS. Cancer Incidence in Five Continents. Comparability and quality of data. IARC Sci Publ. 1992;(120):45–173. | ||
Chi DS, Eisenhauer EL, Zivanovic O, et al. Improved progression-free and overall survival in advanced ovarian cancer as a result of a change in surgical paradigm. Gynecol Oncol. 2009;114(1):26–31. | ||
Herrin VE, Thigpen JT. High-dose chemotherapy in ovarian carcinoma. Semin Oncol. 1999;26(1):99–105. | ||
Sonoda Y. Management of early ovarian cancer. Oncology (Williston Park). 2004;18(3):343–356. | ||
McGuire WP, Hoskins WJ, Brady MF, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med. 1996;334(1):1–6. | ||
Neijt JP, Engelholm SA, Tuxen MK, et al. Exploratory phase III study of paclitaxel and cisplatin versus paclitaxel and carboplatin in advanced ovarian cancer. J Clin Oncol. 2000;18(17):3084–3092. | ||
Bookman MA, Brady MF, McGuire WP, et al. Evaluation of new platinum-based treatment regimens in advanced-stage ovarian cancer: a Phase III Trial of the Gynecologic Cancer Intergroup. J Clin Oncol. 2009;27(9):1419–1425. | ||
Katsumata N, Yasuda M, Isonishi S, et al. Japanese Gynecologic Oncology Group. Long-term results of dose-dense paclitaxel and carboplatin versus conventional paclitaxel and carboplatin for treatment of advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer (JGOG 3016): a randomised, controlled, open-label trial. Lancet Oncol. 2013;14(10):1020–1026. | ||
Howell SB, Pfeifle CL, Wung WE, et al. Intraperitoneal cisplatin with systemic thiosulfate protection. Ann Intern Med. 1982;97(6):845–851. | ||
Markman M, Rowinsky E, Hakes T, et al. Phase I trial of intraperitoneal taxol: a Gynecoloic Oncology Group study. J Clin Oncol. 1992;10(9):1485–1491. | ||
Armstrong DK, Bundy B, Wenzel L, et al. Gynecologic Oncology Group. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354(1):34–43. | ||
Tewari D, Sill M, Monk B, et al. Phase III randomized clinical trial of cisplatin plus paclitaxel vs the non-platinum chemotherapy doublet of topotecan plus paclitaxel in women with recurrent, persistent, or advanced cervical carcinoma: A Gynecologic Oncology Group study. Gynecol Oncol. 2013;1:e2. | ||
Jaaback K, Johnson N, Lawrie TA. Intraperitoneal chemotherapy for the initial management of primary epithelial ovarian cancer. Cochrane Database Syst Rev. 2011;(11):CD005340. | ||
Vaughan S, Coward JI, Bast RC Jr, et al. Rethinking ovarian cancer: recommendations for improving outcomes. Nat Rev Cancer. 2011;11(10):719–725. | ||
Burger RA, Sill MW, Monk BJ, Greer BE, Sorosky JI. Phase II trial of bevacizumab in persistent or recurrent epithelial ovarian cancer or primary peritoneal cancer: a Gynecologic Oncology Group Study. J Clin Oncol. 2007;25(33):5165–5171. | ||
Cannistra SA, Matulonis UA, Penson RT, et al. Phase II study of bevacizumab in patients with platinum-resistant ovarian cancer or peritoneal serous cancer. J Clin Oncol. 2007;25(33):5180–5186. | ||
Burger RA, Brady MF, Bookman MA, et al. Gynecologic Oncology Group. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–2483. | ||
Perren TJ, Swart AM, Pfisterer J, et al; ICON7 Investigators. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–2496. | ||
Ledermann JA, Raja FA, Fotopoulou C, et al. ESMO Guidelines Working Group. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24 Suppl 6:vi24–vi32. | ||
Gourley C, McCavigan A, Perren T, et al. Molecular subgroup of high-grade serous ovarian cancer (HGSOC) as a predictor of outcome following bevacizumab. Presented at: 2014 ASCO Annual Meeting; May 30–June 3, 2014; Chicago, Illinois. Abstract 5502. | ||
Aghajanian C, Blank SV, Goff BA, et al. OCEANS: a randomized, double-blind, placebo-controlled phase III trial of chemotherapy with or without bevacizumab in patients with platinum-sensitive recurrent epithelial ovarian, primary peritoneal, or fallopian tube cancer. J Clin Oncol. 2012;30(17):2039–2045. | ||
Pujade-Lauraine E, Hilpert F, Weber B, et al. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: The AURELIA open-label randomized phase III trial. J Clin Oncol. 2014;32(13):1302–1308. | ||
Hall M, Gourley C, McNeish I, et al. Targeted anti-vascular therapies for ovarian cancer: current evidence. Br J Cancer. 2013;108(2):250–258. | ||
Leary A. The PI3K/Akt/mTOR Pathway in Ovarian Cancer: Biological Rationale and Therapeutic Opportunities. Rijeka, Croatia: Intech; 2013. Available from: http://dx.doi.org/10.5772/54170. Accessed December 1, 2014. | ||
Aravantinos G, Pectasides D. Bevacizumab in combination with chemotherapy for the treatment of advanced ovarian cancer: a systematic review. J Ovarian Res. 2014;7:57. | ||
Coleman RL, Duska LR, Ramirez PT, et al. Phase 1–2 study of docetaxel plus aflibercept in patients with recurrent ovarian, primary peritoneal, or fallopian tube cancer. Lancet Oncol. 2011;12(12):1109–1117. | ||
Gotlieb WH, Amant F, Advani S, et al. Intravenous aflibercept for treatment of recurrent symptomatic malignant ascites in patients with advanced ovarian cancer: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Oncol. 2012;13(2):154–162. | ||
Ledermann JA, Hackshaw A, Kaye S, et al. Randomized phase II placebo-controlled trial of maintenance therapy using the oral triple angiokinase inhibitor BIBF 1120 after chemotherapy for relapsed ovarian cancer. J Clin Oncol. 2011;29(28):3798–3804. | ||
Du Bois A, Kristensen G, Ray-Coquard I, et al. AGO-OVAR 12: a randomized placebo-controlled GCIG/ENGOT-Intergroup phase III trial of standard frontline chemotherapy/-nintedanib for advanced ovarian cancer [abstract]. Int J Gynecol Cancer. 2013;23(suppl 1):LBA1. | ||
Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–257. | ||
Monk BJ, Poveda A, Vergote I, et al. A phase III, randomized, double-blind trial of weekly paclitaxel plus the angiopoietin 1 and 2 inhibitor, trebananib, or placebo in women with recurrent ovarian cancer [abstract]: TRINOVA-1. Eur J Cancer. 2013;49(3):LBA:41. | ||
Davidson BA, Secord AA. Profile of pazopanib and its potential in the treatment of epithelial ovarian cancer. Int J Womens Health. 2014 13;6:289–300. | ||
Friedlander M, Hancock KC, Rischin D, et al. A Phase II, open-label study evaluating pazopanib in patients with recurrent ovarian cancer. Gynecol Oncol. 2010;119(1):32–37. | ||
du Bois A, Floquet A, Kim JW, et al. Incorporation of pazopanib in maintenance therapy of ovarian cancer. J Clin Oncol. 2014;32(30):3374–3382. | ||
Mendel DB, Laird AD, Xin X, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9(1):327–337. | ||
Biagi JJ, Oza AM, Chalchal HI, et al. A phase II study of sunitinib in patients with recurrent epithelial ovarian and primary peritoneal carcinoma: an NCIC Clinical Trials Group Study. Ann Oncol. 2011;22(2):335–340. | ||
Baumann KH, du Bois A, Meier W, et al. A phase II trial (AGO 2.11) in platinum-resistant ovarian cancer: a randomized multicenter trial with sunitinib (SU11248) to evaluate dosage, schedule, tolerability, toxicity and effectiveness of a multitargeted receptor tyrosine kinase inhibitor monotherapy. Ann Oncol. 2012;23(9):2265–2271. | ||
Campos SM, Penson RT, Matulonis U, et al. A phase II trial of Sunitinib malate in recurrent and refractory ovarian, fallopian tube and peritoneal carcinoma. Gynecol Oncol. 2013;128(2):215–220. | ||
Matei D, Sill MW, Lankes HA, et al. Activity of sorafenib in recurrent ovarian cancer and primary peritoneal carcinomatosis: a gynecologic oncology group trial. J Clin Oncol. 2011;29(1):69–75. | ||
Herzog TJ, Scambia G, Kim BG, et al. A randomized phase II trial of maintenance therapy with Sorafenib in front-line ovarian carcinoma. Gynecol Oncol. 2013;130(1):25–30. | ||
Thompson DS, Dudley BS, Bismayer JA, et al. Paclitaxel/carboplatin with or without sorafenib in the first-line treatment of patients with stage III/IV epithelial ovarian cancer: A randomized phase II study of the Sarah Cannon Research Institute. Presented at: 2013 ASCO Annual Meeting; May 31–June 4, 2014; Chicago, Illinois. Abstract 5513. | ||
Matulonis UA, Berlin S, Ivy P, et al. Cediranib, an oral inhibitor of vascular endothelial growth factor receptor kinases, is an active drug in recurrent epithelial ovarian, fallopian tube, and peritoneal cancer. J Clin Oncol. 2009;27(33):5601–5606. | ||
Ledermann JA, Perren TJ, Raja FA, et al. Randomized double-blind phase III trial of cediranib (AZD 2171) in relapsed platinum sensitive ovarian cancer: Results of the ICON6 trial. European J of Cancer. 2013;49(Suppl 3):Abstract 10. | ||
Cho KR, Shih IeM. Ovarian cancer. Annu Rev Pathol. 2009;4(1):287–313. | ||
Gershenson DM, Sun CC, Bodurka D, et al. Recurrent low-grade serous ovarian carcinoma is relatively chemoresistant.Gynecol Oncol. 2009;114(1):48–52. | ||
Schmeler KM, Sun CC, Bodurka DC, et al. Neoadjuvant chemotherapy for low-grade serous carcinoma of the ovary or peritoneum. Gynecol Oncol. 2008;108(3):510–514. | ||
Li J, Fadare O, Xiang L, Kong B, Zheng W. Ovarian serous carcinoma: recent concepts on its origin and carcinogenesis. J Hematol Oncol. 2012;5:8. | ||
Bell D, Berchuck A, Birrer M, et al. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. | ||
Farley J, Brady WE, Vathipadiekal V, et al. Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: an open-label, single-arm, phase 2 study. Lancet Oncol. 2013;14(2):134–140. | ||
Gershenson DM. The life and times of low-grade serous carcinoma of the ovary. Presented at: 2013 ASCO Annual Meeting; May 31–June 4, 2014; Chicago, Illinois. | ||
Bast RC Jr. Molecular approaches to personalizing management of ovarian cancer. Ann Oncol. 2011;22 (Suppl 8):viii5–viii15. | ||
Kojima K, Konopleva M, McQueen T, O’Brien S, Plunkett W, Andreeff M. Mdm2 inhibitor Nutlin-3a induces p53-mediated apoptosis by transcription-dependent and transcription-independent mechanisms and may overcome Atm-mediated resistance to fludarabine in chronic lymphocytic leukemia. Blood. 2006;108(3):993–1000. | ||
Kim A, Ueda Y, Naka T, Enomoto T. Therapeutic strategies in epithelial ovarian cancer. Journal of experimental and clinical cancer research. CR (East Lansing, Mich). 2012;31:14. | ||
Kurman RJ, Shih IeM. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer – shifting the paradigm. Hum Pathol. 2011;42(7):918–931. | ||
Anglesio MS, George J, Kulbe H, et al. IL6-STAT3-HIF signaling and therapeutic response to the angiogenesis inhibitor sunitinib in ovarian clear cell cancer. Clin Cancer Res. 2011;17(8):2538–2548. | ||
Stany MP, Vathipadiekal V, Ozbun L, et al. Identification of novel therapeutic targets in microdissected clear cell ovarian cancers. PLoS ONE. 2011;6(7):e21121. | ||
Alexandre J, Ray-Coquard I, Selle F, et al. GINECO. Mucinous advanced epithelial ovarian carcinoma: clinical presentation and sensitivity to platinum-paclitaxel-based chemotherapy, the GINECO experience. Ann Oncol. 2010;21(12):2377–2381. | ||
Anglesio MS, Kommoss S, Tolcher MC, et al. Molecular characterization of mucinous ovarian tumours supports a stratified treatment approach with HER2 targeting in 19% of carcinomas. J Pathol. 2013;229(1):111–120. | ||
Syrios J, Banerjee S, Kaye SB. Advanced epithelial ovarian cancer: from standard chemotherapy to promising molecular pathway targets – where are we now? Anticancer Res. 2014;34(5):2069–2077. | ||
Bookman MA, Darcy KM, Clarke-Pearson D, Boothby RA, Horowitz IR. Evaluation of monoclonal humanized anti-HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: a phase II trial of the Gynecologic Oncology Group. J Clin Oncol. 2003;21(2):283–290. | ||
McAlpine JN, Wiegand KC, Vang R, et al. HER2 overexpression and amplification is present in a subset of ovarian mucinous carcinomas and can be targeted with trastuzumab therapy. BMC Cancer. 2009;9(1):433. | ||
Behbakht K, Sill MW, Darcy KM, et al. Phase II trial of the mTOR inhibitor, temsirolimus and evaluation of circulating tumor cells and tumor biomarkers in persistent and recurrent epithelial ovarian and primary peritoneal malignancies: a Gynecologic Oncology Group study. Gynecol Oncol. 2011;123(1):19–26. | ||
Farmer H, McCabe N, Lord CJ, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–921. | ||
Bryant HE, Schultz N, Thomas HD, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–917. | ||
Virág L, Szabó C. The therapeutic potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev. 2002;54(3):375–429. | ||
Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J Clin Oncol. 2008;26(22):3785–3790. | ||
Antoniou A, Pharoah PD, Narod S, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117–1130. | ||
Walsh T, Casadei S, Lee MK, et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108(44):18032–18037. | ||
Bell D, Berchuck A, Birrer M, et al. Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–615. | ||
Tan DS, Rothermundt C, Thomas K, et al. “BRCAness” syndrome in ovarian cancer: a case-control study describing the clinical features and outcome of patients with epithelial ovarian cancer associated with BRCA1 and BRCA2 mutations. J Clin Oncol. 2008;26(34):5530–5536. | ||
Pennington KP, Walsh T, Harrell MI, et al. Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin Cancer Res. 2014;20(3):764–775. | ||
Murai J, Huang SY, Das BB, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012;72(21):5588–5599. | ||
Fong PC, Boss DS, Yap TA, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. | ||
Fong PC, Yap TA, Boss DS, et al. Poly(ADP)-ribose polymerase inhibition: frequent durable responses in BRCA carrier ovarian cancer correlating with platinum-free interval. J Clin Oncol. 2010;28(15):2512–2519. | ||
Audeh MW, Carmichael J, Penson RT, et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: a proof-of-concept trial. Lancet. 2010;376(9737):245–251. | ||
Gelmon KA, Tischkowitz M, Mackay H, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–861. | ||
Kaye SB, Lubinski J, Matulonis U, et al. Phase II, open-label, randomized, multicenter study comparing the efficacy and safety of olaparib, a poly(ADP-ribose) polymerase inhibitor, and pegylated liposomal doxorubicin in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer. J Clin Oncol. 2012;30(4):372–379. | ||
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med. 2012;366(15):1382–1392. | ||
Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–861. | ||
Evers B, Drost R, Schut E, et al. Selective inhibition of BRCA2-deficient mammary tumor cell growth by AZD2281 and cisplatin. Clin Cancer Res. 2008;14(12):3916–3925. | ||
Rottenberg S, Jaspers JE, Kersbergen A, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proc Natl Acad Sci U S A. 2008;105(44):17079–17084. | ||
Rajan A, Carter CA, Kelly RJ, et al. A phase I combination study of olaparib with cisplatin and gemcitabine in adults with solid tumors. Clin Cancer Res. 2012;18(8):2344–2351. | ||
Khan OA, Gore M, Lorigan P, et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer. 2011;104(5):750–755. | ||
Samol J, Ranson M, Scott E, et al. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: a phase I study. Invest New Drugs. 2012;30(4):1493–1500. | ||
Lee JM, Hays JL, Annunziata CM, et al. Phase I/Ib study of olaparib and carboplatin in BRCA1 or BRCA2 mutation-associated breast or ovarian cancer with biomarker analyses. J Natl Cancer Inst. 2014;106(6):dju089. | ||
Liu J, Fleming GF, Tolaney SM, et al. A phase I trial of the PARP inhibitor olaparib (AZD2281) in combination with the antiangiogenic cediranib (AZD2171) in recurrent ovarian or triple-negative breast cancer. Presented at: 2011 ASCO Annual Meeting; June 3–7, 2014; Chicago, Illinois. Abstract 5028. | ||
Sandhu SK, Schelman WR, Wilding G, et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: a phase 1 dose-escalation trial. Lancet Oncol. 2013;14(9):882–892. | ||
Lord CJ, Ashworth A. Mechanisms of resistance to therapies targeting BRCA-mutant cancers. Nat Med. 2013;19(11):1381–1388. | ||
Tebbutt N, Pedersen MW, Johns TG. Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer. 2013;13(9):663–673. | ||
Lafky JM, Wilken JA, Baron AT, Maihle NJ. Clinical implications of the ErbB/epidermal growth factor (EGF) receptor family and its ligands in ovarian cancer. Biochim Biophys Acta. 2008;1785(2):232–265. | ||
Secord AA, Blessing JA, Armstrong DK, et al. Phase II trial of cetuximab and carboplatin in relapsed platinum-sensitive ovarian cancer and evaluation of epidermal growth factor receptor expression: a Gynecologic Oncology Group study. Gynecol Oncol. 2008;108(3):493–499. | ||
Schilder RJ, Sill MW, Chen X, et al. Phase II study of gefitinib in patients with relapsed or persistent ovarian or primary peritoneal carcinoma and evaluation of epidermal growth factor receptor mutations and immunohistochemical expression: a Gynecologic Oncology Group Study. Clin Cancer Res. 2005;11(15):5539–5548. | ||
Vergote IB, Jimeno A, Joly F, et al. Randomized phase III study of erlotinib versus observation in patients with no evidence of disease progression after first-line platin-based chemotherapy for ovarian carcinoma: a European Organisation for Research and Treatment of Cancer-Gynaecological Cancer Group, and Gynecologic Cancer Intergroup study. J Clin Oncol. 2014;32(4):320–326. | ||
Bookman MA, Darcy KM, Clarke-Pearson D, Boothby RA, Horowitz IR. Evaluation of monoclonal humanized anti-HER2 antibody, trastuzumab, in patients with recurrent or refractory ovarian or primary peritoneal carcinoma with overexpression of HER2: a phase II trial of the Gynecologic Oncology Group. J Clin Oncol. 2003;21(2):283–290. | ||
Farley J, Fuchiuji S, Darcy KM, et al. Associations between ERBB2 amplification and progression-free survival and overall survival in advanced stage, suboptimally-resected epithelial ovarian cancers: a Gynecologic Oncology Group Study. Gynecol Oncol. 2009;113(3):341–347. | ||
Gordon MS, Matei D, Aghajanian C, et al. Clinical activity of pertuzumab (rhuMAb 2C4), a HER dimerization inhibitor, in advanced ovarian cancer: potential predictive relationship with tumor HER2 activation status. J Clin Oncol. 2006;24(26):4324–4332. | ||
Kaye SB, Poole CJ, Dańska-Bidzińska A, et al. A randomized phase II study evaluating the combination of carboplatin-based chemotherapy with pertuzumab versus carboplatin-based therapy alone in patients with relapsed, platinum-sensitive ovarian cancer. Ann Oncol. 2013;24(1):145–152. | ||
Davies S, Holmes A, Lomo L, et al. High incidence of ErbB3, ErbB4, and MET expression in ovarian cancer. Int J Gynecol Pathol. 2014;33(4):402–410. | ||
Tanner B, Hasenclever D, Stern K, et al. ErbB-3 predicts survival in ovarian cancer. J Clin Oncol. 2006;24(26):4317–4323. | ||
Schoeberl B, Faber AC, Li D, et al. An ErbB3 antibody, MM-121, is active in cancers with ligand-dependent activation. Cancer Res. 2010;70(6):2485–2494. | ||
Sheng Q, Liu X, Fleming E, et al. An activated ErbB3/NRG1 autocrine loop supports in vivo proliferation in ovarian cancer cells. Cancer Cell. 2010;17(3):298–310. | ||
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–545. | ||
Madhusudan S, Muthuramalingam SR, Braybrooke JP, et al. Study of etanercept, a tumor necrosis factor-alpha inhibitor, in recurrent ovarian cancer. J Clin Oncol. 2005;23(25):5950–5959. | ||
Brown ER, Charles KA, Hoare SA, et al. A clinical study assessing the tolerability and biological effects of infliximab, a TNF-alpha inhibitor, in patients with advanced cancer. Ann Oncol. 2008;19(7):1340–1346. | ||
Coward J, Kulbe H, Chakravarty P, et al. Interleukin-6 as a therapeutic target in human ovarian cancer. Clin Cancer Res. 2011 15;17(18):6083–6096. | ||
Hamanishi J, Mandai M, Ikeda T, et al. Efficacy and safety of anti-PD-1 antibody (Nivolumab: BMS-936558, ONO-4538) in patients with platinum-resistant ovarian cancer. Presented at: 2014 ASCO Annual Meeting; May 30–June 3, 2014; Chicago, Illinois. Abstract 5511. | ||
Brahmer JR, Tykodi SS, Chow LQ, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–2465. | ||
Miotti S, Canevari S, Ménard S, et al. Characterization of human ovarian carcinoma-associated antigens defined by novel monoclonal antibodies with tumor-restricted specificity. Int J Cancer. 1987;39(3):297–303. | ||
Kalli KR, Oberg AL, Keeney GL, et al. Folate receptor alpha as a tumor target in epithelial ovarian cancer. Gynecol Oncol. 2008;108(3):619–626. | ||
Chen YL, Chang MC, Huang CY, et al. Serous ovarian carcinoma patients with high alpha-folate receptor had reducing survival and cytotoxic chemo-response. Mol Oncol. 2012;6(3):360–369. | ||
Toffoli G, Russo A, Gallo A, et al. Expression of folate binding protein as a prognostic factor for response to platinum-containing chemotherapy and survival in human ovarian cancer. Int J Cancer. 1998;79(2):121–126. | ||
Armstrong DK, White AJ, Weil SC, Phillips M, Coleman RL. Farletuzumab (a monoclonal antibody against folate receptor alpha) in relapsed platinum-sensitive ovarian cancer. Gynecol Oncol. 2013;129(3):452–458. | ||
Elit L, Konner J, Armstrong DK, et al. A randomized, double-blind, placebo-controlled phase II study of the efficacy and safety of farletuzumab (MORAb-003) in combination with weekly paclitaxel in subjects with platinum-resistant or refractory relapsed ovarian cancer. Presented at: 2013 ASCO Annual Meeting; May 31–June 4, 2014; Chicago, Illinois. Abstract TPS255. | ||
Naumann RW, Coleman RL, Burger RA, et al. PRECEDENT: A randomized phase II trial comparing EC145 and pegylated liposomal doxorubicin (PLD) in combination, versus PLD alone, in subjects with platinum resistant ovarian cancer. J Clin Oncol. 2011;29:343s. Abstract 5045. | ||
Werner H, Weinstein D, Bentov I. Similarities and differences between insulin and IGF-I: structures, receptors, and signalling pathways. Arch Physiol Biochem. 2008;114(1):17–22. | ||
Fürstenberger G, Senn HJ. Insulin-like growth factors and cancer. Lancet Oncol. 2002;3(5):298–302. | ||
Bruchim I, Werner H. Targeting IGF-1 signaling pathways in gynecologic malignancies. Expert Opin Ther Targets. 2013;17(3):307–320. | ||
Buck E, Gokhale PC, Koujak S, et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): rationale for cotargeting IGF-1R and IR in cancer. Mol Cancer Ther. 2010;9(10):2652–2664. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.