Back to Archived Journals » Neuroscience and Neuroeconomics » Volume 6
Neuroprotective effects of estrogen in CNS injuries: insights from animal models
Authors Raghava N ![]() , Das BC, Ray SK
, Das BC, Ray SK
Received 5 November 2016
Accepted for publication 23 May 2017
Published 4 July 2017 Volume 2017:6 Pages 15—29
DOI https://doi.org/10.2147/NAN.S105134
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 6
Editor who approved publication: Professor SH Annabel Chen
Narayan Raghava,1 Bhaskar C Das,2 Swapan K Ray1
1Department of Pathology, Microbiology, and Immunology, University of South Carolina School of Medicine, Columbia, SC, USA; 2Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY, USA
Abstract: Among the estrogens that are biosynthesized in the human body, 17β-estradiol (estradiol or E2) is the most common and the best estrogen for neuroprotection in animal models of the central nervous system (CNS) injuries such as spinal cord injury (SCI), traumatic brain injury (TBI), and ischemic brain injury (IBI). These CNS injuries are not only serious health problems, but also enormous economic burden on the patients, their families, and the society at large. Studies from animal models of these CNS injuries provide insights into the multiple neuroprotective mechanisms of E2 and also suggest the possibility of translating the therapeutic efficacy of E2 in the treatment SCI, TBI, and IBI in humans in the near future. The pathophysiology of these injuries includes loss of motor function in the limbs, arms and their extremities, cognitive deficit, and many other serious consequences including life-threatening paralysis, infection, and even death. The potential application of E2 therapy to treat the CNS injuries may become a trend as the results are showing significant therapeutic benefits of E2 for neuroprotection when administered into the animal models of SCI, TBI, and IBI. This article describes the plausible mechanisms how E2 works with or without the involvement of estrogen receptors and provides an overview of the known neuroprotective effects of E2 in these three CNS injuries in different animal models. Because activation of estrogen receptors has profound implications in maintaining and also affecting normal physiology, there are notable impediments in translating E2 therapy to the clinics for neuroprotection in CNS injuries in humans. While E2 may not yet be the sole molecule for the treatment of CNS injuries due to the controversies surrounding it, the neuroprotective effects of its metabolite and derivative or combination of E2 with another therapeutic agent are showing significant impacts in animal models that can potentially shape the new treatment strategies for these CNS injuries in humans.
Keywords: estrogen, neuroprotection, spinal cord injury, traumatic brain injury, ischemic brain injury
Introduction
Estrogen is an important steroidal sex hormone that is biosynthesized and released for its participation in many signaling pathways in the human body.1 It is released by the adrenal cortex, which is under the control of the hypothalamus. The hypothalamus releases the adrenocorticotropic hormone, which triggers the adrenal cortex for the biosynthesis and release of the steroidal sex hormones such as estrogen, progesterone, and testosterone. Estrogen is a well-known steroidal sex hormone in women. In women, four natural estrogens are biosynthesized: estrone (E1), estradiol (E2), estriol (E3), and estetrol (E4). E1, which is the weakest form of estrogen, is predominant when women arrive at menopause. E2, which is the strongest form of estrogen, is produced during menstrual cycle and before pregnancy or menopause. E3 is the predominant estrogen when women are pregnant and it replaces E2. E4 is only present in women during their pregnancy. Thus, the four types of estrogen are at low or high levels in women, based on their age and status of menstrual cycle, pregnancy, and menopause. Among these, E2 is the best estrogen to provide neuroprotective effects in the central nervous system (CNS) injuries such as spinal cord injury (SCI), traumatic brain injury (TBI), and ischemic brain injury (IBI) in various animal models.2–4 The biosynthesis of E2 occurs via multiple steps beginning from cholesterol (Figure 1), and E2 has significant physiological roles not only in females but also in males.5 It should be clearly emphasized here that E2 is mainly a female sex hormone and, thus, is biosynthesized at higher levels in females than in males. In addition to its reproductive roles in females, E2 has nonreproductive effects in the CNS in both females and males. The precise molecular mechanisms for the neuroprotective effects of E2 still remain elusive. While there have been many speculations and experiments, the generally known neuroprotective effects of E2 result from both its genomic (receptor-dependent) and nongenomic (receptor-independent) mechanisms.6 The neuroprotective effects of E2 (Figure 2) via its genomic signaling require binding of E2 to its receptors, while these effects of E2 via nongenomic signaling, antioxidant pathways, or bioenergetic pathways do not involve binding of E2 to its receptors.7 Genomic signaling of E2 to provide neuroprotective effects works via three main receptors (Table 1). The two main receptors, which are E2 receptor alpha (ERα) and E2 receptor beta (ERβ), are known to be highly involved in providing neuroprotective effects of E2 (Table 1). E2 and other ligands, which bind to these receptors, are known as ER agonists that are highly regarded for their roles in attenuation of neuroinflammation and neurodegeneration.8,9
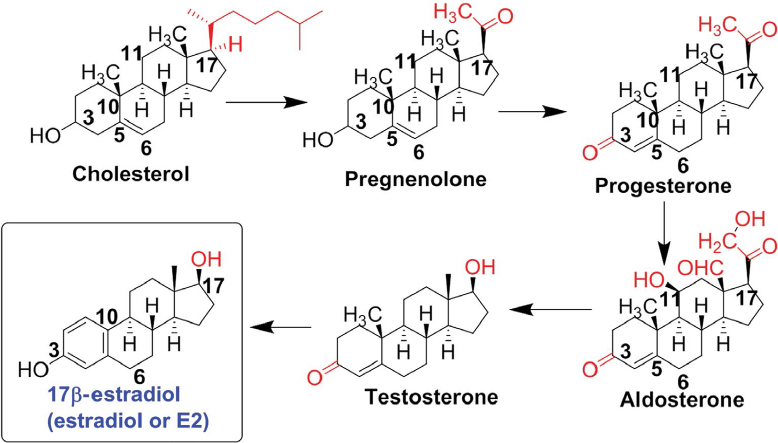 | Figure 1 Biosynthesis of E2 from cholesterol. Note: The intermediate products are also important steroidal sex hormones and some of them have shown notable neuroprotective actions. Abbreviation: E2, estradiol. |
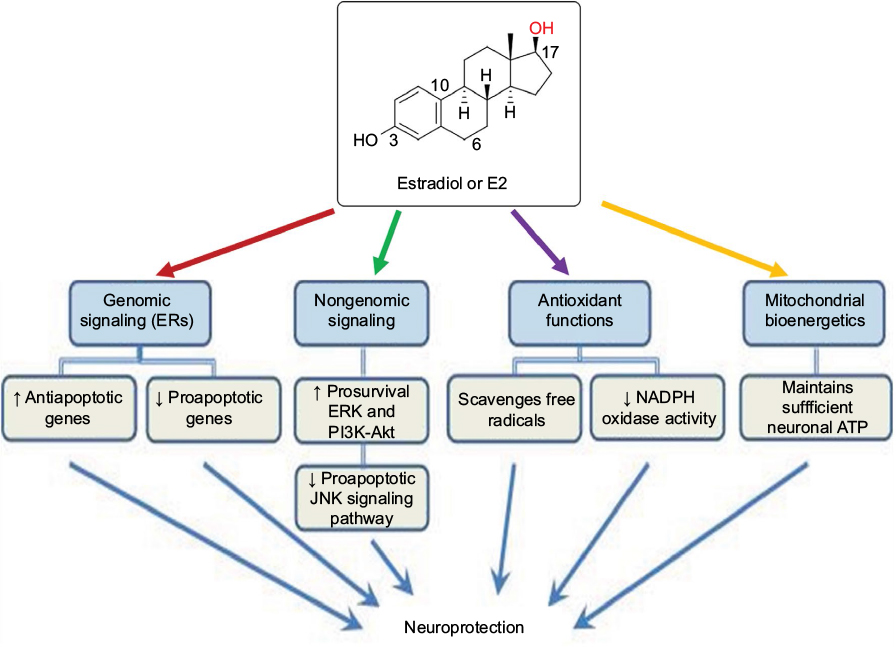 | Figure 2 Various molecular mechanisms of E2 for its neuroprotective effects. Notes: Genomic and nongenomic signaling mechanisms are mostly involved in modulation of expression or activity of multiple molecular components for inhibition of apoptosis and promotion of cell survival. Its antioxidant functions are useful for scavenging reactive free radicals, which are heavily produced following a CNS injury. Moreover, E2 plays an important role in maintaining mitochondrial bioenergetics for the optimal production of ATP for supporting cellular metabolism. Abbreviations: ATP, adenosine triphosphate; CNS, central nervous system; E2, estradiol; ER, estrogen receptor; ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal kinase. |
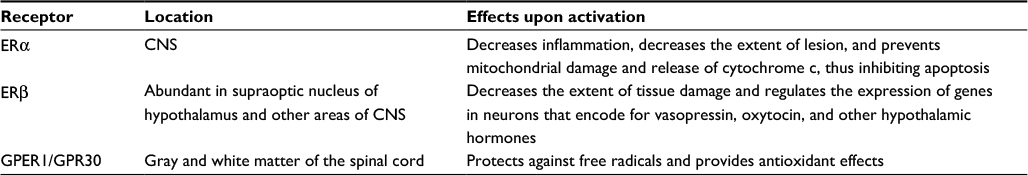 | Table 1 The three main E2 receptors, their CNS locations, and their effects upon ligand binding Abbreviations: CNS, central nervous system; E2, estradiol; ER, estrogen receptor; GPER1/GPR30, G protein-coupled estrogen receptor 1 / G protein-coupled receptor 30 . |
The genomic signaling mechanisms of E2 involve activation of ERs and modulation of expression of the target genes (Figure 3) for providing neuroprotective effects.10–13 There have been many experiments to determine which ER is more important than others in providing the neuroprotective effects of E2. One investigation used homozygous ERα- and homozygous ERβ-knockout mice (C57BL/6J×129) to determine the efficacy of E2 and concluded that it was the ERα that played a key role in providing the best neuroprotection following E2 therapy in IBI.14 The results from this study demonstrated that deletion of ERα in mice completely obliterated the neuroprotective effect of E2 in all regions of the brain, while the capability of E2 was totally preserved for neuroprotection against this brain injury in the absence of ERβ. These results clearly showed that ERα played a critical mechanistic link in mediating the protective effects of E2 in this brain injury. A recent study in rats with SCI reported that E2 therapy upregulated ERα expression and also antioxidant capacity for inhibition of neuroinflammation and promotion of neuroprotection to the surrounding regions of the spine.15 Another study used the ERα-deficient mice (C57BL/6J background strain) and determined that the deficiency of ERα did not increase tissue damage and the neuroprotective effects of E2 in IBI were provided by other mechanisms that were independent of activation of this ERα.16 This study did not explore the other mechanisms of E2-mediated neuroprotective effects; however, such protective effects could be due to the ERβ-dependent and/or nonreceptor signaling mechanisms of E2.
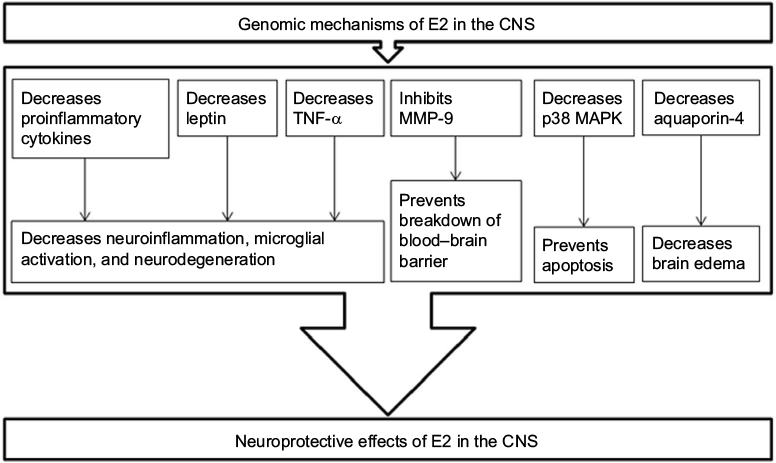 | Figure 3 E2 has genomic signaling mechanisms for modulation of expression of various proteins for its neuroprotective effects in the CNS. Notes: Activation ERs by E2 modulates the expression of the target genes, so as to decrease the detrimental pathways, thus leading to neuroprotection. Abbreviations: CNS, central nervous system; E2, estradiol; ERs, estrogen receptors; MMP-9, matrix metalloprotease-9; TNF-α, tumor necrosis factor-alpha. |
The argument of which ER is the most effective can be resolved with a reasonable statement that both ERα and ERβ work effectively in providing the neuroprotective effects of E2. However, it has been established that ERβ is present in most parts of the brain and spinal cord where ERα is not abundant, implying that ERβ plays an important role in governing the physiological responses of neuronal populations in the CNS and also in peripheral nervous system.17 It was also settled experimentally that E2 coupled with one of the ER modulators could lead to an increase in neurogenesis, which would promote the amount of spinal density that was depleted due to injury. While ERα and ERβ serve as the two main receptors associated with the neuroprotective effects of E2, a third receptor has also emerged as a plausible route for E2 to take when exerting its neuroprotective effects on the body. The third receptor is a G protein-coupled ER (GPER1/GPR30), as shown in Table 1, which is found in the gray matter as well as in the white matter of the spinal cord.18 Experiments showed that GPER1/GPR30 was mainly distributed in the ventral horn and white matter of the spinal cord and specifically expressed in neurons, oligodendrocytes, and microglial cells, but not in astrocytes.18 Moreover, E2 or the GPER1/GPR30 agonist G-1 could upregulate the GPER1/GPR30 protein expression to enhance the estrogenic effects for neuroprotection in SCI.18
The nongenomic signaling and other mechanisms of E2 for neuroprotection have also been evaluated. E2 promoted local neuroregeneration through the nongenomic signaling pathway with activation of the prosurvival extracellular signal-regulated kinase in lumbar spinal cord following sciatic nerve crush injury in ovariectomized mice.19 The neuroprotective effects of E2, in part, may also result from a rapid nongenomic signaling with upregulation of the prosurvival phosphoinositide 3-kinase/Akt response in male rats.20 The nongenomic signaling mechanism of E2 not only activates prosurvival phosphoinositide 3-kinase /Akt pathway, but also inhibits the proapoptotic c-Jun N-terminal kinase 1/2 pathway for neuroprotection in rats.20 Another investigation showed that changes in subcellular localization of the active p38α isoform were needed for neuronal apoptosis in cerebral ischemia, but E2 therapy provided a robust neuroprotection due to dual inhibition of activation and subcellular trafficking of p38α.12 Further, E2 has been shown to have antioxidant properties. Substantial experimental evidence now strongly indicates that intrinsic free radical scavenging contributes to the ER-independent neuroprotective effects of E2. Studies indicated that E2 provided neuroprotection via ER-independent antioxidant mechanism in the brain tissue after permanent occlusion of the middle cerebral artery in male mice and also in the primary neurons exposed to reactive oxygen species (ROS).21 This study showed that the ER antagonist tamoxifen influenced neither the neuroprotective effect nor the free radical scavenging properties of E2, indicating that E2 protected primary neurons from damage by radical scavenging mechanism rather than through ER activation. Another investigation demonstrated that interaction of E2 with ER was not involved in inhibiting the increases in intracellular Na+ and Ca2+ concentrations during metabolic inhibition in mouse cardiomyocytes; however, E2 reduced free radical-induced impairment of Na+–K+ ATPase in the cardiomyocytes, and this effect reduced intracellular Ca2+ concentration for cytoprotection by an ER-independent antioxidant mechanism.22 What specific signaling pathway that E2 takes when it is protecting the CNS cells and other cells from injuries is still debatable; however, it is found that the hydroxyl group in the phenolic steroid ring is what produces the antioxidant effects of E2.21–23 It is now well established that quinol-based cyclic antioxidant mechanism of E2 is responsible for its neuroprotective effects.24 Exciting recent studies show that most of the neurosteroids including E2 are highly capable of improving mitochondrial bioenergetics in neurons by increasing the ATP levels, mitochondrial membrane potential, and basal mitochondrial respiration.25
Neuroecomonics of CNS injuries
Spinal cord injury
Among all the CNS injuries, SCI is of the highest importance as it has huge epidemiological and economic impacts in our society.26,27 In the US, consistent with all other industrialized nations, majority of the SCI cases result from motor vehicle accidents followed by falls, gunshot wounds and stabbings, and sporting activities. As expected, there are statistically significant relationships between the etiology of SCI and factors such as age, sex, race, and geography. The incidence of SCI most frequently occurs in persons between 15 and 20 years of age. The US National SCI Database, which is maintained by the Department of Rehabilitation Medicine at the University of Alabama at Birmingham, shows that the mean age of SCI victims is 29.7 years, the median age is 25 years, and the mode (i.e., the most frequent age of SCI victims) is 19 years. The US National SCI Database also suggests that SCI occurs more frequently in males than females (4:1) and the annual SCI rates are slightly higher in blacks than whites. The recent estimate showed about 88 cases of acute SCI per million adult population per year in the US.27 Another recent study indicated that there were about 12,000 hospitalizations per year due to SCI in the US.28 A quirk of fate is that ~12% of the patients with SCI are being diagnosed with diabetes that imposes an additional economic burden on the patients with SCI, who are already vulnerable with high health care costs.29 The economic burden is highly noticeable in minority populations suffering from the devastating consequences of SCI.
About 20 years ago, a study reported that the US spent ~ $8 billion annually for medical care, household assistance, environmental modifications, and other support services for the victims of SCI.30 As expected, the current amount of spending for SCI victims in the US is significantly higher. Besides the health care costs, SCI often wreaks an individual with permanent neurologic impairments that may prevent him or her from getting back to school, job, or other activities for living. If neurodisability management after such an insidious SCI is denied or discontinued due to economic reasons, the patients with SCI discharged from the treatment and rehabilitation may feel emotionally exhausted, socially segregated, and professionally unprepared for facing the entirely different reality of life. In the wake of fast-increasing health care expenditures, decisions are often made to reduce the costs of care and increase the efficiency of health care delivery, especially by reducing the length of stay (LOS) in the hospital or rehabilitation.31 However, rehabilitation providers need to be concerned that LOS is becoming too short that may put the patients with SCI at grave risk of subsequent complications. LOS is often considered as an indicator of the efficiency of health care delivery. It is argued that the shorter stays are the result of effective management of care and proper arrangement of resources. However, LOS is a function of multiple interacting factors such as medical perception of the patients, health care practices, insurance type, reimbursement pattern, social resources available to the patients with SCI and their families, psychologic status of the patient with SCI, and other issues. Ideally, discharge should occur when the patient with SCI gains substantial neurologic functions. It still remains unclear how short the LOS can be for a specific patient with SCI before undesirable outcomes escalate, causing lower efficiency and higher health care cost.
So far, methylprednisolone (MP), not E2, is the only drug recommended by the US Food and Drug Administration (FDA) for treatment of acute SCI in humans (Table 2). The results have been reported from the studies using MP and E2 in animal models of SCI, indicating several limitations of MP and promising neuroprotective effects of E2 in the treatment of both acute and chronic SCI.4,32–34 Moreover, MP has shown serious side effects in humans and it is now considered to be unsuitable for treatment of even the patients with acute SCI.35 So, there is an urgent need to find out a suitable therapeutic agent for treatment of both acute and chronic SCI in humans.
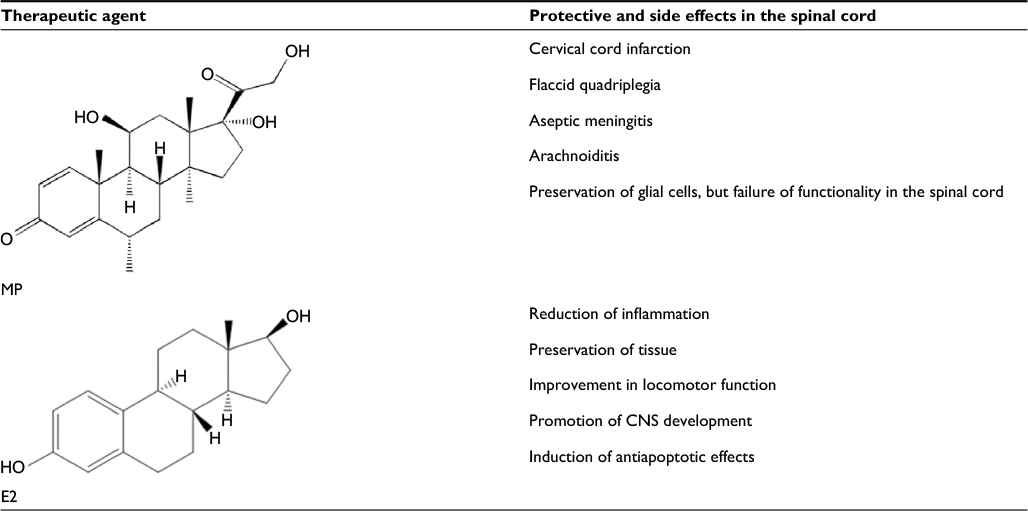 | Table 2 Effects of MP, the current drug used for SCI treatment, and E2 in the spinal cord Abbreviations: CNS, central nervous system; E2, estradiol; MP, methylprednisolone; SCI, spinal cord injury. |
Traumatic brain injury
TBI is a serious CNS injury in the military and civilian populations in the US and other nations. TBI received wide public and media attention during the wars in Afghanistan and Iraq. It causes significant number of death and also lifelong disability in the victims. Management of TBI requires involvement of major health care services and resources leading to enormous economic burden on the patients, their families, and the society at large.36 A study showed significant variation in utilization of health care services and costs of TBI treatments, depending on the specific population and also seriousness of TBI.37 This study also indicated the existence of race, sex, insurance, and geographic disparities in the US and other distinctive disparities worldwide, leading to different outcomes from TBI management. There is no doubt that insurance and racial disparities will continue to dictate the outcomes for the persons who suffered a TBI. However, insurance category appears to have more impact on both the short- and long-term outcomes than the race of the person who suffered a TBI.38 Recently, a study has been conducted to predict the costs of TBI, so as to assist planning for hospital, medical, paramedical, and long-term care costs for the first 10 years following a TBI.39 This study performed a comprehensive analysis of various factors to predict the types of costs following TBI, indicating that combination of severity of TBI and demography could predict 23%–44% of the costs. Data from this study may be used for planning and managing the TBI individuals. Comprehensive economic evaluations seem to be a foundation that gives an opportunity to identify the best possible plans for the diagnosis and management of TBI, which brings so much uncertainty from the beginning affecting the mental health and enforces extra economic burden on the health care system.40 Studies also indicate some of the big challenges that brain injury survivors face in managing their finances and the adjustment associated with the loss of financial independence.37,41 To overcome these challenges, health care teams play important roles in interprofessional communication and also in recognition of needs and financial burdens of the persons who have suffered TBI when rendering service to them.
There is no pharmacotherapy yet approved by the US FDA for treatment of TBI in humans.42 Because progressive pathogenesis in TBI occurs via multiple cellular and molecular mechanisms, a multiactive compound such as E2 alone or in combination with another suitable therapeutic agent may be used to target simultaneously the multiple mechanisms of pathogenesis. An effective pharmacotherapeutic strategy should block harmful cellular and molecular mechanisms in the brain, resulting in some improvements in persons who suffered a TBI. However, combination of drugs should work at least additively and the best combination of drugs should work synergistically.43 Combination of drugs is, therefore, considered to be the best option of having a larger therapeutic efficacy than any individual drug for the treatment of TBI.
Ischemic brain injury
Acute IBI is the third leading cause of death in the US. Each year, there are over 500,000 IBI cases, which are associated with a blockage of blood flow in the brain. This blockage of blood flow causes tissue injury and damages specific portions of the brain. Also, it is the leading cause of adult disability that directly and indirectly costs >$60 billion annually in the US.44 As approved by the US FDA, administration of intravenous tissue plasminogen activator (tPA) is the first treatment for acute IBI.45 However, it is essential that the emergency physicians understand the pathophysiology of IBI, the molecular basis and rationale for its treatment, and the long-term therapeutic strategies.46 As IBI is associated with premature death, disability, and health care costs, it imposes a huge burden on our society. Obviously, the efficacy and cost-effectiveness of the interventions need to be evaluated before their widespread implementation in IBI patients.47 As clinical trials cannot measure the long-term impact of new interventions in IBI, a modeling approach is essential. A study reported that new interventions were cost-effective or health beneficial at an acceptable additional cost.48 IBI patients generally choose their care providers based on hospital expertise. Socioeconomic status of the IBI patients has little bearing on the distance traveled for treatments. IBI, whether acute or chronic, may result in major neurologic impairment and disability to the individuals. Following IBI, individuals experience brain tissue damage and, thus, restrictive physical and mental health. The economic burden on the IBI individuals and society is significant and it is increasing every year. The IBI costs may be reduced by appropriate health care planning, accurate diagnosis, effective therapy, and preventive measures.49 The development of innovative and novel diagnostic techniques and therapeutic strategies will have major positive impacts on reducing the costs of IBI in the future.
The main early goal of IBI treatment is bringing back the blood flow to the distressed brain area as soon as possible, so as to prevent further damage and preserve brain function. There are two main early treatment options for IBI: 1) intravenous thrombolytic or clot-buster therapy with tPA and 2) intra-arterial mechanical thrombectomy or opening of blocked artery with a catheter. Both tPA and catheter have some inherent problems in treating the patients who are suffering from IBI.50,51 So, better therapeutic options are needed not only to restore the blood flow, but also to promote formation of new blood vessels or angiogenesis in the brain of the IBI victim. Probably, E2 therapy alone or in combination with another therapeutic agent will be an alternative option for treatment of IBI.
There is an urgent need to develop effective strategies to prevent SCI, TBI, and IBI. Unfortunately, these CNS injuries do occur in spite of preventive measures. So, neuroeconomics of these CNS injuries should be considered to determine their appropriate treatments and long-term health care services with effectiveness and efficiency.
Neuroprotective mechanisms of E2 in animal models
Neuroprotective effects of E2 in SCI in animal models
Over the past few years, the amount of surgical treatment has shown only a slight increase compared to the pharmacologic therapy to treat SCI, which has so far shown no real change in the treatment outcome. While surgical treatments have advanced, the risk of progression of pathogenesis of SCI still remains high, and thus, a safer treatment option is necessary. Untreated acute SCI is associated with progressive pathogenesis and eventually becomes a chronic SCI leading to sustained neurodegeneration and spinal cord damage with activation of cysteine proteases including calpain.52 E2, unlike its counterpart MP, may not be dangerous when administered at low doses, and thus, E2 can serve as an effective neuroprotective agent without the dangerous side effects of MP in the treatment of chronic SCI in rats.34 There have been some studies showing the pros and cons of using MP (Table 2) in the treatment of SCI in vitro and in vivo.53,54 When comparing male and female rats post-SCI following a treatment, it is seen that the female rats have improved motor function with less tissue damage, implying that E2 plays a vital role in neuroprotection following SCI in female rats.55 Comparing the male and female rats on a weekly basis, it is found that the female rats have faster and larger amounts of improvement in the locomotor function following SCI, when compared with their male counterparts.38 When comparing male rats in the control group with those in the E2 treatment group following SCI, the E2-treated group showed significant improvement in transmission of neural impulses while the control group displayed a significant decrease in the number of neurons in the histologic findings.56
While E2 has its benefits, it must be carefully administered at an optimal dose; otherwise, it may cause a significant negative impact on the physiology of the host.9 Investigations in our and other laboratories demonstrated that supraphysiologic high dose of E2 worked very effectively due to its anti-inflammatory and antioxidant effects in the treatment of SCI in animal models.15,57,58 The results from these studies are provocative for using supraphysiologic high dose of E2 for its antioxidant effects and high therapeutic efficacy in SCI in humans. However, administration of high dose of E2 for a persistent period may be associated with the risks of reduced fertility or infertility59,60 and several cancers in females61 and also the possibility of conspicuous feminine feature (gynecomastia) and development of cancer in males.62 For these concerns, a high dose of E2 should not be used in the clinics for treatment of SCI in humans. Administration of E2 at low doses (2.5–25 µg/kg) displayed efficient anti-inflammatory response by decreasing interleukin, leptin, and tumor necrosis factor alpha (TNF-α) in SCI rats, showing a significant amount of beneficial effects.10 Other than its anti-inflammatory effects, E2 has been shown to prevent the breakdown of the blood–spinal cord barrier by decreasing the expression of SUR1/TrpM4 and by inhibiting the activity of matrix metalloprotease-9 (MMP-9) following SCI.11 An important preclinical study, which should have profound clinical implication, has been reported showing that post-injury E2 treatment can prevent spinal cord damage and improve locomotor function in chronic SCI in rats.58
The results from an E2 dose-dependent study indicate that continuous administration of E2 alone at high dose (3 mg E2 capsule to give rise to pregnancy level of 55–630 pg/mL E2 in plasma), but not at low dose (10 µg E2 to achieve the normal estrous cycle of 5–45 pg/mL E2 in plasma), can promote formation of mammary tumor in intact, but not in ovariectomized August-Copenhagen-Irish rats.63 A unique feature of August-Copenhagen-Irish rats is that the ovary-intact females develop high incidence of mammary tumors upon prolonged exposure to high levels of E2 alone. This study implies that treatment of SCI with E2 alone, even at a low dose (10 µg E2/animal), may remain risky in ovary-intact rats. Very low dose of E2 in combination with another therapeutic agent may provide desirable amount of neuroprotection without the risk of tumor growth in SCI in animal models. Indeed, combination therapy seems to be a promising future treatment strategy for functional repair and regeneration in SCI.64 Combination of low doses of E2 and another therapeutic agent should be explored to increase the therapeutic effects of E2 in SCI. Combination therapy makes it easy to use two different drugs at low doses for additive or synergistic effects, target diverse detrimental pathways, discourage expression and encourage degradation of harmful factors, and thereby improve overall therapeutic outcomes in SCI. Recently, we demonstrated enhancement of neuroprotective efficacy of the combination of miR-7-1 and E2 for protection in the ventral spinal cord 4.1 motoneurons in the cell culture model of SCI.65
Neuroprotective effects of E2 in TBI in animal models
The pathogenesis in TBI is associated with enormous amounts of oxidative stress that damages the brain tissue. The Fenton reaction in the brain following TBI is what produces the free hydroxyl radical (•OH), which is extremely harmful in the brain. The hydroxyl group (-OH) in the phenol ring of E2 (Figure 1) can act as a chemical shield and protect against the free radical produced in the Fenton reaction, creating a quinol form of E2.24 Rats were tested following induction of TBI to see the efficacy of ERα and ERβ, and it was determined that there was no significant difference between the neuroprotective efficacy of these two receptors; E2 reduced the brain edema caused by the TBI and protected the permeability of the blood–brain barrier (BBB).66
Premarin (Figure 4), a mixture of conjugated estrogens (about 50% sodium estrone sulfate and other conjugated estrogens) purified from the urine of pregnant mares, improved the brain–blood gradient in rats following a TBI, allowing for reuptake of excess glutamate produced by the presynaptic neurons, which were otherwise damaged by the TBI.67 Another study showed that E2 also worked to reduce brain edema induced by TBI by decreasing the expression of aquaporin-4 and interleukin-6, both of which heavily contributed to the water content in the brain.13 A similar study concluded that E2 therapy not only decreased brain edema, but also decreased the expression of interleukin-1β, interleukin-6, and TNF-α in TBI in the ovariectomized female rats.68 Genistein (an estrogenic compound of plant origin, also called a phytoestrogen), a major isoflavone of soy, reduced brain edema and BBB permeability, inhibited the escalation of intracranial pressure, and prevented a few deficits in neurobehavioral skills following TBI in the male Wistar rats.69 When E2 was administered within an hour of induction of TBI, it resulted in a decrease in intracranial pressure and brain edema. The salutary effects of E2 were tested in TBI in Sprague-Dawley rats, where they showed significant decreases in intracranial pressure and brain edema size but increases in cerebral perfusion pressure, partial brain oxygen pressure, and cerebral glycolysis, when compared with the vehicle-treated TBI animals.70
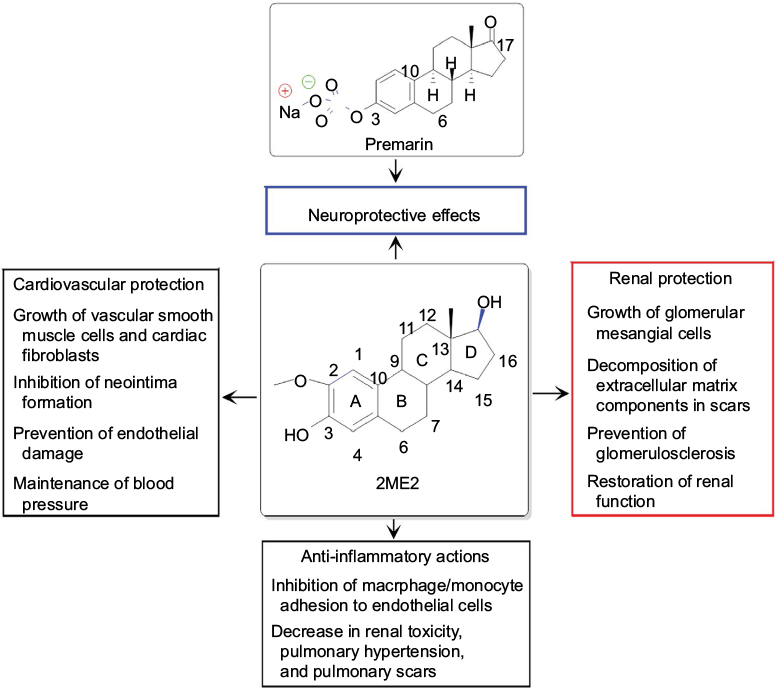 | Figure 4 Chemical structures of the main component in Premarin and of 2ME2 and their therapeutic effects. Notes: Both Premarin and 2ME2 provide significant amounts of neuroprotection. In addition to its neuroprotective effects, 2ME2 has other beneficial effects in the body. The different color boxes were used to indicate different beneficial effects of 2ME2. Abbreviation: 2ME2, 2-methoxyestradiol. |
2-Methoxyestradiol or 2ME2 (Figure 4), a metabolite of E2, is an inhibitor of hypoxia-inducible factor-1 alpha (HIF-1α). It has been reported that 2ME2 provides neuroprotection due to inhibition of a maladaptive HIF-1α response after TBI in mice.71 When 2ME2 was administered in mice following TBI, it not only inhibited a maladaptive HIF-1α response, but also decreased the expression of TNF-α, the overexpression of which caused neuropathology; 2ME2 further decreased the expression of BNIP3, a proapoptotic protein, thereby displaying diverse neuroprotective capabilities of this E2 metabolite in the treatment of TBI in mice.71 It should be noted here that 2ME2 may show neurotoxicity and genotoxicity under specific conditions. For example, a study demonstrated that 2ME2, at pharmacologically and also physiologically relevant concentrations, increased nuclear localization of neuronal nitric oxide synthase that resulted in DNA strand breaks and genomic instability in hippocampal HT22 cell line.72 Nevertheless, 2ME2 has other beneficial effects in the body (Figure 4). When used with caution, E2 and its metabolite are capable of providing significant amounts of neuroprotection in TBI in animal models.
It should be mentioned here that E2 may not be a therapeutic option for treatment of infant brain injury, because the results from a study show that E2 therapy aggravates hippocampal damage in a rat model of preterm infant brain injury due to a biphasic action of E2 involving an early nongenomic protection followed by a late receptor-mediated destruction in the brain.73 However, combination of drugs needs to be explored as the promising therapeutic option for treatment of adult TBI, owing to clinical failure of many monotherapies for treatment of TBI in adults. A combination of E2 and memantine appeared to be significantly more neuroprotective than either agent alone in an organotypic brain slice culture model of TBI, and this synergistic effect of the combination therapy could result from memantine-mediated blockage of a deleterious E2-mediated enhancement of N-methyl-d-aspartate receptors.74
Neuroprotective effects of E2 in IBI in animal models
The results from different animal model studies show promising neuroprotective effects of E2 in IBI. E2 treatment of the animals following an IBI could improve the recovery of the contralateral forelimb sensorimotor function and also reduce cyst formation, leading to an increase in cerebral blood flow that was severely reduced due to the IBI.75 When E2 was combined with progesterone (Figure 1), another female steroidal sex hormone, the combination therapy reduced the infarct size and was quite effective in restoring many behavioral aspects that had been lost following IBI in rats.76 Another therapy for IBI in rats is the treatment with one of the selective ER modulators (SERMs). Bazedoxifene is a SERM, which is used as monotherapy or in combination therapy for treatment of menopausal osteoporosis and also moderate to severe hot flushes. E2, which is most effective when given in the acute stage of IBI, shows neuroprotective effects mainly in the subcortical level, while bazedoxifene shows its effects mainly in the cortical level; and both E2 and bazedoxifene significantly decreased total infarct volumes in IBI in male Wistar rats.77
Activation of GPER1/GPR30 by E2 is neither necessary nor sufficient for acute neuroprotection in IBI, as demonstrated in an organotypic hippocampal slice culture model.78 Also, it should be noted that the neuroprotective capabilities of E2 are dependent on its dose, and its administration above the physiologic levels can significantly increase phosphorylation of Akt and extracellular signal-regulated kinase, which are the well-known downstream effects of GPER1/GPR30 activation in this model of IBI. After an IBI in rats, it was demonstrated that the E2 levels spiked in the astrocytes in hippocampus, drawing the conclusion that dramatically increased E2 could provide important anti-inflammatory and neuroprotective effects in this region of the brain.79 It was also determined that E2 primarily used the ERβ for neuroprotection following an IBI to show significant recovery in the neurologic functions in the animals.80
It needs to be mentioned here that E2 therapy in brain injury may have limitations under specific conditions. For example, E2 showed neurotoxic effects on hypothalamic β-endorphin neurons in a model of chronic estrogenization, leading to reproductive senescence.81 Also, E2 caused neurotoxic effects in IBI in older female rats due to age-dependent loss of insulin-like growth factor-1 in these animals.82 Therefore, E2 therapy for treatment of IBI should be avoided in female animals, which are under chronic estrogenization or just old. Combination of low-dose E2 with another therapeutic agent continues to be an active research area to enhance the therapeutic outcomes in IBI. Ovariectomized female rats following induction of IBI were treated with E2 and recombinant tPA alone and also in combination and they showed the following results: no protection was observed upon treatment with E2 alone, side effects (expression and activation of urokinase plasminogen activator, MMP-2, and MMP-9) were enhanced due to administration of recombinant tPA alone, and combination of E2 and recombinant tPA provided protective action.83 Hence, this study suggested that E2 could be a candidate for combination therapy with recombinant tPA to attenuate its side effect and also to increase its short therapeutic window for treatment of IBI.
Neuroprotective effects of E2 metabolite and derivatives in animal models of CNS injuries
Not only E2, but also different E2 metabolites and derivatives are now considered to be highly important neuroprotective agents in the treatment of SCI, TBI, and IBI in animal models. 2ME2 is an endogenous metabolite of E2, while Premarin is important derivative of E2 (Figure 4). Physiologically, 2ME2 is formed by the sequential hydroxylation of E2 by cytochrome P450 enzymes followed by O-methylation by catechol-O-methyltransferase. 2ME2 interacts with ERs and microtubules, but 2ME2 shows a low affinity for ERs. 2ME2 significantly increases progesterone receptor transcripts both in vitro and in vivo, suggesting that the estrogenic activities of 2ME2 may be mediated via the ER and/or progesterone receptor signaling pathway.84 Premarin is also known to possess profound neuroprotective effects. Its neuroprotective effects have been useful for saving neurons from irreversible injury in the CNS. Premarin (1 mg/kg of body weight) administration prevented neuronal apoptosis and promoted angiogenesis and neurogenesis in SCI in male rats.85 Premarin (1 mg/kg of body weight) also attenuated cerebral infarction, motor and cognitive function deficits, neuronal apoptosis in cortical and hippocampal regions, and neuroinflammation, while it promoted both angiogenesis (increased numbers of endothelial and vascular endothelial growth factor-positive cells) and neurogenesis (increased numbers of both 5-bromodeoxyuridine/neuronal-specific nuclear protein double-positive and glial cell line-derived neurotrophic factor-positive cells) through stimulation of ERα to protect against TBI in male rats.86
As upregulation of HIF-1α is correlated with the expression of aquaporin-4 (AQP4) and aquaporin-1 (AQP1) that cause edema in SCI, therapy targeted to inhibition of HIF-1α is highly desirable to decrease edema in SCI. An investigation using adult rat SCI model has shown that 2ME2, which can post-transcriptionally inhibit the expression of HIF-1α, significantly inhibited the expression of AQP4 and AQP1 for reduction in edema.87 In the CNS, HIF-1α acts as a regulator of both prosurvival and prodeath signaling pathways. The results of a study showed that HIF-1α promoted brain injury in IBI and administration of 2ME2, an inhibitor of HIF-1α, immediately or 3 hours after induction of IBI exhibited dose-dependent (1.5, 15, and 150 mg/kg) neuroprotection by preserving BBB integrity and by decreasing the infarct volume and attenuating brain edema.88 Another study showed that 2ME2 dramatically suppressed the development of mouse experimental autoimmune encephalomyelitis, a rodent model of multiple sclerosis, by specifically reducing the nuclear translocation and transcriptional activity of the nuclear factor of activated T cells and subsequently inhibiting lymphocyte activation.89 A more recent study suggested that 2ME2 may mediate its neuroprotective actions by inhibiting microglia proliferation and activation via an ER-independent mechanism.90
It is of further interest to mention that 2ME2 is currently undergoing Phase I and Phase II clinical trials under the commercial name Panzem for breast cancer treatment.91 Early results suggested that 2ME2 is a bioavailable drug following administration of an oral formulation and it causes selective inhibition of cancer cell proliferation without any of the usual chemotherapy-associated toxicities. However, the molecular mechanisms of action of 2ME2 for cancer-specific growth inhibition have not yet been clearly understood. 2ME2 is a multiactive metabolite of E2 and it has further shown therapeutic activities for renal protection,92 anti-inflammatory actions,93 and cardiovascular protection.94,95
It should be noted that there are other estrogens that act as neuroprotectants in the context of CNS diseases and injuries.96 The “phenolic ring hypothesis” has been formulated to account for the neuroprotective effects of estrogens.97,98 Recently, a variety of SERMs have been developed to retain the desirable effects and eliminate or minimize the undesirable side effects of estrogens.99 Seemingly, raloxifene and arzoxifene are two specific SERMs that have well-known neuroprotective effects in CNS injuries. However, their mechanisms of action for neuroprotection are not yet clearly understood and it also remains unclear whether the neuroprotective effects of SERMs are due to activation of ERs. There are also noteworthy endeavors for developing the nonfeminizing estrogen therapeutic strategy for neuroprotection while eliminating the undesirable off-target effects of chronic estrogen treatment.100,101
Progressively, soy isoflavones are rising in popularity as alternative to estrogen therapy, because the Women’s Health Initiative has shown increases in the risks of breast cancer, stroke, and heart attacks following treatment with estrogen and progesterone. Isoflavones are heterocyclic polyphenols; they are structurally similar to E2 and SERMs and show therapeutic actions depending on the target tissue, level of endogenous estrogen, and receptor status of the tissue. Clinical studies revealed that consumption of soy-based diets significantly decreased total cholesterol, low-density lipoprotein, and triglyceride levels, showed lower rates of breast cancer, and relieved menopausal symptoms by reducing hot flashes; most importantly, there are no known adverse effects due to the short- or long-term use of soy proteins in humans.102 A recent study showed that genistein, which is a soy isoflavone as mentioned earlier, provides neuroprotection by inhibition of mitochondria-dependent apoptosis pathways and reactive oxygen-induced NF-κB activation in a mouse model of IBI.103 Overall, our current understanding of the neuroprotective and other beneficial effects of the steroid hormones such as estrogen, progesterone, and testosterone and their metabolites and derivatives is based on both in vitro and in vivo studies. Further studies on the roles of these fascinating molecules under physiologic and pathologic conditions will help us design novel and effective therapeutic strategies for the treatment of CNS injuries.
Conclusion and future directions
Studies indicate that E2 can produce quite favorable results in treating the CNS injuries in animal models, as both protective and reparative mechanisms of E2 act on different cellular and molecular targets in the CNS (Figure 5). While these preclinical studies have come up with some breakthrough results, the fact how exactly E2 mechanistically works is still up for debate. Results from E2-treated CNS injuries have raised additional questions that need to be addressed before E2 therapy can move to the translational phase. While there are still many questions regarding the practice of administering E2 as a neuroprotective agent, one thing is for certain that it has opened the door for a new era in the pharmaceutical world of drug design and synthesis. It has given important insights into the innovative and novel treatment options for the CNS injuries. As the hydroxyl group of phenyl ring in E2 is the real transaction point for its neuroprotective effect by scavenging the ROS including hydroxyl radicals,24,104 we believe that the development of boron-based E2 derivatives is an innovative and novel concept to trap the ROS more efficiently for further increasing the neuroprotective effects of E2 in the CNS injuries. To enhance the ROS scavenging mechanism of E2, our group is now devoted to developing boron-based E2 derivatives (Figure 6). Both E2 and its derivatives, which may be more natural compounds than many artificially synthesized drugs, keep the hope alive for successful neuroprotection in different CNS injuries without major side effects. Any nonphysiologic high dose of E2 may cause notable side effects such as feminization and tumorigenesis,61 which will prevent E2 from its translation to the clinics for treatment of CNS injuries in humans. Combination of low doses of E2 and another therapeutic agent will probably be suitable to increase the desirable neuroprotective effects of E2 without its undesirable side effects. In fact, it is now highly regarded that combination of therapeutic agents should be the future treatment strategy for functional repair in CNS injuries.64 Combination therapy can provide an opportunity to use two different therapeutic agents at very low doses for additive effects or even synergistic effects to improve the overall therapeutic outcomes in CNS injuries.105,106 If a more concrete understanding of E2 and its intricate workings in the neurologic system soon comes to light, the possibilities of E2 or its new derivative for treatment of CNS injuries start to become endless.
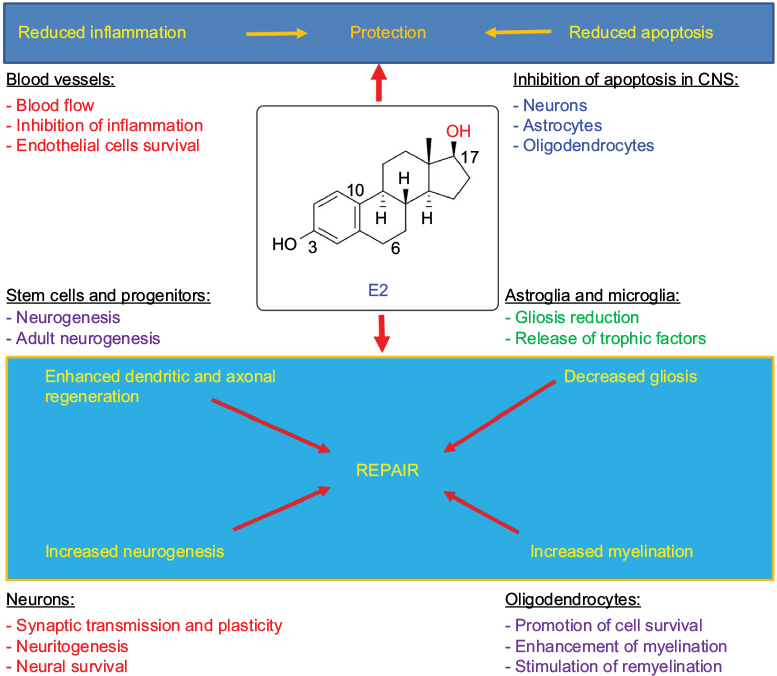 | Figure 5 E2 activates both protective and reparative mechanisms in CNS injuries in vivo. Note: Different color fonts and arrows indicate diverse biological activities of E2. Abbreviations: CNS, central nervous system; E2, estradiol. |
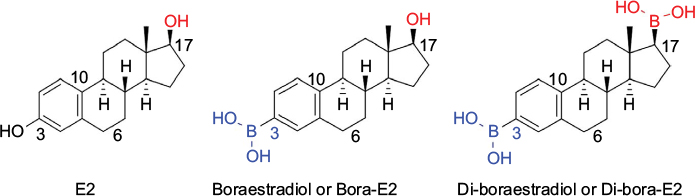 | Figure 6 Boron-containing E2 molecules may be more effective than E2 for neuroprotection. Abbreviation: E2, estradiol. |
Acknowledgments
This work was supported in part by the Incentive Award from the United Soybean Board (USB, Chesterfield, MO, USA), the Spinal Cord Injury Research Fund (SCIRF-2015-I-0) grant from the State of South Carolina (Columbia, SC, USA), and also the earlier R01 grants (CA91460 and NS057811) from the National Institutes of Health (Bethesda, MD, USA).
Disclosure
The authors report no conflicts of interest in this work.
References
Vrtačnik P, Ostanek B, Mencej-Bedrač S, Marc J. The many faces of estrogen signaling. Biochem Med (Zagreb). 2014;24(3):329–342. | ||
Sribnick EA, Ray SK, Banik NL. Estrogen as a multi-active neuroprotective agent in traumatic injuries. Neurochem Res. 2004;29(11):2007–2014. | ||
Chakrabarti M, Das A, Samantaray S, Smith JA, Banik NL, Haque A, Ray SK. Molecular mechanisms of estrogen for neuroprotection in spinal cord injury and traumatic brain injury. Rev Neurosci. 2016;27(3):271–281. | ||
Samantaray S, Das A, Matzelle DC, et al. Administration of low dose estrogen attenuates gliosis and protects neurons in acute spinal cord injury in rats. J Neurochem. 2016;136(5):1064–1073. | ||
Simpson E, Rubin G, Clyne C, et al. The role of local estrogen biosynthesis in males and females. Trends Endocrinol Metab. 2000;11(5):184–188. | ||
Siddiqui AN, Siddiqui N, Khan RA, et al. Neuroprotective role of steroidal sex hormones: an overview. CNS Neurosci Ther. 2016;22(5):342–350. | ||
Marin R, Guerra B, Alonso R, Ramírez CM, Díaz M. Estrogen activates classical and alternative mechanisms to orchestrate neuroprotection. Curr Neurovasc Res. 2005;2(4):287–301. | ||
Chakrabarti M, Haque A, Banik NL, Nagarkatti P, Nagarkatti M, Ray SK. Estrogen receptor agonists for attenuation of neuroinflammation and neurodegeneration. Brain Res Bull. 2014;109:22–31. | ||
Ray SK, Samantaray S, Banik NL. Future directions for using estrogen receptor agonists in the treatment of acute and chronic spinal cord injury. Neural Regen Res. 2016;11(9):1418–1419. Erratum in: Neural Regen Res. 2017;12 (2):266. | ||
Cox A, Varma A, Barry J, Vertegel A, Banik N. Nanoparticle estrogen in rat spinal cord injury elicits rapid anti-inflammatory effects in plasma, cerebrospinal fluid, and tissue. J Neurotrauma. 2015;32(18):1413–1421. | ||
Lee JY, Choi HY, Na WH, Ju BG, Yune TY. 17β-estradiol inhibits MMP-9 and SUR1/TrpM4 expression and activation and thereby attenuates BSCB disruption/hemorrhage after spinal cord injury in male rats. Endocrinology. 2015;156(5):1838–1850. | ||
Han D, Scott EL, Dong Y, Raz L, Wang R, Zhang Q. Attenuation of mitochondrial and nuclear p38α signaling: a novel mechanism of estrogen neuroprotection in cerebral ischemia. Mol Cell Endocrinol. 2015;400:21–31. | ||
Soltani Z, Khaksari M, Shahrokhi N, et al. Effect of estrogen and/or progesterone administration on traumatic brain injury-caused brain edema: the changes of aquaporin-4 and interleukin-6. J Physiol Biochem. 2016;72(1):33–44. | ||
Dubal DB, Zhu H, Yu J, et al. Estrogen receptor alpha, not beta, is a critical link in estradiol-mediated protection against brain injury. Proc Natl Acad Sci U S A. 2001;98(4):1952–1957. | ||
Mosquera L, Colón JM, Santiago JM, et al. Tamoxifen and estradiol improved locomotor function and increased spared tissue in rats after spinal cord injury: their antioxidant effect and role of estrogen receptor alpha. Brain Res. 2014;1561:11–22. | ||
Sampei K, Goto S, Alkayed NJ, et al. Stroke in estrogen receptor-alpha-deficient mice. Stroke. 2000;31(3):738–743. | ||
Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J Comp Neurol. 1997;388(4):507–525. | ||
Hu R, Sun H, Zhang Q, et al. G-protein coupled estrogen receptor 1 mediated estrogenic neuroprotection against spinal cord injury. Crit Care Med. 2012;40(12):3230–3237. | ||
Islamov RR, Hendricks WA, Katwa LC, et al. Effect of 17 beta-estradiol on gene expression in lumbar spinal cord following sciatic nerve crush injury in ovariectomized mice. Brain Res. 2003;966(1):65–75. | ||
Zhu C, Wang S, Wang B, et al. 17β-Estradiol up-regulates Nrf2 via PI3K/AKT and estrogen receptor signaling pathways to suppress light-induced degeneration in rat retina. Neuroscience. 2015;304:328–339. | ||
Culmsee C, Vedder H, Ravati A, et al. Neuroprotection by estrogens in a mouse model of focal cerebral ischemia and in cultured neurons: evidence for a receptor-independent antioxidative mechanism. J Cereb Blood Flow Metab. 1999;19(11):1263–1269. | ||
Sugishita K, Li F, Su Z, Barry WH. Anti-oxidant effects of estrogen reduce [Ca2+ ]i during metabolic inhibition. J Mol Cell Cardiol. 2003;35(3):331–336. | ||
Qian M, Engler-Chiurazzi EB, Lewis SE, Rath NP, Simpkins JW, Covey DF. Structure-activity studies of non-steroid analogs structurally-related to neuroprotective estrogens. Org Biomol Chem. 2016;14(41):9790–9805. | ||
Prokai-Tatrai K, Perjesi P, Zharikova AD, Perez EJ, Liu R, Simpkins JW. Quinol-based cyclic antioxidant mechanism in estrogen neuroprotection. Proc Natl Acad Sci U S A. 2003;100(20):11741–11746. | ||
Grimm A, Schmitt K, Lang UE, Mensah-Nyagan AG, Eckert A. Improvement of neuronal bioenergetics by neurosteroids: implications for age-related neurodegenerative disorders. Biochim Biophys Acta. 2014;1842(12 Pt A):2427–2438. | ||
Fiedler IG, Laud PW, Maiman DJ, Apple DF Jr. Economics of managed care in spinal cord injury. Arch Phys Med Rehabil. 1999;80:1441–1449. | ||
Selvarajah S, Hammond ER, Haider AH, et al. The burden of acute traumatic spinal cord injury among adults in the United States: an update. J Neurotrauma. 2014;31(3):228–238. | ||
Mahabaleshwarkar R, Khanna R. National hospitalization burden associated with spinal cord injuries in the United States. Spinal Cord. 2014;52(2):139–144. | ||
Dismuke CE, Egede LE, Saunders L, Krause JS. Diabetes increases financial burden of individuals with traumatic spinal cord injury (TSCI). Spinal Cord. 2015;53(2):135–138. | ||
DeVivo MJ. Causes and costs of spinal cord injury in the United States. Spinal Cord. 1997;35(12):809–813. | ||
Eastwood EA, Hagglund KJ, Ragnarsson KT, Gordon WA, Marino RJ. Medical rehabilitation length of stay and outcomes for persons with traumatic spinal cord injury– 1990–1997. Arch Phys Med Rehabil. 1999;80:1457–1463. | ||
Pereira JE, Costa LM, Cabrita AM, et al. Methylprednisolone fails to improve functional and histological outcome following spinal cord injury in rats. Exp Neurol. 2009;220(1):71–81. | ||
Wu Y, Hou J, Collier L, et al. The administration of high-dose methylprednisolone for 24 hour reduced muscle size and increased atrophy-related gene expression in spinal cord-injured rats. Spinal Cord. 2011;49(8):867–873. | ||
Samantaray S, Das A, Matzelle DC, et al. Administration of low dose estrogen attenuates persistent inflammation, promotes angiogenesis, and improves locomotor function following chronic spinal cord injury in rats. J Neurochem. 2016;137(4):604–617. | ||
Kim YH, Ha KY, Kim SI. Spinal cord injury and related clinical trials. Clin Orthop Surg. 2017;9(1):1–9. | ||
Griesbach GS, Kreber LA, Harrington D, Ashley MJ. Post-acute traumatic brain injury rehabilitation: effects on outcome measures and life care costs. J Neurotrauma. 2015;32(10):704–711. | ||
Dismuke CE, Walker RJ, Egede LE. utilization and cost of health services in individuals with traumatic brain injury. Glob J Health Sci. 2015;7(6):156–169. | ||
Schiraldi M, Patil CG, Mukherjee D, et al. Effect of insurance and racial disparities on outcomes in traumatic brain injury. J Neurol Surg A Cent Eur Neurosurg. 2015;76(3):224–232. | ||
Spitz G, McKenzie D, Attwood D, Ponsford JL. Cost prediction following traumatic brain injury: model development and validation. J Neurol Neurosurg Psychiatry. 2016;87(2):173–180. | ||
Wei W, Sambamoorthi U, Crystal S, Findley PA. Mental illness, traumatic brain injury, and Medicaid expenditures. Arch Phys Med Rehabil. 2005;86(5):905–911. | ||
Alali AS, Burton K, Fowler RA, et al. Economic evaluations in the diagnosis and management of traumatic brain injury: a systematic review and analysis of quality. Value Health. 2015;18(5):721–734. | ||
Diaz-Arrastia R, Kochanek PM, Bergold P, et al. Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma. 2014;31(2):135–158. | ||
Margulies S, Hicks R; Combination Therapies for Traumatic Brain Injury Workshop Leaders. Combination therapies for traumatic brain injury: prospective considerations. J Neurotrauma. 2009;26(6):925–939. | ||
Flynn RW, MacWalter RS, Doney AS. The cost of cerebral ischaemia. Neuropharmacology. 2008;55(3):250–256. | ||
Lewandowski C, Barsan W. Treatment of acute ischemic stroke. Ann Emerg Med. 2001;37(2):202–216. | ||
Goyal N, Male S, Al Wafai A, Bellamkonda S, Zand R. Cost burden of stroke mimics and transient ischemic attack after intravenous tissue plasminogen activator treatment. J Stroke Cerebrovasc Dis. 2015;24(4):828–833. | ||
Brinjikji W, Rabinstein AA, Cloft HJ. Hospitalization costs for acute ischemic stroke patients treated with intravenous thrombolysis in the United States are substantially higher than medicare payments. Stroke. 2012;43(4):1131–1133. | ||
Chambers MG, Koch P, Hutton J. Development of a decision-analytic model of stroke care in the United States and Europe. Value Health. 2002;5(2):82–97. | ||
Dion JE. Management of ischemic stroke in the next decade: stroke centers of excellence. J Vasc Interv Radiol. 2004;15(1 Pt 2): S133–S141. | ||
Chen HS, Qi SH, Shen JG. One-Compound-Multi-Target: combination prospect of natural compounds with thrombolytic therapy in acute ischemic stroke. Curr Neuropharmacol. 2017;15(1):134–156. | ||
de Ridder IR, Fransen PS, Beumer D, et al. Is intra-arterial treatment for acute ischemic stroke less effective in women than in men? Interv Neurol. 2016;5(3–4):174–178. | ||
Ray SK, Samantaray S, Smith JA, Matzelle DD, Das A, Banik NL. Inhibition of cysteine proteases in acute and chronic spinal cord injury. Neurotherapeutics. 2011;8(2):180–186. | ||
Sámano C, Kaur J, Nistri A. A study of methylprednisolone neuroprotection against acute injury to the rat spinal cord in vitro. Neuroscience. 2016;315:136–149. | ||
MacMahon PJ, Huang AJ, Palmer WE. Spine injectables: What is the safest cocktail? AJR Am J Roentgenol. 2016;207(3):526–533. | ||
Farooque M, Suo Z, Arnold PM, et al. Gender-related differences in recovery of locomotor function after spinal cord injury in mice. Spinal Cord. 2006;44(3):182–187. | ||
Letaif OB, Cristante AF, Barros Filho TE, et al. Effects of estrogen on functional and neurological recovery after spinal cord injury: an experimental study with rats. Clinics (Sao Paulo). 2015;70(10):700–705. | ||
Sribnick EA, Wingrave JM, Matzelle DD, Wilford GG, Ray SK, Banik NL. Estrogen attenuated markers of inflammation and decreased lesion volume in acute spinal cord injury in rats. J Neurosci Res. 2005;82(2):283–293. | ||
Sribnick EA, Samantaray S, Das A, et al. Postinjury estrogen treatment of chronic spinal cord injury improves locomotor function in rats. J Neurosci Res. 2010;88(8):1738–1750. | ||
Hendriks AE, Laven JS, Valkenburg O, et al. Fertility and ovarian function in high-dose estrogen-treated tall women. J Clin Endocrinol Metab. 2011;96(4):1098–1105. | ||
Hendriks AE, Drop SL, Laven JS, Boot AM. Fertility of tall girls treated with high-dose estrogen, a dose-response relationship. J Clin Endocrinol Metab. 2012;97(9):3107–3114. | ||
Benyi E, Kieler H, Linder M, et al. Risks of malignant and non-malignant tumors in tall women treated with high-dose estrogen during adolescence. Horm Res Paediatr. 2014;82(2):89–96. | ||
Wibowo E, Schellhammer P, Wassersug RJ. Role of estrogen in normal male function: clinical implications for patients with prostate cancer on androgen deprivation therapy. J Urol. 2011;185(1):17–23. | ||
Blank EW, Wong PY, Lakshmanaswamy R, Guzman R, Nandi S. Both ovarian hormones estrogen and progesterone are necessary for hormonal mammary carcinogenesis in ovariectomized ACI rats. Proc Natl Acad Sci U S A. 2008;105(9):3527–3532. | ||
Tohda C, Kuboyama T. Current and future therapeutic strategies for functional repair of spinal cord injury. Pharmacol Ther. 2011;132(1):57–71. | ||
Chakrabarti M, Banik NL, Ray SK. miR-7–1 potentiated estrogen receptor agonists for functional neuroprotection in VSC4, 1 motoneurons. Neuroscience. 2014;256:322–333. | ||
Naderi V, Khaksari M, Abbasi R, Maghool F. Estrogen provides neuroprotection against brain edema and blood brain barrier disruption through both estrogen receptors α and β following traumatic brain injury. Iran J Basic Med Sci. 2015;18(2):138–144. | ||
Zlotnik A, Leibowitz A, Gurevich B, et al. Effect of estrogens on blood glutamate levels in relation to neurological outcome after TBI in male rats. Intensive Care Med. 2012;38(1):137–144. | ||
Khaksari M, Abbasloo E, Dehghan F, Soltani Z, Asadikaram G. The brain cytokine levels are modulated by estrogen following traumatic brain injury: Which estrogen receptor serves as modulator? Int Immunopharmacol. 2015;28(1):279–287. | ||
Soltani Z, Khaksari M, Jafari E, Iranpour M, Shahrokhi N. Is genistein neuroprotective in traumatic brain injury? Physiol Behav. 2015;152(Pt A):26–31. | ||
Kim H, Cam-Etoz B, Zhai G, Hubbard WJ, Zinn KR, Chaudry IH. Salutary effects of estrogen sulfate for traumatic brain injury. J Neurotrauma. 2015;32(16):1210–1216. | ||
Schaible EV, Windschügl J, Bobkiewicz W, et al. 2-Methoxyestradiol confers neuroprotection and inhibits a maladaptive HIF-1α response after traumatic brain injury in mice. J Neurochem. 2014;129(6):940–954. | ||
Gorska M, Zmijewski MA, Kuban-Jankowska A, Wnuk M, Rzeszutek I, Wozniak M. Neuronal nitric oxide synthase-mediated genotoxicity of 2-methoxyestradiol in hippocampal HT22 cell line. Mol Neurobiol. 2016;53(7):5030–5040. | ||
Nuñez JL, McCarthy MM. Estradiol exacerbates hippocampal damage in a model of preterm infant brain injury. Endocrinology. 2003;144(6):2350–2359. | ||
Lamprecht MR, Morrison B 3rd. A combination therapy of 17β-estradiol and memantine is more neuroprotective than monotherapies in an organotypic brain slice culture model of traumatic brain injury. J Neurotrauma. 2015;32(17):1361–1368. | ||
Carpenter RS, Iwuchukwu I, Hinkson CL, et al. High-dose estrogen treatment at reperfusion reduces lesion volume and accelerates recovery of sensorimotor function after experimental ischemic stroke. Brain Res. 2016;1639:200–213. | ||
Hoffmann S, Beyer C, Zendedel A. Comparative analysis of gonadal steroid-mediated neuroprotection after transient focal ischemia in rats: route of application and substrate composition. J Mol Neurosci. 2015;56(1):12–16. | ||
Castelló-Ruiz M, Torregrosa G, Burguete MC, et al. The selective estrogen receptor modulator, bazedoxifene, reduces ischemic brain damage in male rat. Neurosci Lett. 2014;575:53–57. | ||
Lamprecht MR, Morrison B 3rd. GPR30 activation is neither necessary nor sufficient for acute neuroprotection by 17β-estradiol after an ischemic injury in organotypic hippocampal slice cultures. Brain Res. 2014;1563:131–137. | ||
Zhang QG, Wang R, Tang H, et al. Brain-derived estrogen exerts anti-inflammatory and neuroprotective actions in the rat hippocampus. Mol Cell Endocrinol. 2014;389(1–2):84–91. | ||
Madinier A, Wieloch T, Olsson R, Ruscher K. Impact of estrogen receptor beta activation on functional recovery after experimental stroke. Behav Brain Res. 2014;261:282–288. | ||
Desjardins GC, Brawer JR, Beaudet A. Estradiol is selectively neurotoxic to hypothalamic beta-endorphin neurons. Endocrinology. 1993;132(1):86–93. | ||
Selvamani A, Sohrabji F. The neurotoxic effects of estrogen on ischemic stroke in older female rats is associated with age-dependent loss of insulin-like growth factor-1. J Neurosci. 2010;30(20):6852–6861. | ||
Liu R, Liu Q, He S, Simpkins JW, Yang SH. Combination therapy of 17beta-estradiol and recombinant tissue plasminogen activator for experimental ischemic stroke. J Pharmacol Exp Ther. 2010;332(3):1006–1012. | ||
Lee JS, Kim YK, Yang H, Kang HY, Ahn C, Jeung EB. Two faces of the estrogen metabolite 2-methoxyestradiol in vitro and in vivo. Mol Med Rep. 2015;12(4):5375–5382. | ||
Chen SH, Yeh CH, Lin MY, et al. Premarin improves outcomes of spinal cord injury in male rats through stimulating both angiogenesis and neurogenesis. Crit Care Med. 2010;38(10):2043–2051. | ||
Chen SH, Chang CY, Chang HK, et al. Premarin stimulates estrogen receptor-alpha to protect against traumatic brain injury in male rats. Crit Care Med. 2009;37(12):3097–3106. | ||
Wang YF, Fan ZK, Cao Y, Yu DS, Zhang YQ, Wang YS. 2-Methoxyestradiol inhibits the up-regulation of AQP4 and AQP1 expression after spinal cord injury. Brain Res. 2011;1370:220–226. | ||
Chen W, Jadhav V, Tang J, Zhang JH. HIF-1 alpha inhibition ameliorates neonatal brain damage after hypoxic-ischemic injury. Acta Neurochir Suppl. 2008;102:395–399. | ||
Duncan GS, Brenner D, Tusche MW, et al. 2-Methoxyestradiol inhibits experimental autoimmune encephalomyelitis through suppression of immune cell activation. Proc Natl Acad Sci U S A. 2012;109(51):21034–21039. | ||
Schaufelberger SA, Rosselli M, Barchiesi F, Gillespie DG, Jackson EK, Dubey RK. 2-Methoxyestradiol, an endogenous 17β-estradiol metabolite, inhibits microglial proliferation and activation via an estrogen receptor-independent mechanism. Am J Physiol Endocrinol Metab. 2016;310(5):E313–E322. | ||
James J, Murry DJ, Treston AM, et al. Phase I safety, pharmacokinetic and pharmacodynamic studies of 2-methoxyestradiol alone or in combination with docetaxel in patients with locally recurrent or metastatic breast cancer. Invest New Drugs. 2007;25(1):41–48. | ||
Jennings BL, Moore JA, Pingili AK, et al. Disruption of the cytochrome P-450 1B1 gene exacerbates renal dysfunction and damage associated with angiotensin II-induced hypertension in female mice. Am J Physiol Renal Physiol. 2015;308(9):F981–F992. | ||
Shand FH, Langenbach SY, Keenan CR, Ma SP, Wheaton BJ, Schuliga MJ, Ziogas J, Stewart AG. In vitro and in vivo evidence for anti-inflammatory properties of 2-methoxyestradiol. J Pharmacol Exp Ther. 2011;336(3):962–972. | ||
Tofovic SP, Salah EM, Dubey RK, Melhem MF, Jackson EK. Estradiol metabolites attenuate renal and cardiovascular injury induced by chronic nitric oxide synthase inhibition. J Cardiovasc Pharmacol. 2005;46:25–35. | ||
Dubey RK, Jackson EK. Potential vascular actions of 2-methoxyestradiol. Trends Endocrinol Metab. 2009;20(8):374–379. | ||
Engler-Chiurazzi EB, Brown CM, Povroznik JM, Simpkins JW. Estrogens as neuroprotectants: estrogenic actions in the context of cognitive aging and brain injury. Prog Neurobiol. 2016 Feb 15. pii: S0301–0082(15)30,063–0. | ||
Green PS, Simpkins JW. Neuroprotective effects of estrogens: potential mechanisms of action. Int J Dev Neurosci. 2000;18(4–5):347–358. | ||
Green PS, Yang SH, Simpkins JW. Neuroprotective effects of phenolic A ring oestrogens. Novartis Found Symp. 2000;230:202–213; discussion 213–220. | ||
Littleton-Kearney MT, Ostrowski NL, Cox DA, Rossberg MI, Hurn PD. Selective estrogen receptor modulators: tissue actions and potential for CNS protection. CNS Drug Rev. 2002;8(3):309–330. | ||
Yi KD, Perez E, Yang S, Liu R, Covey DF, Simpkins JW. The assessment of non-feminizing estrogens for use in neuroprotection. Brain Res. 2011;1379:61–70. | ||
Petrone AB, Gatson JW, Simpkins JW, Reed MN. Non-feminizing estrogens: a novel neuroprotective therapy. Mol Cell Endocrinol. 2014;389(1–2):40–47. | ||
Song WO, Chun OK, Hwang I, Shin HS, Kim BG, Kim KS, Lee SY, Shin D, Lee SG. Soy isoflavones as safe functional ingredients. J Med Food. 2007;10(4):571–580. | ||
Qian Y, Guan T, Huang M, Cao L, Li Y, Cheng H, Jin H, Yu D. Neuroprotection by the soy isoflavone, genistein, via inhibition of mitochondria-dependent apoptosis pathways and reactive oxygen induced-NF-κB activation in a cerebral ischemia mouse model. Neurochem Int. 2012;60(8):759–767. | ||
Prokai-Tatrai K, Perjesi P, Rivera-Portalatin NM, Simpkins JW, Prokai L. Mechanistic investigations on the antioxidant action of a neuroprotective estrogen derivative. Steroids. 2008;73(3):280–288. | ||
Sharma HS. Neurotrophic factors in combination: a possible new therapeutic strategy to influence pathophysiology of spinal cord injury and repair mechanisms. Curr Pharm Des. 2007;13(18):1841–1874. | ||
Liu J, Zhou CX, Zhang ZJ, Wang LY, Jing ZW, Wang Z. Synergistic mechanism of gene expression and pathways between jasminoidin and ursodeoxycholic acid in treating focal cerebral ischemia-reperfusion injury. CNS Neurosci Ther. 2012;18(8):674–682. |
 © 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2017 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.