Back to Archived Journals » Gastrointestinal Cancer: Targets and Therapy » Volume 4
New developments in management of gastrointestinal stromal tumors: regorafenib, the new player in the team
Authors Boichuk S, Rausch J, Duensing A
Received 17 October 2013
Accepted for publication 31 October 2013
Published 12 December 2013 Volume 2014:4 Pages 1—10
DOI https://doi.org/10.2147/GICTT.S20679
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Sergei Boichuk,1,2 Jessica L Rausch,1 Anette Duensing1,3
1Cancer Virology Program, University of Pittsburgh Cancer Institute, Hillman Cancer Center, Pittsburgh, PA, USA; 2Department of Pathology, Kazan State Medical University, Kazan, Russia; 3Department of Pathology, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
Abstract: Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract and the most frequent single type of sarcoma, at least in some geographical regions. They arise from the interstitial cells of Cajal (or a common progenitor cell). The vast majority of GISTs are characterized by oncogenically activating mutations in the KIT or platelet-derived growth factor receptor alpha (PDGFRA) receptor tyrosine kinase genes. This molecular feature has been successfully exploited for therapeutic purposes, and as of a decade ago, GISTs have become the prototype of a solid tumor that can be targeted with small molecule kinase inhibitors. Imatinib mesylate (Gleevec®/Glivec®) benefits more than 85% of patients with unresectable and/or metastatic GIST. Unfortunately, the majority of patients develop resistance to imatinib within the first 2 years of treatment and new therapeutic options are needed. Although the broad-range kinase inhibitor sunitinib malate (Sutent®) has been the second-line therapy approved by the US Food and Drug Administration since 2006, it was not until recently (February 2013) that regorafenib (Stivarga®) was approved as a third-line therapeutic agent for GIST. This review summarizes the development process of regorafenib for GIST and highlights its biochemical, pharmacologic, and clinical properties.
Keywords: gastrointestinal stromal tumors, GIST, regorafenib
Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the gastrointestinal tract, and new epidemiologic data suggest that GISTs may be the most common single type of sarcoma, at least in some geographical regions.1,2 GISTs are believed to arise from the interstitial cells of Cajal or a common progenitor cell.3 Interstitial cells of Cajal are part of the myenteric plexus in the muscular wall of the gastrointestinal tract and comprise the pacemaker cells of the gut that coordinate peristalsis. Based on this biological feature, GISTs can generally occur anywhere in the gastrointestinal tract, from the esophagus to the rectum. Nevertheless, they are most commonly found in the stomach (50%–60%). Less frequent locations include the small intestines (30%–35%), colon and rectum (5%), as well as the esophagus (<1%).4 In addition, GISTs are rarely diagnosed in extra-gastrointestinal locations, such as the omentum, mesentery, or retroperitoneum.
The incidence of GISTs is approximately 5,000 newly diagnosed cases per year. At the time of diagnosis, most patients are older than 50 years (median 63 years), although GISTs can occur at any age. There is a slight predilection for male individuals.4 The median size at time of detection is 5–6 cm, but the tumors can be much larger. Ten percent to 20% of patients present with metastases at the time of diagnosis, and the main metastatic sites for GISTs are the liver and the peritoneal cavity.5 Lymph nodes and other distant sites are rarely involved.
Symptoms at diagnosis are often nondescript and can range from irregular bowel movements (constipation, diarrhea), nausea, and pain, to bleeding, resulting in anemia or overt hemorrhage. Bowel perforation or obstruction by the tumor mass may lead to emergency surgery. On the other side, it is not uncommon to detect a GIST as an incidental finding during an examination or surgical procedure for other reasons. In fact, recent findings suggest that up to 30% of the general population carry small GISTs (up to 1 cm in diameter), ie, so-called micro-GISTs or GISTlets.6,7 The malignant potential of GISTs ranges from essentially benign-acting to highly aggressive. Several risk stratification schemes used to assess the risk for recurrence following surgery are available, and rely mainly on the size, mitotic rate, and location of the tumor.8–11 Environmental risk factors for developing a GIST are not known,12–14 but familial GISTs and GIST syndromes exist.
GISTs have distinct molecular features that are crucial for their correct diagnosis and that were imperative in identifying current treatment strategies. The diagnosis of a GIST is made based on histopathologic appearance and immunohistochemical analysis. The majority of GISTs (95%) stain positive for the transmembrane receptor tyrosine kinase (RTK) KIT (CD117).15,16 Many tumors are also positive for DOG1 (ANO1) and PKC-theta (PKCQ), which is important for the small percentage of GISTs negative for KIT immunohistochemistry.17
On a molecular level, the majority of GISTs (70%–85%) carry mutations in the KIT RTK gene.12,18 These genomic changes lead to constitutive, ligand-independent activation of the transmembrane receptor and hence constitutive activation of downstream signaling cascades that promote survival and proliferation.19 Mutations preferentially occur in KIT exon 11 (65%), thus relieving the physiologic autoinhibitory function of the juxtamembrane domain of the receptor. Other regions affected by activating mutations are parts of the extracellular domain encoded by exon 9 (6%–10%) as well as the ATP-binding pocket and activation loop of the kinase domain (exons 13 and 17, respectively; together approximately 2%). Twenty percent to 25% of GISTs do not carry KIT mutations, but in approximately one third of them (5%–10%), mutations in the gene coding for platelet-derived growth factor receptor alpha (PDGFRA) can be found.20 It is of note that PDGFRA mutations in GIST occur in domains that are homologous to those detected in the KIT gene. GISTs that harbor no mutation in KIT or PDGFRA (referred to as wild-type) can, however, carry mutations in other genes, such as succinate dehydrogenase subunits (SDHA, SDHB, SDHC or SDHD) or BRAF.21,22
Clinical management of GIST
Most GISTs present as localized tumors at diagnosis.23,24 Hence, the primary therapeutic goal is their complete surgical removal (R0) with clear microscopic margins and without rupturing the tumor pseudocapsule.25–27 In the case of a primarily unresectable and/or metastatic GIST, the standard therapy is imatinib mesylate (Gleevec®/Glivec®, Novartis Pharma, Basel, Switzerland).25,26 Imatinib is an orally available small molecule tyrosine kinase inhibitor that served as proof of principle of targeted therapy with chemical inhibitors in chronic myeloid leukemia as well as solid tumors (eg, GISTs). In GISTs, imatinib targets the oncogenically altered KIT/PDGFRA kinase and effectively inhibits their constitutive activation (Table 1).
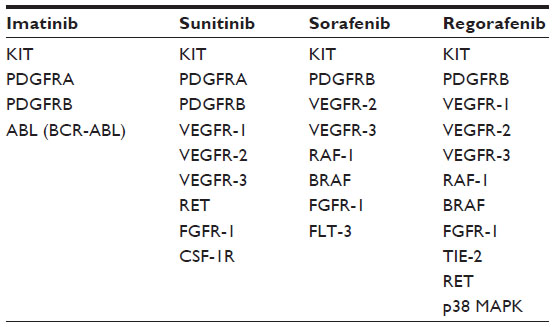 | Table 1 Comparison of the kinase inhibitory profiles of imatinib, sunitinib, sorafenib, and regorafenib |
The majority of patients with inoperable/metastatic GIST (more than 85%) respond to therapy with imatinib; however, complete remissions are rare and most patients only experience partial response or disease stabilization.28,29 Nevertheless, these response rates are quite dramatic, especially when considering the fact that, historically, chemotherapy and radiotherapy have not proven to be effective in GISTs. Notably, some ongoing responses are now exceeding a decade.30 A minor proportion of patients experience primary resistance to imatinib (defined as no response to the treatment at all or progression within the first 6 months of treatment) and approximately 50% of patients relapse within the first 2 years of treatment (secondary resistance).29 Several mechanisms of resistance development have been identified, but most cases involve secondary mutations in the primarily mutated kinase (KIT or PDGFRA) affecting either the ATP-binding or activation loops so that the binding of imatinib to the protein is impaired. Hence, identification of additional therapeutic strategies to overcome imatinib resistance in GIST has been the focus of intense research in university-based laboratories and in the pharmaceutical industry.
Overcoming imatinib resistance: existing and emerging therapeutic strategies
More than ten years after the approval of imatinib for GIST, there is still a very limited number of standard treatment options approved by the US Food and Drug Administration (FDA) available to overcome imatinib resistance.
GISTs that progress under the standard treatment of 400 mg imatinib per day can sometimes be controlled by dose escalation to 600 mg or 800 mg.31 Up to one third of patients achieve disease stabilization using this strategy, which is especially beneficial for patients with KIT exon 9 mutations. For patients with localized progression of only a single or very few lesions, surgical excision or radiofrequency ablation can be considered and has been shown to be beneficial.32 For generalized progression, however, sunitinib malate (Sutent®, Pfizer, New York City, NY, USA) is the standard second-line therapy after imatinib failure.
Sunitinib is an oral, broad-range kinase inhibitor that targets vascular endothelial growth factor receptors (VEGFR) 1–3, RET (rearranged during transformation), colony-stimulating factor-1 receptor (CSF-1R), and FMS-related tyrosine kinase 3 (FLT3) in addition to KIT and PDGFRA/B (Table 1).33 It was approved by the FDA in 2006 after a placebo-controlled study in imatinib-refractory patients showed a significant increase in time to disease progression when compared with the placebo arm. It is of note that sunitinib is especially beneficial for GIST patients who carry a KIT exon 9 mutation or are KIT/PDGFRA wild-type.34
Development of additional therapeutic agents, especially in the pharmaceutical industry, has mainly focused on further KIT/PDGFRA kinase inhibitors. The main approaches are either novel broad-spectrum inhibitors similar to sunitinib or tyrosine kinase inhibitors with a narrower kinase inhibitory spectrum, but with the goal of better and/or more specific binding to the mutated KIT/PDGFRA kinase (such as nilotinib).35 Both approaches have value and a number of advantages and disadvantages, although tyrosine kinase inhibitors with a broader range of inhibited kinases seem to result in better responses in GIST. Another focus has been the development of non-RTK inhibitors with the aim of targeting downstream signaling kinases, such as members of the PI3K/AKT/mTOR and RAS/RAF/mitogen-activated protein kinase (MAPK) pathways. While some of these downstream inhibitors are being used as single agents, there is the possibility to use them in conjunction with each other or in combination with RTK inhibitors.
In addition, other avenues for therapeutic strategies are being explored. Many of these focus on not directly targeting the mutated KIT/PDGFRA kinases, with an aim to bypass secondary resistance mutations. These approaches include, but are not limited to, heat-shock protein (HSP) 90 and HSP chaperone pathway inhibitors (such as CDC37),36,37 histone deacetylase inhibitors,38 proteasome inhibitors,39 DNA damaging agents,40 agents that target tumor cell quiescence,41 and humanized antibodies targeting KIT.42
A list of agents that are currently under evaluation in clinical trials can be found in Table 2. As a result of these concerted efforts, regorafenib (BAY 73-4506; Stivarga®, Bayer Schering Pharma AG, Berlin, Germany) has recently been approved by the FDA as a third-line therapy for GIST after failure of imatinib and sunitinib (see below).
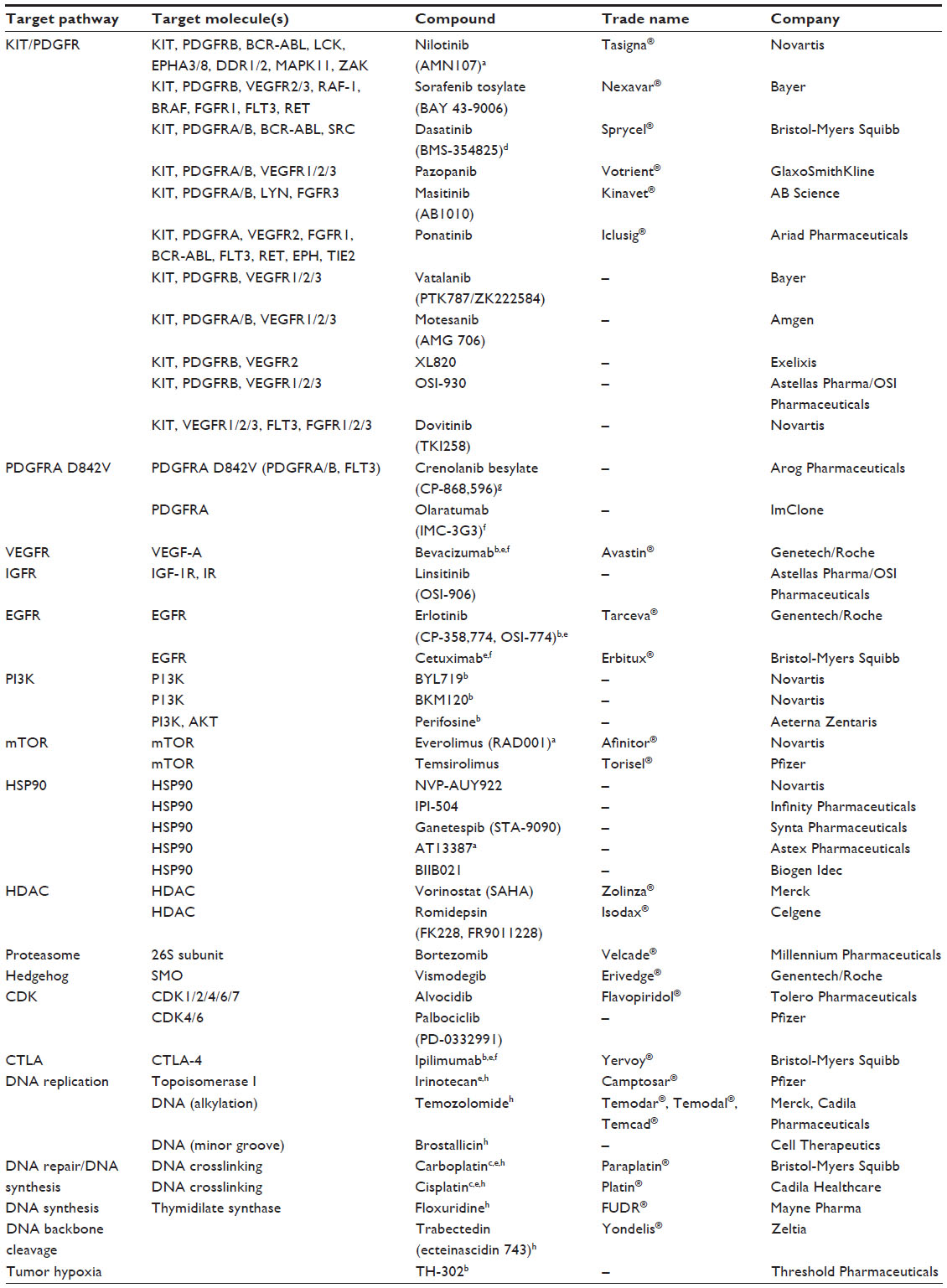 | Table 2 Emerging therapies for GIST |
Pharmacology, mode of action, and pharmacokinetics of regorafenib
Pharmacology
Regorafenib is a novel oral multikinase inhibitor that belongs to the group of biaryl urea chemicals. It was identified in a program aiming to optimize the potency and drug-like properties of known urea class compounds, such as sorafenib (Nexavar®, Bayer Healthcare Pharmaceuticals, Montville, NJ and Onyx Pharmaceuticals, South San Francisco, CA, USA).43 Regorafenib is structurally very similar to sorafenib, differing only by a fluorine atom in the center phenyl ring (Figure 1).44,45 Nevertheless, this minimal structural difference results in a generally higher inhibitory potency and a broader spectrum of kinases inhibited (see below and Table 1).46,47
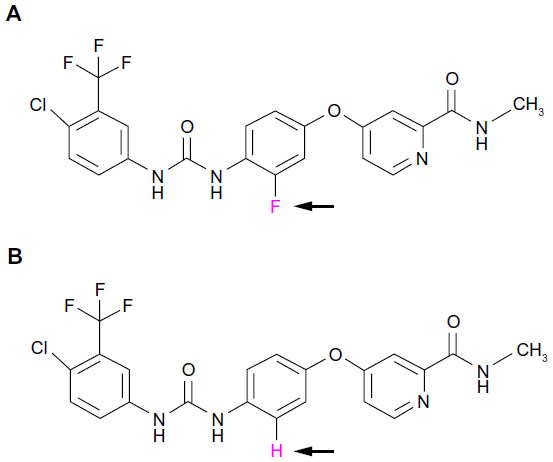 | Figure 1 Chemical structure of regorafenib and sorafenib. (A) Regorafenib (Stivarga®). (B) Sorafenib (Nexavar®). Note the difference of only one fluorine/carbon atom in the central phenyl ring (pink and arrows). Regorafenib is sometimes referred to as “fluoro-sorafenib”. |
Just like sorafenib, which was originally developed as a RAF inhibitor,46 regorafenib inhibits several forms of RAF (RAF-1 and BRAF) as well as mutant BRAF V600E in the low nanomolar range. It may be speculated, however, that the main antitumor activity of both drugs, which are effective in several tumor entities with varying molecular pathogenesis, does not primarily stem from the inhibition of RAF family members, but rather from the entire inhibitory spectrum of regorafenib and sorafenib targeting RTKs involved in the regulation of tumor angiogenesis and the tumor microenvironment, such as VEGFR 1–3 and platelet-derived growth factor receptor beta (PDGFRB), respectively. Compared with sorafenib, the IC50 of regorafenib towards most of these kinases is 2–20-fold lower (see below). Other kinases that are inhibited by regorafenib include TIE-2 (tyrosine kinase with immunoglobulin and EGF factor homology domains 2; also called TEK, endothelial tyrosine kinase), fibroblast growth factor receptor 1 (FGFR1), and KIT (the latter two are also inhibited by sorafenib) as well as the non-receptor p38 MAPK. Although preclinical studies of sorafenib in GIST were quite promising48 and Phase II clinical trials in GIST patients resistant to imatinib and sunitinib reported clinical benefit from treatment with sorafenib in up to 75% of cases,49–51 it was regorafenib that was approved by the FDA for the treatment of imatinib-resistant and sunitinib-resistant GIST in February 2013. One reason for this may be the lack of data from randomized Phase III clinical trials testing sorafenib. In addition, it is likely that the particular effectiveness of regorafenib in GISTs can be attributed to the fact that it inhibits wild-type and mutated KIT (KIT K642E) almost ten times more potently than does sorafenib.52
Mode of action
Based on structural studies, the mechanism of action of regorafenib is similar to that of sorafenib. Like sorafenib, regorafenib is considered to be a type II kinase inhibitor that binds the inactive form of the target kinase.52 In contrast to type I kinase inhibitors, type II inhibitors exploit the position of the DFG residues (aspartate [D]-phenylalanine [F]-glycine [G]) of the activation loop being folded away from the ATP-binding pocket (“DFG out”) in addition to using the ATP-binding site of the target kinase. This allows the inhibitor to make additional hydrophobic and hydrogen bonding interactions and typically results in potent cellular activity. In addition, type II inhibitors only indirectly compete with ATP. Interestingly, imatinib is another known type II kinase inhibitor. In contrast, type I kinase inhibitors (such as sunitinib and dasatinib) target the ATP-binding site of the enzyme in the open (ie, active) conformation.
Preclinical studies: kinase inhibition
In preclinical biochemical assays, regorafenib effectively inhibited a number of angiogenic and tumorigenic kinases, as mentioned above. In these assays, using recombinant human or murine kinases, RET (IC50 1.5 nM), RAF-1 (IC50 2.5 nM), VEGFR2 (IC50 4.2 nM), and KIT (IC50 7 nM) were the kinases most potently inhibited by regorafenib.52 In contrast, when measuring kinase activity in a cellular system in vitro, VEGFR2 expressed in NIH-3T3 cells was the most effectively inhibited kinase tested (IC50 3 nM). Kinase inhibition in primary tumor cell lines showed potent inhibition of mutant KIT K642E (in GIST882 cells, IC50 22 nM), mutant RET C643W (in human medullary thyroid carcinoma TT cells, IC50 10 nM), and ERK1/2 (in MDA-MB-231 breast cancer cells, IC50 43 nM). Importantly, strong KIT inhibitory activity of regorafenib (IC50 values 12–130 nM) was also observed in imatinib-resistant KIT double-mutants containing a primary deletion of amino acids 557–558 in the KIT juxtamembrane region (exon 11) and a secondary mutation in the ATP-binding pocket (T670I or V654A) or the activation loop (D816G, N882K, or Y832D).52
Consistent with the effective biochemical inhibition of oncogenic and angiogenic kinases indicated above, regorafenib significantly decreased proliferative capacity in a broad spectrum of tumor cell lines with different activating oncogenic mutations. Although the antiproliferative activity of regorafenib varied according to the cell line used, it was most effective in the GIST 882 cell line (IC50 42 nM) and thyroid TT cells.52 The antitumor effects of regorafenib were confirmed in vivo in a wide range of preclinical human tumor xenografts derived from colon, lung, pancreatic, and ovarian cell lines. Unfortunately, no GIST xenograft model was included in these studies. Treatment resulted in growth inhibition and reduction of the proliferative index as measured immunohistochemically by the percentage of Ki67-positive cells.52 The effective dose of regorafenib in these models varied between 10 to 30 mg/kg, which corresponds to clinically effective doses. Given that no animal lethality or significant weight loss was observed in mice treated with up to 100 mg/kg of regorafenib, the therapeutic index of regorafenib was considered to be high.
Antiangiogenic effects and preclinical pharmacodynamics
Further preclinical tests evaluated the antiangiogenic properties of regorafenib. Consistent with its ability to inhibit VEGFRs, regorafenib inhibited the proliferation of various vascular cells in vitro.52 Proliferation of VEGF-stimulated human umbilical vein endothelial cells was inhibited at an IC50 value of 2.6 nM, whereas growth of FGF2-stimulated or PDGF-BB-stimulated human aortic smooth muscle cells was affected at 127 nM and 146 nM (IC50), respectively.
Furthermore, the pharmacodynamic effect of regorafenib on tumor vasculature was tested in vivo using dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI) in rat GS9L glioblastoma tumor xenografts.52 It was shown that a single oral dose of regorafenib (10 mg/kg) effectively inhibited tumor perfusion and extravasation of the macromolecular contrast agent, Gadomer-17. This effect was seen 10 hours after a single treatment and persisted for up to 2 days. Similar results were obtained after multiple treatments in a daily dosing scheme. The antiangiogenic activity of regorafenib correlated very well with its therapeutic antitumor activity in this short-term model. The authors reported that no tumor regrowth was observed 4 days after discontinuation of regorafenib.
Clinical pharmacokinetics and pharmacodynamics
Regorafenib is metabolized by the cytochrome P450 3A4 and glucuronosyltransferase 1A9 enzymatic systems in the liver and its main metabolites, M-2 (N-oxide metabolite; BAY 75-7495) and M-5 (N-oxide/N-desmethyl metabolite; BAY 81-8752), have been shown to be pharmacologically active in preclinical studies.52 Pharmacokinetic data obtained in a Phase I dose-escalation study in patients with advanced solid tumors showed that both regorafenib and its metabolites exhibited a similar steady-state profile.53 A dose-dependent increase in exposure of regorafenib reached a plateau at 140–160 mg.53,54 The terminal half-life of regorafenib was 20–40 hours, thus supporting a once-daily treatment schedule. Compared with the parent compound, M-2 and M-5 had similar or even higher systemic exposure, and similar or slower elimination. For example, the terminal half-life for M-2 was comparable with that of the parent compound (26–28 hours), whereas elimination of the M-5 metabolite was much slower, with an estimated half-life of 51–64 hours. Given that the M-2 and M-5 metabolites were shown to be pharmacologically active, as mentioned above, they can be considered to contribute to the clinical activity of regorafenib.
The pharmacodynamic effects of regorafenib were evaluated in the Phase I trial mentioned above by measuring plasma angiogenic cytokines as well as by analyzing tumor perfusion using DCE-MRI.53 A dose-dependent reduction of soluble VEGFR2 as well as an increase in plasma VEGF concentration was observed. In addition, tumor perfusion after 21 days of treatment was decreased according to DCE-MRI assessments.
Safety, tolerability, and efficacy
Safety and tolerability
The safety and tolerability of regorafenib were determined in a Phase I dose-escalation study in patients with advanced solid tumors.53 Fifty-three patients were enrolled and received 10–220 mg of regorafenib once daily in repeating 28-day cycles. Regorafenib was given for 21 days followed by 7 days off treatment. Based on the data obtained from this trial, 160 mg once daily was determined as the maximum tolerated dose and was recommended for future studies.53 The most common adverse effects observed in this study were hoarseness, hand-foot skin reaction (HFSR), mucositis, diarrhea, and hypertension. Eighty-three percent of the patients experienced at least one treatment-related adverse effect and 49% had a grade 3 or 4 adverse reaction.
This safety profile for regorafenib held up for GIST patients in two subsequent Phase II and Phase III studies.55,56 Both studies were conducted in patients with metastatic and/or unresectable GIST that was refractory to imatinib and sunitinib. The Phase II study, which enrolled a total of 34 patients, reported HFSR (85%), fatigue (79%), hypertension (67%), and diarrhea (61%) as the most frequent toxicities at any grade, while hypertension (36%), HFSR (24%), hyperuricemia (6%), and thrombosis (3%) were the most common grade 3 and 4 adverse effects.55 Dose reduction based on toxicity was necessary in 27 (82%) patients, and most patients were eventually treated with 120 mg of regorafenib or less (80 mg or 60 mg) per day. Results from the larger placebo-controlled, double-blind Phase III study (enrolling 199 patients) reported that the most common drug-related side effects in regorafenib-treated patients versus placebo-treated patients were HFSR (56% versus 14%), hypertension (49% versus 17%), diarrhea (40% versus 5%), fatigue (39% versus 27%), and oral mucositis (38% versus 8%).56
Of note, although similar adverse effects were seen in studies treating other tumor entities with regorafenib, their incidence (in particular HFSR) varied significantly according to tumor type. For instance, HSFR was observed in 71.4% and 60.2% of patients with renal cellular carcinoma and GIST, respectively, whereas the frequency of HFSR in patients with hepatocellular carcinoma and metastatic colorectal cancer was much less (50.0% and 46.6%, respectively).57
Efficacy
Evaluation of drug efficacy in the initial dose-escalation study showed promising results.53 Forty-seven of the 53 enrolled patients were evaluable, and 35 patients (66%) achieved disease control (ie, partial response or stable disease). Amongst these were patients with colorectal cancer, renal cancer, malignant melanoma, or sarcoma. Three patients achieved a partial response (one patient each with colorectal cancer, renal cell carcinoma, and osteosarcoma); however, no complete response was seen. No GIST patients were enrolled in this study.
Based on its potent KIT inhibitory activity and preclinical efficacy with regard to inhibition of growth of GIST cell lines (see above), a multicenter Phase II trial of regorafenib was performed in patients with advanced GIST who failed standard therapy.55 The trial enrolled 34 patients with metastatic GIST refractory to imatinib and sunitinib on a 160 mg/day oral 3 weeks on/one week off cycle. The primary endpoint of this trial was the clinical benefit rate, which was defined by the composite of complete responses, partial responses, and stabilization of disease for longer than 16 weeks. Thirty-three of the 34 patients were evaluable, and clinical benefit was shown in 26 (79%): a partial response was achieved in four patients (12%), whereas stabilization of the disease for longer than 16 weeks was seen in 22 patients (67%). In addition, four patients had stabilization of the disease for less than 16 weeks. However, no complete response was reported. Secondary endpoints in this trial included progression-free survival and an evaluation of the safety and tolerability of regorafenib. Median progression-free survival was calculated at 10 months, and toxicities of any grade occurred in at least 25% of patients. Severe (grade 3 and higher) toxicities were observed in less than 5% of patients, and included HFSR, hypertension, and hypophosphatemia (also see above). There were six deaths observed in this trial; five were secondary to disease progression and one was unrelated to the underlying malignancy. Median overall survival was not reached.
Since then, the efficacy and safety profile of regorafenib in GIST patients was examined in the GRID (GIST – Regorafenib In progressive Disease) trial, a randomized, double-blind, placebo-controlled, Phase III study56 that randomized 199 patients with advanced GIST which had progressed despite treatment with imatinib and sunitinib. Patients were randomized in a 2:1 ratio to receive the drug or placebo plus best supportive care (in both groups), respectively. The regorafenib group (n=133) received the drug once daily (160 mg) for the first 3 weeks of each 4-week cycle (see above). The trial protocol included the possibility to unmask treatment assignment in the event of tumor progression, and patients in the placebo group were then allowed to cross over to receive regorafenib. The primary endpoint of this trial was progression-free survival as assessed by modified Response Evaluation Criteria In Solid Tumors (RECIST)58 including a central, blinded radiology review. Secondary endpoints included overall survival, time to progression, tumor response rate, disease control rate, and duration of response. The GRID trial met its primary endpoint of improving progression-free survival in GIST: the median progression-free survival was 4.8 months in the regorafenib-treated patients versus 0.9 months in the placebo group (P<0.0001) based on the central review. Interestingly, investigator-assessed response showed a median progression-free survival of 7.4 months in the regorafenib-treated patients and 1.7 months in the placebo group. Very few patients achieved a partial response under regorafenib (4.5%) and no complete response in either treatment arm was seen. However, treatment with regorafenib led to disease stabilization (at any time for any duration) in more than 71% of the patients (33.3% in the placebo group). There was no statistically significant difference in an interim overall survival analysis between the two groups, while the median overall survival had not been reached at publication of the study. However, most of the patients in the placebo group (56 of 66, 85%) crossed over to regorafenib after disease progression, hence masking a potential statistical difference in this parameter between the two groups.
Almost all assessable patients treated with regorafenib (98%) in this trial had at least one drug-related adverse effect. The toxicity profile of regorafenib was very similar to that in previous studies, with HFSR being the most common adverse effect, seen in 56% of patients. The most common high grade (at least grade 3) unwanted effects were hypertension, HFSR, and diarrhea. Most adverse effects, however, were manageable with dose modification, which was necessary in 72% of regorafenib-treated patients. Discontinuation of treatment due to adverse effects was noted in only 6% of regorafenib-treated patients.
Patient-focused perspectives
In light of the very high frequency of drug-related adverse effects in patients treated with regorafenib (98% of patients in the regorafenib arm of the GRID trial had “any adverse event”), one is tempted to ask about its effect on quality of life. Unfortunately, no data regarding this issue have been published as yet for GIST patients, although health-related quality of life data and health utility values were collected in the GRID trial with a plan to be published at a later time.56
By contrast, quality of life data were included in the initial report of the CORRECT (COloRectal cancer treated with REgorafenib or plaCebo after failure of standard Therapy) trial in metastatic colorectal cancer.59 Values were measured using the European Organization for Research and Treatment of Cancer QLQ-C30 and EQ-5D, in which higher scores represent a higher level of functioning and better health-related quality of life, respectively.60,61 The recorded scores dropped for both systems while the patients were on study, indicating that treatment with regorafenib did not lead to an improvement in quality of life. On the other hand, there was no significant difference between the regorafenib and placebo group with respect to quality of life score, pointing to the fact that the treatment itself was not associated with detrimental effects on quality of life.62
According to both the GRID and CORRECT trials, drug-related adverse effects were generally deemed to be manageable and comprised of typical reactions associated with tyrosine kinase inhibitors, especially those with antiangiogenic properties.55,56,59
Correlative science studies
Only limited data exist on mechanistic and correlative aspects of the effectiveness of regorafenib in GIST. George et al55 investigated the role of the underlying KIT mutation in the response to regorafenib. Mutational data were available for the majority of patients (30 of 34 in total) enrolled in the Phase II trial testing regorafenib in imatinib-resistant and sunitinib-resistant GIST patients.55 Amongst this small group, there was no difference in the clinical benefit rate among genotypes. Kaplan–Meier estimates showed a significantly longer progression-free survival in patients with KIT exon 11 mutations when compared with KIT exon 9 mutant GIST, but only three patients with the latter genotype were included in the study. In addition, immunoblotting of serial tumor biopsies before and after treatment showed that target inhibition as measured by KIT phosphorylation correlated with clinical outcome, while inhibition of signaling kinases downstream of KIT (AKT and MAPK) was variable.
Conclusion
Based on the data detailed above, Bayer submitted a New Drug Application for regorafenib to the FDA on August 30, 2012. It contained the recommendation for the use of regorafenib in the treatment of metastatic and/or unresectable GIST in patients whose disease has progressed despite prior treatment with imatinib and sunitinib. On February 25, 2013, regorafenib was approved for the treatment of patients with advanced GIST that cannot be surgically removed and no longer responds to other treatments approved by the FDA for this disease. Regorafenib is thus the only approved third-line treatment in patients with locally advanced, unresectable, or metastatic GIST who have failed imatinib and sunitinib therapy.56,63
Taken together, imatinib mesylate (Gleevec) as treatment for GIST was the first small molecule kinase inhibitor to be approved for a solid tumor in 2002. While the search for second-line compounds has resulted in FDA approval of sunitinib malate (Sutent) in 2006, it took another 7 years to identify and for the FDA to approve a third-line treatment, regorafenib (Stivarga), for GIST. This highlights the fact that although many target compounds for tyrosine kinase inhibitor-resistant GIST are being studied, effective treatments may become harder to identify with each treatment level. It also calls for a more personalized approach that targets specific patient entities that may be specifically sensitive to a given treatment. This approach, however, will be difficult to pursue in GIST because of the relative rarity of the disease. Nevertheless, our current knowledge on GIST pathogenesis and tyrosine kinase inhibitor resistance mechanisms will help overcome at least part of these obstacles.
Disclosure
The authors report no conflicts of interest in this work.
References
Ducimetière F, Lurkin A, Ranchère-Vince D, et al. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. PLoS One. 2011;6(8):e20294. | |
Joensuu H, Hohenberger P, Corless CL. Gastrointestinal stromal tumour. Lancet. 2013;382(9896):973–983. | |
Min KW. Gastrointestinal stromal tumor: an ultrastructural investigation on regional differences with considerations on their histogenesis. Ultrastruct Pathol. 2010;34(3):174–188. | |
Joensuu H, Vehtari A, Riihimäki J, et al. Risk of recurrence of gastrointestinal stromal tumour after surgery: an analysis of pooled population-based cohorts. Lancet Oncol. 2012;13(3):265–274. | |
DeMatteo RP, Lewis JJ, Leung D, Mudan SS, Woodruff JM, Brennan MF. Two hundred gastrointestinal stromal tumors: recurrence patterns and prognostic factors for survival. Ann Surg. 2000;231(1):51–58. | |
Agaimy A, Wünsch PH, Dirnhofer S, Bihl MP, Terracciano LM, Tornillo L. Microscopic gastrointestinal stromal tumors in esophageal and intestinal surgical resection specimens: a clinicopathologic, immunohistochemical, and molecular study of 19 lesions. Am J Surg Pathol. 2008;32(6):867–873. | |
Kawanowa K, Sakuma Y, Sakurai S, et al. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum Pathol. 2006;37(12):1527–1535. | |
Fletcher CDM, Berman JJ, Corless C, et al. Diagnosis of gastrointestinal stromal tumors: a consensus approach. Hum Pathol. 2002;33(5):459–465. | |
Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol. 2006;23(2):70–83. | |
Joensuu H. Risk stratification of patients diagnosed with gastrointestinal stromal tumor. Hum Pathol. 2008;39(10):1411–1419. | |
Duensing A. Closing in on accurate risk prediction and disease management for patients with operable GIST. Lancet Oncol. 2012;13(3):220–221. | |
Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865–878. | |
Andersson J, Sihto H, Meis-Kindblom JM, Joensuu H, Nupponen N, Kindblom LG. NF1-associated gastrointestinal stromal tumors have unique clinical, phenotypic, and genotypic characteristics. Am J Surg Pathol. 2005;29(9):1170–1176. | |
Carney JA. Gastric stromal sarcoma, pulmonary chondroma, and extra-adrenal paraganglioma (Carney triad): natural history, adrenocortical component, and possible familial occurrence. Mayo Clin Proc. 1999;74(6):543–552. | |
West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT or PDGFRA mutation status. Am J Pathol. 2004;165(1):107–113. | |
Duensing A, Joseph NE, Medeiros F, et al. Protein kinase C theta (PKCtheta) expression and constitutive activation in gastrointestinal stromal tumors (GISTs). Cancer Res. 2004;64(15):5127–5131. | |
Medeiros F, Corless CL, Duensing A, et al. KIT-negative gastrointestinal stromal tumors: proof of concept and therapeutic implications. Am J Surg Pathol. 2004;28(7):889–894. | |
Hirota S, Isozaki K, Moriyama Y, et al. Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science. 1998;279(5350):577–580. | |
Duensing A, Medeiros F, McConarty B, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumors (GISTs). Oncogene. 2004;23(22):3999–4006. | |
Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003; 299(5607):708–710. | |
Pasini B, McWhinney SR, Bei T, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet. 2008;16(1):79–88. | |
Agaram NP, Wong GC, Guo T, et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2008;47(10):853–859. | |
Woodall CE, Brock GN, Fan J, et al. An evaluation of 2537 gastrointestinal stromal tumors for a proposed clinical staging system. Arch Surg. 2009;144(7):670–678. | |
Emile JF, Brahimi S, Coindre JM, et al. Frequencies of KIT and PDGFRA mutations in the MolecGIST prospective population-based study differ from those of advanced GISTs. Med Oncol. 2012;29(3):1765–1772. | |
Demetri GDG, Mehren von MM, Antonescu CRC, et al. NCCN Task Force report: update on the management of patients with gastrointestinal stromal tumors. J Natl Compr Canc Netw. 2010;8 Suppl 2:S1–S4. | |
Casali PG, Blay JY, ESMO/CONTICANET/EUROBONET Consensus Panel of Experts. Gastrointestinal stromal tumours: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2010;21 Suppl 5:v98–v102. | |
Hohenberger P, Ronellenfitsch U, Oladeji O, et al. Pattern of recurrence in patients with ruptured primary gastrointestinal stromal tumour. Br J Surg. 2010;97(12):1854–1859. | |
Blanke CD, Demetri GD, Mehren von M, et al. Long-term results from a randomized phase II trial of standard- versus higher-dose imatinib mesylate for patients with unresectable or metastatic gastrointestinal stromal tumors expressing KIT. J Clin Oncol. 2008;26(4):620–625. | |
Verweij J, Casali PG, Zalcberg J, et al. Progression-free survival in gastrointestinal stromal tumours with high-dose imatinib: randomised trial. Lancet. 2004;364(9440):1127–1134. | |
Nilsson B, Bümming P, Meis-Kindblom JM, et al. Gastrointestinal stromal tumors: the incidence, prevalence, clinical course, and prognostication in the preimatinib mesylate era – a population-based study in western Sweden. Cancer. 2005;103(4):821–829. | |
Hislop J, Mowatt G, Sharma P, et al. Systematic review of escalated imatinib doses compared with sunitinib or best supportive care, for the treatment of people with unresectable/metastatic gastrointestinal stromal tumours whose disease has progressed on the standard imatinib dose. J Gastrointest Cancer. 2012;43(2):168–176. | |
Jones RL, McCall J, Adam A, et al. Radiofrequency ablation is a feasible therapeutic option in the multi modality management of sarcoma. Eur J Surg Oncol. 2010;36(5):477–482. | |
Demetri GD, Van Oosterom AT, Garrett CR, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–1338. | |
Heinrich MC, Maki RG, Corless CL, et al. Primary and secondary kinase genotypes correlate with the biological and clinical activity of sunitinib in imatinib-resistant gastrointestinal stromal tumor. J Clin Oncol. 2008;26(33):5352–5359. | |
Reichardt P, Montemurro M. Clinical experience to date with nilotinib in gastrointestinal stromal tumors. Semin Oncol. 2011;38 Suppl 1:S20–S27. | |
Bauer S, Yu LK, Demetri GD, Fletcher JA. Heat shock protein 90 inhibition in imatinib-resistant gastrointestinal stromal tumor. Cancer Res. 2006;66(18):9153–9161. | |
Marino-Enriquez A, Ou W-B, Cowley G, et al. Genome-wide functional screening identifies CDC37 as a crucial HSP90-cofactor for KIT oncogenic expression in gastrointestinal stromal tumors. Oncogene. April 15, 2013. [Epub ahead of print.] | |
Mühlenberg T, Zhang Y, Wagner AJ, et al. Inhibitors of deacetylases suppress oncogenic KIT signaling, acetylate HSP90, and induce apoptosis in gastrointestinal stromal tumors. Cancer Res. 2009;69(17):6941–6950. | |
Bauer S, Parry JA, Mühlenberg T, et al. Proapoptotic activity of bortezomib in gastrointestinal stromal tumor cells. Cancer Res. 2010;70(1):150–159. | |
Boichuk S, Lee J, Mehalek KR, et al. A medium-throughput compound screen identifies novel treatment options for gastrointestinal stromal tumors (GIST). Presented at the 18th Annual Meeting of the Connective Tissue Oncology Society, November 14–17, 2012, Prague, Czech Republic. | |
Boichuk S, Parry JA, Makielski KR, et al. The DREAM complex mediates GIST cell quiescence and is a novel therapeutic target to enhance imatinib-induced apoptosis. Cancer Res. 2013;73(16):5120–5129. | |
Edris B, Willingham SB, Weiskopf K, et al. Anti-KIT monoclonal antibody inhibits imatinib-resistant gastrointestinal stromal tumor growth. Proc Natl Acad Sci U S A. 2013;110(9):3501–3506. | |
Duensing S, Duensing A. Targeted therapies of gastrointestinal stromal tumors (GIST) – the next frontiers. Biochem Pharmacol. 2010;80(5):575–583. | |
Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64(19):7099–7109. | |
Fabian MA, Biggs WH, Treiber DK, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23(3):329–336. | |
Wilhelm S, Carter C, Lynch M, et al. Discovery and development of sorafenib: a multikinase inhibitor for treating cancer. Nat Rev Drug Discov. 2006;5(10):835–844. | |
Strumberg DD, Schultheis BB. Regorafenib for cancer. Expert Opin Investig Drugs. 2012;21(6):879–889. | |
Heinrich MC, Marino-Enriquez A, Presnell A, et al. Sorafenib inhibits many kinase mutations associated with drug-resistant gastrointestinal stromal tumors. Mol Cancer Ther. 2012;11(8):1770–1780. | |
Kindler HL, Campbell NP, Wroblewski K. Sorafenib (SOR) in patients (pts) with imatinib (IM) and sunitinib (SU)-resistant (RES) gastrointestinal stromal tumors (GIST): Final results of a University of Chicago Phase II Consortium trial. J Clin Oncol. 2011;29(Suppl 15):Abstr 10009. | |
Park SH, Ryu MH, Ryoo BY, et al. Sorafenib in patients with metastatic gastrointestinal stromal tumors who failed two or more prior tyrosine kinase inhibitors: a phase II study of Korean gastrointestinal stromal tumors study group. Invest New Drugs. 2012;30(6):2377–2383. | |
Montemurro M, Gelderblom H, Bitz U, et al. Sorafenib as third- or fourth-line treatment of advanced gastrointestinal stromal tumour and pretreatment including both imatinib and sunitinib, and nilotinib: a retrospective analysis. Eur J Cancer. 2013;49(5):1027–1031. | |
Wilhelm SM, Dumas J, Adnane L, et al. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011;129(1):245–255. | |
Mross K, Frost A, Steinbild S, et al. A Phase I dose-escalation study of regorafenib (BAY 73-4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. 2012;18(9):2658–2667. | |
Shimizu T, Tolcher AW, Patnaik A. Phase I dose-escalation study of continuously administered regorafenib (BAY 73-4506), an inhibitor of oncogenic and angiogenic kinases, in patients with advanced solid tumors. J Clin Oncol. 2010;28(Suppl 15):Abstr 3035. | |
George S, Wang Q, Heinrich MC, et al. Efficacy and safety of regorafenib in patients with metastatic and/or unresectable GI stromal tumor after failure of imatinib and sunitinib: a multicenter phase II trial. J Clin Oncol. 2012;30(19):2401–2407. | |
Demetri GD, Reichardt P, Kang Y-K, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):295–302. | |
Belum VR, Wu S, Lacouture ME. Risk of hand-foot skin reaction with the novel multikinase inhibitor regorafenib: a meta-analysis. Invest New Drugs. 2013;31(4):1078–1086. | |
Mehren von M, Benjamin RS, Bui MM, et al. Soft tissue sarcoma, version 2.2012: featured updates to the NCCN guidelines. J Natl Compr Canc Netw. 2012;10(8):951–960. | |
Grothey A, Van Cutsem E, Sobrero A, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381(9863):303–312. | |
Fayers P, Bottomley A; EORTC Quality of Life Group; Quality of Life Unit. Quality of life research within the EORTC – the EORTC QLQ-C30. Eur J Cancer. 2002;38 Suppl 4:S125–S133. | |
Pickard AS, Neary MP, Cella D. Estimation of minimally important differences in EQ-5D utility and VAS scores in cancer. Health Qual Life Outcomes. 2007;5:70. | |
Waddell T, Cunningham D. Evaluation of regorafenib in colorectal cancer and GIST. Lancet. 2013;381(9863):273–275. | |
Lyseng-Williamson KA. Regorafenib: a guide to its use in advanced gastrointestinal stromal tumor (GIST) after failure of imatinib and sunitinib. BioDrugs. 2013;27(5):525–531. |
 © 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2013 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.