Back to Journals » ImmunoTargets and Therapy » Volume 15
New Insights on IgA Nephropathy from the Perspective of Active Lesions and Systemic Inflammatory Responses
Received 30 November 2025
Accepted for publication 28 March 2026
Published 21 April 2026 Volume 2026:15 585437
DOI https://doi.org/10.2147/ITT.S585437
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Flavio Salazar-Onfray
Yang Yang, Gaosi Xu
Department of Nephrology, The Second Affiliated Hospital, Jiangxi Medical College, Nanchang University, Nanchang, People’s Republic of China
Correspondence: Gaosi Xu, The Second Affiliated Hospital, Jiangxi Medical College, Nanchang University, No. 1, Minde Road, Donghu District, Nanchang, 330006, People’s Republic of China, Tel/Fax +86 791 86312770, Email [email protected]
Abstract: Immunoglobulin A nephropathy (IgAN) is a chronic disease. Active disease typically presents with massive proteinuria and gross hematuria, along with renal pathological features such as mesangial cell proliferation, endothelial cell proliferation, and crescents. Active lesions may achieve remission following treatment but also can relapse at any stage of the patient’s life. Systemic inflammatory markers are significantly associated with IgAN disease activity indicators. Research has found that patients with high disease activity and low chronicity are more likely to benefit from immunosuppressive therapy. We speculated that “four-hit” hypothesis in IgAN may exist as the background for the recurrence of the disease. Bacterial or viral infections and vaccinations are common mucosal triggers that may induce active lesions by activating systemic inflammatory responses. The clinical manifestations triggered by these factors are diverse, which likely reflects the complex interplay between the nature of the injury and individual patient susceptibility. In this narrative review, we explore new insight on IgAN from the perspective of active disease and systemic inflammatory responses.
Keywords: IgA nephropathy, MEST-C classification, active lesion, systemic inflammation, innate immunity
Introduction
Immunoglobulin (Ig) A nephropathy (IgAN) is the highest incidence of primary glomerulonephritis, predominantly affecting young adults with a mean age at diagnosis of 34 to 45 years.1 Its clinical manifestations are diverse, ranging from intermittent episodes of gross hematuria to microscopic hematuria and proteinuria, accompanied by impaired renal function.2 Renal biopsy remains the gold standard for diagnosis, with histology systematically classified according to the Oxford MEST-C score,3 active lesions are typically characterized by mesangial hypercellularity (M1), endocapillary proliferation (E1) or crescents (C1/C2), whereas chronic lesions reflect irreversible renal damage, including segmental glomerulosclerosis (S) and tubular atrophy/interstitial fibrosis (T).4–6 It is reported that the epidemiology of IgAN varies significantly across different regions, with the highest incidence observed in East Asia population.7 Approximately 40% to 50% of patients develop renal failure within 10 years of IgAN diagnosis.1,2
“Four-hit” hypothesis is the recognized pathogenesis of IgAN, and the production of galactose-deficient IgA1 (Gd-IgA1) is the first pathogenic event of IgAN.7 The discovery of several IgAN susceptibility loci has confirmed the role of genetic factors in IgAN.8,9 Although it was found that high levels of Gd-IgA1 were also observed in relatives of IgAN patients, most of relatives were asymptomatic,10 suggesting that other cofactors are required for the development of IgAN. Increasing evidence suggests that infections, food antigens, dysbiosis, and abnormal mucosal immune responses are associated with the production of Gd-IgA.7 After entering the systemic circulation, Gd-IgA1 is recognized by autoantibodies, forming circulating immune complexes. These complexes subsequently deposit in the glomerular mesangial region and trigger the release of pro-inflammatory and pro-fibrotic cytokines and complement activation, ultimately leading to renal injury7 (Figure 1). However, in clinical practice, IgAN typically presents as a chronic disease with acute exacerbations. Both innate and adaptive immune responses contribute to its pathogenesis, and disease activity and fluctuations may be associated with elevated systemic inflammation levels. Clinical observations indicated that events such as bacteria or viruses infections or vaccination, can trigger systemic inflammatory responses and immune activation, resulting in the recurrence or exacerbation of IgAN.11,12 Several indicators of systemic inflammation have been identified as being associated with a poor prognosis in IgAN patients.13–15
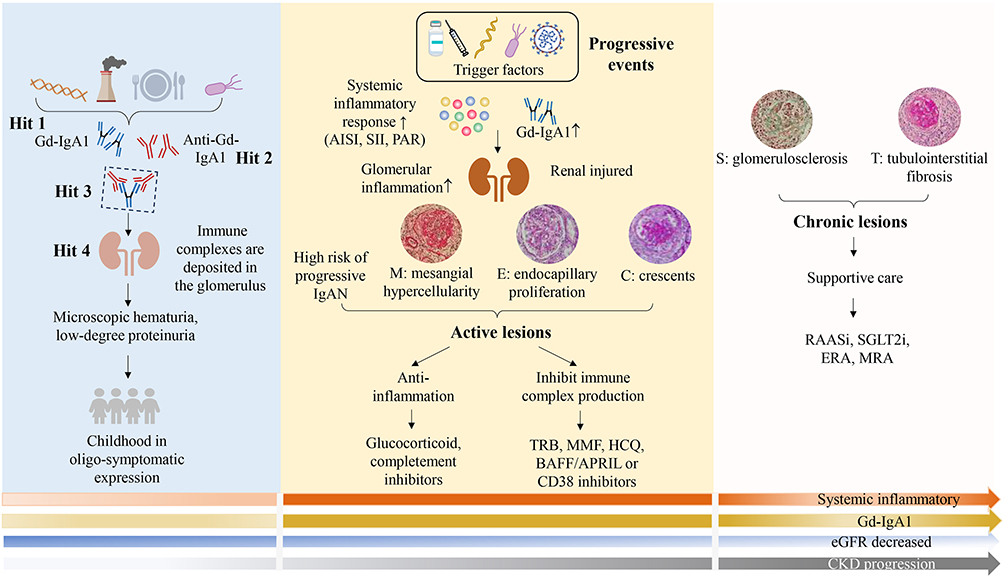 |
Figure 1 Onset, progression, and treatment of IgA nephropathy. The interaction between genetic and environmental factors induces the production of Gd-IgA1. Subsequent antibody generation and immune complex deposition in the kidneys lead to the development of IgAN. In the early stage, the disease may present with only oligo-symptomatic. Infections and vaccination are the triggers of disease progression, manifesting as enhanced systemic inflammatory (AISI, SII, PAR) and immune responses (Gd-IgA1), with active lesions (mesangial hypercellularity, endocapillary proliferation or crescents) representing the predominant pathological finding. Treatment in the active phase primarily focuses on reducing inflammation and inhibiting immune complex formation. With recurrent episodes or persistent slow progression, the disease ultimately manifests as chronic lesions, primarily represented by glomerulosclerosis and tubulointerstitial fibrosis lesions. At this stage, the prime therapeutic strategy shifts to supportive care. Abbreviations: APRIL, a proliferation-inducing ligand; AISI, aggregate index of systemic inflammation; BAFF, B-cell activating factor; ERA, endothelin-1 receptor antagonism; Gd-IgA1, galactose-deficient IgA1; IgAN, IgA nephropathy; MMF, mycophenolate mofetil; MRA, mineralocorticoid receptor antagonists; PAR, platelet-to-albumin ratio; RAASi, renin-angiotensin system antagonists; SGLT2i, sodium-glucose transporter-2 inhibitors; SII, systemic immune-inflammation index; TRB, targeted-release budesonide. |
The 2025 KDIGO guidelines recommend comprehensive lifestyle interventions in IgAN patients, along with strict blood pressure control.3 For those at risk of disease progression, active treatment is required, with therapeutic approaches primarily focused on three key areas: anti-glomerular inflammation (eg: glucocorticoid [CS], iptacopan), suppression of pathogenic IgA production (eg: targeted-release budesonide and telitacicept), and slow chronic kidney disease progression (eg: renin-angiotensin system antagonists, sodium-glucose transporter-2 inhibitors and endothelin-1 receptor antagonism).1,16
Notably, active lesions may subside after treatment and can remerge at any time in a patient’s lifetime in response to different triggers. It typically manifests as increased proteinuria, with histological features showing M1, E1, or C1/2 lesions.4–6 Patients with high activity and little chronicity who are treated with immunosuppressive therapy (IST) demonstrate a significantly lower risk of end-stage renal disease (ESRD).17
IgAN’s status as a lifelong managed disease requires us to understand the major mechanisms of injury that may play a role throughout life and to implement more specific measures to reduce the global impact of IgAN. Although previous reviews have thoroughly discussed the Oxford classification or clinical risk factors for IgAN, there are limited reviews linking systemic inflammation to renal histological activity. This study summarizes the latest evidence on the treatment and prognosis of IgAN based on pathological scoring, and preliminarily explores the hypothesis linking systemic inflammatory responses, infections, and vaccinations to active lesions, offering new insights for deepening the understanding of IgAN mechanisms.
In this narrative review, we searched relevant literatures published before 21 December 2025 through electronic databases, including PubMed, EMBASE and Web of Science, using the following keywords: (“immunoglobulin A nephropathy” OR “IgA nephropathy”) AND (“MEST” OR “MEST-C” OR “Oxford” OR “Oxford Classification”) AND (“inflammation” OR “innate immunity” OR “adaptive immunity”). Then, proceed to read, categorize and summarize the literature.
Evaluation of Treatment and Prognosis in IgAN Based on MEST-C Score
KDIGO 2025 guideline suggested that histological data at the time of biopsy can be used for the initial assessment of IgAN patients.3 The correlation between baselines total MEST-C scores and renal outcomes has been confirmed,18 with T lesions are consistently associated with poorer renal outcomes.19–24 There were also reported that M1 and S1 lesions were risk factors affecting proteinuria remission, requiring more intensive treatment strategies.25 The antiproteinuric effect of iptacopan was independent of pathological scores, with consistent efficacy observed across various subgroups.26 We summarized relevant studies on the relationship between pathological scores and disease prognosis over the past five years (Table 1).
 |
Table 1 Summarize the Relevant Studies of MEST-C Score with Treatment Response or Prognosis in Patients with IgAN Over the Past Five years |
Histopathological findings have been used to assess disease activity and chronicity.4 Itami et al found that patients with M1, E1, S1, or C1/2 scores demonstrate higher renal survival rates with CS therapy. Therefore, researchers added the scores of these lesions to create a “steroid responder score” (SRS) ranging from 0 to 4 points (point 0: low, 1–2: medium, and 3–4: high), while T lesions were classified separately as “steroid non-responder score” (SNRS, 0 or 1 point). Subsequently, by combining these two sets of scores, six new risk combinations were derived. The results showed that patients with a high SRS (more active lesions) benefited from CS treatment, regardless of SNRS score, and CS treatment was ineffective for moderate SRS/low or high SNRS.17 Guo et al classified the presence of crescents and at least one other active lesion (fibrillar necrosis or endocapillary hypercellularity) as severe glomerular activity. IST (CS ± cyclophosphamide [CYC], mycophenolate mofetil [MMF] or calcineurin inhibitors) was a more significant protective factor for renal outcomes in patients with severe glomerular activity and/or those with combined T0 lesions.27 Recently, Keskinis et al subdivided 37 IgAN patients based on combinations of active (M and E) and chronic (S and T) histological lesions. Six (75.0%) patients with severe activity and mild chronicity and 10 (71.4%) patients with severe activity and severe chronicity achieved a reduction in proteinuria of over 30% with stable estimated glomerular filtration rate (eGFR) three months after targeted-release budesonide treatment. Authors considered that presence of tubular atrophy, combined with M1 and/or E1 cannot limit the potential benefit of targeted-release budesonide administration.6 Another study demonstrated that extended treatment with telitacicept was more effective in reducing proteinuria among subgroup of patients with severe pathological changes (M1, E1, T1/2 and C1/2).32 These studies suggested that high active lesions may be the most important parameter for determining IST treatment response.
Specially, the relationship between crescentic lesions and prognosis and treatment response in IgAN remains controversial.33,34 Chen et al used MEST score to predict composite renal outcomes, which achieved an area under curve of 0.76, with a sensitivity of 60.2%, and specificity of 83.6%. The addition of C lesion did not alter the area under curve and resulted in only a slight increase in sensitivity and specificity (63.0% and 84.8%).35 When the proportion of crescentic glomeruli was below 50%, the intensive regimen of CS combined with IST (CYC, MMF, tacrolimus) did not demonstrate superior renal protection compared with CS alone.31 Similarly, no evidence was found in three other studies that the presence of crescents influenced prognostic assessment.19,21,28 However, a prospective (TESTTING) study of 279 IgAN patients showed that 17.6% were classified as C1/2 lesions. These patients experienced a significant reduction in the risk of renal outcomes following CS treatment, though this did not reach statistical significance due to the small sample size, there was a statistically difference in the entire cohort of 379 patients.29 In contrast, another study found that IST therapy (CS ± MMF, leflunomide, calcineurin inhibitors) was significantly associated with a reduced risk of composite outcomes in IgAN patients with C1 lesions.30 In addition, presence of C2 lesions was an independent risk factor for composite renal endpoint in patients without IST and in patients with T0 lesions (with or without IST). The risks were 1.85 times and 3.61 times higher than those in patients with C0 lesions, respectively.27
IgAN patients with endocapillary proliferation had a higher level of proteinuria. A large number of differentially expressed genes have been found in their glomeruli, and these genes are mainly involved in innate immunity and complement activation.36 Additionally, the M2 macrophage marker CD163 was highly expressed in the glomerular of this lesion,36 all of which stimulate glomerular inflammation. Subsequent study further confirmed that the level of urinary soluble CD163 was positively correlated with the scores of E and C lesions, and the reduction in its levels following CS therapy was also correlated with the remission of proteinuria and a reduced risk of renal progression events during follow-up.37 It has been confirmed that active glomerular lesions in IgAN patients can be reversed by IST treatment, accompanied by a significant reduction in proteinuria and hematuria.38 IgAN patients with elevated level of systemic inflammation have also been found to be more prone to exhibiting E and C lesions.13,39 It means that the presence of this active proliferative lesion may indicate high systemic inflammatory level and glomerular inflammation.
Of course, the activity and chronicity phenotypes in IgAN are not yet well defined, MEST-C scores exist geographic variations,5 therefore, the efficacy prediction for each pathological lesion cannot be completely determined. Additionally, the selection of IST is influenced by racial disparities and adverse events. Limited follow-up periods constrain the assessment of long-term efficacy, and existing evidence primarily stems from observational studies, lacking randomized controlled trials to validate its long-term efficacy and safety. Future efforts are needed to develop more accurate predictive tools for assessing disease prognosis and treatment efficacy.
Systemic Inflammatory Response and the Progression of IgAN
During the early stages of IgAN, the kidneys may be only mildly involved, manifesting as microscopic hematuria or low-degree proteinuria. We speculated that the disease state of some adult IgAN patients probably originates in childhood in oligo-symptomatic expression, with unaware renal involvement due to lack of medical examination at a young age. In fact, majority of IgAN patients are diagnosed by chance.
The mucosal origin of pathogenic Gd-IgA1 in IgAN has been recognized, and episodes of disease activity accompanied by gross hematuria and proteinuria are often associated with acute respiratory or gastrointestinal infections, recurrent mucosal infections provoke episodic surges in aberrant IgA production.12 Bacterial or viral infections, or vaccination, are factors that trigger mucosal immune response, and the activation of mucosal immunity is one of the key mechanisms including systemic inflammatory responses. Inflammation can cause changes in blood components such as neutrophils, macrophage, and lymphocyte. The aggregate index of systemic inflammation is an indicator that more thoroughly assesses systemic inflammatory by integrating this information. It was significantly positively correlated with serum creatinine, 24-hour proteinuria, total cholesterol, and triglyceride, and negatively correlated with serum albumin and eGFR.13 Similarly, systemic immune-inflammation index is another novel inflammatory marker. Patients with higher scores exhibit more severe clinical manifestations at baseline, stronger disease activity, and a higher incidence of renal adverse outcomes, potentially reflecting a pronounced systemic inflammatory level in IgAN patients.14,15 Platelet-to-albumin ratio, which is positively correlated with C-reactive protein, also indicates systemic inflammatory response in IgAN. Patients with high platelet-to-albumin ratio levels have more severe proteinuria, more obvious E and C lesions, and more aggressive treatment.39 Based on the above evidence, it is reasonable to assume that systemic inflammatory markers are significantly associated with IgAN disease activity indicators, suggesting that systemic inflammatory responses may play a role in the pathogenesis and progression of the disease. In the absence of acute infection, systemic inflammatory responses may remain quiescent. However, due to the retrospective nature of this study, establishing these markers as surrogate indicators of disease activity requires further evidence from longitudinal studies and rigorous validation methods. Treatment during active lesions primarily on the anti-inflammation (CS and complement inhibitors) and inhibit immune complex production, including budesonide, telitacicept, MMF or hydroxychloroquine.40
Subsequently, renal reserve function is activated and compensates for the early loss of glomerular function, eventually leading to chronic lesions characterized by glomerulosclerosis, tubulointerstitial fibrosis, and persistent low-grade inflammatory and immune response. At this point treatment shifts to support care represented by renin-angiotensin system antagonists, sodium-glucose transporter-2 inhibitors, endothelin-1 receptor antagonism, and mineralocorticoid receptor antagonists16 (Figure 1).
Innate Immunity and Adaptive Immunity in IgAN
Innate immunity provides a rapid but nonspecific defense against pathogens and danger signals, it is primarily mediated by macrophages, neutrophils, dendritic cells (DCs), and granulocytes.41 The innate immune system plays a vital role in IgAN, particularly through toll-like receptors (TLRs), which are responsible for detecting pathogen-associated molecular patterns or damage-associated molecular patterns.42,43 In patients with IgAN, proteinuria levels are associated with the mRNA expression of TLR2, TLR3, TLR5, and TLR9 as well as TLR7 protein intensity, while serum IgA levels are correlated with TLR4 mRNA expression.44,45
Infection-induced TLR activation may exacerbate IgAN by stimulating innate immune cells. Lipopolysaccharides from gram-negative bacteria walls and glycoproteins on the virus surface were recognized by TLR4. Subsequently, TLR4 interact with toll-interleukin-1 receptor domain adaptor molecule 1 and MyD88 adaptor molecule, activating the interferon regulatory factors and NF-κB pathway, respectively, producing inflammatory cytokines, such as interferon-γ and interferon-β, interleukin (IL)-6, IL-1, tumor necrosis factor-α, a proliferation-inducing ligand (APRIL) and B-cell activating factor (BAFF).46,47 The high expression of these cytokines induced B-cell IgA class switching and participated in abnormal IgA1 galactosylation.48,49 TLR9 stimulate interactions between DCs and B-cell in tonsils-associated lymphoid tissue.43 DNA sequences (CpG-ODN) commonly found in bacteria and viruses are ligands for TLR9 and can induce innate immune response. They activate MyD88 signaling and promote adaptive immune responses and B-cell activation through IL-6 and APRIL pathways, including the production of Gd-IgA1 and corresponding immune complexes.50 TLR7 is located in the endosome, where it can recognize single-stranded RNA derived from viruses and mRNA vaccines,46,51 and is also involved in the generation of inflammation cytokines and Gd-IgA145,52 (Figure 2).
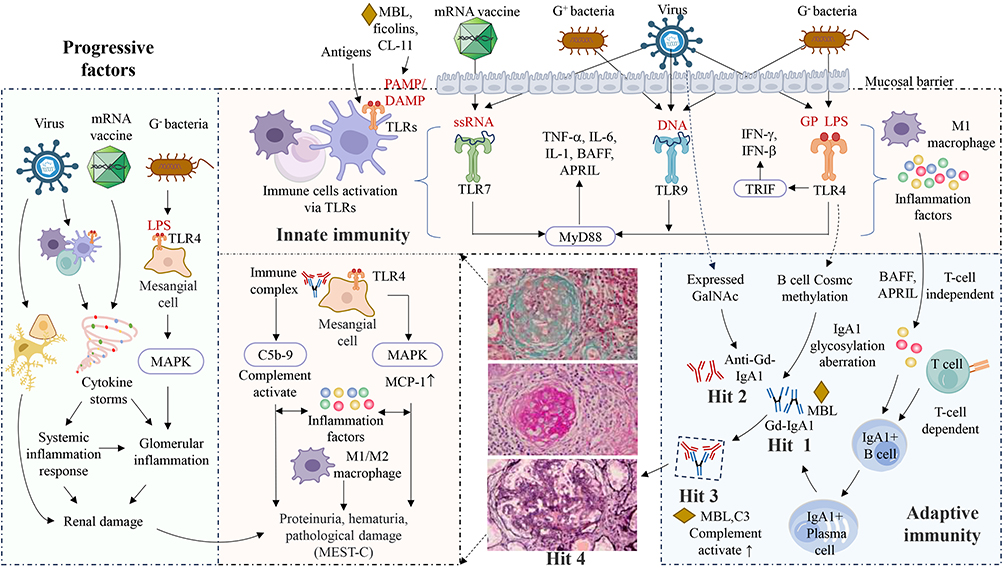 |
Figure 2 Innate immunity and adaptive immunity in IgAN. Pathogens invasion serves as the initiating signal for the activation of innate immune responses at mucosal sites. Upon recognition of pathogen-associated ligands, TLRs located on innate immune cells are activated, inducing the production of a large number of cytokines (IL-1, IL-6, TNF-α, BAFF, April, INF-γ, INF-β).46 Activated TLRs mediate the maturation of dendritic cells, thereby establishing them as a bridge between innate and adaptive immune responses, ultimately producing into IgA-secreting plasma cells. BAFF and APRIL can induce B-cell IgA class switching and participated in production of Gd-IgA1.49 The substantial production of Gd-IgA1 can induce autoantibody generation, leading to the formation of immune complexes and deposit in the kidneys and activate the complement system.7 Complement system is also an important component of innate immunity, MBL, ficolin, and collectin-11, can also trigger immune responses by recognizing DAMP and PAMP,53,54 synergizing with TLRs to promote the release of pro-inflammatory cytokines,55 all of which are the multiple pathogenetic hits identified in IgAN. Additionally, Gd-IgA1 immune complexes can also activate complement directly as they bind C3 and MBL.7 Reinfection with pathogens may contribute to the progression of IgAN, especially for individuals with immune dysregulation. Bacteria damage the kidneys by inducing cytokine production through activation of TLR4 on mesangial cells. Viruses may affect the kidneys either through direct stimulation or by indirectly inducing cytokine storms. The receptor-binding domain of vaccines can act as a superantigen, triggering cytokine storms that damage renal tissue. Different pathogens may engage distinct mechanisms to initiate disease, leading to heterogeneity in disease severity, which ultimately necessitates different treatment strategies. Abbreviations: APRIL, a proliferation-inducing ligand; BAFF, B-cell activating factor; CL-11, collectin-11; Cosmc, core I β3-gal-T-specific molecular chaperone; DAMP, damage-associated molecular patterns; GP, glycoprotein; IL, interleukin; LPS, lipopolysaccharide; MAPK, p38 MAP kinase; MBL, mannan-binding lectin; MCP, monocyte chemoattractant protein-1; PAMP, pathogen-associated molecular patterns; ssRNA, single-stranded RNA; TLR, toll-like receptors; TNF-α; INF, interferon; TRIF, toll-interleukin-1 receptor domain adaptor molecule 1; ↑, increased; ↓, decreased. |
TLRs induce DCs maturation, promoting their migration to lymph nodes, where DCs become potent activators of naive T cells and act as a bridge between innate and adaptive immunity.56 A higher proportion of DCs were observed in lymphocytes from IgAN mice that had undergone fecal microbiota transplantation.57 B-cell trigger class switching through T cell-dependent and T cell-independent pathways, ultimately differentiating into IgA-secreting plasma cells.56 The activation of innate and adaptive immunity may partially amplify the systemic inflammatory response, driving disease progression. Activation of TLR4 by lipopolysaccharide promotes peripheral B lymphocytes core I β3-gal-T-specific molecular chaperone (Cosmc) methylation, thereby reducing the glycosylation level of IgA1 molecules, which is basis of the pathogenesis of IgAN.58,59 N-acetylgalactosamine (GalNAc) is expressed on the surface structures of pathogens including respiratory syncytial virus, Epstein–Barr virus, herpes simplex virus, and streptococci, and may induce the production of cross-reactive antibodies.60,61 The MTMR3/HORMAD2/LIF/OSM locus has been reported to be associated with genetic susceptibility to IgAN, and this region may influence serum IgA levels by increasing MTMR3 expression.62 Furthermore, animal experiments have demonstrated that MTMR3 can enhance IgA production in B cells, glomerular IgA deposition, and mesangial cell proliferation in a T cell-independent manner by mediating the activation of the TLR9 pathway.62
The complement system is involved in the pathogenesis of IgAN and is also an important component of innate immunity. Multiple soluble molecules in the complement system, such as mannan-binding lectin (MBL), ficolin, and collectin-11, can also trigger immune responses by recognizing pathogen-associated molecular patterns or damage-associated molecular patterns.53,54 Complement and TLRs are co-activated in response to microbial infection, complement anaphylatoxin receptor signaling stimulated by C3a or C5a synergizes with TLR-MyD88 signaling to promote the activation of p38 MAP kinases and transcription factors, leading to elevated expression levels of proinflammatory cytokines and costimulatory molecules.55 Of note, MBL can also recognize carbohydrate structures on antibodies, including the common IgG glycosylation variant IgG-G0 and polymeric IgA.63 In IgAN, Gd-IgA1 revealed abnormal carbohydrate structures within the hinge region O-glycans. These aberrant structures may serve as potential ligands for MBL, activating MBL-associated serine proteases and initiating the lectin pathway. This process also establishes a complex network that bridges adaptive and innate immunity. Additionally, Gd-IgA1 immune complexes can also activate complement directly as they bind C3 and MBL.7 Similarly, alternative pathway is also a key effector pathway leading to glomerular injury in IgAN. Upon complement activation, C3 is cleaved to form C5 convertase, which subsequently cleaves C5 into the anaphylatoxins C5a and C5b fragment.54 C5b initiates the formation of the membrane attack complex (C5b-9), induces mesangial cell activation and facilitates the release of inflammatory mediators and extracellular matrix components, thereby exacerbating glomerular injury and driving disease progression. Multiple clinical studies demonstrated that complement activation fragments such as MBL, C3, C5b-9, and factor H-related protein are elevated in patient serum or deposited in the kidneys, and are associated with disease activity and prognosis.54,64 Targeted therapies for specific complement components continue to achieve new breakthroughs. Compared with placebo, iptacopan (a B-factor inhibitor) reduced the 24-hour urine protein-to-creatinine ratio by an average of 38.3% at month 9.26
In addition, in vitro studies showed that stimulation of mesangial cells with human secretory IgA and lipopolysaccharide elevates TLR4 mRNA and protein expression. TLR4 activation promotes p38 MAP kinase and enhances monocyte chemoattractant protein-1 secretion, which participate in the injury of mesangial cells in IgAN.65 In addition, cytokines promote the polarization of macrophages into M1 and M2. M1 macrophages recruited to the glomerular facilitate inflammatory response, while M2 macrophages accelerate mesangial cell proliferation and interstitial fibrosis, showing positive correlations with proteinuria and fibrotic progression.66 Further research confirmed by the correlation between urinary soluble CD163 levels and eGFR, proteinuria, and serum albumin.37 Increased glomerular inflammation conversely exacerbates systemic inflammatory responses and immune dysregulation.
IgAN usually onset or relapse immediately after an infection and may also be triggered by vaccination, but the severity of the disease induced by these triggers differs, necessitating distinct therapeutic approaches.11 We propose a hypothesis that some individuals may have pre-existing low levels of Gd-IgA1, anti-Gd-IgA1 antibodies, or even immune complexes. The immune dysregulation within these latent individuals, analogous to the “four-hit” hypothesis in IgAN, may constitute the pathological basis for susceptibility to IgAN. Upon re‑exposure to the pathogen, different triggers may activate systemic inflammation and immune responses through distinct immune pathways, thereby leading to varying degrees of IgAN. TLR4 is expressed in both mesangial cells and podocytes in the kidney,42 lipopolysaccharide activation of TLR4 may directly stimulate kidney damage. A retrospective study found that microscopic hematuria prior to SARS-CoV-2 mRNA vaccination is the primary predictor of gross hematuria following vaccination in IgAN patients.67 Except for hematuria and proteinuria, vaccination may also cause systemic symptoms such as fever and pain,11 reflecting a widespread inflammatory response. The mechanism linking vaccines to IgAN onset remains speculative at present. Some studies suggested that the receptor-binding domain of the vaccine itself can also act as a superantigen, triggering a cytokine storm and systemic inflammatory response that directly or indirectly lead to the occurrence of IgAN.11 However, direct evidence for this pathway in vaccine-associated IgAN remains insufficient. Whether this systemic reaction directly triggers disease activity or merely coincides with it requires further investigation. While virus can directly infect the kidneys, or they can cause cytokine storms through cytokine-mediated effects, which stimulate kidney damage68,69 (Figure 2).
Limitations
This review has certain limitations. First, most of the treatment-related clinical studies incorporating the Oxford classification scores that we included were non-randomized controlled trials with small sample sizes. Furthermore, the association between pathological scores and treatment in IgAN patients was merely temporal, and we are unable to infer a causal relationship between the two. Second, the mechanisms we have elucidated regarding the association between systemic inflammatory responses, infections, vaccinations, and active IgAN lesions are based solely on hypotheses in the literature and remain unconfirmed.
Conclusion
The pathogenesis of IgAN is complex and multifactorial, involving the combined effects of various factors. Environmental factors such as bacterial or viral infections or vaccinations may act as potential catalysts, inducing disease activity in susceptible individuals. MEST-C scores and systemic inflammatory responses may partially reflect disease status, providing clinically meaningful information for disease assessment. Further research is warranted to elucidate the mechanisms underlying IgAN disease activation and progression. Specifically, prospective studies are needed to validate the utility of specific inflammatory markers in predicting disease onset or progression and assessing prognosis. Integrating these dynamic biomarkers with MEST-C scores into a comprehensive predictive model may enhance the ability to implement personalized treatment for patients. A longitudinal study integrating biopsy and multi-omics analysis elucidates the relationship between different triggering factors and long-term clinical trajectories. Resolving these issues is crucial for understanding IgAN disease progression and guiding individualized treatment strategies.
Abbreviations
APRIL, a proliferation-inducing ligand; BAFF, B-cell activating factor; CS, glucocorticoid; DCs, dendritic cells; eGFR, estimated glomerular filtration rate; ESRD, end-stage renal disease; Gd-IgA1, galactose-deficient IgA1; IgAN, immunoglobulin A nephropathy; IL, interleukin; IST, immunosuppressive therapy; TLRs, toll-like receptors.
Funding
This work was supported by the Key Project of Jiangxi Provincial Nature Science Foundation (No. 20224ACB206008), the “Thousand Talents Plan” project of introducing and training high-level talents of innovation and entrepreneurship in Jiangxi Province (No. JXSQ2023201030), and Key Clinical Research Project of the Second Affiliated Hospital of Nanchang University (No. 2022efyB01).
Disclosure
The authors declare that they have no competing interests.
References
1. Stoneman S, Teh JW, O’Shaughnessy MM. IgA nephropathy in adults: a review. JAMA. 2026;335:799. doi:10.1001/jama.2025.25020
2. Konda R, Rajasekaran A, Rizk DV. What do clinical trials teach us about the pathophysiology of human IgA nephropathy? Nephrol Dial Transplant. 2025. doi:10.1093/ndt/gfaf144
3. Rovin BH, Barratt J, Cook HT, et al. KDIGO 2025 clinical practice guideline for the management of Immunoglobulin A Nephropathy (IgAN) and Immunoglobulin A Vasculitis (IgAV). Kidney Int. 2025;108(4s):S1–12. doi:10.1016/j.kint.2025.04.004
4. Radhakrishnan J, Lafayette RA. Active glomerular inflammation versus chronicity and fibrosis: the role of targeted therapies in IgA nephropathy. Nephrol Dial Transplant. 2025;40(10):1811–1814. doi:10.1093/ndt/gfaf059
5. Roberts ISD. Pathology of IgA nephropathy: a global perspective. Nephrology. 2024;29 Suppl 2:71–74. doi:10.1111/nep.14343
6. Keskinis C, Moysidou E, Kapsia E, et al. Factors influencing early response of IgA nephropathy following targeted-release budesonide (TRB) treatment: preliminary results from a multicenter study. Clin Kidney J. 2025;18(1). doi:10.1093/ckj/sfae364
7. Cheung CK, Alexander S, Reich HN, et al. The pathogenesis of IgA nephropathy and implications for treatment. Nat Rev Nephrol. 2025;21(1):9–23. doi:10.1038/s41581-024-00885-3
8. Bisceglia L, Cerullo G, Forabosco P, et al. Genetic heterogeneity in Italian families with IgA nephropathy: suggestive linkage for two novel IgA nephropathy loci. Am J Hum Genet. 2006;79(6):1130–1134. doi:10.1086/510135
9. Paterson AD, Liu XQ, Wang K, et al. Genome-wide linkage scan of a large family with IgA nephropathy localizes a novel susceptibility locus to chromosome 2q36. J Am Soc Nephrol. 2007;18(8):2408–2415. doi:10.1681/asn.2007020241
10. Gharavi AG, Moldoveanu Z, Wyatt RJ, et al. Aberrant IgA1 glycosylation is inherited in familial and sporadic IgA nephropathy. J Am Soc Nephrol. 2008;19(5):1008–1014. doi:10.1681/asn.2007091052
11. Ma Y, Xu G. New-onset IgA nephropathy following COVID-19 vaccination. Qjm. 2023;116(1):26–39. doi:10.1093/qjmed/hcac185
12. Zhou XJ. Immunoglobulin A nephropathy: molecular pathogenesis and targeted therapy. MedComm. 2025;6(9). doi:10.1002/mco2.70382
13. Liu H, Tang G, Yu D, et al. The Aggregate Index of Systemic Inflammation (AISI) is a novel IgA nephropathy prognosis predictor. J Inflamm Res. 2025;18:5031–5046. doi:10.2147/jir.S512574
14. Hu Y, Huang Z, Zhao W, et al. Clinical significance of the time-average systemic immunoinflammatory index in primary immunoglobulin A nephropathy: a bicentric retrospective cohort study. Kidney Res Clin Pract. 2025. doi:10.23876/j.krcp.24.260
15. Zhai Y, Sun S, Zhang W, et al. The prognostic value of the systemic immune inflammation index in patients with IgA nephropathy. Ren Fail. 2024;46(2):2381613. doi:10.1080/0886022X.2024.2381613
16. Floege J, Bernier-Jean A, Barratt J, et al. Treatment of patients with IgA nephropathy: a call for a new paradigm. Kidney Int. 2025;107(4):640–651. doi:10.1016/j.kint.2025.01.014
17. Itami S, Moriyama T, Miyabe Y, et al. A novel scoring system based on Oxford classification indicating steroid therapy use for IgA nephropathy. Kidney Int Rep. 2022;7(1):99–107. doi:10.1016/j.ekir.2021.10.007
18. Saleem N, Nasir H, Anwar F, et al. To evaluate the utility of Oxford classification in predicting renal outcome in IgA nephropathy patients. Int Urol Nephrol. 2024;56(1):345–353. doi:10.1007/s11255-023-03685-z
19. Alza-Arcila L, Echeverri-Fernandez E, Restrepo-Escobar M, et al. Prognostic utility of the MEST-C score combined with clinical parameters in hispanic patients with IgA nephropathy. Int J Nephrol. 2025;2025:6974280. doi:10.1155/ijne/6974280
20. Chen CH, Wu MJ, Tsai SF. Validating the association of Oxford classification and renal function deterioration among Taiwanese individuals with Immunoglobulin A nephropathy. Sci Rep. 2023;13(1):21904. doi:10.1038/s41598-023-49331-7
21. Haaskjold YL, Bjørneklett R, Bostad L, et al. Utilizing the MEST score for prognostic staging in IgA nephropathy. BMC Nephrol. 2022;23(1). doi:10.1186/s12882-021-02653-y
22. Kang D, Ban TH, Chin HJ, et al. Prognostic value of chronicity grading on renal outcomes in patients with IgA nephropathy. Front Med. 2022;9. doi:10.3389/fmed.2022.952050
23. Thapa S, Sigdel MR. MEST C score and treatment response in IgA nephropathy in a tertiary care hospital: a descriptive cross-sectional study. J Nepal Med Assoc. 2024;62(276):536–541. doi:10.31729/jnma.8707
24. Zoccali C, Mallamaci F, Egido J, et al. Is there long-term value of pathology scoring in immunoglobulin A nephropathy? A validation study of the Oxford classification for IgA nephropathy (VALIGA) update. Nephrol Dial Transplant. 2020;35(6):1002–1009. doi:10.1093/ndt/gfy302
25. Shen X, Chen P, Liu M, et al. Long-term outcomes of IgA nephropathy in China. Nephrol Dial Transplant. 2025;40(6):1137–1146. doi:10.1093/ndt/gfae252
26. Perkovic V, Barratt J, Rovin B, et al. Alternative complement pathway inhibition with Iptacopan in IgA nephropathy. N Engl J Med. 2025;392(6):531–543. doi:10.1056/NEJMoa2410316
27. Guo Y, Shi S, Zhou X, et al. The predictive value and response to immunosuppressive therapy of IgA nephropathy patients with crescents in a large retrospective Chinese cohort. Clin Kidney J. 2023;16(12):2417–2428. doi:10.1093/ckj/sfad134
28. Satirapoj B, Chueaboonchai T, Nata N, et al. Kidney outcomes with corticosteroid treatment in IgA nephropathy according to the Oxford-MEST-C classification. Glomerular Dis. 2025;5(1):191–199. doi:10.1159/000545382
29. Shi S, Roberts ISD, Wang Z, et al. Predictive value of the oxford classification for the effect of glucocorticoid therapy in IgA nephropathy. J Am Soc Nephrol. 2026;37(1):150–159. doi:10.1681/asn.0000000796
30. Tang Y, Liao Y, Su L, et al. Impact of crescents on the immunosuppression treatment of IgA nephropathy. Ren Fail. 2025;47(1):2593690. doi:10.1080/0886022X.2025.2593690
31. Wang Z, Zhou X, Shi S, et al. Clinical implications of C2 lesion in IgA nephropathy: a cohort study. Am J Nephrol. 2024;55(5):529–538. doi:10.1159/000540268
32. Weng Q, Ouyang Y, Chen Z, et al. Efficacy and safety of telitacicept in IgA nephropathy: real-world study outcomes. Clin Kidney J. 2025;18(6):sfaf154. doi:10.1093/ckj/sfaf154
33. Haas M, Verhave JC, Liu ZH, et al. A multicenter study of the predictive value of crescents in IgA nephropathy. J Am Soc Nephrol. 2017;28(2):691–701. doi:10.1681/asn.2016040433
34. Park S, Baek Chung H, Park S-K, et al. Clinical significance of crescent formation in IgA nephropathy – a multicenter validation study. Kidney Blood Pressure Res. 2019;44(1):22–32. doi:10.1159/000497808
35. Chen C-H, Wu M-J, Tsai S-F. Validating the association of Oxford classification and renal function deterioration among Taiwanese individuals with Immunoglobulin A nephropathy. Sci Rep. 2023;13(1). doi:10.1038/s41598-023-49331-7
36. James LR, Hodgin JB, Berthier CC, et al. The molecular phenotype of endocapillary proliferation: novel therapeutic targets for IgA nephropathy. PLoS One. 2014;9(8). doi:10.1371/journal.pone.0103413
37. Li J, Lv J, Wong MG, et al. Correlation of urinary soluble CD163 levels with disease activity and treatment response in IgA nephropathy. Kidney Int Rep. 2024;9(10):3016–3026. doi:10.1016/j.ekir.2024.07.031
38. Shen X-H, Liang -S-S, Chen H-M, et al. Reversal of active glomerular lesions after immunosuppressive therapy in patients with IgA nephropathy: a repeat-biopsy based observation. J Nephrol. 2015;28(4):441–449. doi:10.1007/s40620-014-0165-x
39. Tan J, Song G, Wang S, et al. Platelet-to-albumin ratio: a novel IgA nephropathy prognosis predictor. Front Immunol. 2022;13:842362. doi:10.3389/fimmu.2022.842362
40. Del Vecchio L, Allinovi M, Comolli S, et al. Drugs in development to treat IgA nephropathy. Drugs. 2024;84(5):503–525. doi:10.1007/s40265-024-02036-1
41. Basset C, Holton J, O’Mahony R, et al. Innate immunity and pathogen-host interaction. Vaccine. 2003;21 Suppl 2:S12-23. doi:10.1016/s0264-410x(03)00195-6
42. Coppo R, Amore A, Peruzzi L, et al. Innate immunity and IgA nephropathy. J Nephrol. 2010;23(6):626–632. doi:10.1093/joneph/23.6.626
43. Chang S, Li X-K. The role of immune modulation in pathogenesis of IgA nephropathy. Front Med. 2020;7. doi:10.3389/fmed.2020.00092
44. Saito A, Komatsuda A, Kaga H, et al. Different expression patterns of Toll-Like Receptor mRNAs in blood mononuclear cells of IgA nephropathy and IgA vasculitis with nephritis. Tohoku J Exp Med. 2016;240(3):199–208. doi:10.1620/tjem.240.199
45. Zheng N, Xie K, Ye H, et al. TLR7 in B cells promotes renal inflammation and Gd-IgA1 synthesis in IgA nephropathy. JCI Insight. 2020;5(14). doi:10.1172/jci.insight.136965
46. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. 2009;22(2):240–273. doi:10.1128/cmr.00046-08
47. Häcker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006(357):re13. doi:10.1126/stke.3572006re13
48. Suzuki H, Raska M, Yamada K, et al. Cytokines alter IgA1 O-Glycosylation by dysregulating C1GalT1 and ST6GalNAc-II enzymes. J Biol Chem. 2014;289(8):5330–5339. doi:10.1074/jbc.M113.512277
49. Vincent FB, Saulep-Easton D, Figgett WA, et al. The BAFF/April system: emerging functions beyond B cell biology and autoimmunity. Cytokine Growth Factor Rev. 2013;24(3):203–215. doi:10.1016/j.cytogfr.2013.04.003
50. Makita Y, Suzuki H, Kano T, et al. TLR9 activation induces aberrant IgA glycosylation via April- and IL-6–mediated pathways in IgA nephropathy. Kidney Int. 2020;97(2):340–349. doi:10.1016/j.kint.2019.08.022
51. Chen R, Zou J, Chen J, et al. Pattern recognition receptors: function, regulation and therapeutic potential. Signal Transduct Target Ther. 2025;10(1):216. doi:10.1038/s41392-025-02264-1
52. Lee M, Suzuki H, Ogiwara K, et al. The nucleotide-sensing Toll-Like Receptor 9/Toll-Like Receptor 7 system is a potential therapeutic target for IgA nephropathy. Kidney Int. 2023;104(5):943–955. doi:10.1016/j.kint.2023.08.013
53. Ricklin D, Hajishengallis G, Yang K, et al. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–797. doi:10.1038/ni.1923
54. Medjeral-Thomas NR, Cook HT, Pickering MC. Complement activation in IgA nephropathy. Semin Immunopathol. 2021;43(5):679–690. doi:10.1007/s00281-021-00882-9
55. Reis ES, Mastellos DC, Hajishengallis G, et al. New insights into the immune functions of complement. Nat Rev Immunol. 2019;19(8):503–516. doi:10.1038/s41577-019-0168-x
56. Carroll SL, Pasare C, Barton GM. Control of adaptive immunity by pattern recognition receptors. Immunity. 2024;57(4):632–648. doi:10.1016/j.immuni.2024.03.014
57. Liu J, Chen Y, Wan Q. Immune cell characteristics in a gut-kidney axis-induced mouse model of IgA nephropathy: the upregulated dendritic cells and neutrophils. J Inflamm Res. 2025;18:8579–8592. doi:10.2147/jir.S519521
58. Qin W, Zhong X, Fan JM, et al. External suppression causes the low expression of the Cosmc gene in IgA nephropathy. Nephrol Dial Transplant. 2008;23(5):1608–1614. doi:10.1093/ndt/gfm781
59. Tsilibary EC, Sun Q, Zhang J, et al. DNA methylation in cosmc promoter region and aberrantly glycosylated IgA1 associated with pediatric IgA nephropathy. PLoS One. 2015;10(2). doi:10.1371/journal.pone.0112305
60. Tomana M, Novak J, Julian BA, et al. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and antiglycan antibodies. J Clin Invest. 1999;104(1):73–81. doi:10.1172/jci5535
61. Mestecky J, Tomana M, Moldoveanu Z, et al. Role of aberrant glycosylation of IgA1 molecules in the pathogenesis of IgA nephropathy. Kidney Blood Press Res. 2008;31(1):29–37. doi:10.1159/000112922
62. Wang YN, Gan T, Qu S, et al. MTMR3 risk alleles enhance toll like receptor 9-induced IgA immunity in IgA nephropathy. Kidney Int. 2023;104(3):562–576. doi:10.1016/j.kint.2023.06.018
63. Murugaiah V, Varghese PM, Beirag N, et al. Complement proteins as soluble pattern recognition receptors for pathogenic viruses. Viruses. 2021;13(5):824. doi:10.3390/v13050824
64. Teh JW, Stoneman S, O’Shaughnessy MM. Complement inhibition in immunoglobulin A nephropathy: a mini-review. Am J Kidney Dis. 2026;87(3):412–421. doi:10.1053/j.ajkd.2025.07.023
65. Lim BJ, Lee D, Hong SW, et al. Toll-like Receptor 4 signaling is involved in IgA-stimulated mesangial cell activation. Yonsei Med J. 2011;52(4):610. doi:10.3349/ymj.2011.52.4.610
66. Liu Y, Gong Y, Xu G. The role of mononuclear phagocyte system in IgA nephropathy: pathogenesis and prognosis. Front Immunol. 2023;14. doi:10.3389/fimmu.2023.1192941
67. Yokote S, Tsuboi N, Shimizu A, et al. Predictors of gross hematuria after SARS-CoV-2 mRNA vaccination in patients with IgA nephropathy. Kidney360. 2023;4(7):943–950. doi:10.34067/kid.0000000000000192
68. Han H, Ma Q, Li C, et al. Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerging Microbes Infect. 2020;9(1):1123–1130. doi:10.1080/22221751.2020.1770129
69. Kudose S, Batal I, Santoriello D, et al. Kidney biopsy findings in patients with COVID-19. J Am Soc Nephrol. 2020;31(9):1959–1968. doi:10.1681/ASN.2020060802
 © 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2026 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 4.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.