Back to Journals » Therapeutics and Clinical Risk Management » Volume 10
New orally active anticoagulant agents for the prevention and treatment of venous thromboembolism in cancer patients
Authors Gerotziafas G, Mahé I, Elalamy I
Received 16 October 2013
Accepted for publication 31 January 2014
Published 13 June 2014 Volume 2014:10 Pages 423—436
DOI https://doi.org/10.2147/TCRM.S49063
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Grigoris T Gerotziafas,1,2 Isabelle Mahé,3 Ismail Elalamy1,2
1Service d'Hématologie Biologique, Hôpital Tenon, Hôpitaux Universitaires Est Parisien Assistance Publique Hôpitaux de Paris, Paris, France; 2ER2UPMC, Faculté de Médecine Pierre et Marie Curie, Université Paris VI, Paris, France; 3Service de Médecine interne, Hôpital Louis Mourier, Universite´ Paris 7, Assistance Publique Hôpitaux de Paris, EA REMES Université Paris Diderot, Sorbonne Paris Cité, Paris,France
Abstract: Patients with cancer have a 6–7-fold higher risk of venous thromboembolism (VTE) as compared with non-cancer patients. Effective and safe anticoagulation for the prevention and treatment of VTE is the cornerstone of the management of patients with cancer, aiming to decrease morbidity and mortality and to improve quality of life. Unfractionated heparin, low molecular weight heparins, fondaparinux and vitamin K antagonists (VKAs) are used in the prevention and treatment of VTE in cancer patients. Heparins and fondaparinux are administered subcutaneously. VKAs are orally active, but they have a narrow therapeutic window, numerous food and drug interactions, and treatment requires regular laboratory monitoring and dose adjustment. These limitations among others have important negative impact on the quality of life of patients and decrease adherence to the treatment. New orally active anticoagulant (NOAC) agents are specific inhibitors of activated factor Xa (FXa) (rivaroxaban and apixaban) or thrombin (dabigatran). It is expected that NOACs will improve antithrombotic treatment. Cancer patients are a particular group that could benefit from treatment with NOACs. However, NOACs present some significant interactions with drugs frequently used in cancer patients, which might influence their pharmacokinetics, compromising their efficacy and safety. In the present review, we analyzed the available data from the subgroups of patients with active cancer who were included in Phase III clinical trials that assessed the efficacy and safety of NOACs in the prevention and treatment of VTE. The data from the Phase III trials in prophylaxis of VTE by rivaroxaban or apixaban highlight that these two agents, although belonging to the same pharmacological group (direct inhibitors of factor Xa), have substantially different profiles of efficacy and safety, especially in hospitalized acutely ill medical patients with active cancer. A limited number of patients with VTE and active cancer were included in the Phase III trials (EINSTEIN, AMPLIFY, and RE-COVER) which evaluated the efficacy and safety of NOACs in the acute phase and secondary prevention of VTE. Although, from a conceptual point of view, NOACs could be an attractive alternative for the treatment of VTE in cancer patients, the available data do not support this option. In addition, due to the elimination of the NOACs by the liver and renal pathway as well as because of their pharmacological interactions with drugs which are frequently used in cancer patients, an eventual use of these drugs in cancer patients should be extremely cautious and be restricted only to patients presenting with contraindications for low molecular weight heparins, fondaparinux, or VKAs. The analysis of the available data presented in this review reinforces the request for the design of new Phase III clinical trials for the assessment of the efficacy and safety of NOACs in specific populations of patients with cancer.
Keywords: rivaroxaban, apixaban, dabigatran, antithrombotic treatment
Introduction
Cancer is linked to hypercoagulability and risk of thrombosis, and this close association was recognized in 1865 by Armand Trousseau.1,2 The relation between cancer and blood coagulation is in fact reciprocal: cancer induces a hypercoagulable state and is a major risk factor for venous thromboembolism (VTE). Activated platelets and factors of blood coagulation and fibrinolysis interfere with tumor cells and tumor growth, angiogenesis, and metastatic process and are thus involved in cancer progression. Patients with cancer have a 6–7-fold higher risk of VTE as compared with non-cancer patients.3,4 According to Shen and Pollak,5 one in every seven hospitalized cancer patients presents with pulmonary embolism (PE), and 60% of all hospitalized patients who die of massive PE have localized cancer or limited metastatic disease which would have allowed for a reasonably long survival in the absence of lethal PE. Idiopathic recurrent VTE is considered as an early clinical manifestation of cancer; it may reveal a tumor in 10%–25% of cases. The risk of cancer is multiplied by ten after a recurrent episode of idiopathic VTE.6–9 Metastasis increases VTE risk 3.2-fold. The increase of VTE risk is even higher in metastasis of aggressive types of cancer (eg, pancreatic cancer). Cancer doubles the risk of postoperative deep vein thrombosis (DVT) and triples the risk of postoperative fatal PE.10 Upper-limb DVT is also a frequent (7%) serious complication in patients with cancer.11 In summary, the risk of VTE in patients with cancer depends on the histological type of tumor, the time since diagnosis of the cancer, its stage, the therapeutic interventions, and the presence of intrinsic risk factors that are identified in each patient (ie, obesity, comorbidities, other medications, and previous personal or family history of VTE).
Effective and safe anticoagulation for prevention and treatment of VTE is the cornerstone of the management of patients with cancer, aiming to decrease morbidity, improve quality of life, and contribute to the decrease of mortality. Low molecular weight heparins (LMWHs) and the synthetic pentasaccharide (fondaparinux) are the main antithrombotic drugs used for the prevention of VTE in cancer patients.12,13 Unfractionated heparin (UFH), LMWHs, fondaparinux, and vitamin K antagonists (VKAs) are recommended for the treatment of the acute phase of VTE.13 The LMWHs enoxaparin, dalteparin, or tinzaparin rather than VKAs are recommended for long-term treatment (3–6 months) in cancer patients with VTE.12,14 In addition, in some groups of patients, treatment with LMWHs has a favorable effect on the progression of the disease and cancer-related mortality.15
Recurrent VTE rates of 9%–17% occur despite the use of therapeutic anticoagulation. LMWHs afford several advantages over warfarin. In LMWH-treated patients, in contrast to those treated with VKAs, routine laboratory monitoring and dose adjustment is not necessary. Nevertheless, up to 9% of cancer patients treated with LMWHs and 20% of those treated with warfarin develop recurrent VTE.16 Complete resolution and partial resolution of DVT occurs in up to 38% and 54%, respectively, after 6 months of anticoagulant treatment.17 Thrombi remain detectable in half of non-cancer patients after a year.18 The treatment with LMWHs presents several limitations such as the need for daily subcutaneous injections and the risk of heparin-induced thrombocytopenia. VKAs also have well known limitations, which increase the risk of major bleeding or recurrence of VTE and stem from drug and food interactions and the need for regular laboratory monitoring and dose adjustment.
Patients with cancer are at particularly high risk of VTE but also present several bleeding risk factors (ie, chemotherapy-induced thrombocytopenia, disseminated intravascular coagulation, and renal or liver impairment). The limitations of heparins and VKAs may negatively influence the benefit–risk ratio of antithrombotic treatment. Moreover, daily subcutaneous injections and skin hematoma following the administration of LMWHs and fondaparinux negatively affect compliance and adherence to antithrombotic treatment. Quality of life is compromised because of the need for frequent dose adjustment of VKAs, regular international normalized ratio (INR) measurement, dietary restrictions, and interactions with drugs which are used in cancer patients.
Important concerns on the management of treatment with LMWHs and VKAs, particularly in cancer patients, stem from the complexity of their mechanism of action and the heterogeneity of their structure. Programmed and designed synthesis of new oral anticoagulants (NOACs), which are homogeneous pharmacological preparations specifically targeting selected factors in the blood coagulation process, is expected to improve the quality of antithrombotic treatment, and this will be of particular importance for cancer patients.
Many expectations have arisen since the development of NOACs. Among numerous new anticoagulant agents being in clinical development, the orally active direct inhibitors of activated factor Xa (FXa), rivaroxaban and apixaban, and the orally active direct inhibitor of thrombin, dabigatran, are in the most advanced stage of clinical development.19–23 Apixaban, rivaroxaban, and dabigatran have been approved by health authorities in Europe and North America for the prevention of VTE during major orthopedic surgery and the prevention of stroke in patients with atrial fibrillation. In addition, rivaroxaban has been approved for the treatment of VTE. NOACs might offer a therapeutic option for anticoagulation in cancer patients. However the lack of clinical trials designed specifically for cancer patients is an important drawback for their use in oncology.
In this review, we will present the available data of Phase III clinical studies that assess the efficacy and safety of NOACs in VTE prophylaxis in hospitalized patients and in the treatment of VTE. We will present the results, although limited, from subgroups of patients with cancer.
Pharmacological and pharmacokinetic properties of NOACs
The target serine protease of blood coagulation is the basic criterion for the classification of NOACs. Accordingly, rivaroxaban and apixaban are direct inhibitors of FXa, and dabigatran is a direct inhibitor of thrombin. The direct NOACs, in contrast to UFH, LMWHs, and fondaparinux can inhibit both free and prothrombinase-bound FXa as well as fibrin-bound FXa. Direct FIIa inhibitors, in contrast to UFH and LMWHs inhibit free thrombin as well as fibrin-bound thrombin. The administration route, the delay of onset of the anticoagulant activity, the duration of the antithrombotic activity after the last dose, and the elimination route are essential characteristics for the classification of NOACs and their use in cancer patients. The “pro-drug” concept must also be taken into account when dabigatran is used, since dabigatran etexilate is the pro-drug.
Rivaroxaban
Rivaroxaban is an orally active, direct, selective, rapid, reversible, and competitive inhibitor of FXa. Rivaroxaban inhibits thrombin generation in a concentration-dependent manner. Rivaroxaban has high bioavailability after oral administration and presents predictable pharmacokinetic and pharmacodynamic properties, allowing administration of a single oral daily dose. Rivaroxaban is principally eliminated by the kidneys (66%). Of note, 36% of rivaroxaban exerted by the renal pathway is in active form, and 30% is in the form of inactive metabolites. Rivaroxaban is also eliminated by the liver, intestinal, and fecal pathways. Elimination of rivaroxaban is delayed in elderly patients (>75 years of age). The principal pharmacokinetic properties of rivaroxaban are summarized in Table 1.
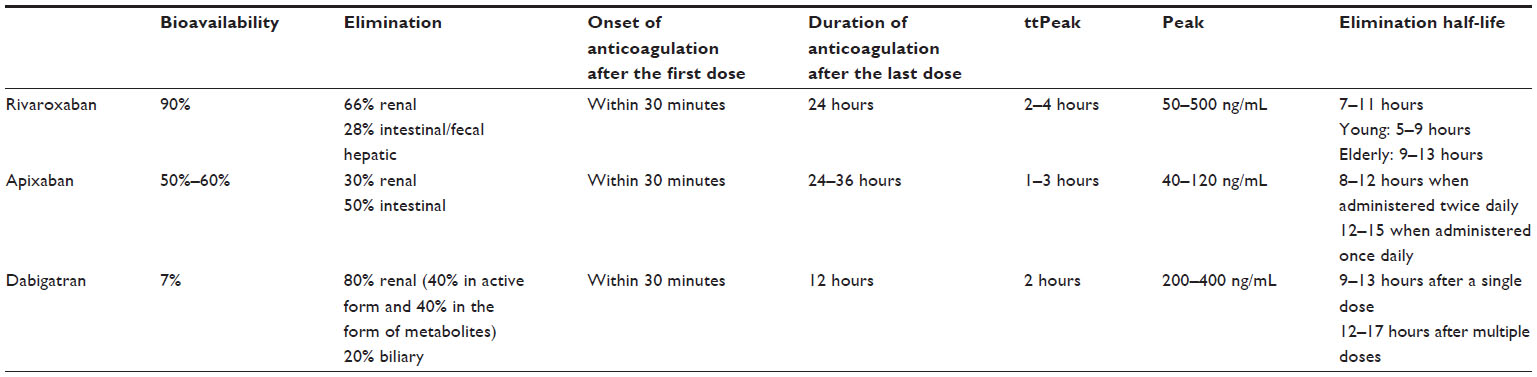 | Table 1 Principal pharmacokinetic properties of rivaroxaban, apixaban, and dabigatran |
Rivaroxaban is a minor substrate for ABCB1 P-glycoprotein (P-gp) transporters and is metabolized by CYP3A4 and CYP2J2 and also by CYP-independent mechanisms prior to elimination.24 Thus, it interacts with inhibitors of CYP3A4 and P-gp, resulting in an increase in the concentration of rivaroxaban in plasma and a potential increased risk of bleeding. In contrast, CYP3A4 inducers result in a decrease in the concentration of rivaroxaban in plasma and a potential reduction in the efficacy of the treatment. Co-administration of rivaroxaban with combined P-gp and strong CYP3A4 inducers (eg, rifampicin and phenytoin) decreased rivaroxaban exposure and also its anticoagulant activity by 27%–50%. The anticoagulant effect of rivaroxaban is not influenced by ranitidine or by co-administration of clopidogrel or aspirin alone or in combination. Rivaroxaban may be taken with or without food. However, administration of rivaroxaban after food intake delays the Tmax (time drug is present a maximum concentration) by about 1.5 hours. Food intake slightly reduces the peak of the maximum concentration in blood. Rivaroxaban does not induce any significant increase of transaminases, at least during the observation period of the Phase III clinical studies. Rivaroxaban exposure is increased by approximately 44%–64% in patients with renal impairment.
Patients with renal impairment receiving full dose rivaroxaban in combination with drugs classified as combined P-gp and weak or moderate CYP3A4 inhibitors (eg, amiodarone, diltiazem, verapamil, quinidine, ranolazine, dronedarone, felodipine, erythromycin, and azithromycin) may present increased exposure to the drug as compared with patients having normal renal function and no inhibitor use, since both pathways of rivaroxaban elimination are affected. In patients with moderate hepatic impairment (Child-Pugh B), exposure to rivaroxaban (in terms of the area under the curve) is increased by 127%. The most frequent drug interactions of rivaroxaban are summarized in Table 2. Rivaroxaban is contraindicated in renal impairment. Dyspepsia or alopecia may appear as a side-effect in some patients treated with rivaroxaban. The pharmacokinetics and pharmacodynamics of rivaroxaban in patients with hepatic impairment (Child-Pugh C) have not been evaluated.
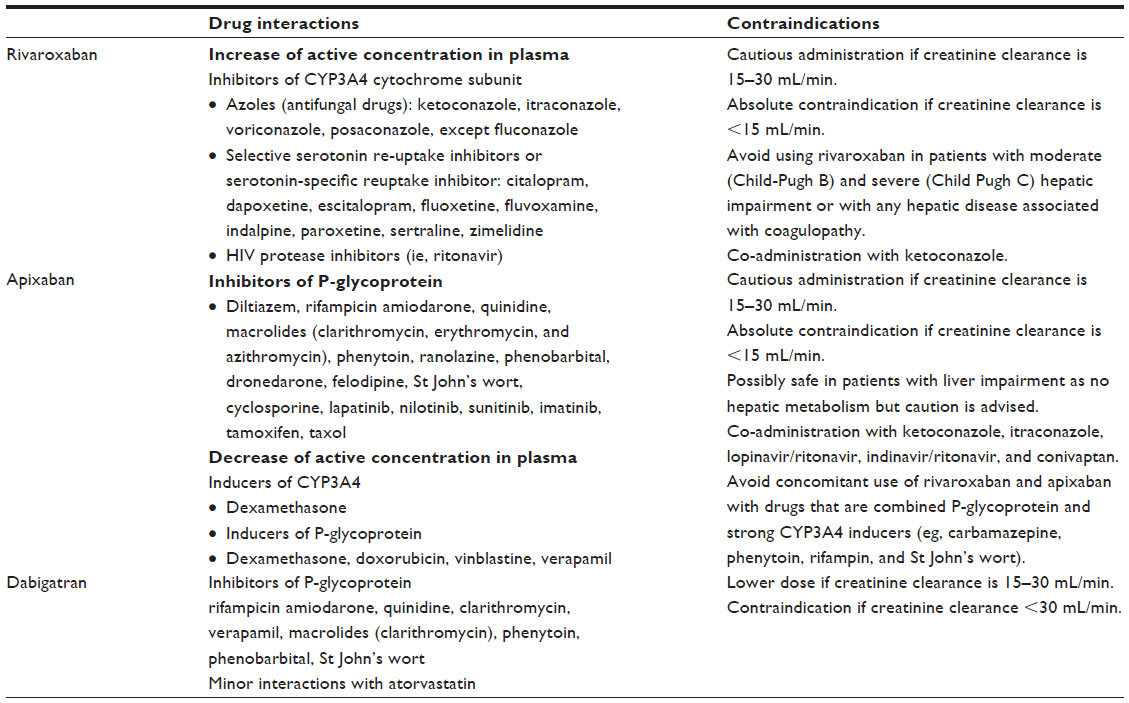 | Table 2 Main drug interactions and contraindications of the new antithrombotic agents |
Apixaban
Apixaban is an orally active, direct, reversible, competitive, and selective inhibitor of FXa. It inhibits thrombin generation in a concentration-dependent manner. Food intake does not have any significant impact on the pharmacokinetic and pharmacodynamic properties of apixaban. Steady-state concentration is achieved at the third day of treatment. The elimination of apixaban involves renal (30%) and liver (50%) pathways. It is metabolized mainly by O-demethylation, forming a phenol metabolite using the CYP3A4 system. Apixaban is administered orally in two daily doses. The principal pharmacokinetic properties of apixaban are summarized in Table 1. Apixaban, similarly to rivaroxaban, shows interactions with other drugs that induce or inhibit CYP3A4 and P-gp. The most frequent drug interactions of apixaban are summarized in Table 2. Apixaban is contraindicated in renal impairment. The absorption of apixaban is not affected by medications that alter gastric pH.
Dabigatran
Dabigatran etexilate, the pro-drug of dabigatran, is a peptidomimetic, reversible, competitive, direct thrombin inhibitor. It is a very hydrophilic molecule, has poor intestinal absorption after oral administration, and has low bioavailability (about 7%). Dabigatran is a pro-drug requiring ester cleavage in order to be transformed into its active form. The binding of dabigatran to plasma proteins is about 35%, and the extent of protein binding does not depend on dabigatran plasma concentration. Renal excretion is the predominant elimination pathway of dabigatran. Elimination of dabigatran is delayed in elderly patients because renal function declines with age. The time to the peak of the circulating concentration of dabigatran is delayed by about 2 hours when it is taken concomitantly with high-fat, high-caloric food, but no difference in the extent of absorption has been noted in comparison with a fasting state. The principal pharmacokinetic properties of dabigatran are summarized in Table 1. Due to esterase- and microsomal carboxylesterase-dependent biotransformation and the non-involvement of CYP450 enzymes, dabigatran seems to have limited drug interactions. The most frequent drug interactions of dabigatran are summarized in Table 2. The most common adverse reactions of dabigatran treatment are dyspepsia and gastritis-like symptoms.
Clinical trials with the new antithrombotics in the prevention of VTE – focus on cancer patients
Rivaroxaban and apixaban have been tested in Phase III clinical trials for VTE prophylaxis in acutely ill medical patients (MAGELLAN25 trial and ADOPT26 trials respectively). The design and the results of these trials are summarized in Table 3.
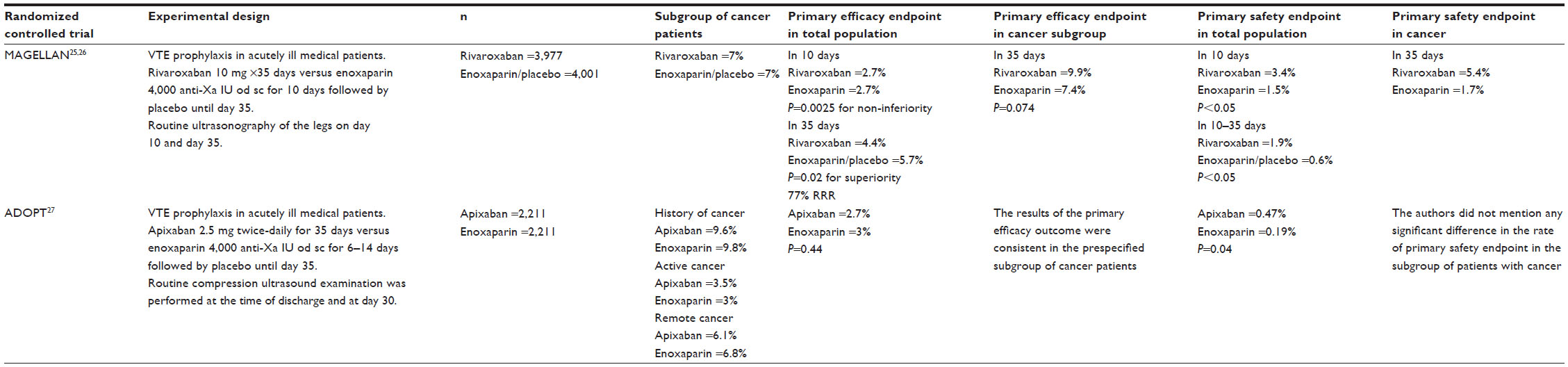 | Table 3 Efficacy and safety of new antithrombotic agents in thromboprophylaxis in acutely ill medical patients – focus on subgroups with cancer |
The MAGELLAN trial studied the efficacy and safety of rivaroxaban in acutely ill hospitalized medical patients.25 The trial was organized in two consecutive phases. The first phase compared rivaroxaban (10 mg given orally once-daily) versus enoxaparin (4,000 anti-Xa IU given subcutaneously once-daily) administered for 10 days. In the second phase, the patients who had received rivaroxaban (n=3,977) continued to receive the same dose of the drug until day 35 post-inclusion in the study. Those stratified in the enoxaparin group (n=4,001) received placebo. Routine ultrasonography of the leg and assessment for symptomatic VTE were performed on day 10 and day 35 post-inclusion. The composite efficacy endpoint included asymptomatic DVT and symptomatic VTE. In each group, 7% of the patients had active cancer. In the first phase of the trial, the incidence of VTE at 10 days was not significantly different between rivaroxaban- and enoxaparin-treated patients (2.7% in each group). The incidence of major and clinically relevant bleeding was significantly higher in the rivaroxaban group as compared with the enoxaparin group (3.4% versus 1.5%, respectively; P<0.05); similarly, the incidence of the primary safety outcome occurred in 2.8% of patients in the rivaroxaban group and in 1.2% of patients in the enoxaparin group (P=0.02). In the second phase of the trial (from day 10 to day 35), the incidence of VTE was 4.4% in the rivaroxaban group versus 5.7% in the placebo group (P=0.02). A significant increase in the incidence of major and clinically relevant bleeding events in the rivaroxaban group (1.9%) as compared with the placebo group (0.6%; P<0.05) compromised the prophylaxis with rivaroxaban. The primary safety outcome occurred in 4.1% of patients in the rivaroxaban group and in 1.7% of patients in the placebo group. In addition, the post hoc analysis of the subgroup of patients with active cancer showed a non-significant trend of less efficacy in patients treated with rivaroxaban as compared with enoxaparin. Indeed, the primary efficacy outcome on day 35 occurred in 9.9% of cancer patients in the rivaroxaban group and in 7.4% of patients in the control group (P=0.074). Of note, rivaroxaban administration in patients with active cancer was associated with a significantly higher bleeding risk as compared with the standard treatment. The primary safety outcome on day 35 occurred in 5.4% of patients in the rivaroxaban group and in 1.7% of patients in the control group (P<0.05; relative risk [RR] 1.34; 95% confidence interval [CI] 0.71–2.54). Descriptive values for the incidence of clinically relevant bleeding consistently favored enoxaparin over rivaroxaban in patients with active cancer.27
The ADOPT trial investigated the efficacy and safety of routine extended thromboprophylaxis with apixaban in acutely ill medical patients.26 The trial was a double-blind, double-dummy, placebo-controlled trial performed on 6,528 patients hospitalized for congestive heart failure, respiratory heart failure, infection, or other medical disorders and at least one additional risk factor for VTE. Only 68% of the patients included in the study could be evaluated for the primary efficacy endpoint. Patients included in the study were randomized to receive apixaban (n=2,211), administered orally at a dose of 2.5 mg twice-daily for 30 days or enoxaparin (n=2,111) administered subcutaneously (40 mg once-daily) for 6–14 days. A systematic compression ultrasound examination was performed at the time of discharge (but no earlier than day 5 and no later than day 14) and at day 30.
The primary efficacy outcome was the 30-day composite of death related to VTE, PE, symptomatic DVT, or asymptomatic proximal-leg DVT. The main safety outcomes were: bleeding, clinically relevant non-major bleeding, and all bleedings reported by investigators; myocardial infarction; stroke; thrombocytopenia; and death from any cause. About 10% of patients enrolled – equally distributed in both groups – had history of cancer, of whom 3.5% in the apixaban group and 3% in the enoxaparin group had active disease. The incidence of VTE was not significantly different between the two groups (2.7% in the apixaban group and 3% in the enoxaparin group; RR with apixaban, 0.87; 95% CI, 0.62–1.23; P=0.44). The rate of symptomatic DVT was lower among patients who received extended thromboprophylaxis with apixaban than among those who received enoxaparin (0.15% versus 0.49%), but this difference did not reach significance. The results of the primary efficacy outcome were consistent in the pre-specified subgroup of cancer patients.
The incidence of major and clinically relevant bleeding events during the 30-day treatment period was significantly higher in the apixaban group (0.47%) as compared with the enoxaparin group (0.19%; RR with apixaban, 2.58; 95% CI, 1.02–7.24; P=0.04). There was no significant difference between the apixaban group and the enoxaparin group regarding the mortality (4.1% in each group). The authors did not mention any significant difference in the rate of primary safety endpoint in the subgroup of patients with cancer.
A Phase II dose-finding, double-blind, randomized study (ADVOCATE) compared the efficacy and safety of thromboprophylaxis with three doses of apixaban (5, 10, and 20 mg once-daily) versus placebo in outpatients with myeloma or selected lymphomas or advanced or metastatic cancer of the lung, breast, gastrointestinal system, bladder, ovary, or prostate, undergoing chemotherapy. Prophylaxis was initiated within 4 weeks of the start of chemotherapy and lasted for 12 weeks.28 None of the patients treated with apixaban experienced VTE. In contrast, three VTE episodes (10.3%) occurred in the placebo group. The proportion of patients remaining free of major or clinically relevant non-major bleeding, VTE, and grade 3 or higher adverse events related to the study drug was 90.3% for apixaban and 82.8% for placebo. The frequency of bleeding was higher in patients receiving 20 mg of apixaban as compared with those receiving lower doses of the drug. These promising results point out the need for well-designed Phase III trials for the assessment of the efficacy and safety of apixaban in homogeneous groups of cancer patients. A Phase II pilot, multicenter, randomized placebo-controlled study, which assessed the efficacy and safety of thromboprophylaxis with apixaban in patients with advanced or metastatic cancer has been recently completed. Patients were randomly stratified to receive either apixaban 5 mg orally twice-daily or placebo for 12 weeks. The trial has been completed, and the publication of the results is expected to elucidate if routine administration of apixaban in cancer patients is a beneficial and safe antithrombotic strategy.
New antithrombotic agents in the treatment of VTE – focus on cancer patients
Specific FXa inhibitors
Rivaroxaban
The EINSTEIN-DVT study was an open-label, randomized Phase III trial, performed on patients with acute symptomatic, objectively confirmed DVT. Patients were allocated to receive rivaroxaban 15 mg twice-daily for 3 weeks, followed by 20 mg once-daily (n=1,731), or standard treatment with 1 mg/kg of enoxaparin administered subcutaneously once-daily, followed by treatment with VKAs at adjusted doses aiming for an INR between 2 and 3 (n=1,718) for 3, 6, or 12 months.29 Active cancer was present in 2.1% of patients in the rivaroxaban group and in 3% of patients in the enoxaparin/VKA group.
Recurrent nonfatal or fatal VTE was the primary efficacy outcome and occurred in 2.1% of patients treated with rivaroxaban and in 3% of patients treated with enoxaparin/VKA. Major and clinically relevant hemorrhagic episodes occurred in 8.1% of patients in each group. Regarding the efficacy endpoints, treatment with rivaroxaban was non-inferior as compared with the treatment with enoxaparin/VKA. At the inclusion, 6.8% of patients in the rivaroxaban group and 5.2% in the enoxaparin/VKA group had cancer. The primary efficacy endpoint occurred in 3.4% of cancer patients treated with rivaroxaban and in 5.6% of cancer patients treated with enoxaparin/VKA (P>0.05). The safety profile was also similar between the two treatments. The primary safety endpoint occurred in 14.4% of cancer patients on rivaroxaban and in 15.9% of cancer patients on enoxaparin/VKA.
The EINSTEIN-DVT extension trial was designed to assess the efficacy and safety of prolonged treatment with rivaroxaban (20 mg orally once-daily) for six additional months after the end of the initial period of treatment. Patients having completed the first phase of the treatment in the EINSTEIN-DVT trial, were randomized to receive rivaroxaban (n=602) or placebo (n=594) for an additional period of 6 or 12 months. At inclusion, 4.5% of patients in the rivaroxaban group and 4.4% in the placebo group had cancer. The incidence of VTE was 1.3% in patients treated with rivaroxaban and 7.1% in patients treated with placebo (P<0.001; RR ratio, 82%). The incidence of major and clinically relevant bleeding was 0.7% in the rivaroxaban group. None of the patients in the placebo group had bleeding episodes (P=0.11). The authors did not report any significant difference on the efficacy and safety of rivaroxaban treatment in the subgroup of cancer patients as compared with the total population included in the study.
EINSTEIN-PE was a randomized, open-label, event-driven, non-inferiority trial involving 4,832 patients who had acute symptomatic and objectively confirmed PE with or without DVT. Patients enrolled in the study received treatment with rivaroxaban 15 mg twice-daily for 3 weeks, followed by 20 mg once-daily (n=2,419), or standard treatment with 1 mg/kg of enoxaparin administered subcutaneously once-daily, followed by VKA, started within 48 hours post-randomization at doses aiming for an INR between 2 and 3 (n=2,413) for 3, 6, or 12 months.30 Active cancer was present in 4.7% of patients in the rivaroxaban group and in 4.5% of patients in the enoxaparin/VKA group.
The primary efficacy outcome was symptomatic recurrent VTE, which was defined as a composite of fatal or nonfatal PE or DVT. The principal safety outcome was clinically relevant bleeding, which was defined as a composite of major or clinically relevant non-major bleeding. VTE occurred in 2.1% of patients treated with rivaroxaban and in 1.8% of patients treated with enoxaparin/VKA (95% CI, 0.75–1.68; P=0.003 for a one-sided non-inferiority margin of 2.0, and P=0.57 for superiority). By day 21, at the end of twice-daily rivaroxaban administration, the primary efficacy outcome had occurred in 0.7% of patients in the rivaroxaban group and in 0.9% of patients in the standard-therapy group. The results of the on-treatment and per-protocol analyses were similar to those of the intention-to-treat analysis. The rates of recurrent VTE among patients with anatomically limited, intermediate, or extensive PE at baseline were 1.6%, 2.5%, and 1.7%, respectively, in the rivaroxaban group and 1.3%, 2.2%, and 1.4%, respectively, in the standard-therapy group. The principal safety outcome occurred in 10.3% of patients in the rivaroxaban group and in 11.4% of patients in the standard-therapy group (hazard ratio [HR], 0.90; 95% CI, 0.76–1.07; P=0.23). The rate of major bleeding was 1.1% in the rivaroxaban group and 2.2% in the standard-therapy group (HR, 0.49; 95% CI, 0.31–0.79; P=0.003). The outcome of a net clinical benefit occurred in 3.4% of patients in the rivaroxaban group and in 4% of patients in the enoxaparin/VKA group (HR, 0.85; 95% CI, 0.63–1.14; P=0.28). Rivaroxaban had similar efficacy as the control treatment in the prevention of the recurrent thromboembolic events. The primary efficacy endpoint occurred in 2.1% of cancer patients treated with rivaroxaban and in 1.8% of cancer patients treated with enoxaparin/VKA (P>0.05). The safety profile was also similar between the two treatments. The primary safety endpoint occurred in 12.3% of cancer patients on rivaroxaban and in 9.3% of cancer patients on enoxaparin/VKA.
The design and the main results of the EINSTEIN-DVT, EINSTEIN-DVT extension, and EINSTEIN-PE trials are summarized in Table 4.
Apixaban
Apixaban has been assessed for the treatment of DVT in a dose-finding study (Botticelli DVT study).31 The Phase III studies (AMPLIFY and AMPLIFY extension), testing apixaban at the doses of 10 and 5 mg twice-daily, have been undertaken.
The AMPLIFY trial, a randomized, double-blind study, included patients with acute VTE and compared apixaban, administered at a dose of 10 mg twice-daily for 7 days, followed by 5 mg twice-daily for 6 months (n=2,609), with conventional therapy by subcutaneous enoxaparin, at a dosage of 1 mg/kg every 12 hours for a median of 7 days followed by warfarin for 6 months at doses aiming a target INR within the range 2.0–3.0 (n=2,635).32 The primary efficacy outcome was recurrent symptomatic VTE or death related to VTE. The principal safety outcomes were major bleeding alone and major bleeding plus clinically relevant non-major bleeding. The treatment with apixaban showed similar efficacy to the standard treatment. The primary efficacy outcome occurred in 2.3% of patients in the apixaban group, and in 2.7% of the patients in the standard therapy group (P>0.05). These results met the study’s pre-specified criterion for apixaban’s non-inferiority to standard treatment. Major bleeding occurred in 0.6% of patients who received apixaban and in 1.8% of those who received standard treatment (RR, 0.31; 95% CI, 0.17–0.55; P<0.001 for superiority). The composite outcome of major bleeding and clinically relevant non-major bleeding occurred in 4.3% of the patients in the apixaban group as compared with 9.7% of those in the standard treatment group (RR, 0.44; 95% CI, 0.36–0.55; P<0.001). Rates of other adverse events were similar in the two groups. Patients with active cancer and VTE were excluded from the study if a long-term treatment with LMWHs was planned. In total, patients with active cancer were 2.5% in the apixaban group and 2.8% in the standard-treatment group. The authors did not report any data on efficacy and safety of the studied treatments in the subgroup of patients with active cancer.
The AMPLIFY-extension study, a randomized double-blind trial, compared the efficacy and safety of two doses of apixaban (2.5 and 5 mg, twice-daily) with placebo in 2,482 patients with VTE who had completed 6–12 months of anticoagulation therapy and for whom there was clinical equipoise regarding the continuation or cessation of anticoagulation therapy.33 The study drugs were administered for 12 months. The rates of symptomatic recurrent VTE or death from VTE were 8.8% in the placebo group (n=829), 1.7% in the group treated with apixaban 2.5 mg (n=840), and 1.7% in patients treated with apixaban 5 mg (n=813). The difference was statistically significant for both comparisons (P<0.001). The rates of major bleeding were 0.5% in the placebo group, 0.2% in the 2.5 mg apixaban group, and 0.1% in the 5 mg apixaban group. The rates of clinically relevant non-major bleeding were 2.3% in the placebo group, 3.0% in the 2.5 mg apixaban group, and 4.2% in the 5 mg apixaban group. Patients with active cancer were 2.2% in the placebo group, 1.8% in the 2.5 mg apixaban group, and 1.1% in the 5 mg apixaban group. The authors did not report any data on the efficacy and safety of apixaban in the group of patients with active cancer.
The design and the main results of the AMPLIFY and AMPLIFY-extension trials are summarized in Table 4.
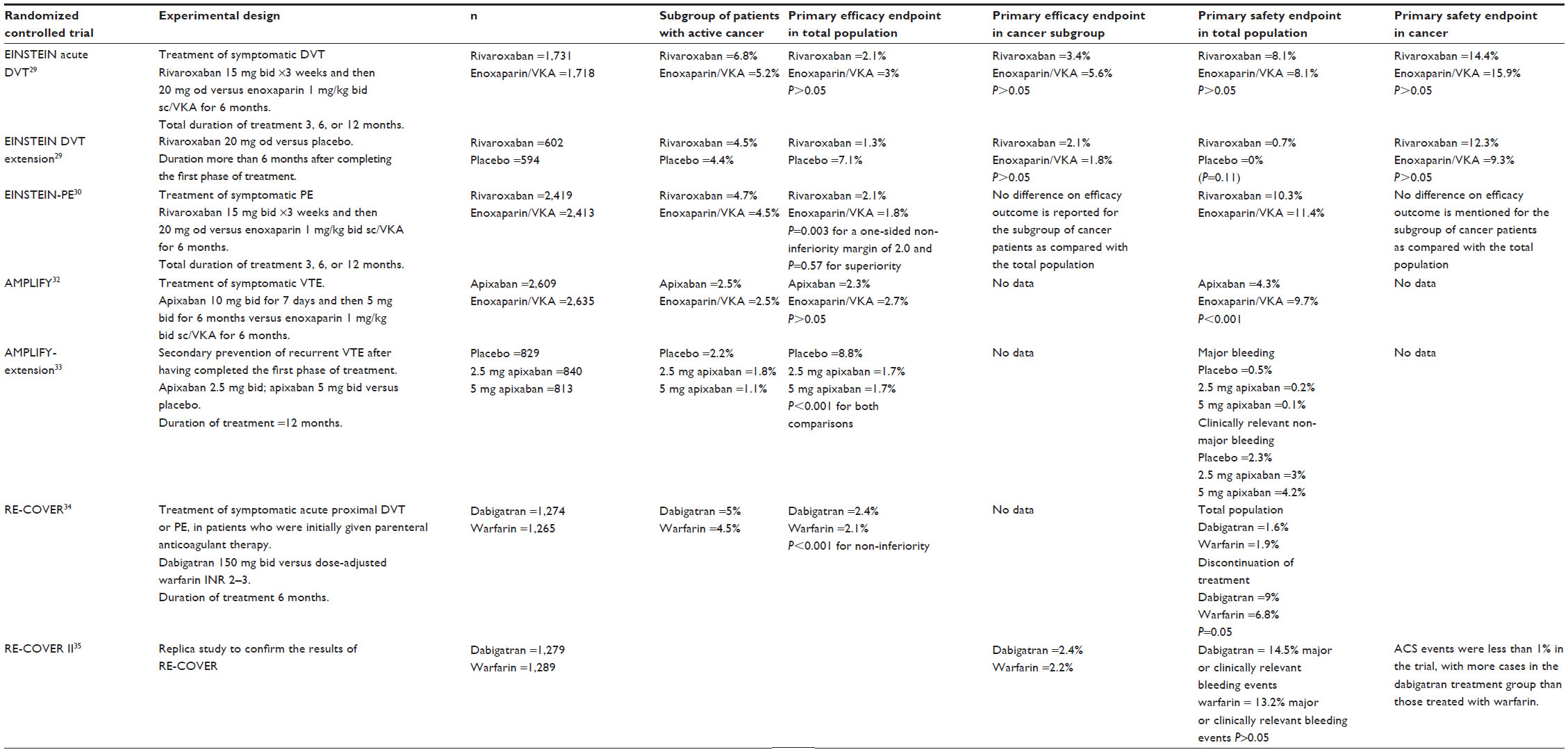 | Table 4 Efficacy and safety of NOACs in the treatment of VTE in patients with active cancer |
Dabigatran
The RE-COVER and the RE-COVER II trials are two randomized studies of a similar design that assessed the efficacy and safety of treatment with dabigatran in patients with symptomatic and objectively confirmed DVT or PE.34,35 Before randomization, all patients eligible for the study were treated with intravenous UFH or subcutaneous LMWHs. In the VKA group, warfarin was started on the day of random assignment, and the dose was adjusted to achieve an INR within the range of 2–3. Administration of dabigatran (150 mg orally twice-daily) or placebo was initiated, and the parenteral anticoagulant was stopped once dabigatran had been given for at least 5 days. The true or sham INR was recorded as 2.0 or higher on 2 consecutive days. The first dose of dabigatran was given within 2 hours, before the time that the next dose of initial parenteral therapy would have been due, or at the time of discontinuation of intravenous UFH. The duration of the treatment was 6 months. Recurrent VTE was the primary efficacy endpoint. Dabigatran was given in 1,274 patients, and warfarin was given to 1,265 patients. About 5% of patients in each of the two groups had active cancer. The incidence of VTE was not significantly different between the two groups (2.4% and 2.2%, respectively; P<0.05). The incidence of major and clinically relevant bleeding was 1.6% in the dabigatran group and 1.9% in the warfarin group (Table 4). The same incidence of VTE and major and clinically relevant bleeding was observed in the RE-COVER II trial. Acute coronary syndrome events were less than 1% in the trial, with more cases in the dabigatran group than those in the warfarin group.
Discussion
Prophylaxis and treatment of VTE in cancer patients is a challenging task, and the use of the new orally active and target-specific anticoagulant agents is an attractive therapeutic option. The clinical trials on the efficacy and safety of the selective FXa inhibitors (rivaroxaban and apixaban) in the primary prevention of VTE in acutely ill medical patients have shown that at least 7% of them have active cancer. The subgroup analysis in patients with cancer, although of limited power, enables the postulation that the two drugs, although belonging to the same class, seem to have different profiles of efficacy and safety in the prevention of VTE. Indeed, the MAGELLAN trial showed that thromboprophylaxis with rivaroxaban in acutely ill hospitalized cancer patients is less effective than enoxaparin. Interestingly, this finding opposes the main results of the trail, which show that thromboprophylaxis with rivaroxaban is as effective and as safe as enoxaparin. Whether the presence of cancer cells or the anticancer treatment modifies the antithrombotic potency of rivaroxaban needs to be studied. The study of the interactions between rivaroxaban and cancer cells could offer useful information required in establishing the optimum dose of rivaroxaban treatment in cancer patients.
The ADOPT trial showed that apixaban in the subgroup of cancer patients was as effective as enoxaparin. According to the results of the dose-finding study performed in outpatients with advanced or metastatic cancer receiving chemotherapy, apixaban appears to be effective and safe for thromboprophylaxis in cancer. Assuming that the characteristics of the patients are similar in the MAGELLAN and ADOPT trials, the difference of the efficacy between rivaroxaban and apixaban might be due to the difference in intensity of anticoagulation and its variability during the 24-hour therapeutic cycle induced by the different regimens of the two compounds (ie, once-daily for rivaroxaban and twice-daily for apixaban). Another parameter that might influence the efficacy of rivaroxaban and apixaban in cancer patients is the co-administration of cancer or other drugs which qre CYP3A4 cytochrome subunit inducers or inhibitors of P-gp. This parameter has not been controlled in the abovementioned trials. An additional reason for this difference on the efficacy and safety profile of rivaroxaban and apixaban, particularly in cancer patients, might be the different pharmacological properties of each drug, ie, the affinity and the reversibility of the binding to FXa and the degree of inhibition of thrombin generation. The potential differences between rivaroxaban and apixaban regarding the interaction with cancer cells should be investigated.
The EINSTEIN trials as well as the AMPLIFY and RE-COVER studies did not report any significant difference for rivaroxaban, apixaban, and dabigatran efficacy in cancer patients as compared with the total studied population of patients. However, less than 5% of patients included in these studies had active cancer; consequently, safe and generalized conclusions cannot be supported by the data published so far.
The data from the subgroup analysis on the efficacy and safety of NOACs in the acute phase and secondary prevention of VTE in patients with active cancer are encouraging. The Twice-daily Oral Direct Thrombin Inhibitor Dabigatran Etexilate in the Long Term Prevention of Recurrent Symptomatic VTE Phase III trial has been completed, but the results have not yet been published. Thus, it is not known whether long-term administration of dabigatran in the secondary prophylaxis of VTE is an effective and safe treatment, especially in cancer patients.
According to the data analyzed in the present review, specific Phase III trials assessing the efficacy and safety of treatment with rivaroxaban, apixaban, or dabigatran in patients with active cancer and VTE should be carried out. According to the pharmacokinetic and pharmacodynamic properties of NOACs and their interactions with drugs which are used in cancer patients, future studies should be carefully designed taking in consideration the type of chemotherapy and the adjuvant anticancer treatments. Since the data published so far on the efficacy and safety of specific FXa inhibitors in the prevention and treatment of VTE in cancer patients are very limited, the need for specific dose-finding clinical trials in cancer patients is highlighted. Taking into consideration that the cancer-related VTE risk is strongly dependent on the histological type of the cancer cells, the burden of the cancer, and the type of the anticancer treatment, the modelization of the antithrombotic efficacy of the specific FXa inhibitors in cancer-induced hypercoagulability might offer substantial information for a more accurate design of future clinical trials.
The specific direct inhibitors of FXa (rivaroxaban and apixaban) or thrombin (dabigatran) bind to plasma proteins. However, they efficiently inhibit thrombin generation at very low concentrations.36,37 Thus, it is less probable that fluctuations of plasma proteins, such as those related to inflammation, could influence the antithrombotic efficiency of NOACs. In addition, the capacity of rivaroxaban and apixaban to inhibit both free and prothrombinase-bound FXa theoretically warrants stable antithrombotic efficiency. These properties of NOACs might have a particular value for the optimization of the antithrombotic treatment in cancer patients who are frequently in an inflammatory state related to the evolution of the cancer, the chemotherapy, or the presence of infections.
Theoretically, the capacity of NOACs to inhibit clot-bound FXa or FIIa could offer a potential advantage over heparins and the indirect specific inhibitors of FXa (fondaparinux), since the former can neutralize the thrombogenic activity of the thrombi formed in the microvascular environment of cancer cells expressing tissue factor or other procoagulant molecules.38 The inhibition of clot-bound FXa or thrombin by NOACs may offer the advantage of in situ inhibition of thrombogenesis in cancer patients who present with chronic compensated disseminated intravascular coagulation.
The data from the Phase III clinical trials presented in this review do not allow any comparison between the specific inhibitors of FXa or thrombin or between apixaban and rivaroxaban. However, we have to highlight that both rivaroxaban and apixaban bring a different concept to the management of VTE as compared with that of dabigatran. The design of the EINSTEIN and AMPLIFY trials simplifies the treatment of VTE. Oral administration of a single antithrombotic drug – rivaroxaban or apixaban – during the acute phase of VTE as well as during the long-term secondary prevention of VTE recurrence is an efficient antithrombotic strategy.
The NOACs presented in this review are rapidly acting anticoagulants after oral administration characterized by predictable pharmacokinetics and pharmacodynamics. Their active concentration in plasma is dose dependent and is not significantly influenced by diet and food intake. In contrast to the treatment with VKAs, treatment with NOACs does not need any routine laboratory monitoring for dose adjustment, at least for the majority of patients. These properties might improve the adherence of the antithrombotic treatment in cancer patients if these drugs prove their efficacy and safety.
LMWHs offer advantages over warfarin for extended VTE treatment in cancer patients, since they induce a more significant reduction of the risk of recurrent VTE as compared with warfarin. In addition, the use of LMWHs simplifies the treatment because they can be administered in weight-adjusted doses without routine coagulation monitoring. Nonetheless, the need for daily subcutaneous injections renders LMWHs less than ideal for long-term treatment of VTE and negatively influences the quality of life of patients.
The oral administration and the needlessness of routine laboratory monitoring and dose adjustment are the major advantages of NOACs over LMWHs and VKAs. These advantages are expected to improve the adherence to the treatment and the quality of life of cancer patients. However, nausea and vomiting are among the most frequent side effects of anticancer treatments and might compromise the efficacy of NOACs. In patients treated with VKAs, the therapeutic hypocoagulability is related to the decreased synthesis of functional vitamin K-dependent clotting factors. One dose omission of VKA, as a consequence of vomiting, has limited effects, since the coagulation status is normalized after 3–5 days of treatment cessation. In contrast, the anticoagulant activity of the orally active antithrombotic agents is dose dependent, and within about 24–30 hours after treatment interruption, coagulation is normalized. Thus, the efficacy of the NOACs might be compromised. The measurement of plasma concentration and the global antithrombotic effect of NOACs might be mandatory in this situation. The NOACs presented herein are substrates of CYP3A4 and/or P-gp, and their active concentration in plasma is influenced by the co-administration of inducers or inhibitors of these enzymes. NOACs present significantly less drug–drug interactions as compared with VKAs. Several anticancer agents or drugs frequently used in cancer patients are metabolized in the liver and are inducers or inhibitors of CYP3A4 or P-gp. Cancer patients are at risk of opportunistic infections or fungal infections, thus they may receive P-gp inhibitors of inducers or inhibitors of CYP3A4. For this reason, administration of orally active NOACs in cancer patients should be particularly cautious. A check-list of potential drug interactions should be carefully applied. The development of easy to use laboratory assays for the monitoring of the NOACs and adaptation of the dose is of particular interest in cancer patients.
LMWHs interfere with cancer cells, resulting in inhibition of their proliferation and in downregulation of their angiogenic and metastatic potential.39–41 Several lines of evidence suggest that in some groups of cancer patients, the administration of LMWHs might improve cancer-related mortality.15 The interactions of NOACs with cancer cells have not yet been studied.
The need for a specific antidote to NOACs is of major importance in cancer patients, who are frequently in hemorrhagic risk due to the effect of anticancer treatment (ie, thrombocytopenia induced by chemotherapy) or the presence of metastasis in organs that participate in hemostasis (ie, liver or bone marrow).
In conclusion, the available – although limited – data from Phase III clinical trials in the prevention and treatment of VTE are encouraging for the development of clinical research of NOACs in the field of cancer patients.
Disclosure
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in, or financial conflict with, the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. No writing assistance was utilized in the production of this manuscript.
References
Trousseau A. Phlegmasia alba dolens. In: Trousseau A, editor. [Clinique Medicale de l’Hotel Dieu de Paris]. Paris: Balllier; 1865:654–712. French. | |
Zwicker JI, Furie BC, Furie B. Cancer-associated thrombosis. Crit Rev Oncol Hematol. 2007;62(2):126–136. | |
Heit JA, Mohr DN, Silverstein MD, Petterson TM, O’Fallon WM, Melton LJ 3rd. Predictors of recurrence after deep vein thrombosis and pulmonary embolism. Arch Intern Med. 2000;160(6):761–768. | |
Blom JW, Doggen CJ, Osanto S, Rosendaal FR. Malignancies, prothrombotic mutations, and the risk of venous thrombosis. JAMA. 2005;293(6):715–722. | |
Shen VS, Pollak EW. Fatal pulmonary embolism in cancer patients: is heparin prophylaxis justified? South Med J. 1980;73(7):841–843. | |
White RH, Chew HK, Zhou H, et al. Incidence of venous thromboembolism in the year before the diagnosis of cancer in 528,693 adults. Arch Intern Med. 2005;165(15):1782–1787. | |
Polite BN, Lamont EB. Are venous thromboembolic events associated with subsequent breast and colorectal carcinoma diagnoses in the elderly? A case-control study of Medicare beneficiaries. Cancer. 2006;106(4):923–930. | |
Rance A, Emmerich J, Guedj C, Fiessinger JN. Occult cancer in patients with bilateral deep-vein thrombosis. Lancet. 1997;350(9089):1448–1449. | |
Sutherland DE, Weitz IC, Liebman HA. Thromboembolic complications of cancer: epidemiology, pathogenesis, diagnosis and treatment. Am J Hematol. 2003;72(1):43–52. | |
Lee AY. Epidemiology and management of venous thromboembolism in patients with cancer. Thromb Res. 2003;110(4):167–172. | |
Monreal M, Munoz FJ, Rosa V, et al. Upper extremity DVT in oncological patients: analysis of risk factors. Data from the RIETE registry. Exp Oncol. 2006;28(3):245–247. | |
Coleman R, MacCallum P. Treatment and secondary prevention of venous thromboembolism in cancer. Br J Cancer. 2010;102 Suppl 1: S17–S23. | |
Kahn SR, Lim W, Dunn AS, et al. American College of Chest Physicians. Prevention of VTE in nonsurgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(Suppl 2):e195S–e226S. | |
Kearon C, Akl EA, Comerota AJ, et al; American College of Chest Physicians. Antithrombotic therapy for VTE disease: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(Suppl 2):e419S–e494S. | |
Gerotziafas GT, Papageorgiou C, Hatmi M, Samama MM, Elalamy I. Clinical studies with anticoagulants to improve survival in cancer patients. Pathophysiol Haemost Thromb. 2008;36(3–4):204–211. | |
Lee AY. Thrombosis in cancer: an update on prevention, treatment, and survival benefits of anticoagulants. Hematology Am Soc Hematol Educ Program. 2010;144–149. | |
Prandoni P, Lensing AW, Prins MH, et al. Residual venous thrombosis as a predictive factor of recurrent venous thromboembolism. Ann Intern Med. 2002;137(12):955–960. | |
Kearon C. Natural history of venous thromboembolism. Circulation. 2003;107(23 Suppl 1):I22–I30. | |
Gerotziafas GT, Samama MM. Heterogeneity of synthetic factor Xa inhibitors. Curr Pharm Des. 2005;11(30):3855–3876. | |
Gómez-Outes A, Suárez-Gea ML, Lecumberri R, Rocha E, Pozo-Hernández C, Vargas-Castrillón E. New parenteral anticoagulants in development. Ther Adv Cardiovasc Dis. 2011;5(1):33–59. | |
Lassen MR, Laux V. Emergence of new oral antithrombotics: a critical appraisal of their clinical potential. Vasc Health Risk Manag. 2008;4(6):1373–1386. | |
Samama MM, Gerotziafas GT. Newer anticoagulants in 2009. J Thromb Thrombolysis. 2010;29(1):92–104. | |
Denas G, Pengo V. Emerging anticoagulants. Expert Opin Emerg Drugs. 2011;16(1):31–44. | |
Ufer M. Comparative efficacy and safety of the novel oral anticoagulants dabigatran, rivaroxaban and apixaban in preclinical and clinical development. Thromb Haemost. 2010;103(3):572–585. | |
Cohen A; Investigators of the MAGELLAN trial. Rivaroxaban compared with enoxaparin for the prevention of venous thromboembolism in acutely ill medical patients. 60th meeting of the American College of Cardiology; Apr 2011; New Orleans, USA. | |
Goldhaber SZ, Leizorovicz A, Kakkar AK, et al. Apixaban versus enoxaparin for thromboprophylaxis in medically ill patients. N Engl J Med. 2011;365(23):2167–2177. | |
Cohen AT, Spiro TE, Buller HR, et al. Rivaroxaban for thromboprophylaxis in acutely ill medical patients. N Engl J Med. 2013;368(6):513–523. | |
Levine MN, Gu C, Liebman HA, et al. A randomized phase II trial of apixaban for the prevention of thromboembolism in patients with metastatic cancer. J Thromb Haemost. 2012;10(5):807–814. | |
EINSTEIN Investigators, Bauersachs R, Berkowitz SD, Brenner B, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med. 2010;363(26):2499–2510. | |
EINSTEIN-PE Investigators, Büller HR, Prins MH, Lensin AW, et al. Oral Rivaroxaban for the Treatment of Symptomatic Pulmonary Embolism. N Engl J Med. 2012;366(14):1287–1297. | |
Botticelli Investigators, Writing Committe, Buller H, Deitchman D, Prins M, Segers A. Efficacy and safety of the oral direct factor Xa inhibitor apixaban for symptomatic deep vein thrombosis. The Botticelli DVT dose-ranging study. J Thromb Haemost. 2008;6(8):1313–1318. | |
Agnelli G, Buller HR, Cohen A, et al; AMPLIFY Investigators. Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369(9):799–808. | |
Agnelli G, Buller HR, Cohen A, et al; AMPLIFY-EXT Investigators. Apixaban for extended treatment of venous thromboembolism. N Engl J Med. 2013;368(8):699–708. | |
Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med. 2009;361(24):2342–2352. | |
Schulman S, Kakkar AK, Schellong SM, et al. A randomized trial of dabigatran versus warfarin in the treatment of acute venous thromboembolism (RE-COVER II); 53rd ASH Annual Meeting and Exposition; December 10–13, 2011; San Diego. | |
Gerotziafas GT, Baccouche H, Sassi M, et al. Optimisation of the assays for the measurement of clotting factor activity in the presence of rivaroxaban. Thromb Res. 2012;129(1):101–103. | |
Samama MM, Guinet C. Laboratory assessment of new anticoagulants. Clin Chem Lab Med. 2011;49(5):761–772. | |
Gerotziafas GT, Galea V, Mbemba E, et al. Tissue factor over-expression by human pancreatic cancer cells BXPC3 is related to higher prothrombotic potential as compared to breast cancer cells MCF7. Thromb Res. 2012;129(6):779–786. | |
Borsig L. Antimetastatic activities of heparins and modified heparins. Experimental evidence. Thromb Res. 2010;125 Suppl 2:S66–S71. | |
Mousa SA, Petersen LJ. Anti-cancer properties of low-molecular-weight heparin: preclinical evidence. Thromb Haemost. 2009;102(2):258–267. | |
Falanga A, Marchetti M. Heparin in tumor progression and metastatic dissemination. Semin Thromb Hemost. 2007;33(7):688–694. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.