Back to Journals » Neuropsychiatric Disease and Treatment » Volume 17
Neuroprotective Function of TNFAIP3 Interacting Protein 2 Against Oxygen and Glucose Deprivation/Reoxygenation-Induced Injury in Hippocampal Neuronal HT22 Cells Through Regulation of the TLR4/MyD88/NF-κB Pathway
Authors Yan Z, Chen Y, Zhang X, Hua L ![]() , Huang L
, Huang L
Received 25 February 2021
Accepted for publication 25 June 2021
Published 8 July 2021 Volume 2021:17 Pages 2219—2227
DOI https://doi.org/10.2147/NDT.S308360
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 4
Editor who approved publication: Professor Taro Kishi
Zhaoxian Yan,1 Yahui Chen,2 Xin Zhang,3 Lin Hua,3 Lifa Huang3
1First Clinical Medical College, Zhejiang Chinese Medical University, Hangzhou, Zhejiang, 310053, People’s Republic of China; 2Department of Rheumatology, Ningbo No.6 Hospital, Ningbo, 315040, Zhejiang, People’s Republic of China; 3Department of Neurosurgery, Zhejiang Provincial Hospital of Traditional Chinese Medicine, The First Affiliated Hospital of Zhejiang Chinese Medical University, Hangzhou, 310006, Zhejiang, People’s Republic of China
Correspondence: Lifa Huang
Department of Neurosurgery, Zhejiang Provincial Hospital of Traditional Chinese Medicine, The First Affiliated Hospital of Zhejiang Chinese Medical University, No. 54 Youdian Road, Shangcheng District, Hangzhou, 310006, Zhejiang, People’s Republic of China
Email [email protected]
Background: Tumor necrosis factor-α (TNF-α)-induced protein 3-interacting protein 2 (TNIP2) has been well demonstrated to act as a principal contributor to the development of inflammatory diseases; however, the role of TNIP2 in cerebral ischemic/reperfusion injury has never been studied.
Methods: Gene expression was examined by using quantitative real-time polymerase chain reaction and Western blot. The functional role of TNIP2 in oxygen and glucose deprivation/reoxygenation (OGD/R)-induced neuronal injury was evaluated using cell counting kit-8, terminal deoxynucleotidyl transferase dutp nick end labeling assay and enzyme-linked immunosorbent assay. Commercial kits were applied to evaluate the activity of NF-kappa-B (NF-κB) and caspase-3, as well as the release of lactate dehydrogenase release (LDH).
Results: TNIP2 expression was substantially declined in HT22 cells following OGD/R stimulation. TNIP2 overexpression attenuated ODG/R-induced inflammation in HT22 cells, as evidenced by reduced levels of TNF-α, interleukin (IL)-1β, and intercellular cell adhesion molecule-1 (ICAM-1), and increased levels of IL-10. TNIP2 overexpression also reduced activity of NF-κB under ODG/R condition. Meanwhile, OGD/R treatment caused a reduction of cell viability and an elevation of cell apoptosis in HT22 cells, as indicated by the increase in LDH and caspase-3 activity. Whereas, OGD/R-induced HT22 cell injury was mitigated by TNIP2 overexpression in HT22 cells. Besides, we found the involvement of toll-like receptor 4 (TLR4)/myeloid differentiation factor 88 (MyD88)/NF-κB pathway in the neuroprotective effect of TNIP2 on OGD/R-induced HT22 cell damage.
Conclusion: TNIP2 overexpression mitigates OGD/R-induced inflammatory response and apoptosis. Moreover, TLR4/MyD88/NF-κB pathway participates in the protective effect of TNIP2 on OGD/R-induced neuronal damage.
Keywords: TNF-α-induced protein 3-interacting protein 2, ischemic reperfusion injury, inflammation, apoptosis, toll-like receptor 4/myeloid differentiation factor 88/NF-kappa-B signaling
Introduction
Ischemic stroke resulting from the blockage of a cerebral vessel is one of the leading causes of death and disability throughout the world.1 The disorder of blood supply leads to the hypoxia and ischemia of brain issues, eventually resulting in cell death.2 Up to date, thrombolysis within 4 h after onset of symptoms is the most efficient treatment for ischemic stroke.3 Unfortunately, recovery of the cerebral circulation may further exacerbate the brain damage induced by ischemic stroke and cause the irreversible damage, otherwise known as the “ischemic/reperfusion (I/R) injury”.4 Remarkably, several clinical trials have demonstrated that cerebral I/R injury is the main pathophysiological mechanism of ischemic stroke.5 Therefore, how to reduce I/R injury has become the key issue in the ischemic stroke research field.
Tumor necrosis factor-α (TNF-α)-induced protein 3-interacting protein interacting protein 2 (TNIP2), also called A20-binding inhibitor of NF-kappa-B (NF-κB) activation 2, has been well demonstrated to be a negative regulator of NF-κB signaling in terms of its capability to bind A20 and dampen inflammatory cytokines-induced NF-κB activation.6 NF-κB is an important regulator of cellular processes, including cell apoptosis and inflammation.7 Abnormal expression of NF-κB has been discovered in various human diseases. Furthermore, targeting NF-κB is considered as a potential therapeutic approach for human diseases.8,9 Based on the literatures regarding the regulatory role of TNIP2 in NF-κB activation, it is not surprising that TNIP2 acts as a crucial player in human diseases. In hepatocellular carcinoma, TNIP2 was downregulated by miR-1180, and its silencing conversed miR-1180-mediated promotion of cell proliferation in HepG2 cells.10 Moreover, TNIP2 was downregulated in the blood of patients with multiple organ dysfunction syndrome, and its overexpression suppressed the inflammation response and oxidative stress induced by multiple organ dysfunction syndrome through inhibiting the activation of NF-κB.11 Besides, TNIP2 upregulation inhibited the apoptosis of endothelial cells and participate in the anti-apoptosis effect of angiopoietin-1.12 However, the functional role of TNIP2 in cerebral I/R injury has not been determined yet.
In this work, we aimed to determine the functional role of TNIP2 in cerebral I/R injury. TNIP2 was overexpressed in HT22 cells to evaluate its effect on inflammation and apoptosis under oxygen and glucose deprivation/reoxygenation (OGD/R) conditions. We identified the neuroprotective function of TNIP2 in cerebral I/R injury, which may provide evidence for the potential therapeutic target role of TNIP2 in cerebral I/R injury.
Materials and Methods
Cell Culture and Transfection
Murine HT22 cells from the American Type Culture Collection (Manassas, VA, USA) and then grown in Dulbecco’s modified Eagle’s medium (DMEM) plus 10% fetal bovine serum (FBS; Solarbio, Beijing, China) and 1% penicillin-streptomycin. HT22 cells were kept in a 5% CO2 humidified incubator at 37°C.
In order to construct an in vitro model of cerebral I/R injury, HT22 cells were cultured in glucose-free DMEM for 4 h under hypoxic condition (37°C, 5% CO2/95% N2), followed by incubation for 6, 12, or 24 h in reoxygenation condition (37°C, 5% CO2/95% air).
Lentiviral TNIP2-overexpressing plasmid (LV-TNIP2) and the empty lentiviral vector (LV-Scramble) were purchased from GenePharma (Shanghai, China). After transfection with LV-TNIP2 or LV-Scramble for 48 h, HT22 cells were transferred to fresh culture medium. The transfection efficiency was verified using Western blot.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
RNA isolation was done using TRIzol kit from Beyotime (Shanghai, China), as directed by the manufacturer’s instruction. The concentration and purity of isolated RNA were assessed using a spectrophotometer. cDNAs were synthesized using PrimeScript™ RT Enzyme Mix I from Takara (Dalian, China). qRT-PCR analysis was carried out using SYBR®-Green PCR Master mix (TaRaRa). The relative expression of TNIP2 was calculated according to the 2–ΔΔCt method with β-actin as an internal control.
Western Blot Analysis
After treatment, total protein from HT22 cells were extracted by using RIPA lysate buffer from Beyotime, and then tested for protein concentration using the BCA protein assay kit from Solarbio. Nuclear and Cytoplasmic Protein Extraction Kit (Boster, Beijing, China) was utilized to isolate the nuclear and cytoplasmic proteins from HT22 cells following the manufacturer’s recommendation. After sodium dodecyl sulphate polyacrylamide gel electrophoresis, the separated protein was transferred onto polyvinylidene fluoride membranes. Subsequently, the members were blocked for 1 h with 5% non-fat milk, and then probed at 4°C overnight with the special primary antibodies against TNIP2 (Novus, Shanghai, China), toll-like receptor 4 (TLR4; Novus), myeloid differentiation factor 88 (MyD88; Novus), NF-кB (Novus), and β-actin (Novus). The membranes were rinsed with TBST for 3 times and then immunoblotted for 1 h with horseradish peroxidase-conjugated secondary antibody (Novus) at 37°C. The immunoblots were visualized using the chemiluminescence reagent from Solarbio, following the product manual.
Enzyme-Linked Immunosorbent Assay (ELISA)
After transfection with LV-TNIP2 or LV-Scramble, HT22 cells were exposed to OGD/R condition. Afterward, the cell supernatants were collected and tested for TNF-α, Interleukin (IL)-1β, intercellular cell adhesion molecule-1 (ICAM-1), and IL-10 levels using ELISA kits (Beyotime) in the light of the manufacturer’s recommendations.
Determination of NF-кB Activity
After treatment, HT22 cells were harvested and tested for NF-кB activity using the TransAMTM NF-κB p65 protein assay (Active Motif, Carlsbad, CA, USA) per manufacturer’s instructions. Nuclear lysates were acquired from HT22 cells by Nuclear Extraction kit (Active Motif). The protein concentration of nuclear lysates was detected using the BCA method (Beyotime). Nuclear extracts (20 μg) were added to each reaction system in 96-well plate, which were immobilized with double-stranded DNA oligonucleotides containing NF-κB consensus sequences. The activated NF-кB p65 and p50 specifically bound to this oligonucleotide was determined with the corresponding specific antibodies. The DNA-binding activity of NF-кB was determined by measurement of the absorbance at 450 nm with a microplate reader.
Cell Viability Assay
After transfection with LV-TNIP2 or LV-Scramble, HT22 cells were exposed to OGD/R, and then treated with cell counting kit-8 (CCK-8) solution (10 µL) from Dojindo (Kumamoto, Japan) for 2 h. Cell viability was evaluated by measurement of the absorbance at 450 nm.
Cytotoxicity was also assessed by determination of lactate dehydrogenase (LDH) release. In brief, HT22 cells were collected after OGD/R exposure, followed by incubation with LDH release reagent (Beyotime) for 1 h. After centrifugation, cell supernatant was treated with LDH detection reagent away from the light for 30 min. The release of LDH was examined by measurement of the absorbance at 490 nm.
Cell Apoptosis Assay
Cell apoptosis was evaluated using one step terminal deoxynucleotidyl transferase dutp nick end labeling (TUNEL) apoptosis assay kit from Beyotime, following the product manual. Briefly, HT22 cells grown on coverslips were rinsed with PBS solution, followed by fixation with 4% paraformaldehyde solution on a shaker for 30 min. Following permeation with 0.3% Triton X-100 in PBS for 5 min at room temperature, cells were incubated with TUNEL solution (50 μL) for 1 h at 37°C in dark condition. Whereafter, the coverslips were sealed with anti-fade mounting medium from Beyotime and analyzed by use of a fluorescence microscope (Olympus, Tokyo, Japan).
The apoptosis of HT22 cells were also evaluated by measurement of caspase-3 activity using GreenNuc™ Caspase-3 Assay Kit for Live Cells (Beyotime). After treatment, HT22 cells were collected and incubated with GreenNuc™ Caspase-3 Substrate in dark condition for 30 min. Thereafter, the samples were analyzed using a microplate reader (excitation: 485 nm; emission: 515 nm).
Statistical Analysis
The data were displayed as mean ± standard deviation. The comparison between groups was analyzed using Students’ t test and one-way analysis of variance by SPSS 20.0 software. P < 0.05 was considered statistically significant.
Results
Expression of TNIP2 in HT22 Cells Subjected to OGD/R
Several researches reported that TNIP2 is a crucial player in inflammatory diseases. However, little is known about the function of TNIP2 in OGD/R-induced inflammatory response. First, we evaluated the expression of TNIP2 in HT22 cells subjected to OGD/R using qRT-PCR and Western blot. As shown in Figure 1A, OGD/R substantially reduced the mRNA expression level of TNIP2 in HT22 cells, with the lowest expression of TNIP2 at 24 h after reoxygenation. Consistently, decreased expression of TNIP2 protein was discovered in HT22 cells subjected to OGD/R (Figure 1B).
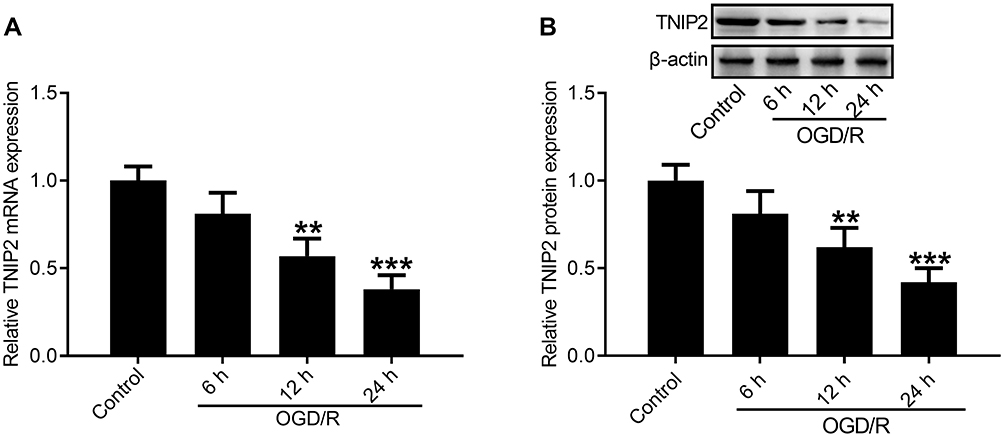 |
Figure 1 Expression of TNIP2 in HT-22 cells subjected to OGD/R. (A) qRT-PCR analysis of TNIP2 expression showed the reduced level of TNIP2 mRNA in HT22 cells exposed to OGD/R (n = 3). (B) Western blot analysis of TNIP2 expression showed the reduced level of TNIP2 protein in HT22 cells exposed to OGD/R (n = 3). **P <0.01 and ***P <0.001, compared with the control group. |
TNIP2 Overexpression Attenuates ODG/R-Induced Apoptosis in HT22 Cells
Since TNIP2 was downregulated following OGD/R, we upregulated the expression of TNIP2 in HT22 cells to study the biological role of TNIP2 in ODG/R-induced neuronal damage. LV-Scramble and LV-TNIP2 were transfected into HT22 cells for 48 h and the overexpression efficiency was identified. As determined by Western blot, the protein levels of TNIP2 were strikingly increased in LV-TNIP2-transfected HT22 cells compared with that in LV-Scramble-transfected HT22 cells (Figure 2A). The mRNA and protein expression levels of TNIP2 in LV-TNIP2-transfected HT22 cells were significantly increased under the condition of OGD/R (Figure 2B and C). To assess the neuroprotective effect of TNIP2 on OGD/R-induced neuronal damage, cell proliferation and cytotoxicity were determined using CCK-8 and LDH assays, respectively. The results showed that OGD/R exposure inhibited the viability of HT22 cells, however, upregulation of TNIP2 could reverse the inhibitory effect of OGD/R on cell viability in HT22 cells (Figure 2D). Consistently, we found that OGD/R promoted the release of LDH in HT22 cells, which was blocked by LV-TNIP2 transfection (Figure 2E). In parallel, TUNEL assay revealed that OGD/R induced the apoptosis of HT22 cells, as evidenced by the increased percentage of TUNEL positive cells. Whereas overexpression of TNIP2 rescued cell survival (Figure 2F). Similarly, caspase-3 activity was strikingly elevated following OGD/R, while overexpression of TNIP2 by LV-TNIP2 could weaken OGD/R-induced elevation of caspase-3 activity (Figure 2G).
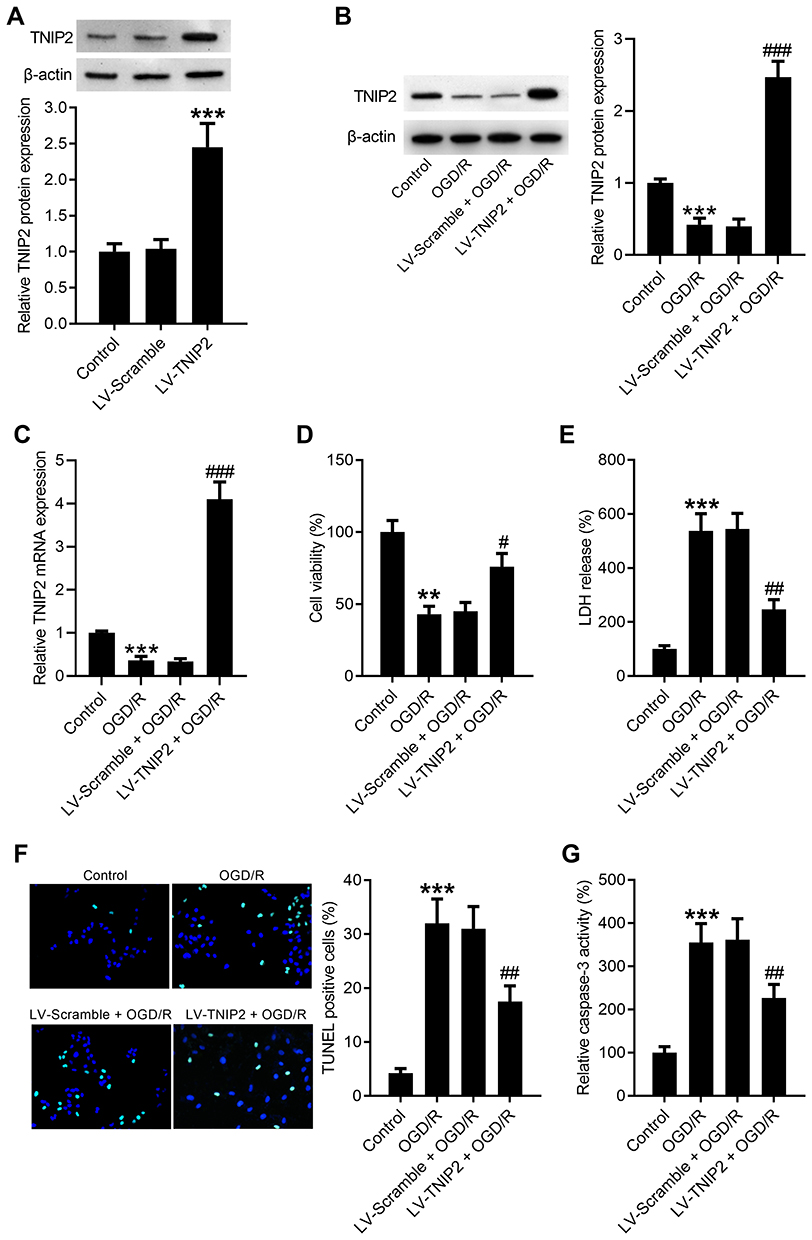 |
Figure 2 TNIP2 overexpression attenuates ODG/R-induced apoptosis in HT22 cells. (A) LV-Scramble and LV-TNIP2 were transfected into HT22 cells for 48 h and the transfection efficiency was confirmed using Western blot (n = 3). HT22 cells were transfected with LV-TNIP2 or LV-Scramble, followed by exposure to OGD/R. (B and C) The expression levels of TNIP2 protein and mRNA were detected by Western blot and qRT-PCR, respectively. (D) The viability of HT22 cells was examined using CCK-8 assay (n = 3). (E) The viability of HT22 cells was determined by measurement of LDH release (n = 3). (F) Representative images of TUNEL staining were shown and the bar graph represents the quantification of TUNEL positive cells (n = 3). (G) Caspase-3 activity assay kit was applied to examine the activity of caspase-3 (n = 3). **P <0.01 and ***P <0.001, compared with the LV-Scramble or control group. #P <0.05, ##P <0.01, and ###P <0.001, compared with the LV-Scramble + OGD/R group. |
TNIP2 Overexpression Attenuates ODG/R-Induced Inflammation in HT22 Cells
As indicated by ELISA assay, the levels of pro-inflammatory cytokines, TNF-α, IL-1β, and ICAM-1, were remarkably elevated in HT-22 cells exposed to OGD/R, whereas TNIP2 overexpression counteracted this tendency (Figure 3A–C). Meanwhile, the level of IL-10 in HT22 cells were decreased after OGD/R and forced expression of TNIP2 abolished this decline (Figure 3D). Besides, a marked increase in NF-кB activity in HT22 cells was discovered at 24 h after OGD/R, and this increase was blocked after LV-TNIP2 transfection (Figure 3E).
 |
Figure 3 TNIP2 overexpression attenuates ODG/R-induced inflammation in HT22 cells. ELISA assays were conducted to evaluate the levels of TNF-α (A), IL-1β (B), ICAM-1 (C), and IL-10 (D) in HT22 cells transfected with LV-TNIP2 or LV-Scramble after OGD/R (n = 3). (E) The activity of NF-кB was determined in HT22 cells transfected with LV-TNIP2 or LV-Scramble after OGD/R (n = 3). **P <0.01 and ***P <0.001, compared with the control group. #P <0.05 and ##P <0.01 compared with the LV-Scramble + OGD/R group. |
TLR4/MyD88/NF-κB Pathway is Implicated in the Protective Effect of TNIP2 on OGD/R-Induced Neuronal Damage
To investigate the possible mechanism involved in TNIP2-mediated protection against OGD/R-induced injury, the expression levels of TLR4, MyD88, NF-κB-cytoplasm, and NF-κB-nucleus were measured using Western blot. As displayed in Figure 4A and B, TLR4 and MyD88 expression levels were upregulated following OGD/R, and this tendency was counteracted following LV-TNIP2 transfection. Besides, the expression level of NF-κB was reduced in the cytoplasm of HT22 cells exposed to OGD/R, but was elevated in the nucleus of HT22 cells exposed to OGD/R. Notably, these changes triggered by OGD/R were abolished when LV-TNIP2 was transfected into HT22 cells.
 |
Figure 4 The TLR4/MyD88/NF-κB pathway is involved in the protective effects of TNIP2 on OGD/R-induced neuronal damage. HT22 cells were transfected with LV-TNIP2 or LV-Scramble, followed by exposure to OGD/R. (A) Representative and (B) quantitative Western blot analysis of TLR4, MyD88, NF-κB-cytoplasm, and NF-κB-nucleus expression in HT22 cells transfected with LV-TNIP2 or LV-Scramble following OGD/R (n = 3). ***P <0.001, compared with the control group. ##P <0.01, compared with the LV-Scramble + OGD/R group. |
Discussion
The mechanism of cerebral I/R injury is complex, which involves many biochemical events, such as oxidative stress, autophagy, and inflammatory response.13 Among them, inflammatory response is recognized as the key step in cerebral I/R injury and it is drawing more and more attention.14 During an ischemic insult, the dynamic balance between pro-inflammatory response and anti-inflammatory response was disrupted and the inflammatory cells were activated and recruited into the ischemic region. Subsequently, ischemic insult induced the production of pro-inflammatory cytokines and cellular adhesion molecules, such as TNF-α, IL-1β, and ICAM-1. Meanwhile, ischemic insult reduced the generation of anti-inflammatory cytokines (IL-10), which leads to brain damage and neuronal death.15 Critically, the reestablishment of cerebral blood flow further aggravated this inflammatory response induced by cerebral ischemia.16,17 So, how to effectively reduce cerebral I/R-induced inflammatory response is a critical question for ischemic stroke therapy.
TNIP2, a suppressor of NF-κB, has been shown to protect against excessive inflammatory response.18 Previously, TNIP2 as an essential protective factor of allergic airway inflammation has been established. Interaction between ABIN-2 and A20 exacerbated house dust mite-induced allergic responses in airway of mice. Deletion of TNIP2 aggravated house dust mite-induced airway inflammation by impeding the binding of ABIN-2 to A20.19 In a recent report from Luo and colleagues, TNIP2 was downregulated in RAW264.7 macrophages treated with lipopolysaccharide (LPS), and participated in miR-15a-5p-mediated regulation of the inflammatory process during sepsis.20 In both murine and cell models of acute pancreatitis, cerulein stimulation induced the downregulation of TNIP2. Upregulation of TNIP2 mitigated acute pancreatitis-induced myocardial injury in in vitro and in vivo.21 TNIP2 was reportedly downregulated in the tissues from patients with lupus nephritis. Moreover, TNIP2 was affirmed as a downstream target gene of miR-663a/miR-423-5p and functioned as a vital contributor to LPS-induced NF-κB activation.22 Additionally, it has been demonstrated that TNIP2 repressed TNF-α-induced NF-κB activation and then regulated the progression of human cancers.23 However, prior to our study, whether TNIP2 performs a function in cerebral I/R-injury has not yet been elucidated. We documented here that TNIP2 was downregulated in HT22 cells stimulated with OGD/R, revealing the involvement of TNIP2 in cerebral I/R injury. Moreover, OGD/R exposure promoted cell apoptosis, inhibited cell viability, and triggered inflammatory process, as evidenced by the elevated levels of TNF-α, IL-1β, and ICAM-1, the reduced levels of IL-10, and the increased activity of NF-кB. Significantly, TNIP2 overexpression could counteract OGD/R-induced HT22 cell injury, revealing the concept that TNIP2 overexpression mitigates OGD/R-induced neuronal damage.
The neuroprotective role of TNIP2 has been demonstrated in our study, but its mechanism is still blanked. The TLR4/MyD88/NF-кB signaling-mediated inflammatory response has been well reported to serve as a principal player in I/R injury.24 When the organism is stimulated by ischemic insult, the innate immunity is triggered, which in turn causes the adaptive immune response by inducing the expression of TLR4.25 Subsequently, TLR4 recruits MyD88 and then causes the translocation and activation of NF-кB, ultimately leading to the overproduction of inflammatory cytokines.26,27 As an example, madecassoside protected BV-2 cells against OGD/R-induced apoptosis and inflammation through inducing the inactivation of the TLR4/MyD88/NF-кB signaling pathway, indicating the involvement of the TLR4/MyD88/NF-кB signaling in cerebral I/R injury.28 In addition, cerebral I/R-induced the upregulation of genes belonged to the TLR4/MyD88/NF-кB signaling and then aggravated cerebral infarction damage, revealing that the TLR4/MyD88/NF-кB signaling may be a promising therapeutic approach for ischemic stroke.29 Interestingly, there is evidence to suggest that TNIP1 expression was downregulated in hepatic I/R mice and its upregulation could alleviate hepatic I/R-induced inflammatory response and apoptosis via the TLR4/MyD88/NF-кB signaling.30 Nevertheless, whether the TLR4/MyD88/NF-кB signaling is involved in the TNIP2-mediated neuroprotective effect still needs to be determined. Herein, we found that TNIP2 overexpression counteracted OGD/R-induced elevation of TLR4, MyD88 and cytoplasmic NF-кB expression, and decrease of nuclear NF-кB expression, implying involvement of the TLR4/MyD88/NF-кB signaling in the protective effect of TNIP2 on OGD/R-induced neuronal damage.
In conclusion, we found that TNIP2 was downregulated in the in vitro model of cerebral I/R injury. Functionally, forced expression of TNIP2 alleviated OGD/R-induced inflammation and apoptosis in HT22 cells. Besides, the TLR4/MyD88/NF-кB signaling was implicated in the neuroprotective effect of TNIP2 on OGD/R-induced neuronal damage. Our work provides a functional assessment of TNIP2 in cerebral I/R injury, which provides a novel potential target for the treatment of cerebral I/R injury.
Funding
This study was funded by the Medical and Health Science and Technology Project of Zhejiang Province (grant No. 2017KY142).
Disclosure
The authors report no conflicts of interest for this work.
References
1. Randolph SA. Ischemic Stroke. Workplace Health Saf. 2016;64(9):444. doi:10.1177/2165079916665400
2. Ekker MS, Boot EM, Singhal AB, et al. Epidemiology, aetiology, and management of ischaemic stroke in young adults. Lancet Neurol. 2018;17(9):790–801. doi:10.1016/s1474-4422(18)30233-3
3. Phipps MS, Cronin CA. Management of acute ischemic stroke. BMJ. 2020;368:l6983. doi:10.1136/bmj.l6983
4. Yuan D, Liu C, Hu B. Dysfunction of membrane trafficking leads to ischemia-reperfusion injury after transient cerebral ischemia. Transl Stroke Res. 2018;9(3):215–222. doi:10.1007/s12975-017-0572-0
5. Schaller B, Graf R. Cerebral ischemia and reperfusion: the pathophysiologic concept as a basis for clinical therapy. J Cereb Blood Flow Metab. 2004;24(4):351–371. doi:10.1097/00004647-200404000-00001
6. Leotoing L, Chereau F, Baron S, et al. A20-binding inhibitor of nuclear factor-kappaB (NF-kappaB)-2 (ABIN-2) is an activator of inhibitor of NF-kappaB (IkappaB) kinase alpha (IKKalpha)-mediated NF-kappaB transcriptional activity. J Biol Chem. 2011;286(37):32277–32288. doi:10.1074/jbc.M111.236448
7. Zhang Q, Lenardo MJ, Baltimore D. 30 years of NF-κB: a blossoming of relevance to human pathobiology. Cell. 2017;168(1–2):37–57. doi:10.1016/j.cell.2016.12.012
8. Giridharan S, Srinivasan M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J Inflamm Res. 2018;11:407–419. doi:10.2147/jir.s140188
9. Shabab T, Khanabdali R, Moghadamtousi SZ, et al. Neuroinflammation pathways: a general review. Int J Neurosci. 2017;127(7):624–633. doi:10.1080/00207454.2016.1212854
10. Zhou X, Zhu HQ, Ma CQ, et al. MiR-1180 promoted the proliferation of hepatocellular carcinoma cells by repressing TNIP2 expression. Biomed Pharmacother. 2016;79:315–320. doi:10.1016/j.biopha.2016.02.025
11. Gong H, Sheng X, Xue J, et al. Expression and role of TNIP2 in multiple organ dysfunction syndrome following severe trauma. Mol Med Rep. 2019;19(4):2906–2912. doi:10.3892/mmr.2019.9893
12. Tadros A, Hughes DP, Dunmore BJ, et al. ABIN-2 protects endothelial cells from death and has a role in the antiapoptotic effect of angiopoietin-1. Blood. 2003;102(13):4407–4409. doi:10.1182/blood-2003-05-1602
13. Wu MY, Yiang GT, Liao WT, et al. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem. 2018;46(4):1650–1667. doi:10.1159/000489241
14. Pundik S, Xu K, Sundararajan S. Reperfusion brain injury: focus on cellular bioenergetics. Neurology. 2012;79(13 Suppl 1):S44–51. doi:10.1212/WNL.0b013e3182695a14
15. Petrovic-Djergovic D, Goonewardena SN, Pinsky DJ. Inflammatory disequilibrium in stroke. Circ Res. 2016;119(1):142–158. doi:10.1161/circresaha.116.308022
16. Mizuma A, Yenari MA. Anti-inflammatory targets for the treatment of reperfusion injury in stroke. Front Neurol. 2017;8:467. doi:10.3389/fneur.2017.00467
17. Tohidpour A, Morgun AV, Boitsova EB, et al. Neuroinflammation and infection: molecular mechanisms associated with dysfunction of neurovascular unit. Front Cell Infect Microbiol. 2017;7:276. doi:10.3389/fcimb.2017.00276
18. Webb LV, Ventura S, Ley SC. ABIN-2, of the TPL-2 signaling complex, modulates mammalian inflammation. Trends Immunol. 2019;40(9):799–808. doi:10.1016/j.it.2019.07.001
19. Ventura S, Cano F, Kannan Y, et al. A20-binding inhibitor of NF-kappaB (ABIN) 2 negatively regulates allergic airway inflammation. J Exp Med. 2018;215(11):2737–2747. doi:10.1084/jem.20170852
20. Lou Y, Huang Z. microRNA-15a-5p participates in sepsis by regulating the inflammatory response of macrophages and targeting TNIP2. Exp Ther Med. 2020;19(4):3060–3068. doi:10.3892/etm.2020.8547
21. Xie H, Yang M, Zhang B, et al. Protective role of TNIP2 in myocardial injury induced by acute pancreatitis and its mechanism. Med Sci Monit. 2017;23:5650–5656. doi:10.12659/msm.904398
22. Liu Y, Zhang J, Wang Y, et al. Apelin involved in progression of diabetic nephropathy by inhibiting autophagy in podocytes. Cell Death Dis. 2017;8(8):e3006. doi:10.1038/cddis.2017.414
23. Van Huffel S, Delaei F, Heyninck K, et al. Identification of a novel A20-binding inhibitor of nuclear factor-kappa B activation termed ABIN-2. J Biol Chem. 2001;276(32):30216–30223. doi:10.1074/jbc.M100048200
24. Tao X, Sun X, Xu L, et al. Total flavonoids from rosa laevigata michx fruit ameliorates hepatic ischemia/reperfusion injury through inhibition of oxidative stress and inflammation in rats. Nutrients. 2016;8(7):418. doi:10.3390/nu8070418
25. Roy A, Srivastava M, Saqib U, et al. Potential therapeutic targets for inflammation in toll-like receptor 4 (TLR4)-mediated signaling pathways. Int Immunopharmacol. 2016;40:79–89. doi:10.1016/j.intimp.2016.08.026
26. Satoh T, Akira S, Gordon S. Toll-like receptor signaling and its inducible proteins. Microbiol Spectr. 2016;4(6). doi:10.1128/microbiolspec.MCHD-0040-2016
27. Vidya MK, Kumar VG, Sejian V, et al. Toll-like receptors: significance, ligands, signaling pathways, and functions in mammals. Int Rev Immunol. 2018;37(1):20–36. doi:10.1080/08830185.2017.1380200
28. Luo Y, Wang C, Li WH, et al. Madecassoside protects BV2 microglial cells from oxygen-glucose deprivation/reperfusion-induced injury via inhibition of the toll-like receptor 4 signaling pathway. Brain Res. 2018;1679:144–154. doi:10.1016/j.brainres.2017.11.030
29. Cheng X, Yang YL, Li WH, et al. Cerebral ischemia-reperfusion aggravated cerebral infarction injury and possible differential genes identified by RNA-Seq in rats. Brain Res Bull. 2020;156:33–42. doi:10.1016/j.brainresbull.2019.12.014
30. Zhang Y, Lei X, Li W, et al. TNIP1 alleviates hepatic ischemia/reperfusion injury via the TLR2-Myd88 pathway. Biochem Biophys Res Commun. 2018;501(1):186–192. doi:10.1016/j.bbrc.2018.04.209
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.