Back to Journals » International Journal of Nanomedicine » Volume 10 » Issue 1
New findings about iron oxide nanoparticles and their different effects on murine primary brain cells
Authors Neubert J, Wagner S, Kiwit J, Bräuer AU, Glumm J
Received 16 September 2014
Accepted for publication 16 October 2014
Published 13 March 2015 Volume 2015:10(1) Pages 2033—2049
DOI https://doi.org/10.2147/IJN.S74404
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Thomas Webster
Jenni Neubert,1 Susanne Wagner,2 Jürgen Kiwit,3 Anja U Bräuer,1,* Jana Glumm1,3,*
1Institute of Cell Biology and Neurobiology, Center for Anatomy, 2Institute for Radiology, Charité-Universitaetsmedizin Berlin, 3Clinic for Neurosurgery, HELIOS Klinikum Berlin-Buch, Berlin, Germany
*These authors contributed equally to this work
Abstract: The physicochemical properties of superparamagnetic iron oxide nanoparticles (SPIOs) enable their application in the diagnostics and therapy of central nervous system diseases. However, since crucial information regarding side effects of particle–cell interactions within the central nervous system is still lacking, we investigated the influence of novel very small iron oxide particles or the clinically approved ferucarbotran or ferumoxytol on the vitality and morphology of brain cells. We exposed primary cell cultures of microglia and hippocampal neurons, as well as neuron–glia cocultures to varying concentrations of SPIOs for 6 and/or 24 hours, respectively. Here, we show that SPIO accumulation by microglia and subsequent morphological alterations strongly depend on the respective nanoparticle type. Microglial viability was severely compromised by high SPIO concentrations, except in the case of ferumoxytol. While ferumoxytol did not cause immediate microglial death, it induced severe morphological alterations and increased degeneration of primary neurons. Additionally, primary neurons clearly degenerated after very small iron oxide particle and ferucarbotran exposure. In neuron–glia cocultures, SPIOs rather stimulated the outgrowth of neuronal processes in a concentration- and particle-dependent manner. We conclude that the influence of SPIOs on brain cells not only depends on the particle type but also on the physiological system they are applied to.
Keywords: microglia, hippocampal neurons, degeneration, morphology, nanoparticles
Introduction
The number of products engineered using nanotechnology for such applications as biomedicine, pharmaceutics, cosmetics, and electronics is continually increasing, which consequently leads to increasing exposure of the environment and humans to nanoscale materials.1 Cellular accumulation of nanoparticles, especially of those that have been designed for systemic injections, has unpredictable consequences on human health.
Superparamagnetic iron oxide nanoparticles (SPIOs) as contrast agents in magnetic resonance imaging (MRI) have proved to be promising tools for visualizing pathological processes.2,3 SPIOs have been optimized to label single cells in vitro and subsequently to visualize tissue alterations or disease progression in vivo.4–7 In addition, SPIOs serve as carriers for targeted drug delivery or in cancer treatment with magnetic hyperthermia.8–10 However, the application of nanoparticles, in particular under disease conditions, raises the important question of how they may potentially cause adverse effects or influence the cell vitality after entering the central nervous system (CNS). For instance, in MRI pilot studies for imaging inflammatory processes within the human brain, the SPIO ferumoxytol was injected in very high doses of 2–10 mg/kg body weight, to achieve a high signal-to-noise ratio. Consequently, ferumoxytol was still detectable after 5 days of initial administration by MRI. Even at 19 days postinjection, Prussian blue staining of the inflamed resected tissue still revealed iron-positive cells.11,12
In neurological diseases with a functionally impaired or disrupted blood–brain barrier, such as traumatic brain injury or multiple sclerosis, the permeation of SPIO-based contrast agents used for diagnostics is facilitated. Therefore, nanometer-size particles can easily be taken up by phagocytic cells or interact with the extracellular matrix and neuronal network.13–15 Furthermore, the respective surface charges of SPIOs determine their pharmacokinetic and physicochemical properties, and could consequently induce particle interactions with the blood–brain barrier and affect its integrity.16,17
In the CNS, 10% of the total glial cell population is comprised of resident and highly phagocytic microglial cells that play a pivotal role in innate immune reaction. Microglia in the so-called resting state exhibit a ramified morphology, and by rapidly extending their processes, survey the local microenvironment to maintain homeostasis. In various neuropathological events, eg, infection, stroke, or neurodegeneration, microglial cells become activated and undergo a transformation from a ramified to an amoeboid morphology.18–20 SPIOs are taken up by activated microglia in primary and mixed cell cultures in a time-, concentration-, and temperature-dependent manner.21,22 This raises the possibility of sustained microglial activation that can prove to be severely disruptive to neural function.23–25 Interestingly, other studies have demonstrated that cellular reactions critically depend on the respective particle properties, including composition, size, and biocompatibility.26–28 Indeed, larger hydrodynamic diameters and larger surfaces with high surface-to-volume ratios cause increased reactivity of SPIOs with surrounding tissue or hamper cellular particle uptake, which is favored for MRI of certain cell types or targets.29–31 Fundamental for inducing cytotoxicity is the process of nanoparticle degradation. The release of free iron ions affects distinct subcellular processes – eg, it can enforce mitochondrial dysfunction through the production of reactive oxygen species – but can also promote neurite outgrowth under stimulus conditions.32,33 Iron deposits in activated microglia have also been found to be a notable feature in neurodegenerative diseases, including multiple sclerosis and Alzheimer’s and Parkinson’s disease, leading to homeostatic imbalance and even cell death.32,34,35 The use of SPIOs as contrast agents in MRI is especially critical, because whether or not particles interact with brain cells, they can potentially induce neurotoxicity. However, conclusive information regarding side effects of particle accumulations on primary brain cells is still lacking.
We investigated the effects of SPIO exposure on murine primary microglia and hippocampal neurons, as well as on neuron–glia cocultures. We systematically explored the exposure effects of the novel very small iron oxide particles (VSOPs), as well as the clinically approved MRI contrast agents ferucarbotran and ferumoxytol.36,37 VSOPs have been tested in Phase II trials.38,39 We first investigated the impact of these SPIOs, which differ in surface coating and size, on microglial morphology, particle accumulation, and viability. Secondly, we analyzed the morphology and vitality of neurons from primary cultures, and finally of neurons from neuron–glia cocultures. We were able to show that influences of SPIOs strongly depend on the particle type, their concentration and exposure time, and furthermore on the respective cell type and cell-culture system.
Materials and methods
Animals
Pregnant and postnatal C57BL/6 mice were obtained from our central animal facility and kept under standard laboratory conditions (12-hour light/dark cycle, 55%±15% humidity, 22°C±1.5°C room temperature [RT], and water ad libitum; enriched and grouped). All experimental procedures were carried out in accordance with the German Animal Welfare Act and European guidelines (2010/63/EU) for the use of laboratory animals, and were approved by the local regulatory authority of Berlin (LAGeSo: T0095/11).
Iron oxide nanoparticles
VSOPs were synthesized by the Charité Institute of Radiology, and consisted of monocrystalline iron oxide cores of magnetite (Fe3O4) and maghemite (γ-Fe2O3) coated with a monomeric citrate layer. We applied two types of these nanoparticles: VSOP-R1, with a diameter of 6.5–7.5 nm, comparable to VSOP-C184, and VSOP-R2, with a diameter of 7.5–8.7 nm, comparable to VSOP-C200.40,41 These VSOPs have already been tested in human clinical trials up to Phase II.38,39 Detailed information about the respective physicochemical properties is listed in Table 1. For synthesis, 14 g ferrous chloride tetrahydrate and 28 g ferric chloride hexahydrate were dissolved in 0.4 M hydrochloric acid and then mixed with 310 mL 1.5 M sodium hydroxide. This mixture was stirred for 30 minutes, and then 22 g (for VSOP-R1) or 18 g (for VSOP-R2) citric acid monohydrate was added. After a further 30 minutes’ stirring at 75°C, the mixture was cooled to RT and magnetically separated. Supernatant was withdrawn and sediment resuspended in Milli-Q® water, acidified with monosodium citrate (< pH 5), and then ultrafiltrated (30 kD). Iron concentration was adjusted to 0.5 M, and the final batch was heat-sterilized for 10 minutes at 100°C, resulting in a stable dispersion containing 60 g/L mannitol. The VSOPs were about three times smaller than the ultrasmall particles described in the following.
 | Table 1 Physicochemical properties of VSOPs |
The ultrasmall SPIOs used were ferucarbotran (Resovist®; Bayer Schering Pharma, Germany) and ferumoxytol (Feraheme®; AMAG Pharmaceuticals, USA). Detailed information regarding the synthesis and characterization of ferucarbotran is described by Reimer and Balzer36 and of ferumoxytol by Balakrishnan et al.42 Briefly, ferucarbotran is a carboxydextran-coated SPIO (27–35 mg/mL with an iron-to-carboxydextran ratio of 1:1) composed of 0.5 mol Fe/L, including 40 mg/mL mannitol and 2 mg/mL lactic acid, adjusted to a pH of 6.5. The overall hydrodynamic diameter of ferucarbotran is around 60 nm. Iron oxide cores of ferumoxytol are coated with carboxymethyldextran (polyglucose sorbitol carboxymethyl ether), exhibiting an overall hydrodynamic diameter of 30 nm.
Preparation of primary cell cultures and neuron–glia cocultures
Preparations of primary microglial cultures were performed as described previously.43 The isolated glial cell population consisted of astrocytes and microglia that attached to and grew on the coated surface of the culture flasks in the incubator for 8–10 days in vitro (DIV) until microglia detached from the feeder layer of confluent astrocytes. Subsequently, primary microglial cells were harvested and seeded at densities of 2×105 cells per well in 12-well plates containing poly-L-lysine-coated glass coverslips in 1 mL of Dulbecco’s Modified Eagle’s Medium (Gibco®; Life Technologies, Germany), supplemented with 10% fetal calf serum (FCS), 100 U/mL penicillin, 100 μg/mL streptomycin (Pan-Biotech, Germany), and 0.1% of 50 mM 2-mercaptoethanol. Microglia were maintained at 37°C in 5% CO2 for 24 hours prior to the start of experiments.
Primary astrocytes for neuron–glia cocultures were prepared from C57BL/6 mice at postnatal days 0–2. After isolation, primary astrocytes were seeded at densities of 5×104 cells per well in poly-L-lysine-coated six-well plates and maintained for 11 DIV until primary hippocampal neurons on poly-L-lysine-coated coverslips were transferred to establish neuron–glia cocultures.
Hippocampal neurons for primary cultures and neuron–glia cocultures were prepared from C57BL/6 mice at embryonic day 18 as described previously.43 For primary neuronal cultures, neurons were seeded at densities of 8×104 neurons per well in 12-well plates containing poly-L-lysine-coated glass coverslips and maintained at 37°C and 5% CO2 for 10 DIV. For neuron–glia cocultures, primary neurons on glass coverslips were transferred to astrocyte cultures on the same day of preparation by placing them upside down on the plated astrocytes and keeping them in culture for 10 DIV. Detailed information about cell preparation is given in the Supplementary materials.
Quantitative real-time PCR of neuron–glia cocultures
The identity and purity of primary cells were analyzed by quantitative real-time polymerase chain reaction (qRT-PCR) with neuron-specific class III β-tubulin (Tuj1) determining neuronal origin, glial fibrillary acidic protein (GFAP) as a marker for astrocytes, and the ionized calcium-binding adaptor molecule 1 (Iba1) as a marker for microglial cells (Figure S1).44 Detailed information about the qRT-PCR procedure is given in the Supplementary materials.
SPIO-exposure conditions in cell cultures
VSOP-R1, VSOP-R2, ferucarbotran, or ferumoxytol were added to well plates of primary microglia, primary hippocampal neurons, and neuron–glia cocultures in 1 mL culture medium per well, yielding final particle concentrations of 0.5 mM, 1.5 mM, or 3.0 mM, respectively. Microglial cells were incubated with SPIOs at 8–9 DIV for 6 or 24 hours, respectively. Primary neurons and neuron–glia cocultures were incubated for 24 hours at 9 DIV.
Well plates of all cell-culture models included wells with respective numbers of cells that were not treated with SPIOs serving as negative controls (referred to in figures and tables as “control”). For primary microglial cultures, well plates additionally contained wells with microglia that were not treated with SPIOs but stressed with 0.1% glutaraldehyde (dilution of 25% glutaraldehyde; Sigma-Aldrich, Germany) to serve as positive controls for propidium iodide (PI) staining and quantification.
Determination of microglial cell viability
The viability of primary microglia in terms of cell-membrane integrity following SPIO exposure was assessed using PI staining (Carl Roth, Germany). As PI only perforates cells upon membrane damage and intercalates into nucleic acids,45 the increase in numbers of PI-positive cells reflects the loss of microglial viability. Primary microglia in densities of 2×105 cells per well were incubated with 500 μg/mL of PI for 30 minutes at 37°C. The fluorescent emission of PI at 630 nm was captured with an F-View II digital camera using Cell-Sens Dimension software (version 1.4.1 XV 3.4, build 8624; Olympus, Germany) and an inverted fluorescence microscope (objective UPLFLN 10×/0.30 PH1; Olympus IX81). Four images per exposure condition, including positive and negative controls, were acquired from four different regions of the respective wells. The number of PI-positive microglia was quantified using the Image-Based Tool for Counting Nuclei plug-in in ImageJ software (National Institutes of Health, USA). From four corresponding images, PI values were averaged, values of negative controls were subtracted, and results were normalized to PI values of positive controls. PI-staining and fluorescence measurements for the 6-hour exposure condition were performed on five independently prepared primary microglial cultures, and for the 24-hour condition on seven independently prepared microglial cultures. Numbers of total determinations per SPIO and exposure time can be found in Table 2.
 | Table 2 Data and sample size of primary microglia used for PI and Prussian blue quantification |
Prussian blue staining, immunocytochemistry, and image processing of microglia
Following PI measurements, supernatants of microglial cultures were removed and cells washed twice with phosphate-buffered saline (PBS) and immediately fixed with 4% paraformaldehyde (Carl Roth) in 15% sucrose for 15 minutes at 4°C. Prussian blue staining was performed as previously described on cytochemically detected iron contents of microglia following SPIO exposure.46 Nuclear fast red staining (Carl Roth) was used to counterstain cell nuclei. For each exposure condition, we captured images from six different regions of Prussian blue-stained coverslips with 40× magnification (LUCPLFLN 40×/0.60 PH2) using light microscopy (Olympus IX81). In order to eliminate color-biased values, images were acquired with 256 colors in gray scale. The presence of iron in respective grayscale images was quantified using ImageJ. The mean gray-value intensities of four cells from six corresponding images were measured by defining regions of interest. We quantified 24 microglial cells in total per SPIO-exposure condition and then averaged the mean gray-value intensities of the corresponding cells. Additionally, mean gray-value intensities of the image background in corresponding images, as well as values of negative controls, were averaged and subtracted from the averaged values of cells. Prussian blue staining and subsequent quantification of cellular iron content for the 6- and 24-hour exposure conditions were performed on six independently prepared primary microglial cultures (see Table 2 for the total number of measurements and exposure times).
For representative visualization of microglial morphology and cell-associated SPIOs, microglia were immunocytochemically stained after fixation using cell-specific CD11b-fluorescein isothiocyanate (CD11b-FITC)-conjugated antibody (Miltenyi Biotec, Germany). Cell nuclei were counterstained using the blue fluorescence nucleic acid stain Hoechst 33258 (Sigma-Aldrich). We used light microscopy to detect cell-associated SPIOs, since these iron particles appear brown in color and Prussian blue and immunocytochemical staining cannot be performed in combination. Light microscopy and immunofluorescence images of microglia were captured with an Olympus BX 51 microscope with narrow-band filters (Olympus) using a MagnaFire digital camera and MagnaFire 2.1B software (Intas, Germany). Light microscopy photographs were inverted and pseudocolored in red, in order to create merged images with corresponding immunofluorescence photographs. All images of primary microglia were processed using Adobe Photoshop (CS3 extended, version 10.0; Adobe Systems).
Transfection of neuronal cultures
For ease of morphological analysis, neurons of primary and cocultures were transfected with enhanced green fluorescent protein (Invitrogen, Germany) 24 hours prior to SPIO exposure at 8 DIV. Transfection was performed using Effectene transfection reagent (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. After incubation for 1 hour at 37°C and 5% CO2, the reagent was removed, cells were washed with sterile PBS, and 1 mL fresh NB+ was added to each well.
Immunocytochemistry of neuronal cultures
Following SPIO exposure of transfected primary and coculture neurons, supernatants were removed and coverslips with neurons from cocultures were transferred to new 12-well plates. Cells were washed twice with PBS and immediately fixed with 4% paraformaldehyde in 15% sucrose for 15 minutes at 4°C. Neurons were washed twice with PBS to remove the fixation solution and permeabilized using 0.1% Triton X-100, 0.1% sodium citrate in PBS for 3 minutes at 4 °C, and washed again three times with PBS. Cells were blocked with 10% FCS for 1 hour at RT and incubated with the neuron-specific class III β-tubulin antibody mouse antimouse Tuj1 (Covance, Germany) in 10% FCS in PBS overnight at RT at a dilution of 1:1,000. For neuron–glia cocultures, we additionally used the astrocyte-specific primary antibody rabbit antimouse anti-GFAP (Dako, Denmark) at a dilution of 1:1,000. Secondary antibodies (Alexa Fluor 488 goat antimouse, Alexa Fluor 568 goat antirabbit; Molecular Probes®; Life Technologies) were diluted 1:1,500 in 5% FCS and PBS and incubated for 90 minutes at RT. Finally, cells were washed three times and coverslips were mounted using Immu-Mount (Thermo Scientific, Germany).
Quantification and analysis of primary hippocampal neurons
To determine if SPIOs lead to the degeneration of neurons and subsequent cell death, transfected Tuj1-positive neurons of primary and cocultures were counted under a fluorescence microscope (Olympus IX81; objective 10×). Numbers of degenerated neurons were calculated as percentages of total numbers of neurons. Neurons were classified as degenerated according to standard morphological criteria, ie, fragmentation of neuronal processes, cell shrinkage, and somatic swelling.47 Quantification of neurons was performed via double determination of three independently prepared primary neuron cultures and four independently prepared neuron–glia cocultures for each exposure condition (see Table 3 for sample size and total number of measurements).
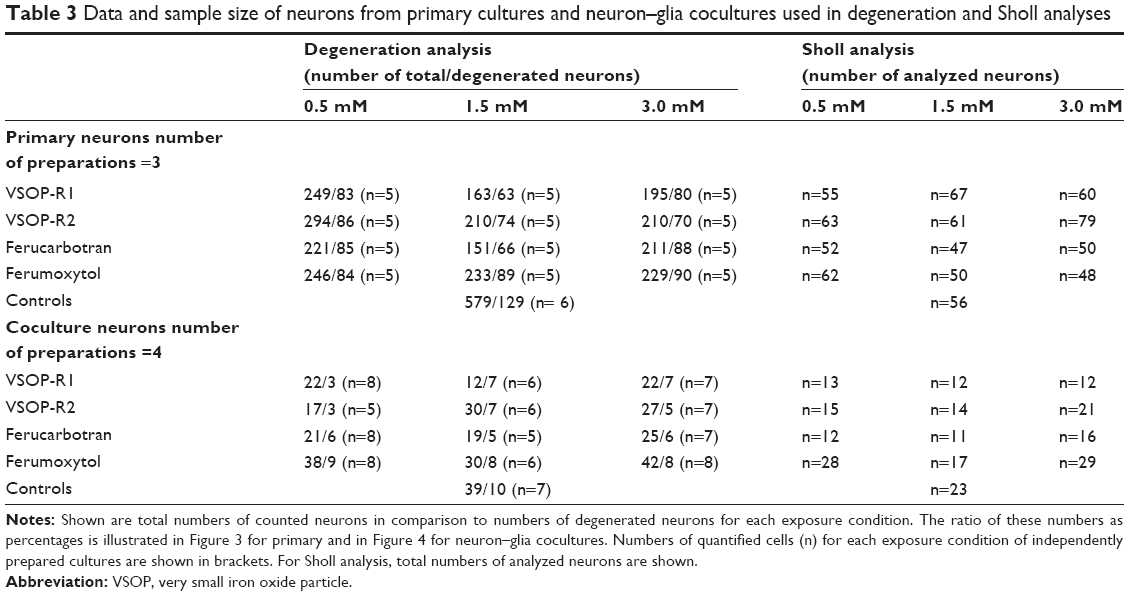 | Table 3 Data and sample size of neurons from primary cultures and neuron–glia cocultures used in degeneration and Sholl analyses |
For morphological Sholl analysis of primary and coculture neurons, images of double-positive-stained neurons, ie, with enhanced green fluorescent protein and red fluorescent Tuj1, were acquired with fluorescence microscopy equipped with CellSens Dimension software and a 10× objective (Olympus IX81). The number of neurites per exposure condition was determined by means of Sholl analysis,48 using 24 20 μm-spaced concentric circles with maximal distances of 480 μm distal to the cell body. The innermost circle with a radius of 40 μm was placed onto each neuron, with its center roughly coinciding with the center of the soma. Neurite intersections at each circle were manually counted using ImageJ. Sholl analysis of neurons was performed via double determination of three independently prepared primary neuron cultures and four independently prepared neuron–glia cocultures for each exposure condition (see Table 3 for sample size and total number of measurements).
Statistics
Unless stated otherwise, data are presented as means ± standard error of the mean. Statistical significance was determined using Kruskal–Wallis one-way analysis of variance followed by Dunn’s multiple comparison post hoc test, at the indicated significance levels.
Results
SPIO accumulation induces alterations of primary microglial morphology
We first addressed the question of whether the applied SPIOs affected primary microglial morphology. For this, we used Prussian blue staining to visualize microglial SPIO accumulation and light microscopy to analyze morphological changes from a ramified toward an amoeboid shape, which can be taken as a reliable indicator of microglial activation.49 Interestingly, microglia incubated for 24 hours with 3.0 mM of only VSOP-R1, VSOP-R2, and ferucarbotran consistently showed intense Prussian blue staining (Figure 1A–C), and appeared to be almost exclusively amoeboid in shape. In comparison, microglia exposed to ferumoxytol were mainly negative for Prussian blue, and typically showed a ramified morphology that is characteristic of resting microglia (Figure 1D). Therefore, ferumoxytol-exposed primary microglia neither accumulated particles nor showed morphological alterations.
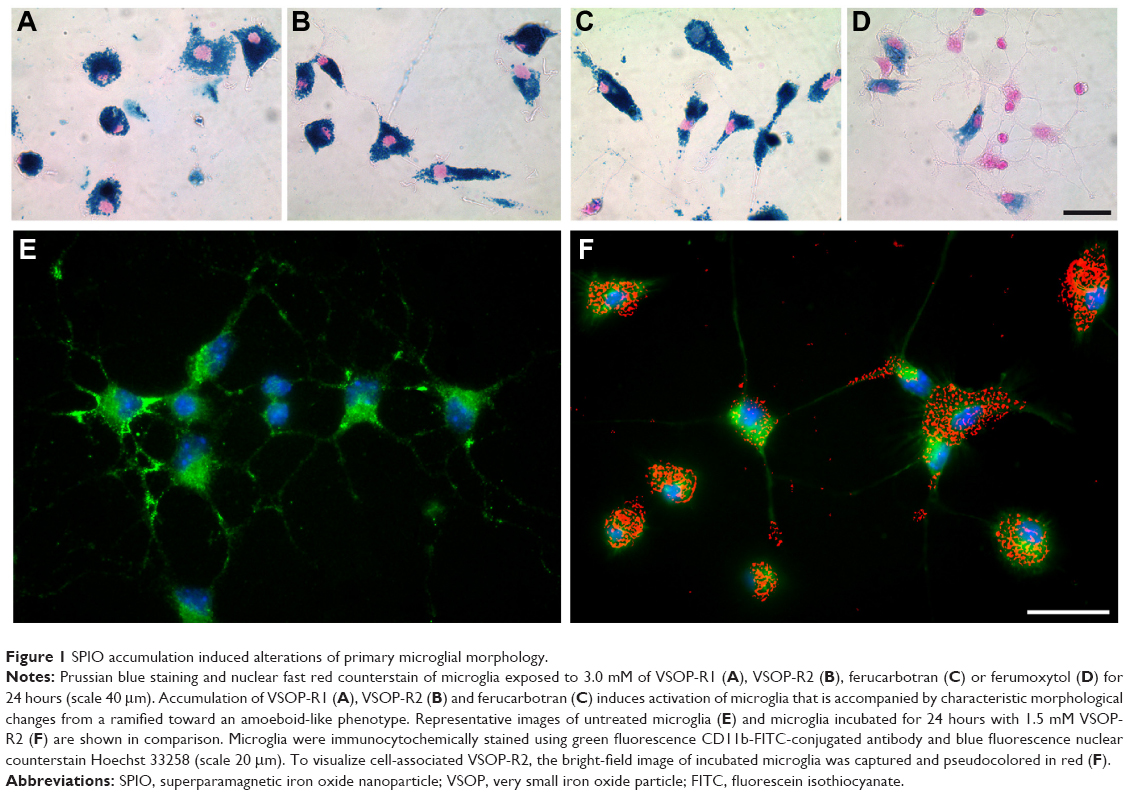 | Figure 1 SPIO accumulation induced alterations of primary microglial morphology. |
Microglial morphology was further investigated with immunocytochemical staining using the microglia-specific CD11b-FITC-conjugated antibody. The representative image of merged fluorescence and red pseudocolored primary microglia after exposure for 24 hours to 1.5 mM VSOP-R1 (Figure 1F) reveals particle accumulation and amoeboid morphology. In comparison, untreated microglia under the same culture conditions showed typical ramified morphology (Figure 1E). Therefore, SPIO accumulation strongly influenced the morphology of primary microglia. However, particle accumulation was dependent on the respective SPIO type.
The viability of primary microglia is SPIO-dependent
Next, we analyzed whether SPIO exposure affected microglial viability. Microglial iron contents quantified by measuring the Prussian blue-staining intensity revealed significantly more accumulated SPIOs by microglia exposed for 6 or 24 hours to VSOPs or ferucarbotran, respectively, than cells exposed to ferumoxytol (Figure 2A–F, blue-framed gray bars). The prolonged incubation from 6 to 24 hours showed only marginal increases in staining intensities (maximum 9.8%) of microglia exposed to all SPIO concentrations, which corresponds with Figure 1. However, incubation with VSOPs or ferucarbotran of concentrations three or six times higher did not result in proportional increases in Prussian blue-staining intensity.
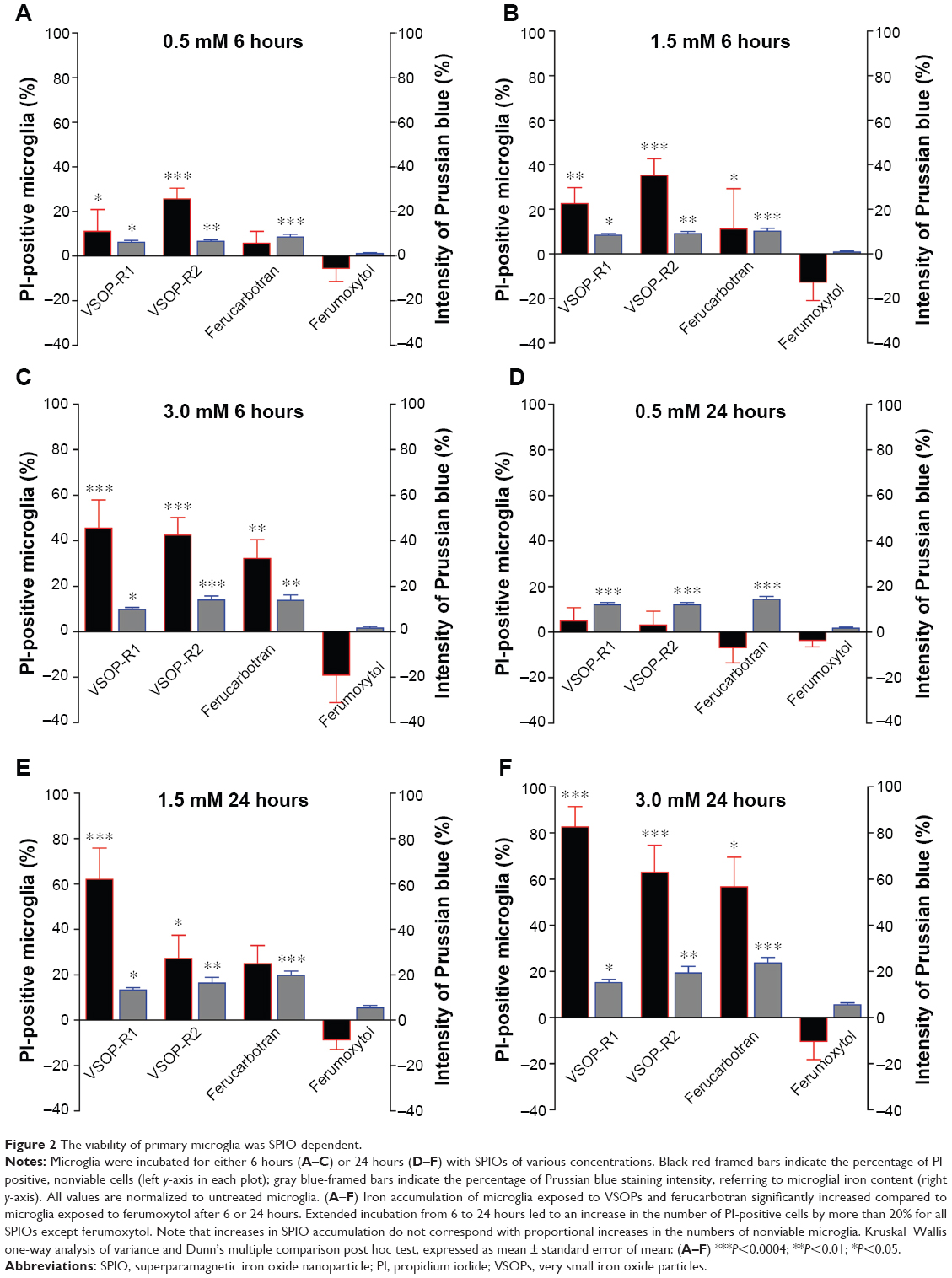 | Figure 2 The viability of primary microglia was SPIO-dependent. |
The decreased microglial viability, as indicated by the percentage of PI-positive cells, was clearly affected after 6 hours’ and strongly affected after 24 hours’ incubation with high concentrations (1.5–3.0 mM) of VSOPs and ferucarbotran, but not ferumoxytol (Figure 2A–F, red-framed black bars). Here, we observed an increase in numbers of PI-positive cells after 6–24 hours of more than 20% (maximum 41.7%) for all SPIOs except ferumoxytol (Figure 2C and F). We noticed the largest increases in PI-positive microglia after exposure to 3.0 mM VSOP-R1, VSOP-R2, or ferucarbotran for 24 hours (Figure 2C and F).
Furthermore, we observed a correlation between cellular iron accumulation and microglial viability. In most experimental conditions, the more nanoparticles were accumulated, the higher numbers of PI-positive microglia were quantified. In contrast, this does not apply for all SPIOs in concentrations of 0.5 mM, for all concentrations of ferumoxytol and for 1.5 mM of VSOP-R2 when comparing 6- and 24-hours exposure (Figure 2).
We also found that while overall Prussian blue-staining intensities were highest after exposure for 24 hours to ferucarbotran (all concentrations; Figure 2C–F), numbers of PI-positive cells were higher after incubating with VSOP-R1 and VSOP-R2 for both the 1.5 mM- and the 3.0 mM-exposure conditions (Figure 2E and F). Interestingly, our data showed concentration-dependent decreases of microglial PI values after incubation for 6 or 24 hours with ferumoxytol (Figure 2A–D). This in turn revealed statistically significant differences when comparing ferumoxytol with the other nanoparticles, but not after incubation with 0.5 mM of all SPIOs for 24 hours (Figure 2D), or with 0.5 mM ferucarbotran for 6 hours and 1.5 mM for 24 hours (Figure 2A and E).
We conclude that extended exposure times and accumulations of VSOPs or ferucarbotran increase numbers of nonviable microglia. However, incubation with ferumoxytol causes completely different effects.
Primary hippocampal neurons degenerate after SPIO exposure
In addition to primary microglia, we investigated primary hippocampal neurons to test the effect of SPIO exposure on neuronal morphology and survival. In contrast to primary microglial cells, both the morphology and viability of primary neurons were severely affected following exposure to all SPIOs tested. Sholl analysis revealed substantially reduced numbers of neuronal processes in distal extensions of exposed primary neurons when compared to untreated controls (Figure 3A). We then raised the question of whether SPIOs not only adversely influenced the differentiation process of neurons but also led to neuronal degeneration or impaired neuronal survival. To investigate this, we quantified total numbers of degenerated neurons and only counted cells fulfilling the morphological criteria of neuronal degeneration, as defined by fragmented neuronal processes, cell shrinkage, and somatic swelling.47 Quantification revealed significant increases in neuronal degeneration after exposing primary neurons to ferucarbotran (all concentrations; Figure 3B–D), VSOP-R2 (1.5 mM and 3.0 mM; Figure 3C and D) and ferumoxytol (1.5 mM; Figure 3C). In contrast to primary microglia (Figure 2), interestingly ferucarbotran and not VSOP-R1 most negatively affected the viability of neurons. Furthermore, while Ferumoxytol did not cause immediate microglial death (Figure 2), it induced severe morphological alterations and increased degeneration of primary neurons.
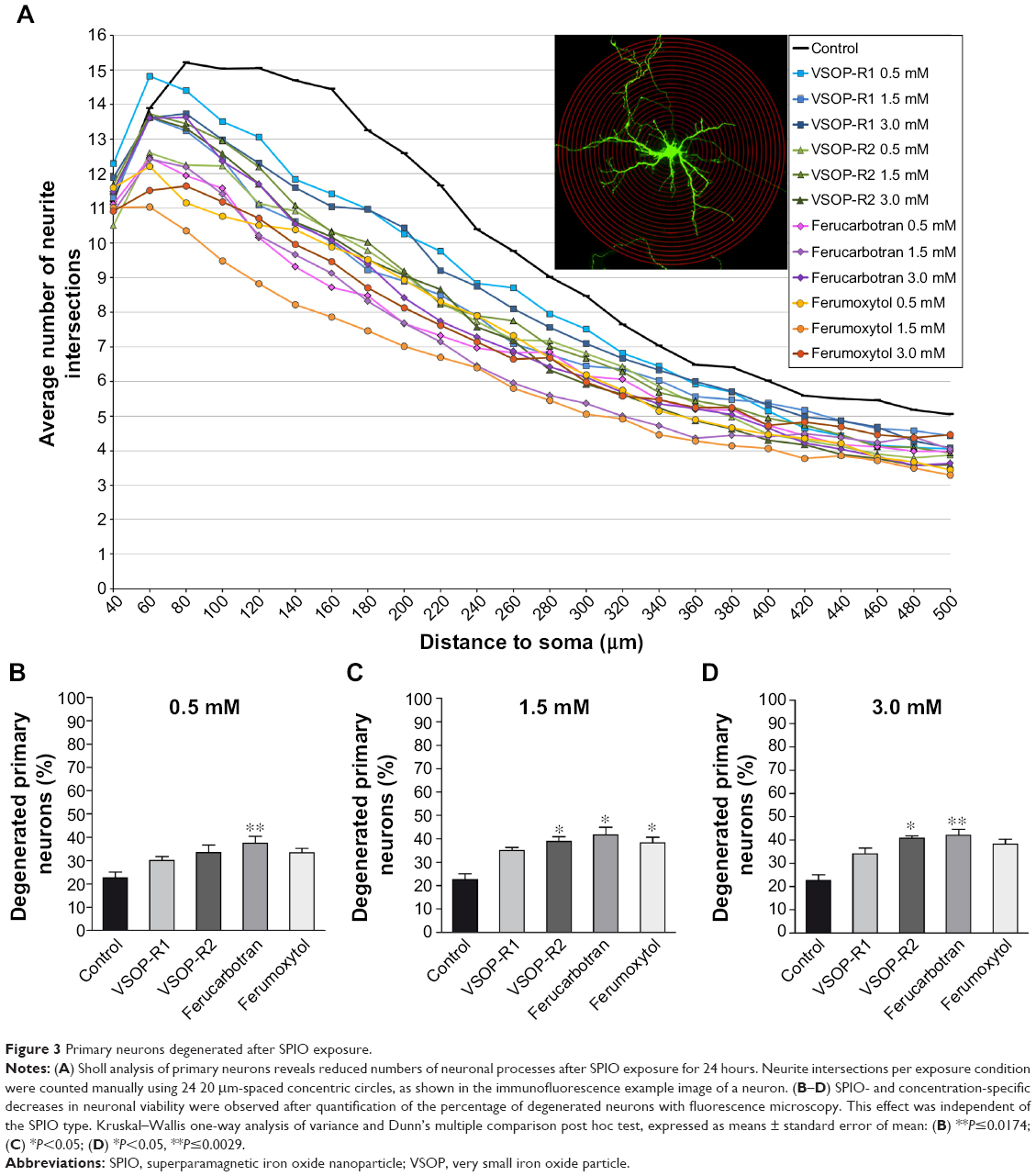 | Figure 3 Primary neurons degenerated after SPIO exposure. |
Since it seemed that different SPIOs caused specific side effects on primary brain cells, we studied the influence of these nanoparticles in a more complex cell-culture model.
Neurons in neuron–glia cocultures do not substantially degenerate after SPIO exposure
To investigate these heterogeneous effects of SPIOs on microglia as well as neurons, we analyzed the neuronal morphology and vitality of neurons from neuron–glia cocultures under equivalent exposure conditions. Our aim was to test whether microglia in neuron–glia cocultures could maintain homeostatic balance and protect neurons from the adverse effects of SPIOs. First of all, we determined the amount of different cell types in the coculture under our experimental condition using qRT-PCR. We quantified equal numbers of microglial cells and neurons in our coculture after 10 DIV (Figure S1).
Sholl analysis of neurons from neuron–glia cocultures revealed contrary effects in both morphological alterations and neuronal degeneration using different SPIO concentrations (Figure 4A, C, and E). Remarkably, we counted more neuronal processes of SPIO-exposed compared to untreated coculture neurons, which decreased with increasing particle concentrations. However, neurons exposed for 24 hours to 0.5 mM VSOP-R1 showed reduced numbers of neurites proximally to cell somata when compared to untreated controls and all other SPIOs (Figure 4A). We also found that the exposure to 1.5 mM VSOP-R2 led to an overall reduction of neurites (Figure 4C, green line), whereas 3.0 mM of this particle resulted in an overall increase (Figure 4E, green line) when compared to untreated controls, respectively. Except for 3.0 mM VSOP-R2, neurons exposed to 3 mM of all other SPIOs possessed similar numbers of neuronal processes as control neurons (Figure 4E). The concentration-dependent increase in neurite branching was also reflected in the degeneration analysis, which did not reveal significant differences for any exposure conditions compared to controls (Figure 4B, D, and F). Consequently, the tested SPIOs seemed rather to stimulate neurite outgrowth of coculture neurons in a concentration- and particle type-dependent manner.
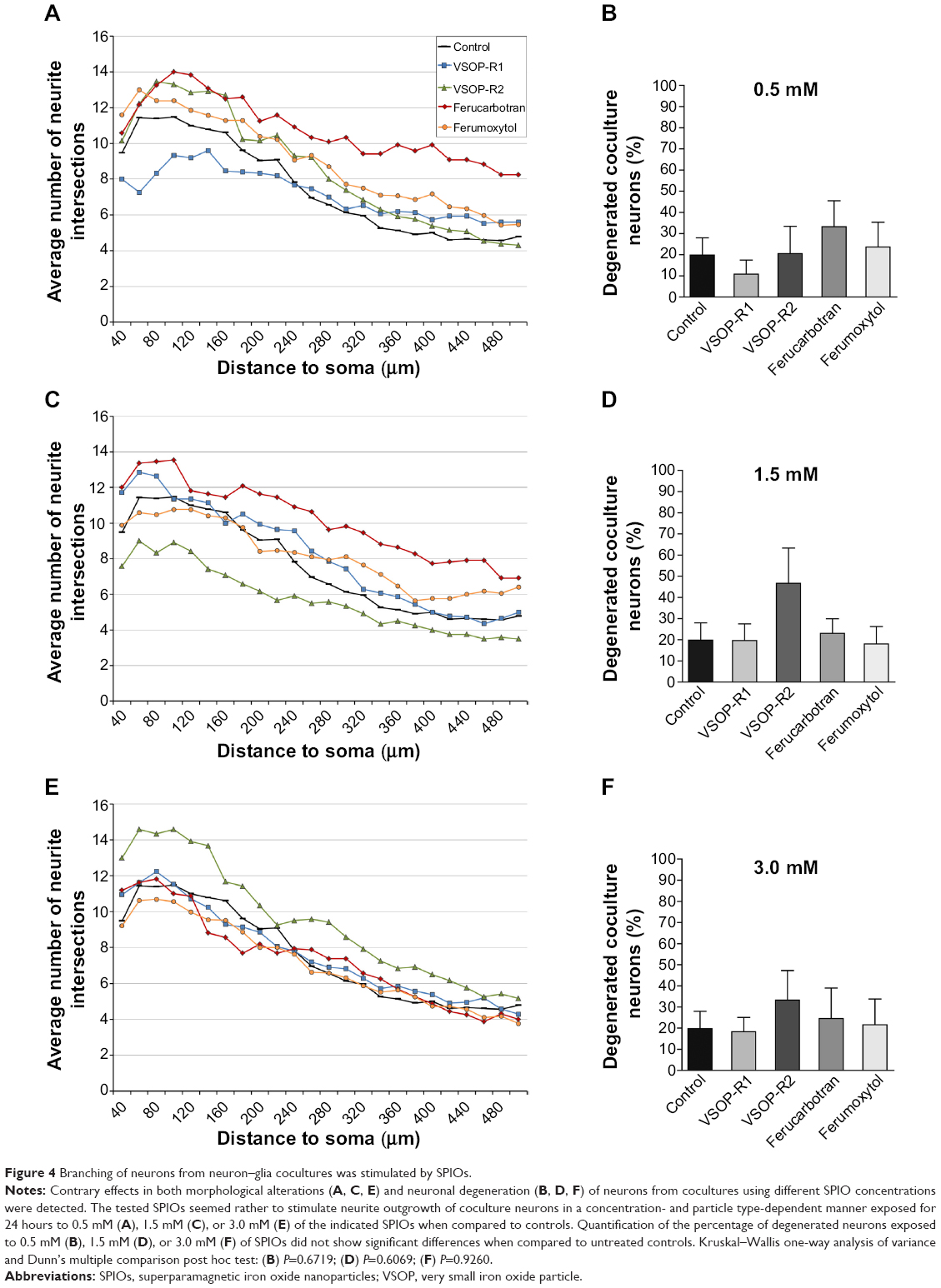 | Figure 4 Branching of neurons from neuron–glia cocultures was stimulated by SPIOs. |
Discussion
Relevance of tested SPIOs
In the present study, we tested different types of SPIOs in terms of their effects on primary microglia and hippocampal neurons, as well as on neurons, from neuron–glia cocultures. We applied the European Medicines Agency-approved ferucarbotran and the US Food and Drug Administration (FDA)-approved ferumoxytol.36,37 The carboxydextran-coated ferucarbotran is a contrast agent for liver imaging in humans, exhibiting a hydrodynamic diameter of 60 nm. Ferucarbotran has been withdrawn by the marketing authorization holder in Europe, but it is still available in pharmaceutical quality from Meito Sangyo in Japan, where the approval for clinical use is still valid. The carboxymethyldextran-coated ferumoxytol with 30 nm hydrodynamic diameter was originally developed for treating iron-deficiency anemia in patients with chronic kidney disease (CKD), and is currently used as a blood pool-contrast agent for visualizing CNS vascular malformations and creating cerebral blood-volume maps with MRI.50–52 It is noteworthy that ferumoxytol can be administered at a single-bolus dose of approximately 0.16 mmol Fe/kg body weight in humans, and the double-bolus dose of 0.320 mmol Fe/kg is currently being tested in clinical trials. In the future, research and clinics will have to deal with extremely high SPIO load and possible brain accumulation. Furthermore, in our study we included two different types of citrate-coated VSOPs that have been tested in human Phase II clinical trials.38,39 These nanoparticles are of special interest, because the small size of around 7 nm prolongs their blood half-life and facilitates their cellular incorporation, which could be beneficial for therapeutic interventions.
So far, conclusive information regarding side effects of SPIOs on primary brain cells is still lacking. Previous studies have often used tumor-cell lines, eg, neuronal PC12 cells, to draw conclusions about cell-labeling efficiency, cytotoxicity, or degradation of SPIOs.53–56 Immortalized cell lines only provide limited evidence about the affected cellular physiology or survival parameters because of their robust proliferation capacity. Therefore, the actual impact of SPIOs on the physiology of primary cells is neglected.
SPIOs strongly affect primary microglia
Fundamental for investigating influences of SPIOs on microglial morphology is that primary microglia show highly ramified morphologies in the resting state in vitro that correspond to the in vivo phenotype.57 Because the morphological criteria for microglia reliably define their state of activation and are used to estimate their influences on the local microenvironment,18 we did not perform common cell-viability measurements, such as 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide or lactate dehydrogenase release assays. The accuracy and reliability with these methods for assessing cytotoxicity is restricted, because SPIOs can physicochemically interfere with the measurement.58
Here, we showed that primary microglia underwent morphological transformation, indicating microglial activation following exposure to VSOPs and ferucarbotran, but not ferumoxytol, which was characterized by progressively less microglial ramification with thickened and enlarged cell bodies.49 However, our data are not in line with previous reports concerning microglial activation following SPIO exposure,22,59 which emphasizes the importance of evaluating SPIO–cell interactions individually according to their physicochemical properties.
So far, the mechanism of microglial SPIO incorporation has been described as endocytosis, in which particles are internalized and transported into lysosomal vesicles after being attached to the extracellular membrane.21,22,60 Nanoparticle internalization is not only dependent on the particle size but also on the surface coating and charge, which in turn determines interactions with extracellular proteins. For instance, positively charged particles interact with the negatively charged glycocalyx, increasing effective and avid particle uptake by mechanisms like clathrin-mediated endocytosis.28,61–63 Furthermore, the respective surface charge can lead to protein adsorption onto nanoparticles in biological media and affect their agglomeration, as well as accumulation by brain cells.61,64–66
In our experiments, VSOPs, as the smallest and electrostatically stabilized particles, and ferucarbotran, as the largest and sterically stabilized particles, were rapidly accumulated by microglial cells. Internalization of ferucarbotran by active phagocytes like microglia has previously been described, demonstrating that ferucarbotran is taken up by macrophages via clathrin receptor-mediated endocytosis and stored in lysosomal vesicles where the low pH facilitates the degradation of the carboxydextran coat.67,68 In addition, the negatively charged carboxyl groups of ferucarbotran enhance particle uptake at higher particle concentrations.7,69 However, cellular uptake of VSOPs appeared to be much faster and quantitatively superior compared to polymer-coated nanoparticles, probably due to their small size (around 7 nm), higher surface-to-volume ratio, and binding to extracellular membrane glycosaminoglycans prior to internalization.30,38,70 On the one hand, citrate-coated particles have been shown to be incorporated by repulsive interaction with the cell membrane, and are subsequently highly concentrated in the cytoplasm.71 On the other hand, within the first 4 hours of incubation, citrate-coated as well as other negatively charged SPIOs are located in early endosomes that migrate toward lysosomes.7,61 Consequently, we suppose that VSOPs and ferucarbotran might rapidly be taken up by activated microglia and stored intracellularly for a period of time, releasing free iron ions via lysosomal degradation. Free iron ions and the formation of intracellular iron deposits most likely contribute to increasing levels of microglial cell death by impairing mitochondrial function.32,34,35 Therefore, the facilitated accumulation of highly concentrated VSOPs or ferucarbotran by microglia is most likely the cause for the consistent decrease of microglial viability over 24 hours, which is caused by intracellular particle localization as well as individual SPIO composition, including size, coating, surface charge, and concentration.
Interestingly, incubating primary microglia with ferumoxytol increased the numbers of viable cells. This effect cannot have been due to highly enhanced microglial proliferation that would lead to elimination of dead cells, because primary microglia in vitro do not actually proliferate after being adherent.72 Ferumoxytol does not exhibit a surface charge due to its coating material, which might diminish the interaction with cell membranes, and thus its uptake in the absence of transfection agents. However, ferumoxytol exhibits a tendency to release free iron ions, which is desired for the treatment of human CKD.50 Ferumoxytol has a relatively high molecular weight and exhibits a poor affinity toward iron oxide, so that the coating material is reversibly adsorbed.42,50,73 Therefore, ferumoxytol was able to degrade rapidly after it was added to cell cultures, resulting in the release of free iron ions and extracellular iron deposits, which might subsequently adversely affect microglial physiology in the long term.34,35 Accordingly, we assume that those elevated iron levels could strongly impair the endocytotic activity of microglia due to altered enzyme function. The exposure to ferumoxytol in high doses might diminish microglial particle accumulation over an extended period of time. In turn, this could explain the increased cell survival within the relatively short ferumoxytol incubation of 24 hours. Whether this actually applies needs to be further investigated, eg, by using much longer exposure times of several days or weeks. Therefore, ferumoxytol as an MRI contrast agent for visualizing CNS pathologies has unpredictable consequences. For instance, in ferumoxytol-enhanced MRI imaging of human brain arteriovenous malformations, signals from these particles are also found in the normal vasculature. Furthermore, differences in ferumoxytol clearance among patients have been reported.11 Even more surprising is the fact that ferumoxytol, as an FDA-approved substance for treating CKD only, is being used for MRI of brain tumors in children.74
Neuronal vitality is modulated by SPIOs
Our data show that primary neurons obviously degenerated following SPIO exposure, which we did not observe for neurons from neuron–glia cocultures. For this reason, we studied how SPIOs affected the vitality of neurons in neuron–glia cocultures to include microglial responses and more closely resemble the in vivo situation. The incubation of neurons in cocultures with SPIOs did not result in a coherent reduction of neurites, but rather induced increased neurite branching in a particle- and dose-dependent manner. For instance, lower concentrations of ferucarbotran and high concentrations of VSOP-R2 stimulated neurite outgrowth. This effect could most likely be due to the composition-dependent SPIO accumulation by microglial cells, determined by the surface coating and charge. As mentioned in the previous section, ferucarbotran and VSOPs are rapidly accumulated by microglial cells using slightly different pathways. Consequently, the cellular mechanisms influencing microglial physiology might differentially affect the vitality of neurons, thereby partially protecting neuronal morphology. However, we cannot exclude that SPIOs per se might also have a stimulatory effect on neurite outgrowth. In this context, it is important to consider that necrotic damage of neurons induces microglial activation, which, in turn leads to neurotoxicity through proinflammatory responses of microglia.23 Activated microglia not only promote neuronal damage by producing a wide range of cytotoxic factors, such as TNFα, nitric oxides, or reactive oxygen species, but also support neuronal survival by releasing trophic and anti-inflammatory factors.24,75 In addition, iron accumulations in neurons and microglia are promoted by inflammatory cytokines like TNFα and IL-6, which are induced by mitochondrial dysfunction and consequently weaken the phagocytic activity of activated microglia.32,35 On the other hand, it has been shown that iron ions from certain nanoparticles enhance neuronal differentiation and survival of immortal PC12 neurons, thereby promoting neurite outgrowth in a dose-dependent manner.33
In our study, we quantified total numbers of neurites of primary neurons, and did not differentiate between dendrites or axons. It would be of certain relevance in terms of therapeutic interventions using specific SPIOs and concentrations if these particles induce dendritic or axonal outgrowth, eg, in the treatment of neurodegenerative diseases. However, more detailed analyses are necessary to consider the cortical layer and target-specificity of applied SPIOs, as well as their fate and degradation.
Conclusion and perspectives
Our study demonstrated that SPIOs partially affect the morphology and viability of essential cell types of the CNS in a manner that depends closely on particle size, surface coating, charge, and concentration. Furthermore, all of these SPIO characteristics induce specific effects while interacting with different cell types and within the physiological system they are applied to. That certainly makes it much more difficult to predict reliably the consequences of nanoparticle exposure on brain cells and the subsequent influences on cell physiology in the long term. In turn, particle–cell interactions, as well as measurement-influencing parameters like SPIO behavior in cell culture, should accurately be validated for individual SPIOs in advance. Additionally, long-term effects of other diagnostically and therapeutically applied SPIOs on cellular metabolism and functionality should be thoroughly investigated, because adverse effects cannot yet be estimated reliably. Even the guidelines of the International Conference for Harmonization for drug development using nanoparticles only assess the survival of animals or cells in culture in order to address biological toxicity.76 In general, studies dealing with nanotoxicology and biocompatibility hardly include cumulative and nanoparticle-associated long-term effects on cell function, which could result in irreversible damage following internalization, especially after entering the CNS. Prospective studies should certainly focus on primary cells to estimate the type of cellular modification, and subsequently on more complex models like slice cultures to test for cell functionality within the biological system.
We conclude that particular caution has to be exercised when administering SPIOs that target the brain. This includes direct SPIO injection into the brain parenchyma, as in particle-based drug delivery, systemic injection into the bloodstream, and nanoscale material entering the organism via any other route. Our results clearly contribute to assessing the prospects and limitations for the application of SPIOs as contrast agents in MRI in vivo.
Acknowledgments
Monika Dulinski, Rike Dannenberg, Anne Heiner, Keerthana Raveendran, and Pascal Fenske are acknowledged for their excellent technical assistance. The authors thank Ulf Strauss for his support with the statistics, and Kimberly Mason and Tim Wanger for their excellent help in editing the manuscript. This work was supported by Deutsche Forschungsgemeinschaft (DFG) grant KFO 213 and the Sonnenfeld-Stiftung, which sponsored technical equipment for AUB.
Author contributions
JN performed all of the experimental work and analyses. SW substantially contributed to the synthesis of VSOPs, performed the pharmacokinetic characterization (Table 1) and provided these nanoparticles for our study. JK considerably helped by analyzing and interpreting data. JG and AUB designed the study. JN together with AUB wrote the manuscript with contributions from all coauthors (JK, JG, SW). All authors read and approved the final manuscript.
Disclosure
The authors report no conflicts of interest in this work.
References
Buzea C, Pacheco II, Robbie K. Nanomaterials and nanoparticles: sources and toxicity. Biointerphases. 2007;2(4):MR17–MR71. | ||
Weinstein JS, Varallyay CG, Dosa E, et al. Superparamagnetic iron oxide nanoparticles: diagnostic magnetic resonance imaging and potential therapeutic applications in neurooncology and central nervous system inflammatory pathologies, a review. J Cerebr Blood F Met. 2010;30(1):15–35. | ||
Rumenapp C, Gleich B, Haase A. Magnetic nanoparticles in magnetic resonance imaging and diagnostics. Pharm Res. 2012;29(5):1165–1179. | ||
Petry KG, Boiziau C, Dousset V, Brochet B. Magnetic resonance imaging of human brain macrophage infiltration. Neurotherapeutics. 2007;4(3): 434–442. | ||
Oude Engberink RD, van der Pol SM, Dopp EA, de Vries HE, Blezer EL. Comparison of SPIO and USPIO for in vitro labeling of human monocytes: MR detection and cell function. Radiology. 2007;243(2): 467–474. | ||
Li L, Jiang W, Luo K, et al. Superparamagnetic iron oxide nanoparticles as MRI contrast agents for non-invasive stem cell labeling and tracking. Theranostics. 2013;3(8):595–615. | ||
Andreas K, Georgieva R, Ladwig M, et al. Highly efficient magnetic stem cell labeling with citrate-coated superparamagnetic iron oxide nanoparticles for MRI tracking. Biomaterials. 2012;33(18):4515–4525. | ||
Wahajuddin, Arora S. Superparamagnetic iron oxide nanoparticles: magnetic nanoplatforms as drug carriers. Int J Nanomedicine. 2012;7: 3445–3471. | ||
Chatterjee DK, Diagaradjane P, Krishnan S. Nanoparticle-mediated hyperthermia in cancer therapy. Ther Deliv. 2011;2(8):1001–1014. | ||
Yang H. Nanoparticle-mediated brain-specific drug delivery, imaging, and diagnosis. Pharm Res. 2010;27(9):1759–1771. | ||
Hasan DM, Amans M, Tihan T, et al. Ferumoxytol-enhanced MRI to image inflammation within human brain arteriovenous malformations: a pilot investigation. Transl Stroke Res. 2012;3(Suppl 1):166–173. | ||
Varallyay CG, Nesbit E, Fu R, et al. High-resolution steady-state cerebral blood volume maps in patients with central nervous system neoplasms using ferumoxytol, a superparamagnetic iron oxide nanoparticle. J Cerebr Blood F Met. 2013;33(5):780–786. | ||
Venneti S, Lopresti BJ, Wiley CA. Molecular imaging of microglia/macrophages in the brain. Glia. 2013;61(1):10–23. | ||
Rausch M, Baumann D, Neubacher U, Rudin M. In vivo visualization of phagocytotic cells in rat brains after transient ischemia by USPIO. NMR Biomed. 2002;15(4):278–283. | ||
Wang Y, Wang B, Zhu MT, et al. Microglial activation, recruitment and phagocytosis as linked phenomena in ferric oxide nanoparticle exposure. Toxicol Lett. 2011;205(1):26–37. | ||
Roohi F, Lohrke J, Ide A, Schütz G, Dassler K. Studying the effect of particle size and coating type on the blood kinetics of superparamagnetic iron oxide nanoparticles. Int J Nanomedicine. 2012;7:4447–4458. | ||
Lockman PR, Koziara JM, Mumper RJ, Allen DD. Nanoparticle surface charges alter blood–brain barrier integrity and permeability. J Drug Target. 2004;12(9–10):635–641. | ||
Napoli I, Neumann H. Microglial clearance function in health and disease. Neuroscience. 2009;158(3):1030–1038. | ||
Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia. 2002;40(2):133–139. | ||
Parkhurst CN, Gan WB. Microglia dynamics and function in the CNS. Curr Opin Neurobiol. 2010;20(5):595–600. | ||
Pinkernelle J, Calatayud P, Goya GF, Fansa H, Keilhoff G. Magnetic nanoparticles in primary neural cell cultures are mainly taken up by microglia. BMC Neurosci. 2012;13:32. | ||
Luther EM, Petters C, Bulcke F, et al. Endocytotic uptake of iron oxide nanoparticles by cultured brain microglial cells. Acta Biomater. 2013; 9(9):8454–8465. | ||
Pais TF, Figueiredo C, Peixoto R, Braz MH, Chatterjee S. Necrotic neurons enhance microglial neurotoxicity through induction of glutaminase by a MyD88-dependent pathway. J Neuroinflamm. 2008;5:43. | ||
Ye L, Huang Y, Zhao L, et al. IL-1β and TNF-α induce neurotoxicity through glutamate production: a potential role for neuronal glutaminase. J Neurochem. 2013;125(6):897–908. | ||
Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69. | ||
Xue Y, Wu J, Sun J. Four types of inorganic nanoparticles stimulate the inflammatory reaction in brain microglia and damage neurons in vitro. Toxicol Lett. 2012;214(2):91–98. | ||
Berry CC, Wells S, Charles S, Aitchison G, Curtis AS. Cell response to dextran-derivatised iron oxide nanoparticles post internalisation. Biomaterials. 2004;25(23):5405–5413. | ||
Zhang S, Li J, Lykotrafitis G, Bao G, Suresh S. Size-dependent endocytosis of nanoparticles. Adv Mater. 2009;21:419–424. | ||
Mahmoudi M, Lynch I, Ejtehadi MR, Monopoli MP, Bombelli FB, Laurent S. Protein-nanoparticle interactions: opportunities and challenges. Chem Rev. 2011;111(9):5610–5637. | ||
Horie M, Fujita K, Kato H, et al. Association of the physical and chemical properties and the cytotoxicity of metal oxide nanoparticles: metal ion release, adsorption ability and specific surface area. Metallomics. 2012;4(4):350–360. | ||
Karlsson HL, Gustafsson J, Cronholm P, Möller L. Size-dependent toxicity of metal oxide particles-a comparison between nano- and micrometer size. Toxicol Lett. 2009;188(2):112–118. | ||
Urrutia PJ, Mena NP, Núñez MT. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front Pharmacol. 2014;5:38. | ||
Kim JA, Lee N, Kim BH, et al. Enhancement of neurite outgrowth in PC12 cells by iron oxide nanoparticles. Biomaterials. 2011;32(11):2871–2877. | ||
Andersen HH, Johnsen KB, Moos T. Iron deposits in the chronically inflamed central nervous system and contributes to neurodegeneration. Cell Mol Life Sci. 2014;71(9):1607–1622. | ||
Mairuae N, Connor JR, Cheepsunthorn P. Increased cellular iron levels affect matrix metalloproteinase expression and phagocytosis in activated microglia. Neurosci Lett. 2011;500(1):36–40. | ||
Reimer P, Balzer T. Ferucarbotran (Resovist): a new clinically approved RES-specific contrast agent for contrast-enhanced MRI of the liver: properties, clinical development, and applications. Eur Radiol. 2003;13(6): 1266–1276. | ||
Lu M, Cohen MH, Rieves D, Pazdur R. FDA report: Ferumoxytol for intravenous iron therapy in adult patients with chronic kidney disease. Am J Hematol. 2010;85(5):315–319. | ||
Taupitz M, Wagner S, Schnorr J, et al. Phase I clinical evaluation of citrate-coated monocrystalline very small superparamagnetic iron oxide particles as a new contrast medium for magnetic resonance imaging. Invest Radiol. 2004;39(7):394–405. | ||
Wagner M, Wagner S, Schnorr J, et al. Coronary MR angiography using citrate-coated very small superparamagnetic iron oxide particles as blood-pool contrast agent: initial experience in humans. J Magn Reson Imaging. 2011;34(4):816–823. | ||
Millward JM, Schnorr J, Taupitz M, Wagner S, Wuerfel JT, Infante-Duarte C. Iron oxide magnetic nanoparticles highlight early involvement of the choroid plexus in central nervous system inflammation. ASN Neuro. 2013;5(1):e00110. | ||
Wuerfel E, Smyth M, Millward JM, et al. Electrostatically stabilized magnetic nanoparticles – an optimized protocol to label murine T cells for in vivo MRI. Front Neurol. 2011;2:72. | ||
Balakrishnan VS, Rao M, Kausz AT, et al. Physicochemical properties of ferumoxytol, a new intravenous iron preparation. Eur J Clin Invest. 2009;39(6):489–496. | ||
Velmans T, Battefeld A, Geist B, Farrés AS, Strauss U, Bräuer AU. Plasticity-related gene 3 promotes neurite shaft protrusion. BMC Neurosci. 2013;14:36. | ||
Drews D, Klar M, Dame C, Bräuer AU. Developmental expression profile of the YY2 gene in mice. BMC Dev Biol. 2009;9:45. | ||
Takano K, Nakamura Y, Yoneda Y. Microglial cell death induced by a low concentration of polyamines. Neuroscience. 2003;120(4):961–967. | ||
Tysiak E, Asbach P, Aktas O, et al. Beyond blood brain barrier breakdown – in vivo detection of occult neuroinflammatory foci by magnetic nanoparticles in high field MRI. J Neuroinflamm. 2009;6:20. | ||
Adamec E, Yang F, Cole GM, Nixon RA. Multiple-label immunocytochemistry for the evaluation of nature of cell death in experimental models of neurodegeneration. Brain Res Brain Res Protoc. 2001;7(3): 193–202. | ||
Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87(4):387–406. | ||
Ling EA, Wong WC. The origin and nature of ramified and amoeboid microglia: a historical review and current concepts. Glia. 1993;7(1): 9–18. | ||
Kowalczyk M, Banach M, Rysz J. Ferumoxytol: a new era of iron deficiency anemia treatment for patients with chronic kidney disease. J Nephrol. 2011;24(6):717–722. | ||
Dosa E, Tuladhar S, Muldoon LL, Hamilton BE, Rooney WD, Neuwelt EA. MRI using ferumoxytol improves the visualization of central nervous system vascular malformations. Stroke. 2011;42(6):1581–1588. | ||
Christen T, Ni W, Qiu D, et al. High-resolution cerebral blood volume imaging in humans using the blood pool contrast agent ferumoxytol. Magn Reson Med. 2013;70(3):705–710. | ||
Soenen SJ, Himmelreich U, Nuytten N, De Cuyper M. Cytotoxic effects of iron oxide nanoparticles and implications for safety in cell labelling. Biomaterials. 2011;32(1):195–205. | ||
Pisanic TR 2nd, Blackwell JD, Shubayev VI, Finones RR, Jin S. Nanotoxicity of iron oxide nanoparticle internalization in growing neurons. Biomaterials. 2007;28(16):2572–2581. | ||
Hong SC, Lee JH, Lee J, et al. Subtle cytotoxicity and genotoxicity differences in superparamagnetic iron oxide nanoparticles coated with various functional groups. Int J Nanomedicine. 2011;6:3219–3231. | ||
Saito S, Tsugeno M, Koto D, et al. Impact of surface coating and particle size on the uptake of small and ultrasmall superparamagnetic iron oxide nanoparticles by macrophages. Int J Nanomedicine. 2012;7: 5415–5421. | ||
Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. | ||
Holder AL, Goth-Goldstein R, Lucas D, Koshland CP. Particle-induced artifacts in the MTT and LDH viability assays. Chem Res Toxicol. 2012;25(9):1885–1892. | ||
Pickard MR, Chari DM. Robust uptake of magnetic nanoparticles (MNPs) by central nervous system (CNS) microglia: implications for particle uptake in mixed neural cell populations. Int J Mol Sci. 2010; 11(3):967–981. | ||
Fleige G, Nolte C, Synowitz M, Seeberger F, Kettenmann H, Zimmer C. Magnetic labeling of activated microglia in experimental gliomas. Neoplasia. 2001;3(6):489–499. | ||
Schweiger C, Hartmann R, Zhang F, Parak WJ, Kissel TH, Rivera Gil P. Quantification of the internalization patterns of superparamagnetic iron oxide nanoparticles with opposite charge. J Nanobiotechnology. 2012;10:28. | ||
Petters C, Irrsack E, Koch M, Dringen R. Uptake and metabolism of iron oxide nanoparticles in brain cells. Neurochem Res. 2014;39(9): 1648–1660. | ||
Sun Z, Yathindranath V, Worden M, et al. Characterization of cellular uptake and toxicity of aminosilane-coated iron oxide nanoparticles with different charges in central nervous system-relevant cell culture models. Int J Nanomedicine. 2013;8:961–970. | ||
Safi M, Courtois J, Seigneuret M, Conjeaud H, Berret JF. The effects of aggregation and protein corona on the cellular internalization of iron oxide nanoparticles. Biomaterials. 2011;32(35):9353–9363. | ||
Hirsch V, Kinnear C, Moniatte M, Rothen-Rutishauser B, Clift MJ, Fink A. Surface charge of polymer coated SPIONs influences the serum protein adsorption, colloidal stability and subsequent cell interaction in vitro. Nanoscale. 2013;5(9):3723–3732. | ||
Sakulkhu U, Mahmoudi M, Maurizi L, Salaklang J, Hofmann H. Protein corona composition of superparamagnetic iron oxide nanoparticles with various physico-chemical properties and coatings. Sci Rep. 2014; 4:5020. | ||
Lunov O, Syrovets T, Rocker C, et al. Lysosomal degradation of the carboxydextran shell of coated superparamagnetic iron oxide nanoparticles and the fate of professional phagocytes. Biomaterials. 2010;31(34): 9015–9022. | ||
Yang CY, Tai MF, Lin CP, et al. Mechanism of cellular uptake and impact of ferucarbotran on macrophage physiology. PloS One. 2011; 6(9):e25524. | ||
Mailänder V, Lorenz MR, Holzapfel V, et al. Carboxylated superparamagnetic iron oxide particles label cells intracellularly without transfection agents. Mol Imaging Biol. 2008;10(3):138–146. | ||
Ludwig A, Poller WC, Westphal K, et al. Rapid binding of electrostatically stabilized iron oxide nanoparticles to THP-1 monocytic cells via interaction with glycosaminoglycans. Basic Res Cardiol. 2013;108(2):328. | ||
Saraswathy A, Nazeer SS, Jeevan M, et al. Citrate coated iron oxide nanoparticles with enhanced relaxivity for in vivo magnetic resonance imaging of liver fibrosis. Colloids Surf B Biointerfaces. 2014;117: 216–224. | ||
Floden AM, Combs CK. Microglia repetitively isolated from in vitro mixed glial cultures retain their initial phenotype. J Neurosci Methods. 2007;164(2):218–224. | ||
Lin MM, Kim do K, El Haj AJ, Dobson J. Development of superparamagnetic iron oxide nanoparticles (SPIONs) for translation to clinical applications. IEEE Trans Nanobioscience. 2008;7(4):298–305. | ||
Thompson EM, Guillaume DJ, Dósa E, et al. Dual contrast perfusion MRI in a single imaging session for assessment of pediatric brain tumors. J Neurooncol. 2012;109(1):105–114. | ||
Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87(1):10–20. | ||
ICH Safety Guidelines [webpage on the Internet]. ICH harmonisation for better health. Available from: http://www.ich.org/products/guidelines/safety/article/safety-guidelines.html. Accessed December 22, 2014. |
Supplementary materials
Preparation of primary cell cultures and neuron-glia cocultures
Preparations of primary microglial cultures were performed as described previously.1 Briefly, C57BL/6 mice at postnatal days 0 to 2 (P 0–2) were decapitated and brains were collected in ice-cold Hank’s Buffered Salt Solution (HBSS+ with Ca, Mg; Gibco®, Life Technologies, Germany). After carefully removing meninges and blood vessels, brains were minced and washed once with HBSS (without Ca, Mg; Gibco®). Subsequently, brains were enzymatically digested at 37°C for 12 minutes using 0.25% trypsin-EDTA (Gibco®) and mechanically homogenized with the addition of 10 mg/mL DNase I (Hoffman-La Roche Ltd., Basel, Switzerland). The culture medium used for resuspending dissociated glial cells was high glucose Dulbecco’s Modified Eagle’s Medium (DMEM+) containing l-glutamine, 4,500 mg/L d-glucose, 110 mg/L sodium pyruvate (Gibco®) supplemented with 10% fetal calf serum (FCS), 100 U/mL penicillin, 100 μg/mL streptomycin (Pan-Biotech, Aidenbach, Germany) and 0.1% of 50 mM 2-Mercaptoethanol. The cell suspension was plated in poly-L-lysine (20 μg/mL; Sigma-Aldrich Co., St Louis, MO, USA) coated 75 cm2 flasks, and DMEM+ was completely replaced 24 hours after cell culture preparation. This plated glial cell population consisted of astrocytes and microglia that attached to and grew on the coated surface of the culture flasks in the incubator for 8 to 10 days in vitro (DIV) until microglia detach from the feeder layer of confluent astrocytes. Primary microglia were harvested by gentle agitation of flasks, collected, centrifuged at room temperature (RT) and resuspended in DMEM+. Microglia were seeded at densities of 2×105 cells per well in 12-well plates containing poly-L-lysine-coated glass coverslips in 1 mL DMEM+. Microglia were maintained at 37°C in 5% CO2 for 24 hours prior to the start of experiments.
Primary astrocytes for neuron-glia cocultures were prepared from C57BL/6 mice at P 0–2. Mice were decapitated and brains were collected in ice-cold HBSS+. Hemispheres were minced and enzymatically digested at 37°C for 5 minutes using 2.5% trypsin and 10 mg/mL DNase I. Dissociated cells were re-suspended in Minimum Essential Medium (MEM+; supplemented with 10% horse serum, 0.6% glucose, 100 U/mL penicillin and 100 μg/mL streptomycin; Gibco) and centrifuged at RT for 5 minutes. The resulting pellet was resuspended in MEM+ and cells were plated in poly-L-lysine-coated 75 cm2 flasks at densities of between 7.5×106 and 12×106 cells per flask and maintained at 37°C in 5% CO2 for 7 DIV, 24 hours after cell preparation MEM was completely replaced. After 7 DIV, primary astrocytes were harvested by mild trypsinization, transferred to poly-L-lysine-coated 6-well plates without coverslips and seeded at densities of 5×104 astrocytes per well in 1 mL MEM+. Forty-eight hours after plating, astrocytes were preconditioned by completely replacing MEM+ with Neurobasal-A medium (NB+; Gibco®) supplemented with 2% B27 (Gibco®), 0.5 mM glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin. From the day of preparation, primary astrocyte cultures were maintained for 11 DIV until primary hippocampal neurons on poly-L-lysine-coated coverslips were transferred to establish neuron-glia cocultures.
Hippocampal neurons for primary cultures and neuron-glia cocultures were prepared from C57BL/6 mice at embryonic day (E) 18 as described previously.1 Briefly, hippocampi of several embryos were collected in ice-cold HBSS+ and enzymatically digested using 2.5% trypsin in HBSS− at 37°C for 15 minutes. Dissociated cells were resuspended in MEM+ and mechanically homogenized after adding 10 mg/mL DNase I. Primary neurons were transferred to 12-well plates containing poly-L-lysine-coated glass coverslips and seeded at densities of 8×104 neurons per well in 1 mL MEM+. Three hours after plating, neurons were washed twice with sterile 0.1 M phosphate buffered saline (PBS), and 1 mL NB+ was added to each well. For primary neuronal cultures, neurons were maintained at 37°C and 5% CO2 for 10 DIV. For neuron-glia cocultures, primary neurons on glass coverslips were transferred to astrocyte cultures the same day of preparation by placing them upside down on the plated astrocytes. After 7 DIV, one third of the NB+ medium was replaced. In total, neuron-glia cocultures were kept in culture for 10 DIV.
Quantitative real-time PCR of neuron-glia cocultures
Identity and purity of primary cells were analyzed by quantitative real-time (qRT-) polymerase chain reaction (PCR) with neuron- specific class III β-tubulin (Tuj1) determining neuronal origin, glial fibrillary acidic protein (GFAP) as a marker for astrocytes, and the ionized calcium-binding adaptor molecule 1 (Iba1) as a marker for microglial cells (Figure S1).2
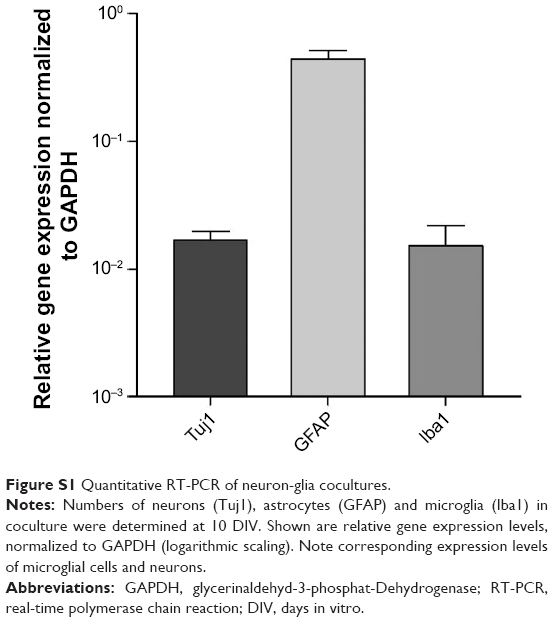 | Figure S1 Quantitative RT-PCR of neuron-glia cocultures. |
For RNA extraction and cDNA synthesis, brains of C57BL/6 mice at P10 were removed and homogenized in TRIzol reagent (Gibco®). Primary neurons, microglial cells or astrocytes were scraped in PBS and centrifuged at 900 rpm for 5 minutes at 4°C. Total RNA was extracted using TRIzol reagent according to the manufacturer’s protocol. The concentration and purity of the total RNA isolated was determined by spectrophotometric analysis (Thermo Scientific* Biomate 3 spectrophotometer, Fisher Scientific, Schwerte, Germany). cDNA was synthesized with 5 μg total RNA using the High-Capacity cDNA Archive Kit (Applied Biosystems, Carlsbad, CA, USA) according to the manufacturer’s protocol. As a control, reaction was performed without MultiScribe reverse transcriptase. cDNA was diluted 1:5 with RNase, DNase-free water and stored at −20°C. The quality of the amplified cDNA (with and without MultiScribe reverse transcriptase) was controlled by β-actin PCR.
Reverse transcriptase qRT-PCR was performed with the following gene expression assays: Tuj1 (assay ID Mm00727586_s1), Iba1 (assay ID Mm00479862_g1), GFAP (assay ID Mm00546086_m1), glycerinaldehyd-3-phosphat-Dehydrogenase (GAPDH) (assay ID 4352932E) and β-actin (assay ID 4352933E) (Applied Biosystems). For HPRT, separate primer and probe were used (Primer Mix for 5′-ATCATTATGCCGAGGATTTGGAA-3′; rev 5′-TTGAGCACACAGAGGGCCA-3′ and probe 5′-TGGACAGGACTGAAAGACTTGCTCGAGATG-3′). The PCR was run on the ABI PRISM™ 7700 Sequence Detection System (Applied Biosystems) and data obtained were processed by ABI PRISM software. Standard curves were created by serial dilutions of cDNA from P10 mouse cortex with amplification efficiency between 90% and 100%. Data were normalized to two different house-keeping genes (β-actin and HTPR), producing similar results. Each result is the average of three separate experiments.
References
Velmans T, Battefeld A, Geist B, Farres AS, Strauss U, Brauer AU. Plasticity-related gene 3 promotes neurite shaft protrusion. BMC Neurosci. 2013;14:36. doi:10.1186/1471-2202-14-36. | ||
Drews D, Klar M, Dame C, Brauer AU. Developmental expression profile of the YY2 gene in mice. BMC developmental biology. 2009;9:45. doi:10.1186/1471-213X-9-45. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.