Back to Journals » OncoTargets and Therapy » Volume 9
New developments in the management of non-small-cell lung cancer, focus on rociletinib: what went wrong?
Authors Van Der Steen N, Caparello C, Rolfo C, Pauwels P, Peters GJ, Giovannetti E
Received 30 June 2016
Accepted for publication 23 August 2016
Published 6 October 2016 Volume 2016:9 Pages 6065—6074
DOI https://doi.org/10.2147/OTT.S97644
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Faris Farassati
Video abstract presented by Van Der Steen et al.
Views: 882
Nele Van Der Steen,1,* Chiara Caparello,2,* Christian Rolfo,3 Patrick Pauwels,4 Godefridus J Peters,1 Elisa Giovannetti5
1Department of Medical Oncology, VU University Medical Center, Amsterdam, the Netherlands; 2Polo Oncologico, Azienda Ospedaliero-Universitaria Pisana, Pisa, Italy; 3Phase I – Early Clinical Trials Unit, Oncology Department, Antwerp University Hospital, Edegem, 4Center for Oncological Research, University of Antwerp, Wilrijk, Antwerp, Belgium; 5Cancer Pharmacology Lab, AIRC Start-Up Unit, University of Pisa, Pisa, Italy
*These authors contributed equally to this work
Abstract: Recently, the development of the third-generation epidermal growth factor receptor-small molecule inhibitor (EGFR-TKI) rociletinib had failed. In this review, the wide-ranging aspects of the evolution of EGFR-TKIs were collected, with a special focus on rociletinib. The influence of different oncogenic mutations on EGFR activity was also discussed. Resistance to the first (erlotinib, gefitinib)- and second (afatinib)-generation EGFR-TKIs provided the rationale behind the development of the third-generation inhibitors (rociletinib, osimertinib). On the basis of these data, a comparison of their efficacy on the different mutated EGFRs and the respective resistance mechanisms is further reported. Moreover, the evolution and results of the clinical trials of rociletinib (TIGER trials) are compared with the trials on osimertinib, another third-generation EGFR-TKI that now has been granted US Food and Drug Administration approval. The reasons behind the arrest in the further development of rociletinib are put in the perspective of future drug development.
Keywords: rociletinib, EGFR, EGFR-TKI, NSCLC, targeted therapies
Introduction
Role of EGFR mutations in NSCLC
Approximately, 10%–20% of non-small-cell lung cancers (NSCLCs) are driven by overactivation of epidermal growth factor receptor (EGFR). This overactivation is caused by mutations in the ATP-binding pocket of the receptor: EGFR exon 19 deletions; exon 20 insertions; L858R, L861Q, or G719S/A/C substitutions.1 The structure of EGFR consists of two lobes lining the ATP-binding pocket, a regulatory C-helix, an activation loop, and a phosphate-coordinating loop.2 In wild-type (WT)-EGFR, the activation loop forms a helix through hydrophobic interactions, which locks the regulatory C-helix in its inactive conformation with the help of the P-loop. Upon dimerization of the receptor, the C-lobe of the one receptor binds the N-lobe of the second one, thus pushing the C-helix inward and leading to activation of the receptor.3
Residue L858 is located in the activation loop and is part of the string of hydrophobic amino acids that form the activation helix. When this leucine is replaced by the hydrophilic arginine, this helix is distorted, resulting in a destabilization of the inactive conformation of EGFR, thus promoting a transition to the active state. The same holds true for residue G719, which is located at the P-loop. Here, the glycine residue is needed to cope with the torsion of the P-loop that contributes hydrophobic residues to the inactive-state helix of the activation loop. When this glycine is replaced by either alanine, serine, or cysteine, it results in a distortion of the P-loop, which in turn affects the activation loop. This also leads to a destabilization of the inactive conformation of EGFR, thus promoting activation of the receptor.4,5 Currently, there are no crystal structures available of mutated EGFR with either deletions or insertions. It is assumed that exon 19 deletions lead to a shortening of the C-helix with usually 1–3 residues. This shortening prevents the C-helix to turn outward to its inactive state. For exon 20 insertions, it is assumed that the addition of 1–4 residues to the C-helix pushes it inward, forcing it into its active state. In vitro studies also show that these insertions lead to a considerable smaller ATP-binding pocket, thus decreasing the affinity for TKIs and explaining the inherent resistance.6,7
Furthermore, it has also been shown that not all mutant forms of EGFR remain dependent on dimerization of the receptor. Cho et al8 have shown that for the WT-EGFR and the L858R mutant, dimerization is required for activation, whereas for the exon 19 deletion, exon 20 insertion, and L858R/T790M double mutant receptor, dimerization is no longer required. These results were also confirmed by in vivo studies with cetuximab, a monoclonal antibody that prevents the dimerization of EGFR. Cetuximab showed the highest inhibitory effect on mice tumors harboring the L858R mutations, whereas it only showed a modest effect on tumors caused by the other EGFR mutations.8
First-generation EGFR-TKIs: erlotinib and gefitinb
Several of these mutations (exon 19 deletions; L858R, L861Q, and G719S/A/C substitutions) confer sensitivity to first-generation EGFR small molecule inhibitors (EGFR-TKIs) (Figure 1) such as erlotinib or gefinitib,9 although the inhibition efficiency depends on the type of sensitizing mutation.10 The results of in vitro studies on the L858R and G719S mutations show that mutant EGFR has an increased affinity for ATP, with a 50-fold and tenfold increase for the L858R and the G719S mutations, respectively. Since the intracellular concentration of ATP is very high, this increase does not lead to a physiological change. Comparing the affinity of both mutated and WT receptors for gefitinib, a 100-fold and sixfold higher affinity was observed for the L858R and G719S mutations, respectively, as compared to the WT receptor,4 thus explaining the lower efficacy of gefitinib in case of the G719S mutation.
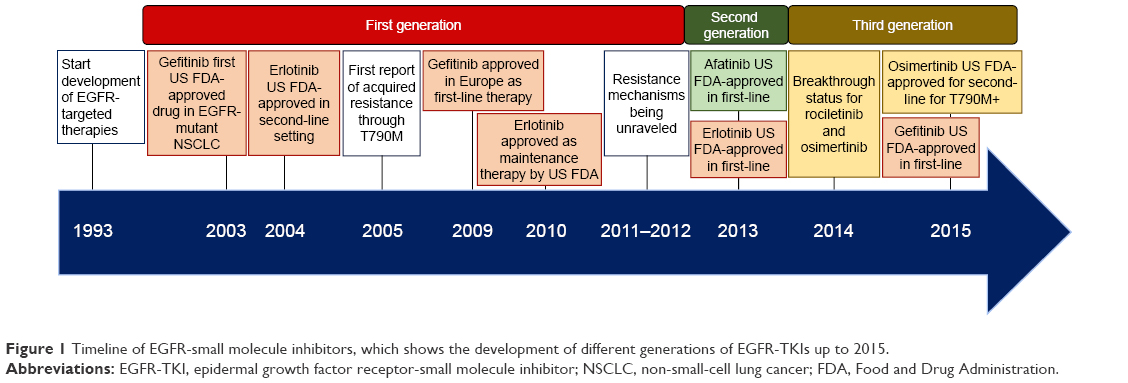 | Figure 1 Timeline of EGFR-small molecule inhibitors, which shows the development of different generations of EGFR-TKIs up to 2015. |
No crystal structures of EGFR in complex with erlotinib have been reported to date. Since brain metastases are common in NSCLC, it is important that these TKIs penetrate the central nervous system, which is observed for both erlotinib and gefitinib.11,12 Overall, both gefitinib and erlotinib show increased affinity for the mutated EGFR as compared to the WT receptor, but since both can bind to WT-EGFR, they result in on-target side effects such as skin rash and diarrhea.
Recent meta-analyses show differential results when comparing the two most common EGFR mutations: exon 19 deletion and L858R. Both erlotinib and gefitinib improved the overall survival of patients with exon 19 deletions, whereas in case of L858R substitution standard chemotherapy was superior over EGFR-TKIs.13 No studies on erlotinib or gefitinib in patients with uncommon EGFR mutants are available till date. The combination of erlotinib with cetuximab showed moderate efficacy against EGFR exon 20 insertions.14 Wheler et al14 described the case of a man with D770<GY insertion in exon 20, who showed an ongoing partial response after 24 months.14 However, the exact mechanism underlying this treatment is not known. One study15 investigated the response of patients with an exon 20 insertion to erlotinib monotherapy. Here, three of eleven patients presenting with an exon 20 insertion (insertion of FQEA or ASV and an unknown variant) showed a partial response, with a time to progression of 3 months, suggesting that standard chemotherapy is the preferred treatment option for these patients.15 However, it has also been shown that these patients might benefit from the Hsp90 inhibitor AUY922, with an ongoing Phase II trial (NCT01854034) working on this approach. Preliminary results on the first ten patients showed partial response in one patient and stable disease in three patients for more than 3 months, with a progression-free survival (PFS) of 6.1 months.16
In rare cases, secondary mutations have been reported that lead to a 3–10-fold decrease in the binding affinity for gefitinib or erlotinib, eg, T854A, L747S, and D761Y.17–20 However, they do not lead to complete resistance against these TKIs.
Resistance against first-generation EGFR-TKIs usually occurs within a year, with the T790M secondary mutation being observed in over 50% of cases. Since the T790M substitution is situated in the back of the ATP-binding cleft, it is often described as a “gatekeeper” mutation. However, crystal structures show that it does not seem to sterically hinder the binding of TKIs to mutant EGFR. This substitution leads to conformational changes that increase the affinity of the mutant receptor for ATP, thus “outcompeting” erlotinib and gefitinib. Yun et al21 showed that for the L858R/T790M double mutant, affinity to gefitinib decreased four times, while affinity to ATP increased 18 times. Furthermore, T790M also increases the catalytic activity of EGFR, thus conferring growth advantage to these cells. In addition to secondary mutations in EGFR, activation of parallel pathways (eg, cMET amplification) can also confer resistance against EGFR-TKIs, but these are beyond the scope of this review.22
Second-generation EGFR-TKIs: afatinib
Similar to erlotinib and gefitinib, afatinib has an aniline-quinazoline structure. It is able to inhibit EGFR and its family members Her2, Her3, and Her4. Unlike the first-generation TKIs, it is able to bind irreversibly to EGFR by forming a covalent bond with C797.23 In vitro studies showed that its efficacy against the L858R- and exon 19 deletion-mutated EGFR is similar as compared to the first generation, but it has a 100-fold higher efficacy against the T790M-mutated receptor.24 It is also able to penetrate the central nervous system reaching a concentration high enough to exert its inhibitory effect. This results in a similar response rate for patients with and without brain metastases.25 Clinical trials in patients with acquired resistance against first-generation EGFR-TKIs showed a similar benefit in patients with and without T790M mutation, suggesting a rather limited benefit of afatinib through the inhibition of T790M-mutant EGFR.26
Afatinib also does not discriminate between WT-EGFR and mutated EGFR. The combination of this non-selectivity for mutant EGFR and the extended binding of afatinib to EGFR can explain the higher on-target toxicities such as skin rash or gastrointestinal problems as compared to erlotinib and gefitinib. Regarding the different mutations in EGFR, in contrast to the LUX-Lung 7 trial which showed no significant differences in afatinib activity between L858R mutation and exon 19 deletion,27 meta-analyses on 3,000 patients reported a limited effect on the L858R mutation as compared to the exon 19 deletion.13 The effect of afatinib on the uncommon EGFR mutations has been analyzed, showing that it is effective against these mutations, with the exception of the exon 20 insertions.28
Third-generation EGFR-TKIs: osimertinib and rociletinib
Rociletinib (CO-1686; Clovis Oncology, Boulder, CO, USA) is a 2,4-disubstituted pyrimidine that covalently binds the C797 of EGFR with its meta-acrylamide group. In vitro tests comparing the effect of rociletinib on the L858R/T790M double mutant with that of WT-EGFR showed that it was 22 times more selective for mutant EGFR as compared to erlotinib.29 NSCLC cell lines with both L858R/T790M and exon 19 del/T790M double mutations were inhibited at concentrations of 7–32 nM, whereas cell lines harboring WT-EGFR (with or without mutated Ras) were inhibited at concentrations of 547–4,275 nM. These results were confirmed using xenograft and transgenic models. Compared to afatinib, rociletinib led to an increased reduction of tumor growth in xenograft models.29 During the clinical trials, the analysis of plasma samples revealed a link between the baseline level of the T790M mutation and the response to rociletinib, suggesting that this might be a useful biomarker for patient selection.30
During the Phase I/II trials, the predominant grade 3 adverse event was hyperglycemia, reported in 22% of the patients.31 The M502 metabolite of rociletinib is able to bind and inhibit the insulin growth factor receptor 1 (IGFR1).32,33 Similar to EGFR, IGFR1 also has a methionine gatekeeper residue in its kinase domain. Overall, dose reduction and metformin treatment were sufficient to overcome this side effect. Although metformin is commonly used as an antidiabetic drug, it has also been shown to reduce tumor growth through a number of different mechanisms.34 First, it reduces the circulating levels of insulin, which functions as a mitogen for tumor cells.35 Second, metformin inhibits gluconeogenesis in the liver, thus reducing circulating glucose levels.36 Third, it inhibits the mitochondrial complex I, thus influencing the electron transport chain and the energy metabolism of the cells.37 These effects of metformin might have influenced the results of the trial given that it was used to treat a large number of patients.
In contrast to rociletinib, osimertinib (AZD9291; AstraZeneca, Cambridge, UK) is especially designed not to target the IGFR1. During in vivo studies, the blood glucose and insulin levels of the rats were monitored to detect possible fluctuations due to treatment, showing no abnormalities.38 In addition to the compound itself, the active metabolites were also tested on a range of EGFR mutants and WT cell lines to assess their activity. The metabolite AZ7750 showed activity and potency similar to osimertinib, whereas the other metabolite AZ5104 showed greater potency but somewhat less selectivity for WT-EGFR.38 Overall, osimertinib showed a 200-fold greater inhibition of mutated EGFR with L858R substitution or exon 19 deletion, with or without the T790M mutation (IC50: 13–54 nM) as compared to the WT receptor (IC50: 480–1,865 nM). The efficacy of erlotinib, afatinib, and osimertinib against rare EGFR mutations was comparable for G719S, L861Q, and the exon 19 insertion mutations, whereas none were as effective against the exon 20 insertion. The activity of osimertinib against EGFRvIII (a splice variant found mainly in glioblastoma) was lower when compared to afatinib. Since this splice variant is structurally closer to WT-EGFR, this was not unexpected.38
Resistance to third-generation EGFR-TKIs
Clones of the H1975 cell line that have been made resistant by continuous exposure to increasing doses of rociletinib showed an EMT phenotype.29 This was confirmed with RNAseq showing an increase in EMT markers such as vimentin, AXL, ZEB1, CDH5, and FN1, whereas epithelial markers such as E-cadherin, MIR200B, CLDN4, EPCAM, and CLDN7 were downregulated. Also, these clones showed cross-resistance to other EGFR-TKIs such as erlotinib and afatinib. When studying the classical EGFR-TKI resistance mechanisms, no extra mutations in EGFR, cMET, BRAF, Her2, Her3, Ras, or PIK3CA were discovered. Also, there was no increase in phosphorylation of cMET, Her2, or Her3 in these clones. However, when combining rociletinib with Akt inhibitors, this resulted in a strong synergistic effect, suggesting a major role for Akt signaling in conferring resistance. The combination of rociletinib with Axl inhibitors resulted only in a partial restoration of sensitivity.29
In contrast, in a study on the rociletinib analog CNX-2006, the EMT phenotype did not play an important role, while NF-κB activation replaced the oncogenic EGFR signaling in NSCLC when effective and persistent inhibition of the target was achieved in the presence of the T790M mutation in cells with CNX-2006 acquired resistance. These data supported the concept of inhibition of members of the NF-κB pathway as a promising therapeutic option to reduce the viability of cells that adapted to third-generation EGFR-TKIs.39
Ercan et al,40 on the other hand, performed a mutagenesis screen in the Ba/F3 cell line with mutant EGFR (L858R, exon 19 deletion with or without T790M). Hereby, they discovered three tertiary mutations in EGFR conferring resistance to one or more third-generation inhibitors. Hereby, the C797S substitution led to resistance against rociletinib, osimertinib, and afatinib. This is not surprising, since this cysteine residue is crucial for the covalent binding of these inhibitors to EGFR. These cells harboring the triple mutation only remained partially sensitive to cetuximab. Cells harboring a primary mutation (L858R or exon 19 deletion) in combination with the C797S mutation remained sensitive to gefitinib, suggesting that this residue is not important for the association of the first-generation EGFR-TKIs. Furthermore, the L718Q and L844V mutations are not oncogenic upon themselves but lead to rociletinib resistance, whereas osimertinib is still able to partially inhibit EGFR. Both the C797S41 and L844V42 mutations have already been reported in patients, whereas for L718Q substitutions to Pro,43 Val,44 or Met45 have been reported but not to Gln.
During the Phase I/II trials of rociletinib, a total of 64 patients were treated, all with a T790M mutation.30 From 12/64 patients, rebiopsies were obtained after rociletinib treatment reported a loss of the T790M mutation in 50% (six) of cases, whereas the original EGFR mutation was retained. Of these six patients, one was intrinsically resistant to rociletinib and five had acquired resistance. A cell line was developed from one of these biopsies. Although these cells were T790 WT, they showed cross-resistance to all EGFR-TKIs by signaling of the Akt-mTOR pathway. Consistent with the previous in vitro results,29 Akt here functions as a bypass pathway. In two cases, a transformation to small-cell lung cancer was observed with loss of the T790M mutation, but retention of the original EGFR mutation, and downregulation of EGFR and loss of RB1.46 In the biopsies that retained the T790M mutation after rociletinib treatment, amplification of EGFR was observed in three cases.30 However, no tertiary mutations in EGFR were detected by next-generation sequencing of the rebiopsies.
Such resistance has not yet been reported for osimertinib. Given the fact that osimertinib also needs C797 to covalently bind to the receptor, it can be expected that C797S also confers resistance against osimertinib. The bypassing pathways also seem to be possible resistance mechanisms against this compound. Although little is known about the sequential use of third-generation EGFR-TKIs, recent data showed that two of four patients with progression after rociletinib experienced a prolonged stabilization of their disease (for more than 200 days) with osimertinib, suggesting that administration of a second EGFR inhibitor after failure of a first TKI of the same generation should be further pursued.47 More research is warranted to confirm these assumptions and to determine the possible unique mechanisms of resistance to osimertinib.
Clinical development of rociletinib
The rationale underlying the clinical development of rociletinib (Table 1) is that most of the EGFR-mutated NSCLC patients treated with a first- or second-generation EGFR-TKI develop acquired resistance due to the acquired T790M EGFR mutation. Therefore, rociletinib was first evaluated in a Phase I–II study32 of patients with EGFR-mutated NSCLC, who had disease progression following first-line treatment with a first- or second-generation EGFR inhibitor. All patients of the TIGER-X trial underwent a tumor biopsy for central assessment of EGFR mutation: in the Phase I part of the trial the enrollment was not restricted to T790M-positive patients, but in the Phase II part of the trial a confirmation of the mutational status was mandatory.
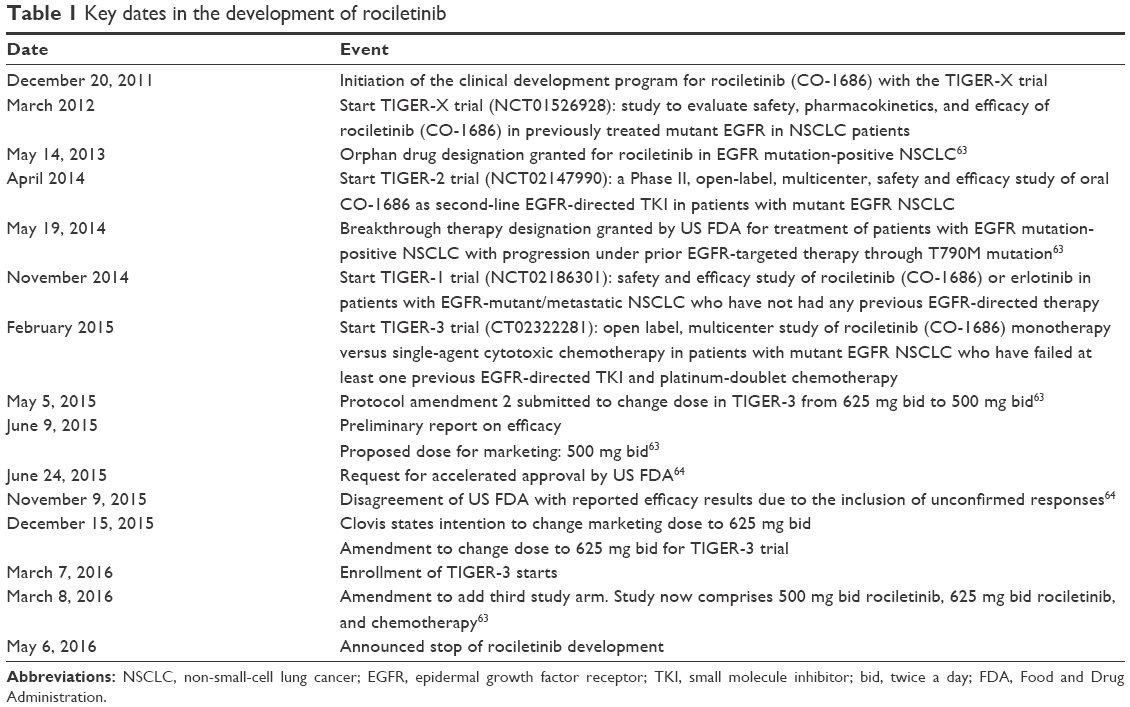 | Table 1 Key dates in the development of rociletinib |
Two forms of rociletinib were developed: a free-base form and a hydrogen bromide salt (Hbr), which were introduced in the second part of the trial. The starting dose of the free-base form was 150 mg once daily, and the highest dose for this form was 900 mg twice a day (bid); the doses of the Hbr form ranged from 500 to 1,000 mg bid. The study consisted of a Phase I dose-escalation part and a Phase II expansion part in order to assess the efficacy of 500, 625, and 750 mg twice-daily dosings.
A total of 130 patients were enrolled at the time of publication: the response rate in the 46 T790M-positive patients was 59%, with a disease control rate of 93% and an estimated PFS of 13.1 months. Among patients who were negative for T790M, the results were significantly different, but some activity was still detected: the response rate was 29%, the disease control rate was 59%, and the PFS was 5.6 months.
Additional data on patients with T790M mutation, detected by plasma genotyping, were presented at the ASCO 2015 Annual Meeting.63 A total of 456 patients received at least one dose of rociletinib Hbr; of these, 243 patients had centrally confirmed T790M-positive tumor tissue and 147 patients were positive for T790M mutation in plasma. The objective response rate for all T790M-positive patients on tissue analysis was 53% and the disease control rate was 85%, while the PFS in patients was 8 months at doses of 500 mg or 625 mg bid. The beaming test was used for the detection of EGFR mutation in plasma: this test showed 81% of positive agreement with tissue as control for T790M mutation and 87% for activating mutations, supporting the use of plasma genotyping as a complementary method to select patients. The overall response rate (ORR) and disease control rate were 53% and 82%, respectively, for plasma T790M-positive patients who received at least one dose of rociletinib. The ORR for T790M-negative patients was 37%.48
At the JP Morgan Healthcare Conference in January 2016, the updated “confirmed” response rate was lowered to 34% at 625 mg bid dose (n=170) and to 28% at 500 mg bid dose (n=79) in T790M-positive patients, with a median duration of response of ~9 months for both doses. These data showed a significant difference from previously reported data of a 59% response rate in centrally confirmed T790M-positive tumors. The data raised many concerns, since none of the three previous press releases disclosed which proportion of responses were confirmed responses according to the usual and customary definition of response.31
Updated results were presented at the ASCO Annual Meeting 2016 after 175 and 242 patients were enrolled in the 500 mg and 625 mg bid dosing groups, respectively. Of these patients, 156 (500 mg bid) and 175 (625 mg bid) had centrally confirmed T790M-positive tumors, in whom the ORR was 25% (21.8% according to the independent radiology review) and 39.4% (31.4% according to the independent radiology review), respectively.49
An additional Phase II trial (TIGER-2) was planned in order to evaluate the safety and efficacy of rociletinib as second-line EGFR-directed therapy in patients with mutant EGFR NSCLC,50 while the confirmatory Phase III open-label randomized study (TIGER-3) was planned to evaluate rociletinib versus investigator-choice single-agent chemotherapy (pemetrexed, docetaxel, or gemcitabine) in patients with EGFR-positive NSCLC progressing on prior EGFR tyrosine kinase inhibitor therapy and platinum-based chemotherapy.51 Of note, the TIGER-1 trial evaluated the safety and efficacy of rociletinib compared to erlotinib in patients with metastatic EGFR-positive NSCLC who have not received any previous EGFR inhibitor.52 To further explore the potential activity of rociletinib in T790M-negative disease, cohorts of patients with this feature have been included in TIGER-2 and TIGER-3 trials.
However, both the trials were stopped when the US Food and Drug Administration (FDA) notified Clovis Oncology that it would receive a complete response letter before the scheduled “Prescription Drug User Fee Act” on June 28, 2016. This letter underlined, indeed, that the FDA would not approve this new drug based on the available data. Therefore, Clovis Oncology terminated patients’ enrollment in all ongoing trials with rociletinib, and has also withdrawn its application for approval by the European Medicines Agency. However, rociletinib will be given to only those patients who are recommended by their clinicians to continue the therapy.
Safety and tolerability of rociletinib
The most common adverse events (>10% of patients) reported in the update of TIGER-X trial during the ASCO 2015 Annual Meeting included hyperglycemia, diarrhea, nausea, fatigue, QT prolongation, and decreased appetite; of note, all these adverse events were less frequent in the 500 mg bid schedule than with a higher dosage. No case of interstitial lung disease was observed in the 500 mg bid schedule, while grade 3 QT prolongation was observed in 2.5% of patients treated with the 500 mg bid schedule. However, grade 3–4 hyperglycemia was observed in 17% of patients treated with the 500 mg bid schedule and in 24%, 36%, and 33% of patients treated with the 625, 750, and 1,000 mg bid schedules, respectively.
Hyperglycemia was not expected in humans as it was not observed in preclinical models; it was found to be related to the rociletinib metabolite (M502), by inhibiting IGF1-R/IR.48 More detailed research revealed that the plasma levels of the metabolites M502 and M460 were much higher in humans than in the preclinical models (Sprague Dawley rats and beagle dogs). These metabolites have been shown to undergo acetylation by the N-acetyltransferase 2 (NAT2) enzyme. The differences in the catalytic activity of NAT2 between the preclinical models and humans caused the differences in plasma levels. The accumulation of these metabolites in humans is also responsible for hyperglycemia and QTc prolongation.33 It is also important to note that genetic variation in humans leads to different activity levels of NAT2, with 40%–60% of Caucasians and populations of African descent being slow acetylators.53 After the observation of this peculiar adverse event, a protocol for observation and monitoring of blood and urine glucose levels and proper treatment with oral antidiabetic agents was introduced, and the adverse event was consequently managed. Thus, the cases of grade 3 or 4 hyperglycemia decreased from 22% to 8%.48
Interestingly, the most common adverse events related to EGFR-TKIs of first and second generations, such as dermatologic toxicity and diarrhea, were limited during rociletinib treatment. This toxicity profile may have a different impact on the patients’ quality of life, since the presence of dermatologic toxicity or diarrhea, although mild, but continuous, could have a negative impact on daily activities and on patients’ adherence to treatment. This aspect was indeed reflected by the discontinuation rate due to adverse events, which was only 2.5%. However, the attempts to position the inhibition of IGFR1 as a positive differentiating attribute of rociletinib, given the preclinical additive/synergistic efficacy of the combination with EGFR-targeted compounds, are in contrast with clinical data from randomized trials showing lack of any incremental clinical benefit of IGFR1-targeted drugs in combination with EGFR inhibitors in NSCLC.
Comparison to osimertinib
While rociletinib was especially designed to inhibit IGFR1, osimertinib was deliberately designed not to bind this receptor. This resulted in only 2.4% of patients with hyperglycemia as adverse event as compared to 22% after rociletinib treatment. No drug reduction or discontinuation was needed in the osimertinib AURA trials. Patients also experienced much less QTc prolongation (4.3% as compared to 11% with rociletinib treatment).54
Osimertinib and rociletinib are metabolized differently. While rociletinib is broken down by NAT2, CYP3A is mainly responsible for osimertinib metabolization.55 No differences between ethnical groups were observed.56 The metabolization process results in two metabolites: AZ5104 and AZ7550. However, in contrast to the metabolites of rociletinib, with osimertinib all metabolites are capable of inhibiting mutant EGFR and have a similar half-life, with 61.2, 55.2, and 82 hours for osimertinib, AZ5104, and AZ7550, respectively.
Pharmacokinetic–pharmacodynamic modeling has shown that osimertinib has a much higher blood–brain barrier penetration as compared to rociletinib. The ratio of drug concentration in brain/plasma levels is 3.41 with osimertinib, whereas for rociletinib the ratio is 0.08, with brain concentration of rociletinib being lower than the quantification limit of the study.57 The blood–brain barrier penetration of osimertinib also leads to responses in patients with brain metastases.58
Overall, the AURA studies showed an ORR of 51%, with T790M-mutated patients showing ORR of 61% and PFS of 9.6 months as compared to ORR of 21% and PFS of 2.8 months in T790M-negative patients. Approximately, 7% of patients needed dose reduction and 6% of patients discontinued treatment due to adverse events.
Conclusion and (no) place in therapy
Clovis Oncology sought accelerated approval of rociletinib based on data of TIGER-X and a cohort of patients from the TIGER-2 study. However, the FDA’s decision was to wait for the Phase III trial results (TIGER-3), expected to complete patient enrollment at the end of 2018/beginning of 2019, as they said that the risk–benefit was not clear at that point and not clearly superior to available therapies. According to the FDA, the most common grade 3/4 adverse events in 400 patients were hyperglycemia and QTc prolongation. Dose reductions were necessary due to hyperglycemia (22%) and QTc prolongation (11%), but dose reductions were not clearly specified in the protocol and were inconsistently used. An additional issue was the Clovis Oncology’s proposed recommended dose: it was 500 mg twice daily initially, but in December 2015, Clovis Oncology notified the FDA that it planned to amend the proposed recommended dose to 625 mg twice daily, arguing a better response rate for this dose, although the pharmacokinetics showed a flat curve for this drug, with higher doses not leading to higher exposure but showing an increased risk for adverse events.
At the time of publication of initial results, TIGER-X had enrolled 130 patients, but the trial ultimately included 612 patients. The median follow-up reported in the initial report was 10.5 weeks, and as a consequence patients underwent computerized tomography scans every 6 weeks. Therefore, half of the patients had received only one response evaluation, and the reported response rate included many unconfirmed responses. An update on the response rate according to RECIST 1.1 criteria was recently published.59 The cases reported in the initial article on TIGER-X were reviewed. All patients except two had progressive disease or died; 23 of 51 T790M-positive patients (45%) had a confirmed response according to RECIST criteria; five patients who were reported to have a partial response did not experience a confirmed response. Of 18 patients with T790M-negative disease, three (17%) had a confirmed partial response.
The development of early brain metastases is a supposed mechanism contributing to the failure of confirming the initial responses. However, of 22 patients with brain metastases before the discontinuation of rociletinib, only one patient had an early appearance of brain metastases. The median PFS in this updated report was 6.1 months for T790M-positive patients and 1.8 months for negative ones. In conclusion, in this most recent report, the updated response rate had dropped from initial 59% to 45%, which was much lower than the previous press release. In contrast, results on osimertinib showed a pooled ORR of 59% (n=411) and a mean duration of response of 12.4 months in 63 T790M-positive patients (AURA Phase I/II trial [NCT01802632]). Dose reductions were necessary in 4.4% of patients and discontinuation due to adverse events was 5.6%. There were no grade 3–4 adverse events in ≥2% of patients. Currently, a randomized, double-blind Phase III trial is organized (FLAURA NCT02296125), comparing the efficacy of osimertinib with that of erlotinib or gefitinib (standard of care).60–62
However, in May 2016, after receiving a negative reply from the FDA’s Oncologic Drugs Advisory Committee, which voted 12–1 against the accelerated approval of rociletinib, Clovis Oncology decided to end the development of rociletinib. The main concerns of the FDA can be summarized in three points: 1) Is the observed ORR and duration of response for patients treated with rociletinib better than available therapies and is it likely to predict clinical benefit? 2) Are the risks of rociletinib (especially with regard to QTc prolongation) acceptable? 3) Is the benefit–risk profile favorable for the proposed patient population?
This negative conclusion of the “rociletinib story” was the consequence of a series of negative developments for the drug, including both the updated data revealing lower response rates and the parallel approval of osimertinib, which was based on both better clinical effects and a favorable risk–benefit profile. As suggested by a recent letter of Dr Dhingra,31 as well as by a Chinese proverb on the futility of hurrying – “A hasty man drinks his tea with a fork”, the failure of rociletinib should provide a lesson for biotechnology companies (and their investors), with respect to the fact that proceeding too quickly can spoil the entire job, despite the still many unmet clinical needs of NSCLC patients. A lesson must also be learned by the regulatory agencies. The approval process for drugs was dramatically changed with the introduction of the new concept of “breakthrough therapy”. This certainly is a great advantage for the patients, allowing quicker access to new drugs. The FDA granted this breakthrough status to rociletinib after achieving important responses in the first clinical trial. However, after extensive analysis, the approval process was stopped. Several questions arose after this process failed. How strong is the review process for a breakthrough designation? Is this failure entirely the fault of the industry or must the blame be shared? Here, it is important to remember that the decision-making process also depends on the transparency of the clinical trials data. With this sad history, we have learned that bad things can happen to both good and bad drugs.
In conclusion, the future development of novel T790 targeted agents should include careful plans for obtaining reliable data from the small datasets of early clinical trials, and evaluate their efficacy in comparison to osimertinib and/or in osimertinib-treated patients, in order to gain marketing approval.
Disclosure
The authors report no conflicts of interest in this work.
References
Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7(3):169–181. | ||
Eck MJ, Yun CH. Structural and mechanistic underpinnings of the differential drug sensitivity of EGFR mutations in non-small cell lung cancer. Biochim Biophys Acta. 2010;1804(3):559–566. | ||
Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277(48):46265–46272. | ||
Yun CH, Boggon TJ, Li Y, et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell. 2007;11(3):217–227. | ||
Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125(6):1137–1149. | ||
Ning J, Wu Q, Liu Z, Wang J, Lin X. Mapping inhibitor response to the in-frame deletions, insertions and duplications of epidermal growth factor receptor (EGFR) in non-small cell lung cancer. J Recept Signal Transduct Res. 2016;36(1):37–44. | ||
Wu JY, Wu SG, Yang CH, et al. Lung cancer with epidermal growth factor receptor exon 20 mutations is associated with poor gefitinib treatment response. Clin Cancer Res. 2008;14(15):4877–4882. | ||
Cho J, Chen L, Sangji N, et al. Cetuximab response of lung cancer-derived EGF receptor mutants is associated with asymmetric dimerization. Cancer Res. 2013;73(22):6770–6779. | ||
Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101(36):13306–13311. | ||
Jiang J, Greulich H, Jänne PA, Sellers WR, Meyerson M, Griffin JD. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 2005;65(19):8968–8974. | ||
Park SJ, Kim HT, Lee DH, et al. Efficacy of epidermal growth factor receptor tyrosine kinase inhibitors for brain metastasis in non-small cell lung cancer patients harboring either exon 19 or 21 mutation. Lung Cancer. 2012;77(3):556–560. | ||
Porta R, Sánchez-Torres JM, Paz-Ares L, et al. Brain metastases from lung cancer responding to erlotinib: The importance of EGFR mutation. Eur Respir J. 2011;37(3):624–631. | ||
Zhang Y, Sheng J, Yang Y, et al. Optimized selection of three major EGFR-TKIs in advanced EGFR-positive non-small cell lung cancer: a network meta-analysis. Oncotarget. 2016;7(15):20093–20108. | ||
Wheler JJ, Tsimberidou AM, Falchook GS, et al. Combining erlotinib and cetuximab is associated with activity in patients with non-small cell lung cancer (including squamous cell carcinomas) and wild-type EGFR or resistant mutations. Mol Cancer Ther. 2013;12(10):2167–2175. | ||
Naidoo J, Sima CS, Rodriguez K, et al. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: clinical outcomes and response to erlotinib. Cancer. 2015;121(18):3212–3220. | ||
Piotrowska Z, Botelho Costa D, Huberman M, et al. Activity of AUY922 in NSCLC patients with EGFR exon 20 insertions. J Clin Oncol. 2015;33(Suppl):abstract 8015. | ||
Bean J, Riely GJ, Balak M, et al. Acquired resistance to epidermal growth factor receptor kinase inhibitors associated with a novel T854A mutation in a patient with EGFR-mutant lung adenocarcinoma. Clin Cancer Res. 2008;14(22):7519–7526. | ||
Costa DB, Schumer ST, Tenen DG, Kobayashi S. Differential responses to erlotinib in epidermal growth factor receptor (EGFR)-mutated lung cancers with acquired resistance to gefitinib carrying the L747S or T790M secondary mutations. J Clin Oncol. 2008;26(7):1182–1184. | ||
Balak MN, Gong Y, Riely GJ, et al. Novel D761Yand common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12(21):6494–6502. | ||
Klughammer B, Brugger W, Cappuzzo F, et al. Examining treatment outcomes with erlotinib in patients with advanced non-small-cell lung cancer whose tumors harbor uncommon epidermal growth factor receptors mutations. J Thorac Oncol. 2016;11(4):545–555. | ||
Yun CH, Mengwasser KE, Toms AV, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105(6):2070–2075. | ||
Engelman JA, Zejnullahu K, Mitsudomi T, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316(5827):1039–1043. | ||
Solca F, Dahl G, Zoephel A, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343(2):342–350. | ||
Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27(34):4702–4711. | ||
Hoffknecht P, Tufman A, Wehler T, et al. Efficacy of the irreversible ErbB family blocker afatinib in epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI)-pretreated non-small-cell lung cancer patients with brain metastases or leptomeningeal disease. J Thorac Oncol. 2015;10(1):156–163. | ||
Landi L, Tiseo M, Chiari R, et al. Activity of the EGFR-HER2 dual inhibitor afatinib in EGFR-mutant lung cancer patients with acquired resistance to reversible EGFR tyrosine kinase inhibitors. Clin Lung Cancer. 2014;15(6):411–417. | ||
Park K, Tan EH, O’Byrne K, et al. Afatinib versus gefitinib as first-line treatment of patients with EGFR mutation-positive non-small-cell lung cancer (LUX-Lung 7): A phase 2B, open-label, randomised controlled trial. Lancet Oncol. 2016;17(5):577–589. | ||
Yang JC, Sequist LV, Geater SL, et al. Clinical activity of afatinib in patients with advanced non-small-cell lung cancer harbouring uncommon EGFR mutations: a combined post-hoc analysis of LUX-Lung 2, LUX-Lung 3, and LUX-Lung 6. Lancet Oncol. 2015;16(7):830–838. | ||
Walter AO, Sjin RT, Haringsma HJ, et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M mediated resistance in NSCLC. Cancer Discov. 2013;3(12):1404–1415. | ||
Piotrowska Z, Niederst MJ, Karlovich CA, et al. Heterogeneity underlies the emergence of EGFRT790 wild-type clones following treatment of T790M-positive cancers with a third-generation EGFR inhibitor. Cancer Discov. 2015;5(7):713–723. | ||
Dhingra K. Rociletinib: has the TIGER lost a few of its stripes? Ann Oncol. 2016;27(6):1161–1164. Epub 2016 Apr 4. | ||
Sequist LV, Soria JC, Goldman JW, et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N Engl J Med. 2015;372(18):1700–1709. | ||
Simmons AD, Jaw-Tsai S, Haringsma HJ, Allen A, Harding TC. Insulin-like growth factor 1 (IGF1R)/insulin receptor (INSR) inhibitory activity of rociletinib (CO-1686) and its metabolites in nonclinical models. In: Proceedings of the 106th Annual Meeting of the American Association for Cancer Research; 2015 Apr 18–22; Philadelphia, PA. Cancer Res. 2015;75(Suppl 15):abstract 793. | ||
Wheaton WW, Weinberg SE, Hamanaka RB, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014;3:e02242. | ||
Gallagher EJ, LeRoith D. The proliferation role of insulin and insulin-like growth factors in cancer. Trends Endocrinol Metab. 2010;21(10):610–618. | ||
Viollet B, Guigas B, Sanz Garcia N, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci (Lond). 2012;122(6):253–270. | ||
Ota S, Horigome K, Ishii T, et al. Metformin suppresses glucose-6-phosphatase expression by a complex I inhibition and AMPK activation-independent mechanism. Biochem Biophys Res Commun. 2009;388(2):311–316. | ||
Cross DA, Ashton SE, Ghiorghiu S, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4(9):1046–1061. | ||
Galvani E, Sun J, Leon LG, et al. NF-κB drives acquired resistance to a novel mutant-selective EGFR inhibitor. Oncotarget. 2015;6(40):42717–42732. | ||
Ercan D, Choi HG, Yun CH, et al. EGFR mutations and resistance to irreversible pyrimidine-based EGFR inhibitors. Clin Cancer Res. 2015;21(17):3913–3923. | ||
Yu HA, Tian SK, Drilon AE, et al. Acquired resistance of EGFR-mutant lung cancer to aT790M-specific EGFR inhibitor: emergence of a third mutation (C797S) in the EGFR tyrosine kinase domain. JAMA Oncol. 2015;1(7):982–984. | ||
Penzel R, Sers C, Chen Y, et al. EGFR mutation detection in NSCLC-assessment of diagnostic application and recommendations of the German Panel for Mutation Testing in NSCLC. Virchows Arch. 2011;458(1):95–98. | ||
Taron M, Ichinose Y, Rosell R, et al. Activating mutations in the tyrosine kinase domain of the epidermal growth factor receptor are associated with improved survival in gefitinib-treated chemorefractory lung adenocarcinomas. Clin Cancer Res. 2005;11(16):5878–5885. | ||
Locatelli-Sanchez M, Couraud S, Arpin D, Riou R, Bringuier PP, Souquet PJ. Routine EGFR molecular analysis in non-small-cell lung cancer patients is feasible: Exons 18–21 sequencing results of 753 patients and subsequent clinical outcomes. Lung. 2013;191(5):491–499. | ||
Liu Y, Wu BQ, Zhong HH, Hui P, Fang WG. Screening for EGFR and KRAS mutations in non-small cell lung carcinomas using DNA extraction by hydrothermal pressure coupled with PCR-based direct sequencing. Int J Clin Exp Pathol. 2013;6(9):1880–1889. | ||
Niederst MJ, Sequist LV, Poirier JT, et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat Commun. 2015;6:6377. | ||
Sequist LV, Piotrowska Z, Niederst MJ, et al. Osimertinib responses after disease progression in patients who had been receiving rociletinib. JAMA Oncol. 2016;2(4):541–543. | ||
Sequist LV, Goldman JW, Wakelee HA, et al. Efficacy of rociletinib (CO-1686) in plasma-genotyped T790M-positive non-small cell lung cancer (NSCLC) patients (pts). J Clin Oncol. 2015;33(Suppl 15):abstract 8001. | ||
Goldman JW, Soria J-C, Wakelee HA, et al. Updated results from TIGER-X, a phase I/II open label study of rociletinib in patients (pts) with advanced, recurrent T790M-positive non-small cell lung cancer (NSCLC). J Clin Oncol. 2016;34(Suppl 15):abstract 9045. | ||
Clovis Oncology, Inc. TIGER-2: A Phase 2, Open-label, Multicenter, Safety and Efficacy Study of Oral CO-1686 as 2nd Line EGFR-directed TKI in Patients With Mutant EGFR Non-small Cell Lung Cancer (NSCLC). NCT02147990. Boulder, CO, USA: Clovis Oncology, Inc. | ||
Clovis Oncology, Inc. TIGER-3: Open Label, Multicenter Study of Rociletinib (CO-1686) Mono Therapy Versus Single-agent Cytotoxic Chemotherapy in Patients With Mutant EGFR NSCLC Who Have Failed at Least One Previous EGFR-Directed TKI and Platinum-doublet Chemothera. NCT02322281. Boulder, CO, USA: Clovis Oncology, Inc. | ||
Clovis Oncology, Inc. TIGER-1: Safety and Efficacy Study of Rociletinib (CO-1686) or Erlotinib in Patients With EGFR-mutant/Metastatic NSCLC Who Have Not Had Any Previous EGFR Directed Therapy. NCT02186301. Boulder, CO, USA: Clovis Oncology, Inc. | ||
Walker K, Ginsberg G, Hattis D, Johns DO, Guyton KZ, Sonawane B. Genetic polymorphism in N-acetyltransferase (NAT): population distribution of NAT1 and NAT2 activity. J Toxicol Environ Health B Crit Rev. 2009;12(5–6):440–472. | ||
Jänne PA, Yang JC, Kim DW, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372(18):1689–1699. | ||
Dickinson PA, Cantarini MV, Collier J, et al. Metabolic disposition of osimertinib in rat, dog, and man: insights into a drug designed to bind covalently to a cysteine residue of EGFR. Drug Metab Dispos. 2016;44(8):1201–1212. | ||
Planchard D, Brown KH, Kim DW, et al. Osimertinib Western and Asian clinical pharmacokinetics in patients and healthy volunteers: implications for formulation, dose, and dosing frequency in pivotal clinical studies. Cancer Chemother Pharmacol. 2016;77(4):767–776. | ||
Ballard P, Yates JW, Yang Z, et al. Preclinical comparison of osimertinib with other EGFR-TKIs in EGFR-mutant NSCLC brain metastases models, and early evidence of clinical brain metastases activity. Clin Cancer Res. Epub 2016 Jul 19. | ||
Ahn MJ, Tsai CM, Yang JC, et al. AZD9291 activity in patients with EGFR-mutant advanced non-small cell lung cancer (NSCLC) and brain metastases: data from Phase II studies. Eur J Cancer. 2015;51(Suppl 3):S625–S626. | ||
Sequist LV, Soria JC, Camidge DR. Update to rociletinib data with the RECIST confirmed response rate. N Engl J Med. 2016;374(23):2296–2297. | ||
Ramalingam SS, Yang JC-H, Lee CK, et al. AZD9291, a mutant-selective EGFR inhibitor, as first-line treatment for EGFR mutation-positive advanced non-small cell lung cancer (NSCLC): Results from a phase 1 expansion cohort. J Clin Oncol. 2015;33(Suppl):abstract 8000. | ||
Janne PA, Ramalingam SS, Yang JC-H, et al. Clinical activity of the mutant-selective EGFR inhibitor AZD9291 in patients (pts) with EGFR inhibitor-resistant non-small cell lung cancer (NSCLC). J Clin Oncol. 2014;32(Suppl):abstract 8009. | ||
Ramalingam SS, Rukazenkov Y, Thomas K, Soria J-C. A randomized, phase III study (FLAURA) of AZD9291, a novel EGFR-TKI, versus gefitinib or erlotinib in treatment-naïve patients with advanced non-small cell lung cancer and an EGFR-TKI-sensitizing mutation. J Clin Oncol. 2015;33(Suppl):abstract TPS8102. | ||
Sequist LV, Goldman JW, Wakelee HA, et al. Efficacy of rociletinib (CO-1686) in plasma-genotyped T790M-positive non-small cell lung cancer (NSCLC) patients (pts). Journal of Clinical Oncology – ASCO Annual meeting. 2015;33(Suppl 15):8001. | ||
FDA. FDA Briefing Document Oncologic Drugs Advisory Committee Meeting NDA 208542 Rociletinib.; 2016. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.