Back to Journals » Cancer Management and Research » Volume 7
New and emerging factors in tumorigenesis: an overview
Authors Kim S
Received 31 December 2014
Accepted for publication 4 May 2015
Published 28 July 2015 Volume 2015:7 Pages 225—239
DOI https://doi.org/10.2147/CMAR.S47797
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Kenan Onel
Suwon Kim1,2
1Department of Basic Medical Sciences, University of Arizona College of Medicine-Phoenix, 2Cancer and Cell Biology Division, Translational Genomics Research Institute, Phoenix, AZ, USA
Abstract: This article provides an overview of the genes and cellular processes that have emerged recently as new key factors in tumorigenesis. We review these in the context of three broad categories. First, genome-scale sequencing studies have revealed a set of frequently mutated genes in cancer. Genes that are mutated in >5% of all cancers across tissue types are discussed, with a highlighted focus on the two most frequently mutated genes, TP53 and PIK3CA. Second, the mechanisms of resistance to targeted therapy are reviewed. These include acquired resistance under targeted therapy selection owing to mutations and amplification of genes in the same or parallel signaling pathways. Importantly, sequencing of primary tumors has revealed that therapy-resistant clones already exist prior to targeted therapy, demonstrating that tumor heterogeneity in primary tumors confers a mechanism for inherent therapy resistance. Third, “metastasis-specific genes”, or rather lack thereof, are discussed. While many genes have been shown to be capable of promoting metastasis in experimental systems, no common genetic alterations have been identified specific to metastatic lesions. Rather, the same gene mutations frequently found in primary tumors are also found prevalent in metastases, suggesting that the genes that drive tumorigenesis may also drive metastasis. In this light, an emerging view of metastatic progression is discussed. Collectively, these recent advances in cancer research have refined our knowledge on cancer etiology and progression but also present challenges that will require innovative new approaches to treat and manage cancer.
Keywords: cancer, genomics, gene mutations, targeted therapy resistance, tumor heterogeneity, metastasis
Introduction
The incidence rate of cancer increases with age.1 This is consistent with the multistep tumorigenesis paradigm – it “takes time” for a normal cell to undergo multiple genetic alterations to become a tumor. How many genetic events are necessary or sufficient to drive tumorigenesis has been debated for over half a century via various investigative methods, from epidemiological studies to experimental model systems.2–4 The discovery of proto-oncogenes by J Michael Bishop and Harold E Varmus, corecipients of the 1989 Nobel Prize in Medicine, marked a conceptual turning point in cancer biology, by providing evidence that our own genes can cause cancer when altered. In the following years, identification of the genes and gene mutations that drive tumorigenesis has been one of the mainstream focuses in cancer research. Efforts have been made on both fronts, searching for deregulated genes in human tumor samples and screening for genes capable of eliciting cell transformation in experimental systems. To date, hundreds of genes have been classified as oncogenes or tumor suppressors. Functional studies have demonstrated that these genes play an integral role in normal cellular processes, including cell proliferation, death, survival, stress response, motility, differentiation, crosstalk between cells, and tissue architecture, indicating that cancer arises from dysregulation of normal cellular processes.5
The goal of identifying oncogenes is not only to decipher the cellular and molecular mechanisms that govern tumorigenesis but also to treat cancer patients with agents that target such genes and pathways, namely molecular targeted therapy. The concept of targeted therapy of cancer came to fruition in the pivotal work of Brian J Druker in 1996, who developed a small-molecule inhibitor STI571, now known as imatinib, for the treatment of chronic myelogenous leukemia (CML).6 CML is characterized by the presence of a unique chromosomal translocation between chromosomes 9 and 22, which results in the production of the Bcr-Abl fusion molecule in leukemic cells. Imatinib binds to and inhibits the Abl kinase. And with imatinib therapy, over 90% of CML patients achieve disease remission. The success story of imatinib energized the cancer research community to identify molecular targets and develop effective targeting agents. As a result of the collective effort of basic scientists, clinicians, and the pharmaceutical industry, we now have several dozen targeted therapy agents approved by the US Food and Drug Administration for cancer treatment, and several dozens more in clinical trials.
While molecular targeted therapy was proven effective in treating some cancers, the use of targeted therapies for other cancers has not been as straightforward, in part due to the following reasons. First, only a few cancers have unique molecular lesions like the Bcr-Abl fusion protein in CML. Another example of unique cancer-specific lesions is the EML4-ALK chromosomal translocation found in ~4% of non-small-cell lung carcinoma (NSCLC). Patients who are treated with crizotinib that targets the ALK kinase have shown a remarkable 90% remission rate.7,8 However, these examples of unique actionable molecular lesions are exceptional than typical in cancer. Instead, the majority of tumor cells express many dysregulated gene products that are not readily discernable as therapy targets. Second, some of the early promising targeted therapy agents, such as gefitinib that targets the EGFR protein often altered or highly expressed in NSCLC, failed to improve patient survival despite dramatic initial tumor regression. This was due to the rampant and rapid insurrection of therapy-resistant tumors within mere weeks of the therapy.9 We now know that therapy resistance is a general phenomenon in cancer, which occurs with the majority, if not with all, of targeted agents. Third, recent genomic studies have revealed that each tumor typically harbors tens to hundreds of mutations that affect protein products.10,11 Since it is impractical to treat patients with tens to hundreds of therapeutic agents simultaneously, the efforts to discern the “Achilles hill” target(s) among the many genes mutated in tumors are ongoing.
This article provides an overview of new factors and intriguing new concepts in tumorigenesis brought to light by recent discoveries in cancer research. We highlight aspects of these new emerging factors to better understand tumorigenesis and strategize innovative approaches in the treatment of cancer going forward. To this end, the subtopics discussed in this article are limited to 1) cancer-driving genes and mutations identified by genome sequencing, 2) targeted therapy resistance and tumor heterogeneity, and 3) lack of metastasis-specific mutations. As there are many excellent and in-depth reviews of each subtopic, we apologize for our limited referencing of the many original papers here.
Cancer-driving genes and mutations identified by genome sequencing
The recent explosion of genomic data over the past decade, enabled by rapid advances in sequencing technology and sophisticated bioinformatics tools, has provided us with the genome-wide view of cancer at single-nucleotide resolution. A general expectation may have been to identify a handful of gene mutations in each tumor, which would point to an actionable therapy target. The whole-genome-sequencing data revealed a more complicated picture of a tumor typically harboring an average of 3,000 mutations, compared to the normal cells of the same person (an average of one mutation per one million nucleotides).10,11 Of these, ~300 mutations are found in the coding sequences (exons), and of these, an average of 30–60 mutations are non-synonymous, which are expected to alter protein products.10 It is notable that the median number of non-synonymous mutations varies depending on the tumor type, ranging from several (eg, acute lymphoblastic leukemia) to hundreds (eg, melanoma, lung cancer). The latter is correlative of known mutagen exposure such as UV and smoking.10 It is fitting that mutagens cause DNA mutations, and therefore result in the accumulation of many mutations in tumors. However, the exact number of mutations required for these mutagen-driven cancers has not been determined. Nevertheless, it is widely accepted that the major portion of these mutations are “bystander” mutations that do not directly contribute to tumorigenesis. By the same token, considering the scale of sequence variations detected in tumors in general, it is thought that the average number of 30–60 non-synonymous mutations found in tumors also includes bystander mutations.
How do we discern cancer-driving mutations from bystander mutations? Studies have analyzed the genome data with various statistical methods and have identified a set of 120–140 genes as “cancer drivers”. These are defined as the genes that are mutated in more than one cancer type. In other words, statistically, all cancers harbor mutations in one or more of these genes, signifying their functional contribution in tumorigenesis. It is estimated that a tumor contains an average of two to eight mutations in these cancer driver genes.10,11 These studies are impressive in their scale and depth and have also been reviewed in equally impressive and thoughtful articles, some of which are cited here.
What are these 120–140 cancer driver genes? These genes are categorized as either oncogenes or tumor suppressors by the distribution pattern of their mutations. Oncogenic mutations are often missense mutations that alter specific amino acid residues that are crucial to the protein function.10,12 These mutations recur in multiple tumors, attesting to their functional importance in driving tumorigenesis. A well-known example is mutations in the KRAS gene found in multiple types of cancers, including colorectal cancer, lung cancer, melanoma, and endometrial cancer.13,14 According to the Catalogue Of Somatic Mutations In Cancer (COSMIC) database, 83% of the KRAS mutations alter the amino acid residue glycine 12 (eg, G12D, G12V), and 14% alter the amino acid residue glycine 13. Functional studies have shown that these amino acid substitutions affect the GTPase activity of k-ras, resulting in constitutively active k-ras in a GTP-bound state.14 Thus, recurring mutations on specific sites are likely gain-of-function mutations and a mark of an oncogene.
In contrast, tumor suppressor gene mutations are typically found distributed along the coding regions as missense or nonsense mutations, suggesting that any mutations that disable the protein product drive tumorigenesis. For example, mutations in the BRCA1 gene, a tumor suppressor gene associated with an increased risk of breast cancer, are found all over the coding region with no specific recurring mutations among the 389 BRCA1 mutations registered in the COSMIC database. Thus, mutations distributed in the coding region of the gene are indicative of a tumor suppressor gene.10,12
By deciphering the distribution pattern of gene mutations in the COSMIC dataset, Vogelstein et al classified 125 “Mut-driver genes” as 71 tumor suppressors and 54 oncogenes. In addition to these “Mut-driver genes”, the authors reported an additional ten frequently amplified (oncogenes) and three frequently deleted (tumor suppressors) genes, concluding a total of 138 cancer driver genes.10 Kandoth et al analyzed the Cancer Genome Atlas (TCGA) dataset and identified 127 “significantly mutated genes (SMGs)”, defined as the genes mutated in multiple tumors in more than one type of cancer.11 Notably, 66 genes are common between the “Mut-driver genes” and SMGs.11 While these 66 genes identified by two independent methods are clearly of importance in tumorigenesis, significance of the other nonoverlapping genes is less clear. The exact set of cancer-driving genes will no doubt be refined in the near future. What the current data do clearly show is that each gene does not have equal potential to drive tumorigenesis, as some of the gene mutations are vastly more prevalent than others.
In keeping with the intent of this article to provide an overview, not a comprehensive review, we discuss the SMGs that are mutated in >5% of all cancers from the data available from Kandoth et al. Using >5% frequency is an arbitrary cutoff, but it translates to one in 20 tumors harboring the gene mutations which we thought should warrant recognition as to represent major pathways involved in tumorigenesis. Only ten genes out of 127 SMGs made the list of genes mutated in >5% of all cancers (Table 1). In fact, if we consider >10% gene mutation frequency as a cutoff (one out of ten tumors harboring the gene mutations), only two genes, TP53 and PIK3CA, make the list, with PTEN trailing behind just shy of 10%.
 | Table 1 Significantly mutated genes in >5% of all cancer and each cancer type |
TP53 and PIK3CA, the two most commonly mutated genes in cancer
It is not surprising, but striking, that TP53 is mutated in 42% of all cancers, attesting to its principal tumor suppressor function in a wide range of tissue types. In ovarian serous carcinoma, over 95% of tumors harbor TP53 mutations in the absence of any other SMG mutations, suggesting that loss of TP53 is a major event that drives tumorigenesis in this tissue type. High-frequency mutations in TP53 are also found in lung squamous cell carcinoma and head and neck squamous cell carcinoma with 80% and 70%, respectively (Table 1).
The TP53 gene product, p53, is a central molecule in DNA damage response, best characterized as a transcription factor that induces expression of cell cycle inhibitors (eg, p21) to allow “time” to repair DNA damage.15–17 When DNA damage persists without repair, p53 induces apoptosis by activating transcription of pro-apoptosis genes (eg, PUMA) and also by a transcription-independent mechanism.18,19 The transcription-independent mechanism of p53 involves direct interaction between p53 and mitochondria, mediating mitochondria outer membrane permeabilization and cytochrome c release, resulting in apoptosis.19 Recently, p53 has also been shown to inhibit autophagy, a cellular process that degrades unnecessary cellular products under stress conditions.20,21 The inhibition of autophagy is considered an oncogenic event that promotes genomic instability and necrotic cell death instead of apoptosis.22 Thus, it is intriguing how p53 balances its tumor suppressor function with a seemingly oncogenic function. Nevertheless, it is evident that p53 is more than a transcriptional factor and regulates multiple pathways involved in the cell death/survival decision under cellular stress.
PIK3CA is the second most frequently mutated gene with a frequency of 18% in all cancers. The high mutation rates in the PIK3CA gene are found in uterine corpus endometrial carcinoma (52%), breast cancer (33%), colon and rectal carcinoma (18%), and bladder urothelial carcinoma (17%) (Table 1). PIK3CA encodes for the p110 catalytic subunit of phosphatidylinositol 3-kinase (PI3k) that phosphorylates phosphatidylinositol-(4,5)-bisphosphate to phosphatidylinositol-(3,4,5)-triphosphate (PIP3).23 Mutations in the PIK3CA gene are found to alter specific amino acid residues, including E545K, E542K, and H1047R.24 These mutations confer an increase in the lipid kinase activity of PI3k, indicating gain-of-function mutations that result in active signaling.24 PIP3 generated by PI3k is a signaling lipid that activates downstream kinases, including phosphatidylinositol-dependent kinase (PDK1) and protein kinase B (PKB, also known as AKT), which in turn inhibit pro-apoptotic molecules (eg, BAD and BIM), thereby mediating cell survival.25 Concordantly, the net result of PI3k pathway activation is enabling cell survival under various cellular stress conditions such as nutrient-restriction and cytotoxic cancer drug treatment.
Why do these two genes stand out as the two most frequently mutated genes in all cancers among many other genes, especially among those that are known to drive cell proliferation such as KRAS, for example? The common function between TP53 and PIK3CA is in the cell death/survival pathway, such that either gene mutations – TP53 loss or PIK3CA activation – result in aberrant cell survival under stress. This implies that TP53 and PIK3CA may mark the crossroads between cell death and survival, crucial decision points in tumorigenesis. Interestingly, we observed that the tumor types with high-frequency mutations in TP53 had relatively low-frequency mutations in PIK3CA. Conversely, the tumor types with high-frequency mutations in PIK3CA had relatively low-frequency mutations in TP53 (Table 1). We graphed the mutation frequencies between TP53 vs PIK3CA in a given tumor type and found an inverse relationship (Figure 1, P=0.0015). This inverse relationship suggests that TP53 and PIK3CA mutations may occur mutually exclusive of one another in a given tumor type; that is, tumors that harbor TP53 mutations do not contain PIK3CA mutations and vice versa. This mutually exclusive relationship between these two gene mutations has also been noted by Kandoth et al. “Mutual exclusivity” of mutations is indicative of a classic genetic interaction between genes that function in the same pathway. For example, KRAS (G12V or G12D) and BRAF(V600E) mutations are mutually exclusive in colorectal cancer, indicating that a mutation in either gene results in the same functional consequence, namely activation of the MAP kinase pathway leading to tumorigenesis.26,27 In this logic, the genome data showing the mutual exclusivity between TP53 and PIK3CA mutations suggest that these two genes may function in the same genetic pathway, the deregulation of which by either gene mutations results in tumorigenesis.
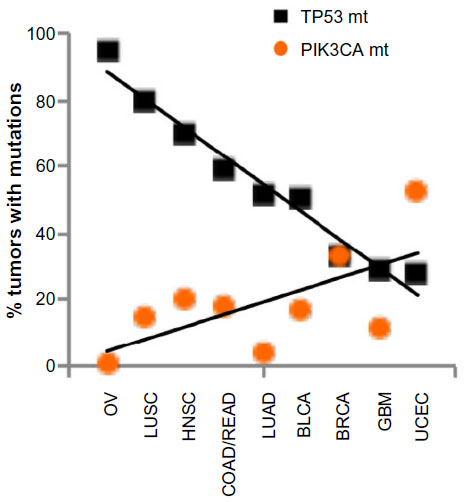 | Figure 1 Inverse relationship of mutation frequencies between TP53 and PIK3CA in cancers. |
Alternatively, TP53 or PIK3CA mutations may drive cancers originating from different cell types. For example, the ovarian cancer found with high-frequency mutations in TP53 (Table 1) is the serous epithelial ovarian carcinoma subtype that makes up >90% of ovarian cancer. Another histologic subtype of ovarian cancer is called clear cell ovarian carcinoma that constitutes 1%–10% of ovarian cancers. The clear cell ovarian-subtype tumors have been shown to contain largely wild-type TP53 but high-frequency mutations in PIK3CA, up to 40%.28 Together, these may suggest that TP53 or PIK3CA mutations occur in different cell types, each driving a different subtype of cancer (serous ovarian cancer vs clear cell ovarian cancer). Interestingly, multiplatform genomic analyses have reported that one of the breast cancer subtypes, basal-like breast cancer, is more closely related to serous ovarian cancer in its genetic footprint, than to other breast cancer subtypes.29 The authors have suggested that these two cancers may have originated from closely related cell types.29 The basal-like subtype of breast cancer has a high frequency of TP53 mutations, 80%, as does serous ovarian cancer with 95%. Thus, it appears to support the argument that TP53 mutation drives cancers originating from similar cell types.
However, molecular classification of 12 cancer types based on cluster-of-cluster assignments using five omics datasets – DNA copy-number variation, DNA methylation, mRNA expression, microRNA profiles, and phosphoprotein levels – has separated basal-like breast cancer and ovarian cancer as two distinct cancer types, with ovarian cancer matching to its own tissue origin.30 Rather, the study points out that some cancers share the “p53 footprint” recognizable by gene expression patterns altered by the loss of p53. Thus, similar genomic profiles between basal-like breast cancer and serous ovarian cancer are likely due to the high-frequency mutation in TP53 in both the cancer types, rather than that they share similar cell type origin. In fact, all four molecular subtypes of breast cancers are found with mutations in TP53 or PIK3CA, albeit with different prevalence but in a mutually exclusive manner within each subtype.29 In addition, the two subtypes of lung cancers, squamous cell carcinoma and adenocarcinoma, also harbor mutations in both TP53 and PIK3CA.31 Thus, the mutual exclusivity between TP53 and PIK3CA mutations is not likely due simply to differences in cell type origin. At any rate, as numerous studies have shown the functions of p53 and PI3k in multiple and diverse cell types, it would be hard-pressed to assign the biological functions of TP53 or PIK3CA to any specific cell types, such that each gene mutation drives different cancer types.
Taken together, it seems tenable that the mutual exclusivity between TP53 and PIK3CA mutations may be due to their function in the same genetic pathway, the deregulation of which leads to tumorigenesis. Considering that the mutation frequency of TP53 is 42%, dominantly higher than the one of PIK3CA, 18% (Table 1), it is tempting to speculate that TP53 is an upstream regulator of PIK3CA; TP53 regulates several major pathways that lead to tumorigenesis, with the PIK3CA pathway being one of the major pathways. In this scenario, TP53 mutations alone could drive tumorigenesis, while PIK3CA mutations result in tumorigenesis if one or more pathways governed by TP53 are deregulated. The genomic data support this postulate, which show that PIK3CA mutations “co-occur” with other gene mutations such as PTEN and AKT, whereas TP53 mutations remain mutually exclusive with these other gene mutations.11 Based on these premises, we put forward a TP53/PIK3CA pathway model in which p53 inhibits PI3k (Figure 2).
 | Figure 2 A proposed genetic pathway model of p53 as an upstream regulator of PI3k in tumorigenesis. |
Whether p53 directly inhibits PI3k is unclear, although some studies have suggested it.32 The better-characterized functional link between p53 and PI3k is via PTEN. The Pten protein is a lipid phosphatase that opposes the lipid kinase activity of PI3k. The p53 protein has been shown to activate Pten in DNA damage response, resulting in the inhibition of PI3k action.33 Conversely, PI3k activates Akt, which in turn activates Mdm2 to inhibit p53.34 Thus, the cross-regulation between p53 and PI3k has already been established. The exact epistatic relationship between the two genes will need further investigation.
Why is it of consequence to place TP53 and PIK3CA in the same genetic pathway? While it is impossible to restore TP53 in tumors that have lost both copies of the gene, there are several agents that target the PI3k pathways already in various clinical trials.25,35 If PIK3CA is functionally downstream of TP53, we could conceivably treat patients with TP53-mutated cancers with PI3k pathway inhibitors and achieve a therapeutic efficacy. Considering that 42% of all human cancers harbor TP53 mutations with no targeted therapy available to date, the TP53/PIK3CA relationship warrants investigative scrutiny.
Genes mutated in specific cancer types
There are several genes in Table 1, whose mutations are prevalent only in a single cancer type: APC in 83% of colorectal adenocarcinoma and VHL and PBRM1 in 52% and 33% of kidney renal cell carcinoma, respectively. These genes may represent the genes relevant to cancers rising from specialized cell types. GATA3 may belong to this group of genes as it is mutated in 11% of breast cancers but less than 5% in all other cancer types. These individual genes have been reviewed extensively elsewhere.36–38 In brief, APC is a regulator of Wnt signaling essential for the maintenance of intestinal epithelium stem cells.39 Thus, the loss of APC results in expansion of aberrant cell population in intestinal epithelium, leading to colon cancer. VHL is best known as a regulator of hypoxia-inducible factor α subunit.37 The exact mechanism of how deregulated hypoxia response specifically drives kidney cancer is not well understood. Interestingly, VHL has been shown to play an essential role in the formation and maintenance of primary cilia in renal epithelial cells.40 As cilia are required for the proper development and maintenance of nephrons in kidney, the absence of cilia due to the loss of VHL apparently results in the formation of lesions in kidney tissue, leading to tumorigenesis.40,41 GATA3 is a transcription factor that is essential in the specification and maintenance of the luminal epithelial cells in mammary gland.38,42 Collectively, the commonality of these gene functions appears to be the maintenance of specific cell types within each tissue type.
Interestingly, the mutations in these genes are loss-of-function mutations, suggesting their tumor suppressor functions. APC has been referred to as a “gate keeper” because its functional loss inevitably leads to colon cancer, evidenced by the fact that patients with familiar adenomatous polyposis syndrome, who inherit a defective copy of APC, develop colon cancer with 100% penetrance.36 Similarly, patients with von Hippel–Lindau syndrome, who inherit a mutated copy of VHL, develop clear cell renal cancer with a high frequency of 70%.43 Thus, APC, VHL, PBRM1, GATA3, and other genes mutated in specific cancer types may qualify as the “gate keepers” of specific tissue type, the loss of which leads to the expansion of aberrant cell population and tumorigenesis.
Acute myeloid leukemia (AML) is a cancer type that contains mutations in a unique set of genes, which includes NPM1 (27%), FLT3 (26%), DNMT3A (25%), and IDH2 (10%).11 Hematologic malignancies often have their unique set of gene mutations, suggesting that these cancers may arise from mutations in the genes involved in determining and/or maintaining specific hematopoietic cell lineage.44 Pediatric AML shares mutations in the NPM1 and FLT3 genes with the adult AML, supporting the role of NPM1 and FLT3 mutations in driving AML regardless of the age of disease onset.45 However, it is notable that DNMT3A and IDH2 are rarely mutated in pediatric AML.45 These implicate that cell lineage may be obtained and/or maintained via additional mechanisms in the adult, relating to the functions of DNMT3A and IDH2.
Both Dnmt3a and Idh2 are shown to function in DNA methylation. DNMT3A encodes a DNA methyltransferase that adds a methyl group to CpG islands in DNA and functions in hematopoietic stem cell differentiation.46 It has also been shown that Dnmt3a mediates the cytotoxic effect of 5-azacytidine, a therapeutic agent to treat various hematopoietic malignancies, suggesting a utility of Dnmt3a as a therapy target.47,48 IDH2 encodes an isocitrate dehydrogenase that converts α-ketoglutarate to isocitrate.49 When mutated, Idh1 or Idh2 produces an aberrant metabolite, 2-hydroxyglutarate, which results in the loss of 5-hydroxymethylcytosine at CpG islands in DNA, resulting in aberrant DNA methylation.49 Both DNMT3A and IDH2 have been found with recurrent mutations such as R882H/C and R140Q, respectively, associated with poor prognosis and disease relapse, indicating their oncogenic function in tumorigenesis.49,50 Temporal and functional regulation of these genes related to hematopoietic cell lineage is not well understood, nor the scope of their effect on genome-wide DNA methylation. Mapping of the CpG sites affected by these gene mutations may provide insight into the biology of adult myeloid cell lineage and AML tumorigenesis.
Chromatin remodelers
The cancer-driving genes brought to light anew by genome sequencing are a group of chromatin remodelers, including MLL2, MLL3, and ARID1A. The fact that these three genes are mutated in >5% of all cancers highlights the importance of epigenetic regulation in tumorigenesis (Table 1).
MLL2/MLL3 are members of the mixed lineage leukemia (MLLs) family, characterized as HOX gene regulators.51 Biochemically, MLLs are methyltransferases responsible for de novo methylation of histone H3 at lysine 4 (H3K4). MLL1, the first member of MLLs, was discovered due to the frequent gene rearrangement in aggressive leukemia, including in-frame chromosomal translocation, partial tandem gene duplication, and gene amplification.52 MLL1 was shown to bind directly to the promoter regions of the HOX genes and upregulate HOX gene expression. As the HOX genes play a critical role in development, MLL1 knockout results in defective hematopoiesis and embryonic lethality.52 While MLL1 gain-of-function mutations drive cell transformation and leukemia, the mutations found in MLL2/MLL3 by genomic sequencing in solid tumors are mostly nonsense or frameshift loss of function mutations.51 How the activation of MLL1 but the loss of MLL2 or MLL3 drives tumorigenesis is unclear. Defining the specificity of each MLL member in histone modification and gene regulation will provide insight into the epigenetic changes that govern tumorigenesis.
While MLL2/MLL3 are histone modifiers, ARID1A (BAF250A) is a subunit of the Brg1 (also known as Smarca4)-containing SWI/SNF complex.53,54 The SWI/SNF complexes utilize ATP hydrolysis to remodel nucleosome configuration, thereby disrupting histone/DNA contacts and “creating” space for the recruitment of transcription factors to mediate gene activation as well as gene repression.53 The mutations found in ARID1A are loss-of-function mutations, suggesting a tumor suppressor function. Consistent with a tumor suppressor function, several in vitro cell studies have shown that knock-down of ARID1A results in enhanced cell proliferation, whereas overexpression results in cell cycle arrest.55,56 Evidence also suggests that ARID1A plays a role in the cellular differentiation program. For example, ARID1A knock-down in embryonic stem (ES) cells results in a differentiation block to certain cell types including cardiomyocytes and adipocytes but permits differentiation of ES cells into other cell types such as neurons.57 These studies suggest that the loss of ARID1A drives tumorigenesis in specific tissue types, the differentiation of which depends on the function of ARID1A. Supportive of this idea, ARID1A is found to be the most frequently mutated in two cancer types, uterine corpus endometrial carcinoma (30%) and bladder urothelial carcinoma (27.6%). The function of ARID1A in differentiation of either tissue type has not been investigated. Nor is well understood how ARID1A-mediated chromatin remodeling facilitates cell differentiation. It is notable that PBRM1 (BAF180), frequently mutated in kidney renal cell carcinoma, is also a subunit of the same SWI/SNF complex, underscoring the importance of chromatin remodeling in tumorigenesis.53
Collectively, MLL2, MLL3, ARID1A, and PBRM1 represent just a few of many chromatin remodelers identified by genome-scale sequencing as new emerging factors in tumorigenesis. Functionally, they were all shown capable of establishing genome-wide epigenetic programs and affecting gene expression. Delineating specific actions, target genes, and/or tissue-specific context activity/regulation of each gene will provide insight into their functions and mechanisms relevant to tumorigenesis and illuminate potential innovative strategies to target tumors harboring such gene mutations.
Emerging mechanisms of targeted therapy resistance
Therapy resistance has been observed with every therapeutic regimen available today, including poly-chemotherapy, radiation therapy, immunotherapy, and molecular targeted therapy. Mechanisms of therapy resistance are numerous from multidrug resistance pump amplification to aberrant DNA repair and apoptosis, to tumor stem cells, to tumor cell niche, and to pharmaco-metabolism that determines drug efficacy. In this overview, the mechanisms specifically related to molecular targeted therapy resistance are discussed.
Acquired therapy resistance: mutations in the target
One of the better understood mechanisms of resistance to targeted therapy is the acquisition of additional mutations in the target molecule. For example, although imatinib is effective in achieving remission in >90% CML patients, those who do not respond or have partial response to imatinib were found to have leukemic cells with additional mutations in Bcr-Abl, specifically in the kinase domain of Abl. Several of these amino acid substitution mutations directly abrogate imatinib binding to Bcr-Abl, thereby rendering imatinib ineffective.58 Similarly, NSCLC patients who become resistant to gefitinib or erlotinib are found to have tumor cells that acquire mutations in Egfr in the kinase domain such as T790M mutation.59 In order to counter this acquired resistance to targeted therapy, second-generation small-molecule inhibitors were developed with molecular configurations that allow binding to the mutated target, which include dasatinib and nilotinib for imatinib-resistant CML.60 In addition, monoclonal antibodies that bind to the extracellular domain of Egfr, such as cetuximab, provide a means to counter gefitinib or erlotinib-resistance, along with other small-molecule inhibitors that target Egfr.61
Antiestrogen therapy resistance in breast cancer
One of the targeted therapies with a long history is the antiestrogen agent, tamoxifen, in the treatment of breast cancer. Tamoxifen is an inhibitor that competes with estrogen for binding to the estrogen receptor (ER). Tamoxifen is proven effective as an adjuvant therapy in preventing recurrence and improving disease-free survival of patients with ER-positive breast cancer. The Early Breast Cancer Trialists’ Collaborative Group reported a meta-analysis of the randomized clinical trials that spanned over 20 years since 1985, which showed 30%–50% reduction in the disease recurrence rate with 5 years of tamoxifen therapy in ER-positive breast cancer patients.62 As ER-positive breast cancer constitutes 70% of all breast cancers, tamoxifen and other antiestrogen therapies have made a significant impact on breast cancer treatment. However, it has also been recognized that 30% or more of patients with ER-positive breast cancer do not benefit from tamoxifen therapy and relapse annually at a steady rate of 1%–5% even during the tamoxifen therapy.63 Thus, studies have focused on the mechanisms of tamoxifen resistance for as long as tamoxifen has been in use.
Using molecular approaches as well as siRNA screening in cell model systems, several genes and pathways have been identified as responsible for tamoxifen resistance. These include overexpression of the MYC oncogene, amplification of ERBB2/HER2, activation of the insulin/Igf receptor signaling, and mutations in the PI3k pathway molecules.64–67 However, genetic alterations of these genes and pathways have been implicated in other drug therapy resistance as well, suggesting that these may confer general cell growth/survival advantage mechanisms under unfavorable conditions, not tamoxifen resistance per se.68–70
More revealing are the specific mutations in the ESR1 gene that encodes ER, recently identified in 20%–50% of recurrent tumor samples from breast cancer patients who were treated with antiestrogen therapy of aromatase inhibitors and/or tamoxifen.71,72 Aromatase inhibitors such as anastrozole are a newer class of antiestrogen therapy agents that block the conversion of androgens to estrogens. This was proven an effective strategy to reduce estrogen in postmenopausal women whose estrogen production relies on local tissues, thereby reducing estrogen action on breast tumor cells. The specific amino acid substitution mutations found in recurrent tumors result in mimicking the active conformation of ER without estrogen bound to it, rendering cancer cells to no longer require estrogen for growth.71,72 Mutations in the ESR1 gene are rarely found in primary tumors, estimated less than 1% according to TCGA data. Thus, the ESR1 mutations found in recurrent tumors are de novo-acquired mutations under the selective pressure of antiestrogen therapy.
Another successful targeted agent in breast cancer is trastuzumab. Trastuzumab is a monoclonal antibody that targets the ERBB2/HER2 receptor amplified and/or overexpressed in the HER2 subtype of breast cancer. It is used in an adjuvant setting like the antiestrogen therapy and has been shown to reduce the recurrence rate of HER2-positive breast cancer by 30%–50%.73,74 With a shorter history of the use, it remains to be determined whether the Her2 receptor too acquires specific amino acid mutations de novo in response to trastuzumab therapy.
Mutations in parallel or the same signaling pathways
As is the case with tamoxifen-resistant breast cancer cells, it has been demonstrated that mutations and amplification of growth receptor genes including IGFR, MET, and FGFR family members confer resistance to various targeted therapies.68,69,75 These suggest that when a cell proliferation signaling pathway is inhibited by targeted agents, tumor cells evolve to activate other growth receptor signaling. Fortunately, some of these signal pathways can be targeted with agents, providing hope for countering resistance.76
Another emerging mechanism of acquired resistance is amplification or reactivation of the same targeted pathway genes. It has been shown that vemurafenib that targets BRAF(V600E) mutation is effective in treating melanoma but has limited success in treating colon cancer despite the same BRAF(V600E) mutation. Subsequently, it was shown that vemurafenib-resistant colon cancer cells reactivate EGFR, an upstream activator of b-Raf, via amplification or overexpression, shortly after vemurafenib treatment.77 Experimental studies showed that the combination treatment with vemurafenib and gefitinib (or erlotinib) was more effective in killing colorectal tumor cells than either agent alone.77 These lines of studies suggest that targeting multiple molecules in the same pathway may be effective in treating cancer by preempting therapy resistance due to the targeted pathway reactivation. These await clinical studies.
Intra-tumor heterogeneity
The time frame that therapy-resistant clones become detectable after initial targeted therapy varies widely from weeks to months (eg, NSCLC with gefitinib or erlotinib therapy) and to years (eg, breast cancer with tamoxifen therapy). It is not clear whether the time to resistance depends on therapeutics (eg, small-molecule kinase inhibitors vs hormone mimetics) or cancer cell types (NSCLC vs breast cancer) or both. The mechanism of acquired targeted therapy resistance – mutations in the target and activation of the same or parallel pathways of the target – is widely accepted as via molecular evolution under selective pressure in a Darwinian sense. However, the time frame in which some of the therapy-resistant diseases arise cannot solely be explained by evolution under selective pressure. For instance, ~2% of CML patients do not respond, and additional 8%–13% have inadequate response to imatinib at disease presentation, suggesting that imatinib-resistant leukemic cells already exist prior to therapy.78 These are the patients who have progressed through the “blasting crisis” characterized as rapid induction and expansion of leukemic cells, suggesting that imatinib-resistant tumor cells may arise during blasting crisis. It is remarkable that the specific mutations in the kinase domain of Abl that confer imatinib resistance arise through seemingly a nonspecific tumor progression stage called blasting crisis without the drug selection. Consistent with this observation in CML, tumor cells with EGFR (T790M) mutation or MET-amplification that confers resistance to Egfr-targeted therapies were found in NSCLC patients prior to targeted therapies.68 These findings show that mutations that confer targeted therapy resistance can be acquired during tumor progression without the targeted therapy selection. Thus, intra-tumor heterogeneity is an emerging mechanism of therapy resistance, not acquired under therapy selection but inherent to tumor progression. Detection of preexisting therapy-resistant mutations in subpopulation within the primary tumor will enable the treatment of patients with multiple targeting agents at the time of initial targeted therapy. Moreover, better understanding of the tumor progression stages that give rise to intra-tumor heterogeneity, such as “blasting crisis” in CML, will be crucial in order to subvert therapy resistance.
Metastasis
The current paradigm of metastasis involves multiple steps in successive progression categorically describing cellular events. These steps include tumor invasion to local tumor parenchyma, intravasation into blood vessels, circulation within the blood stream, extravasation out of blood vessels, and colonization at distant sites.79–82 Utilizing elegant experimental model systems, many genes and factors that contribute to metastatic progression have been identified. The examples are cell migration/motility genes (eg, Ral small GTPase signaling), extracellular matrix-remodeling molecules (eg, MMPs), and cell adhesion molecules (eg, integrins) that enable tumor cell invasion.83–86 The genes that commence epithelial–mesenchymal transition (EMT) were shown to play a role in local tissue invasion and intravasation of tumor cells (eg, Twist, Snail).87,88 In addition, cell adhesion molecules (eg, VCAM-1, ST6GALNAC5) were shown to play a role in mediating the extravasation step in metastasis.89,90 Moreover, contribution of immune cells has been well documented in metastatic processes such as local tissue invasion, intravasation, as well as distant-site colonization.91–95 Collectively, these studies provide a picture of metastasis as an intricate and complex process requiring activation of fine-tuned signaling pathways and also concerted interactions between tumor cells and the tissue microenvironment consisting of stroma, blood vessels, and immune cells. As each of these components in metastatic processes has been reviewed elsewhere, this overview highlights recent findings that reveal several striking features of metastasis.
No common “metastasis genes”
Metastatic cancer has been correlated with large-size and high-grade primary tumors, which has implied that metastasis is a later event in tumorigenesis. Supportive of this, early detection and resection of primary tumors reduce the risk of metastatic recurrence, suggesting that early-stage tumors have less metastatic potential. Thus, it has long been postulated that metastasis arises from tumor cells acquiring “metastasis gene mutations” during later stages in tumor progression. In this postulate, one could compare genetic alterations in metastatic lesions to its primary tumor origin and identify gene mutations uniquely present in metastatic lesions as metastasis-driving gene mutations. Genome-scale studies of primary tumor/metastasis pairs revealed that each metastatic lesion contains the genetic footprint of the primary tumor and also additional gene mutations not present in the primary tumor.96–98 These studies corroborated the postulate that metastases arise from primary tumors by acquiring additional mutations. Strikingly however, no common gene mutations have emerged among metastatic tumor samples. Moreover, comparison of the genome sequences between multiple metastatic lesions from a single patient showed that each metastatic lesion has evolved independently of each other acquiring different gene mutations.96 These findings indicate that there are no prerequisite gene mutations that drive metastasis, and hence no “metastasis genes”.
If there are no “metastasis genes”, what determines metastatic potential of a tumor? Early gene expression studies have identified gene signatures in primary tumors that correlate with and are predictive of metastasis and poor patient survival.99–101 These findings indicated that metastatic potential of a tumor is already established during the development of primary tumors. Consistent with this idea, recent genomic sequencing studies revealed that the same cancer driver mutations found in primary tumors such as KRAS, TP53, and ERBB2/HER2 are highly enriched in metastatic lesions of pancreatic cancer, renal carcinoma, and breast cancer, respectively.97,102,103 These findings suggest that metastasis is driven by the same genetic alterations that drive tumorigenesis, likely by providing the same advantages in cell survival and proliferation through arduous metastatic processes of invasion, intravasation, extravasation, and colonization. The elegant study by Jones et al documented clonal evolution of tumor cells by evaluating genetic signature changes during the progression from adenoma to carcinoma, and to metastasis, in colon cancer.96 The authors assessed the time frame of tumor progression by a mathematical model and reported that it takes 17 years for benign adenoma to progress to carcinoma, whereas it takes only 2 years for carcinoma cells to become metastatic. The short duration of 2 years for the metastatic progression supports the idea that primary tumors have already acquired the genetic mutations that enable metastasis. Collectively, these findings suggest that metastasis, at least the initial steps of metastasis, such as intravasation, is not driven by a set of metastasis-specific gene mutations.
Metastatic lesions do acquire additional mutations albeit different from one lesion to another. These mutations may potentially represent genes that play a role in the later steps of metastasis, such as colonization. “Colonization” refers to the complex process of individual tumor cells adapting to distant tissue environment and becoming overt metastases. The “colonization genes” may be decipherable by comparing de novo mutations found in metastases at the same site from different patients and/or cancer types. While these genes are yet to emerge, since a sufficient number of metastases per distant site has not been sequenced, experimental model studies have provided evidence for such genes. For example, ST6GALNAC5, a surface molecule-modifying enzyme, was found highly expressed specifically in breast cancer cells that metastasize to the brain, whereas VCAM-1, a cell adhesion molecule, was shown to promote lung metastasis by providing survival advantage to tumor cells in the lungs.89,90 It remains to be determined whether a common set of colonization-specific genes emerges per distant metastatic site across different cancer types. It will provide an opportunity to develop therapy targeting such genes and pathways and prevent overt metastases.
Alternatively and/or additionally, metastatic progression including the colonization step may be largely driven by epigenetic changes in tumor cells, extrinsic local factors, and/or systemic state of each patient. These factors have not been evaluated systematically, but identification of such factors presents a formidable task, considering the complexity and diversity that constitute individual tumor and tumor host environment.
Circulating tumor cells
As overt metastases are currently untreatable, considerable efforts have been made to advance technology to detect early metastatic cells disseminated into the blood stream, namely circulating tumor cells (CTCs).104 The presence of CTCs in patients with metastatic breast cancer has been correlated with reduced progression-free survival and overall survival.105 Even in early breast cancer patients, CTCs have been detected in 24% of patients and were predictive of early recurrence and decreased overall survival.106 These provide evidence that CTCs are biologically relevant, the presence of which is indicative of metastatic disease. However, the current difficulty of using CTCs as a biomarker of metastatic disease is due to wide variability in the CTC detection range depending on technology. Reportedly, a method called leukapheresis could detect CTCs in up to 90% of non-metastatic early breast cancer patients, demonstrating that even early-stage tumors release tumor cells into the blood stream albeit in a quantity that may not be readily detectable by conventional methods.107 Taken together, the presence of CTCs is indicative of metastatic disease, while the absence of CTCs may not ensure non-metastatic disease.
Nonetheless, through these technological advances in detecting CTCs, it is now well evidenced that the majority of epithelial-origin cancers release tumor cells into the blood stream.108,109 It is not clear, however, whether a tumor has to progress to a certain definable stage before it starts releasing CTCs. In breast cancer patients, disseminated tumor cells were found in the bone marrow regardless of the size or stage of primary tumors, which included ductal cell carcinoma in situ (DCIS).110 Provocatively, CTCs were detected in a mouse model of pancreatic cancer even before primary tumors became histologically invasive.111 These findings indicate that tumor dissemination is an early event during tumorigenesis, possible preceding tumor cell invasion to local tumor parenchyma. Moreover, genomic studies have shown that some CTCs contain fewer genetic alterations compared to the primary tumor of origin, suggesting that tumor cell dissemination may occur even prior to the establishment of primary tumors.112 These studies indicate that tumor cell release into the blood stream occurs early in tumorigenesis without requiring primary tumors to progress to an “advanced” stage.
These findings challenged the previous views of the intravasation/dissemination step in metastasis that it is neither driven by a set of specific gene mutations nor the result of advanced tumor behavior that includes tumor angiogenesis. The cellular mechanism of intravasation/dissemination is presently unclear. It is possible that the very initial genetic alterations in tumorigenesis, presumably the ones that provide proliferation advantage, may also enable cell dissemination. These early mutations could be identified by genomic studies of CTCs isolated from patients with very early-stage cancer or with precancerous lesions. This is well under way with sequencing of CTCs at a single-cell resolution.112 An alternative explanation may be that cell dissemination is a “normal” cellular process in that even normal cells disseminate at a certain rate. This postulate could be supported by the fact that EMT associated with tumor cell invasion/intravasation/dissemination is in fact a normal cellular process in the development of many tissue types.113 It will be crucial to determine the mechanisms and factors involved in the CTC-release step in order to strategize innovative therapy to target and prevent metastasis. It has been well demonstrated that the number of CTCs detected in the blood stream far exceeds the number of distant metastatic lesions in patients and in animal models alike.109,110 These findings suggest additional events such as mutations in “colonization genes” and/or factors in order for CTCs to become bona fide metastases. Taken together, technologic advances to detect CTCs reliably in very early-stage cancer patients may afford us to determine a therapeutic time window to target “colonization genes” and mitigate metastatic progression of the disease.
Summary and concluding remarks
The recent genomic studies have identified 120–140 cancer-driving genes, consisting of ~65 oncogenes and ~75 tumor suppressor genes. Evidently, tumorigenesis is driven by mutations in two to eight of these cancer-driving genes. These findings support the multistep tumorigenesis paradigm but also present an incredulous number of possible combinations of mutations that could give rise to cancer. This poses fundamental challenges to molecular targeted therapy, portending that one molecule-one targeted therapy is not sufficient to treat cancer. Furthermore, as we strive to target oncogene mutations via therapy development, we urgently need to think of innovative ways to treat cancers driven by the loss of tumor suppressor(s). The importance of addressing tumor suppressors in tumorigenesis is starkly underscored by the fact that more than 40% of cancers across tissue types have the loss of TP53. Considering the “mutual exclusivity” between TP53 and PIK3CA gene mutations, we propose a TP53/PIK3CA common genetic pathway model as one of the critical decision points of cell death/survival in tumorigenesis. If the model is proven valid, we could potentially consider treating TP53-mutated cancers with agents targeting the PI3k pathway and may achieve a therapeutic efficacy. With these therapy implications in mind, genetic interactions between cancer-driving genes especially related to tumor suppressor pathways warrant thorough investigation. New genes found frequently mutated in multiple cancers include histone modifiers and chromatin remodelers, most of which are also tumor suppressors. As mutations in these genes are likely to affect global scale gene transcription, it will require innovative approaches to delineate the mechanisms of these gene mutations in tumorigenesis and even more ingenuity to develop targeted therapy for cancers driven by such gene mutations.
The mechanisms of targeted therapy resistance include acquired de novo mutations in the targets themselves and/or reactivation of the targeted pathway. Additionally, general amplification or activation of powerful growth signals not directly related to the targeted pathway may also provide a mechanism for therapy resistance. Less understood is the intra-tumor heterogeneity that occurs during tumor progression prior to therapy, which confers therapy resistance at disease presentation or shortly after initial therapy. Due to limitations of our current technology (tumor sampling bias and sequencing sensitivity), the scope of heterogeneity in each tumor is unclear. Technological development to assess intra-tumor heterogeneity and evaluate relevant mutations in subpopulation of tumor cells, preferably in early stages of cancer, will enable us to better strategize to manage therapy resistance.
Metastatic progression of cancer remains the most challenging issue facing cancer treatment today. Recent findings of no common metastasis-specific genes have challenged the metastasis paradigm of stepwise evolution by acquiring metastasis-specific gene mutations. In addition, the genetic heterogeneity detected between multiple metastases in a single patient presents an onerous challenge in the development of targeted therapy for metastatic disease. Combination therapy is logical but a daunting task regarding the number of possible combinations. The findings that CTCs are detected even in early-stage non-metastatic cancer patients indicate that tumor dissemination occurs in a much earlier time frame than previously thought, directly challenging the long-standing paradigm that metastatic initiation is a later event in tumorigenesis.
Collectively, recent findings show that tumors gain genetic heterogeneity and systemic dissemination during very early stages of tumorigenesis, which also provide mechanisms for therapy resistance (Figure 3). These present challenges in strategizing cancer treatment and management going forward. First, it will be critical to be able to assess the genetic heterogeneity at the time of diagnosis in considering targeted therapy, or combination thereof. Conceptually, an optimal time frame for effective targeted therapy would be before intra-tumor heterogeneity arises. Investigation of the cellular and molecular mechanisms that give rise to genetic heterogeneity in early tumorigenesis may reveal tractable targets for therapy to treat or prevent tumor progression. Second, technology development of reliably detecting and isolating CTCs in very early-stage cancer patients or precancer patients will provide means to strategize targeted therapy to treat and/or prevent inevitable metastatic progression of the disease. As adjuvant targeted therapy for early-stage breast cancer has proven effective in reducing recurrence, targeted therapy is likely to be more effective in treating early-stage cancers, and more importantly, in preventing tumor progression/metastatic recurrence. Third, although a set of metastasis-specific genes is of no avail, it is still within the scope that we may find “colonization-specific genes”. As genome sequencing becomes more affordable, we will be able to obtain such a dataset representing metastases corresponding to each metastatic site in the near future. Such genes and pathways may provide therapy targets to treat and/or prevent overt metastases to specific organs. Lastly, an optimistic view in cancer treatment comes from the emergence of immunotherapy including ipilimumab, nivolumab, and other agents that target T-cell inhibitory molecules.114 These agents result in unguarded T-cell activation and T-cell-mediated immune response against tumor cells. Eliciting the “host” immune response to eliminate cancer is attractive because of the systemic nature of the effect, which has the potential to eradicate disseminated tumor cells during early stages of tumorigenesis and to prevent metastatic progression. Fundamental basic research of the molecular and cellular mechanisms governing the intricate interplay between tumor and immune cells will instruct us how to optimally modulate the immune response in cancer treatment. Combined use of immunotherapy and targeted therapy upfront in very early stages of cancer utilizing sophisticated CTC detection as a guideline as to the therapy time window may prove to be effective in preventing metastasis and improve cancer mortality.
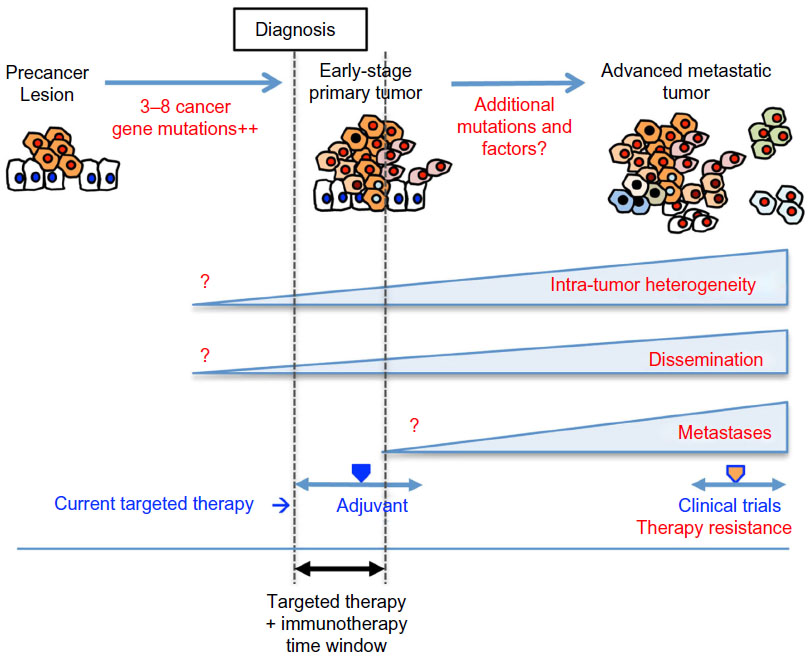 | Figure 3 Schematic illustration of tumorigenesis. |
Acknowledgments
Thanks to Dr Christopher J Yoo for his critical reading of this manuscript and for his helpful discussions on all topics, especially on therapeutics. Also acknowledging Dr Jonathan Keats for his helpful discussions on genomics and Madeline Keenen for her helpful comments on the manuscript.
Disclosure
The author reports no conflicts of interest in this work.
References
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64(1):9–29. | |
Armitage P, Doll R. The age distribution of cancer and a multi-stage theory of carcinogenesis. Br J Cancer. 1954;8(1):1–12. | |
Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. 2004;6(2):171–183. | |
Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–767. | |
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. | |
Druker BJ, Tamura S, Buchdunger E, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2(5):561–566. | |
Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448(7153):561–566. | |
Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363(18):1693–1703. | |
Thatcher N, Chang A, Parikh P, et al. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet. 2005;366(9496):1527–1537. | |
Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. Cancer genome landscapes. Science. 2013;339(6127):1546–1558. | |
Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502(7471):333–339. | |
Watson IR, Takahashi K, Futreal PA, Chin L. Emerging patterns of somatic mutations in cancer. Nat Rev Genet. 2013;14(10):703–718. | |
Neumann J, Zeindl-Eberhart E, Kirchner T, Jung A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol Res Pract. 2009;205(12):858–862. | |
Okudela K, Woo T, Kitamura H. KRAS gene mutations in lung cancer: particulars established and issues unresolved. Pathol Int. 2010;60(10):651–660. | |
Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol. 2008;9(5):402–412. | |
el-Deiry WS, Tokino T, Velculescu VE, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75(4):817–825. | |
el-Deiry WS. Regulation of p53 downstream genes. Semin Cancer Biol. 1998;8(5):345–357. | |
Nakano K, Vousden KH. PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell. 2001;7(3):683–694. | |
Moll UM, Marchenko N, Zhang XK. p53 and Nur77/TR3 – transcription factors that directly target mitochondria for cell death induction. Oncogene. 2006;25(34):4725–4743. | |
Tasdemir E, Maiuri MC, Orhon I, et al. p53 represses autophagy in a cell cycle-dependent fashion. Cell Cycle. 2008;7(19):3006–3011. | |
Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature. 2009;458(7242):1127–1130. | |
Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer. 2007;7(12):961–967. | |
Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424–430. | |
Carson JD, Van Aller G, Lehr R, et al. Effects of oncogenic p110alpha subunit mutations on the lipid kinase activity of phosphoinositide 3-kinase. Biochem J. 2008;409(2):519–524. | |
Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9(8):550–562. | |
Simi L, Pratesi N, Vignoli M, et al. High-resolution melting analysis for rapid detection of KRAS, BRAF, and PIK3CA gene mutations in colorectal cancer. Am J Clin Pathol. 2008;130(2):247–253. | |
Gonsalves WI, Mahoney MR, Sargent DJ, et al; Alliance for Clinical Trials in Oncology. Patient and tumor characteristics and BRAF and KRAS mutations in colon cancer, NCCTG/Alliance N0147. J Natl Cancer Inst. 2014;106(7). | |
del Carmen MG, Birrer M, Schorge JO. Clear cell carcinoma of the ovary: a review of the literature. Gynecol Oncol. 2012;126(3):481–490. | |
TCGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490(7418):61–70. | |
Hoadley KA, Yau C, Wolf DM, et al; Cancer Genome Atlas Research Network. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell. 2014;158(4):929–944. | |
TCGA. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511(7511):543–550. | |
Singh B, Reddy PG, Goberdhan A, et al. p53 regulates cell survival by inhibiting PIK3CA in squamous cell carcinomas. Genes Dev. 2002;16(8):984–993. | |
Ming M, He YY. PTEN in DNA damage repair. Cancer Lett. 2012; 319(2):125–129. | |
Mayo LD, Donner DB. The PTEN, Mdm2, p53 tumor suppressor-oncoprotein network. Trends Biochem Sci. 2002;27(9):462–467. | |
Miller TW, Rexer BN, Garrett JT, Arteaga CL. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13(6):224. | |
Kinzler KW, Vogelstein B. Landscaping the cancer terrain. Science. 1998;280(5366):1036–1037. | |
Shen C, Kaelin WG Jr. The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol. 2013;23(1):18–25. | |
Kouros-Mehr H, Kim JW, Bechis SK, Werb Z. GATA-3 and the regulation of the mammary luminal cell fate. Curr Opin Cell Biol. 2008; 20(2):164–170. | |
Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014;26(3):570–579. | |
Schermer B, Ghenoiu C, Bartram M, et al. The von Hippel-Lindau tumor suppressor protein controls ciliogenesis by orienting microtubule growth. J Cell Biol. 2006;175(4):547–554. | |
Robinson CM, Ohh M. The multifaceted von Hippel-Lindau tumour suppressor protein. FEBS Lett. 2014;588(16):2704–2711. | |
Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell. 2006;127(5):1041–1055. | |
Maher ER, Neumann HP, Richard S. von Hippel-Lindau disease: a clinical and scientific review. Eur J Hum Genet. 2011;19(6):617–623. | |
Simons A, Sikkema-Raddatz B, de Leeuw N, Konrad NC, Hastings RJ, Schoumans J. Genome-wide arrays in routine diagnostics of hematological malignancies. Hum Mutat. 2012;33(6):941–948. | |
Schuback HL, Arceci RJ, Meshinchi S. Somatic characterization of pediatric acute myeloid leukemia using next-generation sequencing. Semin Hematol. 2013;50(4):325–332. | |
Challen GA, Sun D, Jeong M, et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44(1):23–31. | |
Oka M, Meacham AM, Hamazaki T, Rodic N, Chang LJ, Terada N. De novo DNA methyltransferases Dnmt3a and Dnmt3b primarily mediate the cytotoxic effect of 5-aza-2′-deoxycytidine. Oncogene. 2005;24(19):3091–3099. | |
Im AP, Sehgal AR, Carroll MP, et al. DNMT3A and IDH mutations in acute myeloid leukemia and other myeloid malignancies: associations with prognosis and potential treatment strategies. Leukemia. 2014;28(9):1774–1783. | |
Rakheja D, Konoplev S, Medeiros LJ, Chen W. IDH mutations in acute myeloid leukemia. Hum Pathol. 2012;43(10):1541–1551. | |
Ley TJ, Ding L, Walter MJ, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363(25):2424–2433. | |
Ansari KI, Mandal SS. Mixed lineage leukemia: roles in gene expression, hormone signaling and mRNA processing. FEBS J. 2010;277(8):1790–1804. | |
Muntean AG, Hess JL. The pathogenesis of mixed-lineage leukemia. Annu Rev Pathol. 2012;7:283–301. | |
Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11(7):481–492. | |
Wu JN, Roberts CW. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3(1):35–43. | |
Guan B, Wang TL, Shih IeM. ARID1A, a factor that promotes formation of SWI/SNF-mediated chromatin remodeling, is a tumor suppressor in gynecologic cancers. Cancer Res. 2011;71(21):6718–6727. | |
Zang ZJ, Cutcutache I, Poon SL, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44(5):570–574. | |
Gao X, Tate P, Hu P, Tjian R, Skarnes WC, Wang Z. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci U S A. 2008;105(18):6656–6661. | |
Shah NP, Nicoll JM, Nagar B, et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell. 2002;2(2):117–125. | |
Pao W, Miller VA, Politi KA, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2(3):e73. | |
Kimura S, Ando T, Kojima K. Ever-advancing chronic myeloid leukemia treatment. Int J Clin Oncol. 2014;19(1):3–9. | |
Camidge DR, Pao W, Sequist LV. Acquired resistance to TKIs in solid tumours: learning from lung cancer. Nat Rev Clin Oncol. 2014;11(8):473–481. | |
EBCTCG. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005;365(9472):1687–1717. | |
Goss PE, Ingle JN, Pater JL, et al. Late extended adjuvant treatment with letrozole improves outcome in women with early-stage breast cancer who complete 5 years of tamoxifen. J Clin Oncol. 2008;26(12):1948–1955. | |
Miller TW, Balko JM, Ghazoui Z, et al. A gene expression signature from human breast cancer cells with acquired hormone independence identifies MYC as a mediator of antiestrogen resistance. Clin Cancer Res. 2011;17(7):2024–2034. | |
Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011;62:233–247. | |
Fox EM, Miller TW, Balko JM, et al. A kinome-wide screen identifies the insulin/IGF-I receptor pathway as a mechanism of escape from hormone dependence in breast cancer. Cancer Res. 2011;71(21):6773–6784. | |
Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3-kinase and antiestrogen resistance in breast cancer. J Clin Oncol. 2011;29(33):4452–4461. | |
Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14(10):2895–2899. | |
Wilson TR, Fridlyand J, Yan Y, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487(7408):505–509. | |
Rosenzweig SA. Acquired resistance to drugs targeting receptor tyrosine kinases. Biochem Pharmacol. 2012;83(8):1041–1048. | |
Robinson DR, Wu YM, Vats P, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45(12):1446–1451. | |
Toy W, Shen Y, Won H, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45(12):1439–1445. | |
Romond EH, Perez EA, Bryant J, et al. Trastuzumab plus adjuvant chemotherapy for operable HER2-positive breast cancer. N Engl J Med. 2005;353(16):1673–1684. | |
Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al; Herceptin Adjuvant (HERA) Trial Study Team. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353(16):1659–1672. | |
Ware KE, Hinz TK, Kleczko E, et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis. 2013;2:e39. | |
Wong KK, Engelman JA, Cantley LC. Targeting the PI3K signaling pathway in cancer. Curr Opin Genet Dev. 2010;20(1):87–90. | |
Prahallad A, Sun C, Huang S, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483(7387):100–103. | |
Druker BJ, Guilhot F, O’Brien SG, et al; IRIS Investigators. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355(23):2408–2417. | |
Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8(5):341–352. | |
Nguyen DX, Bos PD, Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9(4):274–284. | |
Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147(2):275–292. | |
Vanharanta S, Massague J. Origins of metastatic traits. Cancer Cell. 2013;24(4):410–421. | |
Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406(6795):532–535. | |
Gentry LR, Martin TD, Reiner DJ, Der CJ. Ral small GTPase signaling and oncogenesis: more than just 15minutes of fame. Biochim Biophys Acta. 2014;1843(12):2976–2988. | |
Deryugina EI, Quigley JP. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006;25(1):9–34. | |
Ganguly KK, Pal S, Moulik S, Chatterjee A. Integrins and metastasis. Cell Adh Migr. 2013;7(3):251–261. | |
Yang J, Mani SA, Donaher JL, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117(7):927–939. | |
Kang Y, Massague J. Epithelial-mesenchymal transitions: twist in development and metastasis. Cell. 2004;118(3):277–279. | |
Chen Q, Zhang XH, Massague J. Macrophage binding to receptor VCAM-1 transmits survival signals in breast cancer cells that invade the lungs. Cancer Cell. 2011;20(4):538–549. | |
Bos PD, Zhang XH, Nadal C, et al. Genes that mediate breast cancer metastasis to the brain. Nature. 2009;459(7249):1005–1009. | |
Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124(2):263–266. | |
Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet? Science. 2013;339(6117):286–291. | |
Tan W, Zhang W, Strasner A, et al. Tumour-infiltrating regulatory T cells stimulate mammary cancer metastasis through RANKL-RANK signalling. Nature. 2011;470(7335):548–553. | |
Wolf MJ, Hoos A, Bauer J, et al. Endothelial CCR2 signaling induced by colon carcinoma cells enables extravasation via the JAK2-Stat5 and p38MAPK pathway. Cancer Cell. 2012;22(1):91–105. | |
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–899. | |
Jones S, Chen WD, Parmigiani G, et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci U S A. 2008;105(11):4283–4288. | |
Campbell PJ, Yachida S, Mudie LJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467(7319):1109–1113. | |
Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–1117. | |
van ‘t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415(6871):530–536. | |
van de Vijver MJ, He YD, van’t Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347(25):1999–2009. | |
Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33(1):49–54. | |
Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–892. | |
Shah SP, Morin RD, Khattra J, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461(7265):809–813. | |
Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol. 2011;192(3):373–382. | |
Cristofanilli M, Budd GT, Ellis MJ, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351(8):781–791. | |
Lucci A, Hall CS, Lodhi AK, et al. Circulating tumour cells in non-metastatic breast cancer: a prospective study. Lancet Oncol. 2012;13(7):688–695. | |
Fischer JC, Niederacher D, Topp SA, et al. Diagnostic leukapheresis enables reliable detection of circulating tumor cells of nonmetastatic cancer patients. Proc Natl Acad Sci U S A. 2013;110(41):16580–16585. | |
Nagrath S, Sequist LV, Maheswaran S, et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007;450(7173):1235–1239. | |
Stott SL, Lee RJ, Nagrath S, et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Transl Med. 2010;2(25):25ra23. | |
Husemann Y, Geigl JB, Schubert F, et al. Systemic spread is an early step in breast cancer. Cancer Cell. 2008;13(1):58–68. | |
Rhim AD, Mirek ET, Aiello NM, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1–2):349–361. | |
Schardt JA, Meyer M, Hartmann CH, et al. Genomic analysis of single cytokeratin-positive cells from bone marrow reveals early mutational events in breast cancer. Cancer Cell. 2005;8(3):227–239. | |
Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139(5):871–890. | |
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.