Back to Journals » International Journal of Nanomedicine » Volume 18
Neuroprotective Effects and Therapeutic Potential of Dichloroacetate: Targeting Metabolic Disorders in Nervous System Diseases
Authors Zhang Y, Sun M ![]() , Zhao H, Wang Z, Shi Y, Dong J
, Zhao H, Wang Z, Shi Y, Dong J ![]() , Wang K, Wang X
, Wang K, Wang X ![]() , Li X, Qi H, Zhao X
, Li X, Qi H, Zhao X ![]()
Received 15 September 2023
Accepted for publication 28 November 2023
Published 12 December 2023 Volume 2023:18 Pages 7559—7581
DOI https://doi.org/10.2147/IJN.S439728
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Lijie Grace Zhang
Yue Zhang,1,2,* Meiyan Sun,2,* Hongxiang Zhao,1,2,* Zhengyan Wang,2 Yanan Shi,2 Jianxin Dong,2 Kaifang Wang,3 Xi Wang,4 Xingyue Li,5 Haiyan Qi,6 Xiaoyong Zhao1,2,7
1Department of Radiation Oncology and Shandong Provincial Key Laboratory of Radiation Oncology, Shandong Cancer Hospital and Institute, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, Shandong, People’s Republic of China; 2Laboratory of Anesthesia and Critical Care Medicine in Colleges and Universities of Shandong Province, School of Anesthesiology, Weifang Medical University, Weifang, Shandong Province, People’s Republic of China; 3Department of Anesthesia, Tangdu Hospital, Fourth Military Medical University, Xian, Shanxi Province, People’s Republic of China; 4Department of Anesthesiology, Sir Run Run Shaw Hospital, School of Medicine, Zhejiang University, Hangzhou, Zhejiang Province, People’s Republic of China; 5Department of Hepatobiliary and Pancreatic Surgery, Affiliated Hospital of Weifang Medical University, Weifang, Shandong Province, People’s Republic of China; 6Department of Anesthesiology, Jinan Maternity and Child Care Hospital Affiliated to Shandong First Medical University, Jinan, Shandong Province, People’s Republic of China; 7Department of Anesthesiology, Shandong Cancer Hospital and Institute, Shandong First Medical University and Shandong Academy of Medical Sciences, Jinan, Shandong Province, People’s Republic of China
*These authors contributed equally to this work
Correspondence: Xiaoyong Zhao; Haiyan Qi, Email [email protected]; [email protected]
Abstract: Dichloroacetate (DCA) is an investigational drug used to treat lactic acidosis and malignant tumours. It works by inhibiting pyruvate dehydrogenase kinase and increasing the rate of glucose oxidation. Some studies have documented the neuroprotective benefits of DCA. By reviewing these studies, this paper shows that DCA has multiple pharmacological activities, including regulating metabolism, ameliorating oxidative stress, attenuating neuroinflammation, inhibiting apoptosis, decreasing autophagy, protecting the blood‒brain barrier, improving the function of endothelial progenitor cells, improving mitochondrial dynamics, and decreasing amyloid β-protein. In addition, DCA inhibits the enzyme that metabolizes it, which leads to peripheral neurotoxicity due to drug accumulation that may be solved by individualized drug delivery and nanovesicle delivery. In summary, in this review, we analyse the mechanisms of neuroprotection by DCA in different diseases and discuss the causes of and solutions to its adverse effects.
Keywords: dichloroacetate, neuroprotection, mitochondria, energy metabolism, oxidative stress
Introduction
Neurons are necessary for the brain to perform its normal physiological functions. However, both internal and external factors can damage neurons, which ultimately results in their death.1 Many neurological diseases, such as brain injury and neurodegenerative diseases, cause neuronal damage; therefore, reducing or preventing neuronal damage is especially important in treating or alleviating these diseases.2–5 Currently, many neuroprotective drugs have shown an extraordinary ability to prevent neuronal damage by mechanisms such as limiting mitochondrial dysfunction, oxidative stress, neuroinflammation, apoptosis, excitotoxicity, and protein misfolding, which are leading therapeutic strategies for these diseases.6–8
Dichloroacetate (DCA) inhibits all four pyruvate dehydrogenase kinase (PDK) isoforms found in vivo.9,10 PDK phosphorylates three serine residues (Ser293, Ser300, and Ser232) of the α-subunit of pyruvate dehydrogenase (PDH) to cause a decrease in PDH activity.11,12 PDH is responsible for the irreversible transformation of pyruvate into acetyl coenzyme A, which subsequently engages in the tricarboxylic acid (TCA) cycle to produce energy.13 Thus, DCA promotes the mitochondrial oxidation of pyruvate and increases the rate of glucose oxidation, thereby improving tissue energy metabolism (Figure 1).14 This allows DCA to reverse the Warburg effect in cancer cells, making it a promising anticancer drug that targets cancer cell metabolism.15,16 A large number of studies have been conducted to explore the antitumour effects of DCA or combination therapies of DCA with radiotherapy, chemotherapy, and immunotherapy, which have been developed in phase II clinical trials, in a variety of cancers.17–22 Additionally, DCA has also been used as an investigational drug for lactic acidosis, pulmonary arterial hypertension, sepsis-induced hepatic metabolic dysfunction, and cardiac dysfunction, and a phase III clinical trial involving children with congenital lactic acidosis (NCT00004490) has been completed.23–26
 |
Figure 1 The role of DCA in metabolism. Abbreviations: DCA, dichloroacetate; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; TCA, tricarboxylic acid. Notes: DCA enhances PDH indirectly by inhibiting PDK, thereby enhancing mitochondrial oxidation of glucose. Created with BioRender.com. |
Mitochondria assume a crucial role in upholding cellular physiological activities, encompassing significant functions such as oxidative phosphorylation, phospholipid biogenesis, calcium homeostasis maintenance, and apoptosis regulation.27 Neurons exhibit elevated energy metabolic demands, as the brain accounts for 20% of the body’s resting adenosine triphosphate (ATP) production, despite comprising merely 2% of its overall mass.28 If mitochondrial metabolism is disrupted, severe damage to neurons is likely.13 Disruption of mitochondrial metabolism is considered an important cause of many neurological diseases, such as stroke and neurodegenerative diseases.29–31 Currently, DCA has been shown to modify mitochondrial metabolism in the pathophysiology of several neurological diseases, including ischaemic stroke, perinatal asphyxia-induced hypoxic-ischaemic brain injury, transient cerebral ischaemia, cardiac arrest brain injury, subarachnoid haemorrhage, vascular dementia, hypoglycaemic brain injury, traumatic brain injury, epilepsy, amyotrophic lateral sclerosis, Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease (Table 1). This review comprehensively examines the latest research on the neuroprotective properties of DCA and discusses the molecular mechanisms through which DCA mitigates neuronal death in various neurological illnesses within the context of the corresponding metabolic disorders. Additionally, we discuss some of the newer understandings of the pharmacological effects of DCA. Finally, we discuss the limitations and possible countermeasures of DCA found in clinical trials in other diseases.
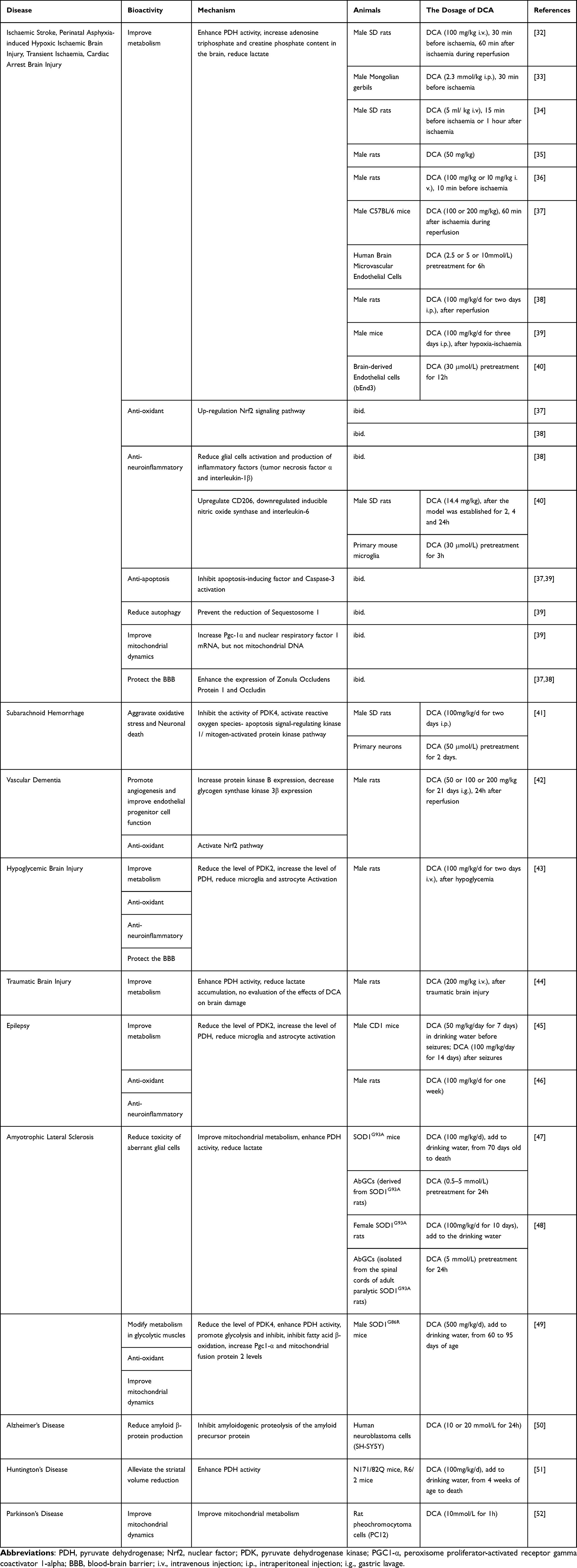 |
Table 1 Neuroprotective effects of Dichloroacetate |
Neuroprotective Effects and Mechanisms of Dichloroacetate
The Effects of Dichloroacetate on the Brain in Different Injuries
Ischaemia‒Reperfusion Injury
Ischaemia‒reperfusion injury (I/RI) is a term used to describe the occurrence of heightened tissue damage and metabolic disturbances following the restoration of blood reperfusion to partially ischaemic tissue.53 Ischaemic stroke, perinatal asphyxia-induced hypoxic-ischaemic brain injury (HIE), cardiac arrest brain injury, and transient cerebral ischaemia are among the diseases in which I/RI plays a role in the pathological processes.54 Insufficient oxygen and glucose supply to brain tissue limits aerobic metabolism and ATP production, which leads to lactate accumulation and ion transport dysfunction, triggering cellular and mitochondrial calcium overload and ultimately leading to neuron death.54–56 More importantly, PDH activity decreases during I/RI.57 DCA has been shown to enhance PDH activity, increase ATP and phosphocreatine levels, and reduce lactate accumulation in the brain, thus reducing lactic acidosis during I/RI.32–35 Peeling et al found that administration of DCA (100 mg/kg i.v.) to rats with normal forebrain ischaemia restored brain pH, lactate, ATP, and phosphocreatine levels after reperfusion, thereby reducing hippocampal neuronal damage.36
Oxidative stress is an important cause of I/RI; moreover, reactive oxygen species (ROS) exacerbate mitochondrial dysfunction and impair cellular structure and function, resulting in a vicious cycle.58 DCA was shown to reduce oxidative stress in subsequent studies.37,38 In middle cerebral artery occlusion mice, the embolus was loosened, and DCA (100 mg/kg, 200 mg/kg) was injected 90 min after embolization, which showed that DCA could activate the nuclear factor erythroid 2-related factor 2 (Nrf2) pathway and reduce ROS production in the brains of mice, reducing the area of cerebral infarction.37 Notably, at low oxidative stress levels, activation of Nrf2 activates downstream antioxidant pathways to exert cytoprotective effects, while excessive activation of Nrf2 stimulates the expression of Kruppel-like factor 9 (Klf9), which, conversely, increases the level of ROS.59 Low doses of sulforaphane have been shown to exert antioxidant effects through activation of the Nrf2 pathway, whereas sulforaphane at toxic doses instead overactivates Klf9, leading to increased oxidative damage in cells.60 Similarly, it remains to be investigated whether DCA also has the same problem of excessive activation of Nrf2 leading to increased oxidative stress. Indeed, DCA may exacerbate oxidative stress during brain injury in some cases. Interestingly, Gao et al found that DCA (100 mg/kg) inhibited PDK4 activity, enhanced PDH activity, increased ROS production and cell death, and conversely aggravated neurological impairment in rats with early brain injury (EBI) due to subarachnoid haemorrhage (SAH).41 EBI is a pathophysiological event that occurs within 72 hours after SAH.61 In contrast to cerebral ischaemia/reperfusion (I/R), cerebral blood flow is suddenly and significantly reduced after SAH, with only a small amount of incomplete reperfusion, and the brain tissue is in a state of persistent hypoxia-ischaemia.62–64 Importantly, DCA increases ROS production in all cells.65 Even in normal cells, the forward electron transfer of the electron transport chain (ETC) will always cause some of the electrons to leak and generate ROS; when the respiratory chain is damaged or inhibited, it may cause more electrons to leak and generate more ROS. The enhancement of aerobic respiration by DCA increases ROS generated by the forward transfer of electrons.66 The cellular shift to glycolysis under sustained hypoxia in EBI is a protective response to alleviate ATP deficiency, and the forced shift from glycolysis to oxidative phosphorylation by DCA in this case is more detrimental to cell survival.67 However, the main cause of ROS production during I/R is reverse electron transfer due to a lack of ATP and metabolite accumulation.68 The reduction of ATP during ischaemia causes cell membrane pump dysfunction, and calcium ions enter the cell to activate calcium-dependent protease, which converts xanthine dehydrogenase (XD) to xanthine oxidase (XO) in large quantities. When reperfusion occurs, under the influence of a large number of oxygen molecules, XO catalyses the conversion of hypoxanthine to xanthine, generating a large amount of ROS.69 On the other hand, the high proton gradient accumulated in the inner mitochondrial membrane and the excessive reduction in coenzyme Q that reverses electron transfer in the ETC also generate large amounts of ROS.70 The gradual decrease in the ATP/adenosine diphosphate (ADP) ratio during ischaemia and the degradation of accumulated adenosine monophosphate (AMP) leads to the depletion of the intracellular adenine nucleotide pools, which inhibits ATP production and the accumulation of protons in the inner mitochondrial membrane.71 In addition, hypoxia leads to a shift in cellular metabolism to glycolysis and β-oxidation, which can result in the accumulation of reduced nicotinamide adenine dinucleotide (NADH), thus causing succinate dehydrogenase to reverse its action to produce succinate.72 At the same time, the lack of coenzyme A and guanosine triphosphate (GTP) prevents the conversion of succinate, and the accumulated succinate is rapidly oxidized to fumarate, which leads to an overreduction of the coenzyme Q pool on complex II.70,73 A possible explanation for the different effects of DCA on oxidative stress is that DCA attenuates metabolite accumulation and reverse electron transfer in the ETC during I/R to reduce ROS production more strongly than it enhances forward electron transfer in the ETC to produce ROS. Oh et al found that DCA (1 g/l) increased cytochrome C oxidase activity and oxygen consumption of renal tubular cells in the kidneys of cisplatin-treated mice, thereby attenuating cisplatin-induced acute kidney injury.74 Increased cytochrome C oxidase activity may favour the continued forward transfer of electrons from complex II and decrease reverse electron transfer.75 Lin et al observed in two human colorectal cancer cell lines that DCA increased the oxidized nicotinamide adenine dinucleotide (NAD)/NADH ratio.76 These studies provide indirect evidence for possible mechanisms for the attenuation of oxidative stress during I/R by DCA, but it has not been studied whether DCA improves XO or succinate accumulation. In addition, the different effects of DCA administration at different times after injury need to be further explored, and administration during the ischaemic or reperfusion phase may produce opposite effects.
It is now widely recognized that neuronal damage in I/RI is the result of a combination of factors, including excitotoxicity, oxidative stress, apoptosis, neuroinflammation, excessive autophagy, cerebrovascular injury, etc.77 Excessive autophagy is a significant cause of neuronal loss during I/RI.78,79 ROS are thought to activate autophagy through various pathways, and cells are needed to eliminate damage from excess ROS through autophagy during I/R; however, excessive autophagy can damage neurons.80,81 Zou et al found that activation of Nrf2 inhibited autophagy to attenuate limb I/R-induced muscle damage, suggesting that attenuating oxidative stress may inhibit excessive autophagy in I/RI.82 Yanyan Sun et al found that DCA (100 mg/kg) prevented an hypoxic-ischaemic-induced decrease in sequestosome 1 (SQSTM1) protein expression, which implies that DCA protects neurons from I/RI by decreasing autophagic activity in the brain.39 During I/R, excess Ca2+ concentration, high levels of inorganic phosphate, and ROS lead to mitochondrial membrane potential alterations, opening mitochondrial permeability transition pores and releasing cytochrome C into the cytoplasm, followed by activation of downstream factors, such as caspase-3, causing apoptosis through a cascade reaction.83,84 Additionally, noncaspase-dependent apoptosis-inducing factor (AIF)-associated apoptosis has been suggested to be associated with excess ROS.85 A study showed that DCA (100 mg/kg) prevented the nuclear translocation of AIF and reduced the activation of caspase-3 after ischaemia and hypoxia in mice, demonstrating that DCA can prevent neuronal apoptosis.39 Injured or dead neurons stimulate the activation of glial cells that release inflammatory factors and cause secondary damage.86 Peng et al found that DCA (80 mg/kg) reduced the mRNA expression of tumour necrosis factor (TNF-α) and interleukin-1β (IL-1β) in the cerebral cortex and hippocampus of rats resuscitated from cardiac arrest, corroborating the regulatory impact of DCA on inflammation.87 Hong et al also found that DCA (100 mg/kg) reduced transient cerebral ischaemia-induced microglial activation.38 Disruption of blood‒brain barrier (BBB) integrity is one of the causes of I/RI, which is triggered by ROS through activation of pathways such as matrix metalloproteinases and the breakdown or modification of tight junction proteins (Occludin and ZO-1, among others).88,89 In addition, inflammatory cytokines (IL-1β and TNF-α) also decrease the expression of Occludin and ZO-1 while disrupting the function of the BBB.90 DCA has also been shown to increase the expression of ZO-1 and Occludin, thereby decreasing the extravasation of dyes in the cerebral vasculature of mice, suggesting a protective effect of DCA on the BBB.37,38 In addition, a study found that DCA increased mRNA levels of peroxisome proliferator-activated receptor-γ coactivator 1α (Pgc-1α) and nuclear respiratory factor-1 (Nrf1), two genes that are associated with mitochondrial biogenesis, in the brains of mice after hypoxic-ischaemic treatment, suggesting that DCA may also modulate mitochondrial dynamics, although the exact mechanism remains to be investigated.39
In conclusion, oxidative stress due to impaired energy metabolism can be regarded as an initiating factor in the pathological process of cerebral I/R that triggers subsequent processes. The above preclinical experiments demonstrated the neuroprotective potential of DCA in its ability to ameliorate energy metabolism disorders, oxidative stress, and a series of downstream pathological processes (Figure 2). One study found that DCA coadministered with pyruvate showed more significant neuroprotective effects than low doses of DCA or pyruvate alone.38 Pyruvate, which also acts to energize cells by facilitating the TCA cycle, has been shown to reduce neuronal death in I/RI.91,92 Guan et al synthesized the rho-associated coiled-coil containing protein kinase (ROCK) inhibitor fasudil and DCA into a new salt, fasudil dichloroacetate (FDCA), and the results showed that FDCA attenuated neuroinflammation and prevented breakdown of the BBB by inhibiting the activation of ROCK and the activity of PDK1, which reduced brain injury in MCAO mice.40 These studies suggest that the combination of DCA with other neuroprotective drugs may be a novel approach to enhance its neuroprotective effects, but the drug ratio and feasible therapeutic window need to be further investigated.
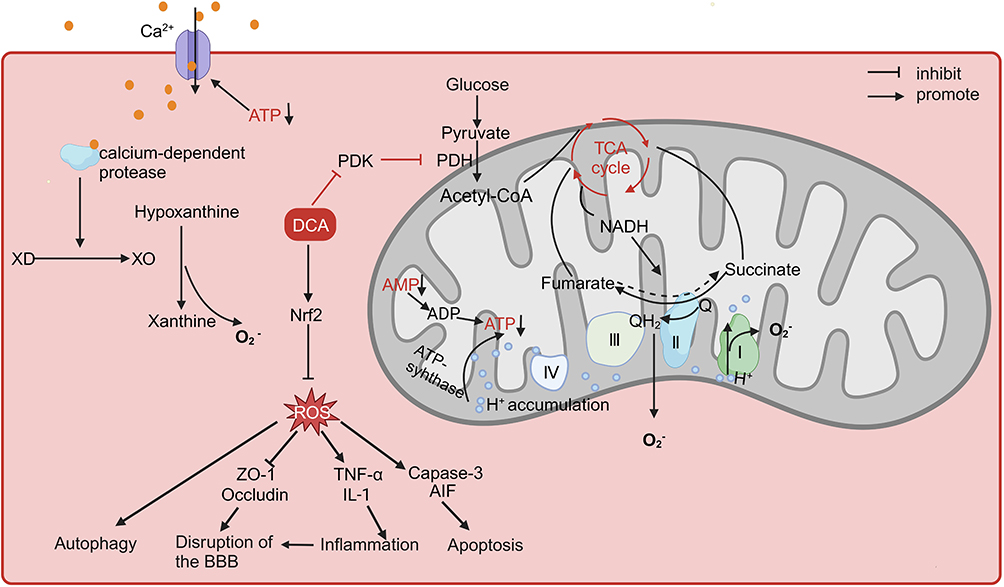 |
Figure 2 The role of DCA in cerebral ischaemia-reperfusion injury. Abbreviations: DCA, dichloroacetate; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; TCA, tricarboxylic acid; XD, xanthine dehydrogenase; XO, xanthine oxidase; Nrf2, nuclear factor erythroid 2-related factor 2; ATP, adenosine triphosphate; ADP, adenosine diphosphate; ROS, reactive oxygen species; AMP, adenosine monophosphate; NADH, reduced nicotinamide adenine dinucleotide; ZO-1, zonula occludens-1; TNF-α, tumor necrosis factor; IL-1, interleukin-1; AIF, apoptosis-inducing factor. Notes: When there is ischaemia-reperfusion injury, ATP deficiency causes the cell membrane ion pump to malfunction and intracellular calcium levels to rise. This triggers the calcium-dependent protease, which in turn catalyzes a sequence of events that result in a significant production of reactive oxygen species. Still, the breakdown of AMP results in the inability to produce new ATP, which causes hydrogen ions to build up in the mitochondrial membrane gap. Similarly, the overabundance of succinate during ischaemia causes the overreduction of CoQ during reperfusion, which causes reverse electron transfer to produce reactive oxygen species. Through the inhibition of PDK activity, DCA regulates mitochondrial metabolism and reduces ROS resulting from reverse electron transfer and ATP deficit. Furthermore, via triggering the Nrf2 pathway, DCA suppresses ROS and lessens ROS-induced cellular autophagy, apoptosis, inflammation, and BBB degradation. Created with BioRender.com. |
Vascular Dementia
Vascular dementia (VD) is a neurocognitive dysfunction caused by cerebrovascular diseases such as stroke.93 Bone marrow-derived endothelial progenitor cells (EPCs) have been demonstrated to promote neovascularization and restoration with a protective role in vascular dementia.94 In MCAO-induced VD rats, DCA (100 mg/kg/day, 200 mg/kg/day) increased serum levels of vascular endothelial growth factor and promoted cerebral angiogenesis by improving the function of EPCs through the protein kinase B (AKT)/glycogen synthase kinase 3β/Nrf2 pathway, which suggested that DCA could enhance the function of EPCs, but the exact mechanism of the regulation of the AKT pathway by DCA is unclear.42 One possible reason is that ROS generated during hypoxia-ischaemia impairs endothelial cell function, and DCA regulation of the AKT pathway may also be mediated through ROS.95 The effects of DCA on vascular endothelial cells in this experiment may provide new potential therapeutic approaches for the treatment of other vascular-related diseases, but the specific mechanisms and efficacy still need more exploration.
Hypoglycaemic Brain Injury, Traumatic Brain Injury, and Epilepsy
The protective effects of DCA on the brain are not only observed in I/RI but also in hypoglycaemic brain injury, traumatic brain injury, and brain injury caused by acute seizures. Kho et al used insulin and glucose to cause hypoglycaemia-induced injury in rats and showed that DCA (100 mg/kg) reduced hypoglycaemia-induced oxidative stress, glial cell activation, BBB disruption, and neuron death.43 DeVience et al assessed the extent of traumatic brain injury in rats by magnetic resonance spectroscopy (MRS) of hyperpolarized [1–13C] pyruvate and found that DCA (200 mg/kg) increased the bicarbonate/lactate ratio on both sides of the brain and attenuated the inhibition of PDH, but they did not assess whether DCA protects neurons.44 DCA (50 and 100 mg/kg/day) was observed to shorten the latency to seizures in a second-hit flurothyl test in the mouse model.45 Lee et al found that DCA (100 mg/kg) and pyruvate (50 mg/kg) improved energy metabolism and attenuated oxidative stress and neuroinflammation, thereby reducing neuronal damage and cognitive dysfunction in seizure rats.46
Protective Effects of Dichloroacetate in Neurodegenerative Diseases
Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is a severe neurodegenerative disease characterized by motor neuron (MN) degeneration leading to progressive muscle atrophy throughout the body, with most patients progressing to respiratory failure and dying within 3–5 years.96 It is currently believed that ALS is caused by the interaction of genetic and environmental factors, and the causative mechanisms include mitochondrial dysfunction, oxidative stress, neuroinflammation, axonal transport disruption, and vesicular transport defects.96–100 In ALS caused by mutations, the gene most commonly affected is superoxide dismutase 1 (SOD1).101 Miquel et al found that oral administration of DCA (100 mg/kg for 10 days) to symptomatic SOD1G93A rats reduced MN degeneration in the spinal cord, attenuated the loss of locomotor activity, and increased survival rates, suggesting that DCA has a therapeutic effect in ALS.47
Astrocytes are essential in the pathology of ALS, and the presence of aberrant glial cells (AbGCs) leads to the death of motor neurons.102,103 AbGCs are a specialized astrocyte A1 phenotype, and it is currently believed that AbGCs cause motor neuronal damage due to the loss of normal astrocyte function and the secretion of some neurotoxic factors.103,104 Miquel et al found that DCA (5 mmol/L, 24 h) enhanced PDH activity in AbGCs and increased the survival of neurons cocultured with AbGCs.47 This suggests that DCA may reduce motor neuron damage by affecting AbGCs, but the exact mechanism remains to be investigated. Martínez-Palma et al found that DCA (5 mmol/L) improved mitochondrial respiration, reduced lactate production, inhibited the growth of AbGCs isolated from SOD1G93A rats, and increased neuronal survival during coculture of AbGCs with neurons but did not alter the expression of typical phenotypic markers of AbGCs, suggesting that the change in the number of AbGCs during coculture was not significant and that the decrease in the toxicity of AbGCs may be due to DCA-induced shifts in their metabolic phenotype.48 SOD1G93A expression in astrocytes leads to a shift in the cellular metabolic phenotype towards glycolysis to produce more lactic acid.101 Lactate released by astrocytes can serve as a metabolic substrate for neurons, and after reaching the neuron, lactate is rapidly metabolized to pyruvate in the cytoplasm and mitochondria, participating in mitochondrial energy metabolism.105 Jia et al found that inhibition of PDH in Schwann cells leads to a sustained and massive accumulation of lactate, which drives a sustained increase in neuronal mitochondrial activity and an increase in ROS, leading to neuronal oxidative stress and axonal destruction.106 Additionally, excessive accumulation of lactic acid can lead to cellular acidosis and neuronal damage.107 This suggests that DCA may protect neurons by reducing lactate production from AbGCs. Another question that remains to be addressed is why DCA inhibits AbGC growth. Mutations in SOD1 result in low glutathione levels and high NO levels, which can lead to oxidative stress and promote the production of peroxynitrite in cells, damaging the ETC.108,109 In addition, aberrant expression of SOD1 disrupts the connection between cytochrome C and the inner mitochondrial membrane, directly leading to ETC damage.110 Inadequate ATP production due to ROS damage to the ETC may be responsible for the forced shift in the metabolic phenotype of AbGCs to glycolysis.111 Stockwin et al found that DCA forces cells to switch to defective oxidative phosphorylation, causing insufficient ATP production and leading to metabolic stress and cell death in ETC-deficient cells.65 Therefore, DCA may inhibit AbGC growth by stimulating the damaged ETC and generating excess ROS, similar to its inhibitory effect on cancer cells.112
Studies in SOD1G86R model mice have shown that altered metabolism of glycolytic muscles may predispose the organism to motor neuron loss.113–115 In the physiological state, both glucose and fatty acids can be used as metabolic raw materials for glycolytic muscles, in which glucose is converted to pyruvate by glycolysis and then enters the TCA cycle or is converted to lactate.116,117 Fatty acids are then converted to lipoyl coenzyme A by lipoyl coenzyme A synthetase and transported to mitochondria via carnitine palmitoyltransferase for β-oxidation.118 However, the production of lipid byproducts from fatty acid β-oxidation increases ROS production.119 In ALS, instability of the neuromuscular junction induces elevated PDK4 expression, which prevents the entry of pyruvate into the TCA cycle.120,121 Energy deficiency leads to more fatty acids entering the glycolytic muscle, which further induces the transcription of the nuclear hormone receptor peroxisome proliferator-activated receptor β/δ (PPARβ/δ) to stimulate forkhead box transcription factor O1 (FOXO1), and high levels of FOXO1, as well as ATP and NADH generated by β-oxidation, can activate the expression of PDK4, which further inhibits glucose oxidation and creates a vicious cycle, with increased oxidative stress leading to neuronal death.49,122,123 Palamiuc et al found that DCA (500 mg/kg) decreased the mRNA expression of PDK4, FOXO1, and glutathione peroxidase 1 and increased the mRNA expression of PPARβ/δ and phosphofructokinase 1 (PFK1) in SOD1G86 mice, suggesting that DCA relieves the inhibition of PFK1 by excess pyruvate through the inhibition of PDK4 activity and the enhancement of mitochondrial oxidation of pyruvate, which enhances glycolysis and reduces oxidative stress by decreasing β-oxidation of fatty acids, thereby reducing motor neuron death and improving locomotor activity and survival rates in mice.49 The results also showed that DCA can increase the mRNA expression of Pgc-1α and mitochondrial fusion protein 2 (Mfn2), which may have a role in improving mitochondrial dynamics.49 Although it was described above that DCA enhances ROS production in all cells, the reason for the attenuation of oxidative stress here may be that on the one hand, glycolytic muscles themselves have a low ETC metabolic flux, and on the other hand, the effect of DCA on attenuating ROS production from β-oxidation is greater than that produced by its increase in ETC function.65
In summary, in ALS, DCA regulates the metabolism of both AbGCs and glycolytic muscles to reduce motor neuron death, but the effects of DCA on motor neurons themselves have not been explored (Figure 3). In addition, an animal model with a single mutation is not fully representative of the complete pathology of ALS; therefore, further preclinical and clinical studies are still necessary to explore the efficacy of DCA.
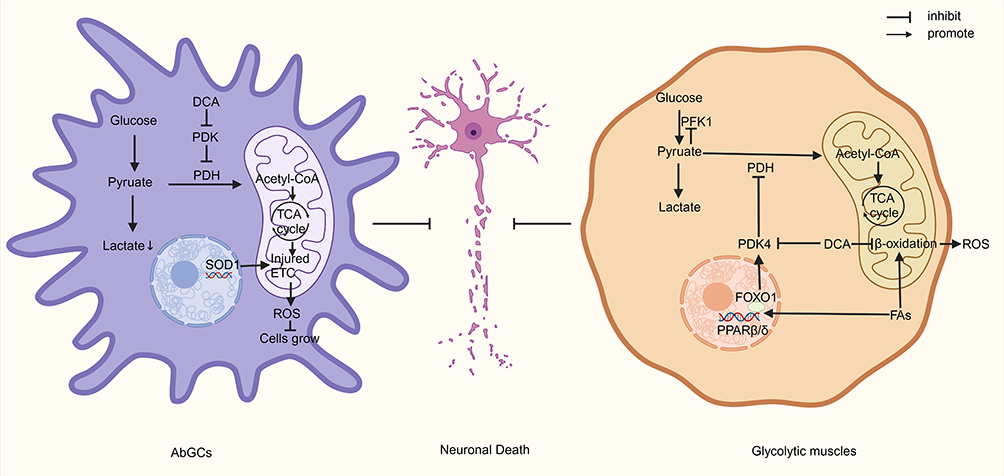 |
Figure 3 DCA attenuates neuronal death in ALS by targeting AbGCs and glycolytic muscles. Abbreviations: DCA, dichloroacetate; PDH, pyruvate dehydrogenase; SOD1, superoxide dismutase 1; ROS, reactive oxygen species; FAs, fatty acids; PFK1, phosphofructokinase 1; PDK, pyruvate dehydrogenase kinase; FOXO1, forkhead box protein O1; PPARβ/δ, peroxisome proliferators-activated receptors β/δ. Notes: In AbGCs, DCA increased PDH activity to cause a change in cellular metabolism to mitochondrial metabolism. This prevented lactate from being harmful to neurons and decreased the amount of lactate produced. Furthermore, DCA stimulated the disrupted electron transport chain in the mitochondria of AbGCs, which raised the degree of oxidative stress and prevented AbGC development. When cellular metabolism is disrupted in glycolytic muscles, fatty acids β oxidation provides cells with additional energy. This process activates PPARβ/δ with FOXO1 and inhibits PDH. By increasing PDH activity, DCA lessens the inhibition of glycolysis caused by pyruvate accumulation and increases energy availability, which lowers the metabolism of fatty acids and the generation of reactive oxygen species. Created with BioRender.com. |
Alzheimer’s Disease
Amyloid β-protein (Aβ) deposition in Alzheimer’s disease (AD) is one of the important pathogenic mechanisms of the disease.124 Amyloid precursor protein (APP) is cleaved by β-site APP cleavage enzyme (BACE1) to generate soluble amyloid precursor protein β (sAPPβ) and β C-terminal fragment (βCTF); βCTF is then cleaved by γ-secretase to generate Aβ.125 In addition, APP can generate neuroprotective soluble amyloid precursor protein α (sAPPα) via metalloproteinase 10 (ADAM10), and this competitive process prevents the cleavage of APP via BACE1 and γ-secretase.126 Parkin et al found in SH-SY5Y cells that DCA (10 and 20 mmol/L) increased the level of sAPPα and decreased the levels of sAPPβ and Aβ but did not change the activity and mRNA expression of ADAM10 and BACE1, which proved that DCA could reduce the generation of Aβ.50 However, since the effect of DCA on the protein expression level of BACE1 was not measured, the mechanism by which DCA affects the generation of Aβ remains to be explored. Velliquette et al found that using drugs (insulin, 2-deoxyglucose, 3-nitropropionic acid, and kainic acid) to inhibit energy metabolism in APP transgenic mice increased BACE1, sAPPβ, and Aβ40 levels, suggesting that reduced energy metabolism increases Aβ production by increasing BACE1 expression levels.127 O’Connor et al demonstrated that energy deprivation leads to phosphorylation of the translation initiation factor eIF2α, which increases BACE1 expression by regulating BACE1 translation rather than transcription, which could also explain why Edward T. Parkin et al detected no change in BACE1 mRNA levels in their experiments.128 In addition, mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations contribute to the pathophysiology of AD.129,130 Some drugs targeting mitochondrial metabolism have been found to have therapeutic potential in AD; for example, a phase 2 clinical trial showed that a combination of metabolic activators (L-serine, nicotinamide ribose, N-acetyl-L-cysteine, and L-carnitine tartrate) improved cognitive function in patients with AD.131 DCA, as a regulator of energy metabolism, may also have therapeutic potential for AD due to its ability to reduce Aβ production by affecting BACE1 expression. In addition, the ability of DCA to modulate oxidative stress, inflammation, and mitochondrial dynamics may also contribute to the reduction of Aβ production, as these factors have all been found to be associated with BACE1.132–134 In conclusion, the current evidence suggests that DCA may have a therapeutic effect on AD, but the pathogenesis of AD is affected by many factors, and further experiments are needed to prove the effects and mechanisms of DCA on AD.135,136
Huntington’s Disease and Parkinson’s Disease
Huntington’s disease (HD) and Parkinson’s disease (PD) are also neurodegenerative diseases, and mitochondrial dysfunction is an important factor in the pathogenesis of both diseases.137,138 DCA has been demonstrated to exert a therapeutic effect in Huntington’s disease animals and Parkinson’s disease cells in vitro. The addition of DCA (100 mg/kg) to drinking water in mice improved body weight, locomotion, and mean survival time in R6/2 versus N171/82Q mice; in R6/2 mice, DCA improved striatal volume reduction and neuronal area reduction.51 In 1-methyl-4-phenyl-pyridinium ion (MPP)-treated PC12 cells, the use of DCA (100 μmol/L) improved mitochondrial dynamics, but the decreased cell viability, increased ROS, altered mitochondrial membrane potential, and reduced oxygen flux after MPP treatment were not reversed.52
DCA and PGC-1α
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) is a transcription factor that is an essential node linking cellular metabolism, mitochondrial dynamics, oxidative stress, and inflammation regulation and has been recognized as an important target for pharmacological intervention in many cardiovascular, metabolic, and neurodegenerative diseases.139–142 The transcriptional activity of PGC-1α is regulated by AMP-activated protein kinase (AMPK) via phosphorylation.143 AMPK is the most direct energy receptor, which directly senses changes in the intracellular AMP/ADP ratio and accordingly regulates mitochondrial function to adapt to energy changes.144 Li et al found that DCA activated the AMPK pathway and ameliorated oxidative stress injury in a mouse model of cardiac ischaemia‒reperfusion injury, providing indirect evidence that DCA activates PGC-1α via AMPK.25 In addition, PGC-1α activity is regulated by acetylation, and sirtuin 1 (SIRT1) activates PGC-1α through deacetylation.145 An increase in the NAD/NADH ratio triggers the activation of SIRT1, which activates PGC-1α.146 Cells with abnormal mitochondrial metabolism are forced to undergo glycolysis and fatty acid β-oxidation to supplement ATP production, but it results in the accumulation of NADH.147,148 Lin et al found that DCA increased the NAD/NADH ratio in human colorectal cancer cell lines.76 However, due to different cellular metabolic phenotypes, the changes in the intracellular AMP/ADP and NAD/NADH ratios induced by DCA under different pathological conditions need to be further investigated. Interestingly, the regulation of the aforementioned Nrf2 pathway, inflammation, apoptosis, mitochondrial dynamics, and Aβ by DCA can also be explained by PGC-1α. Lee et al showed that treatment of dorsal skin cells of nude mice with Galangal activates PGC-1α and further enhances the expression of Nrf2 for antioxidant effects.149 PGC-1α was also found to block the transcription of nuclear factor-kappaB and its downstream genes encoding proinflammatory cytokines, which demonstrated the regulation of anti-inflammation by PGC-1α.150 In addition, Niel et al observed in a mitochondrial creatine kinase knockout model of ageing mice that PGC-1α may affect mitochondrial dynamics by regulating Mfn2.151 Li et al also showed in their study that PGC-1α regulates mitochondrial function through activation of the Nrf1 pathway.152 Liu et al observed that PGC-1α inhibited apoptosis by suppressing caspase activation in heat shock protein 12A knockout mice.153 Wang et al found that PGC-1α directly binds to the promoter of the BACE gene and represses the transcription of BACE1 mRNA, revealing the regulatory role of PGC-1α in Aβ generation.154 In summary, the modulation of PGC-1α by DCA may be a key factor in its multiple pharmacological effects, and mediating the importance of PGC-1α, this hypothesis also opens up pharmacological possibilities for a wider application of DCA (Figure 4).
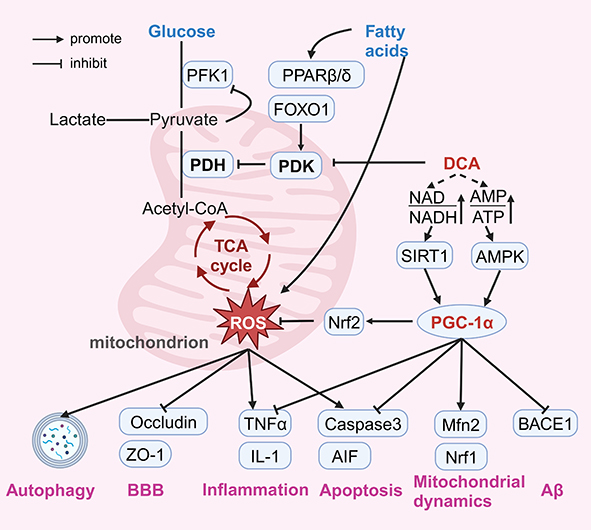 |
Figure 4 Possible mechanisms of neuroprotective effects of Dichloroacetate. Abbreviations: DCA, dichloroacetate; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PFK1, phosphofructokinase 1; PPARβ/δ, nuclear hormone receptor peroxisome proliferator-activated receptor β/δ; FOXO1, forkhead box transcription factor O1; TCA, tricarboxylic acid; ROS, reactive oxygen species; NAD, oxidized nicotinamide adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide; AMP, adenosine monophosphate; ATP, adenosine triphosphate; SIRT1, sirtuin 1; AMPK, AMP-activated protein kinase; PGC1-α, peroxisome proliferator-activated receptor gamma coactivator 1-alpha; Nrf2, nuclear factor erythroid 2-related factor 2; ZO-1, zonula occludens protein 1; BBB, blood-brain barrier; TNFα, tumor necrosis factor α; IL-1β, interleukin-1β; AIF, apoptosis-inducing factor; Mfn2, mitochondrial fusion protein 2; Nrf1, nuclear respiratory factor 1; BACE1, β-site amyloid precursor protein cleavage enzyme; Aβ, amyloid β-protein. Notes: DCA inhibits PDK activity, thus enhancing PDH activity. This allows more glucose to enter the mitochondria for oxidation and reduces fatty acids β-oxidation, which corrects the disturbance of energy metabolism and reduces oxidative stress caused by insufficient ATP production. DCA exerts protective effects by attenuating multiple damages caused by oxidative stress, including: attenuating excessive autophagy, attenuating neuroinflammation, protecting the BBB, and attenuating apoptosis. DCA may increase the intracellular NAD/NADH AMP/ATP ratio by regulating metabolism and thus activating PGC-1α. Activation of PGC-1α has multiple protective effects including: attenuating oxidative stress by activating Nrf2, decreasing the release of inflammatory factors (TNFα, IL-1β), attenuating caspase 3 and AIF-mediated apoptosis, activating Mfn2 and Nrf1 to improve mitochondrial dynamics, and inhibiting BACE1 to reduce Aβ production. Created with BioRender.com. |
Barriers to the Clinical Application of DCA and Possible Coping Strategies
Pharmacokinetics
Oral DCA is quickly absorbed, providing a bioavailability comparable to that of parenteral administration.9,155 Due to the high fat solubility of DCA, it is distributed in the liver, muscle, skin, intestines, kidneys, lungs, heart, brain, and other tissues and organs.156,157 DCA crosses the cell membrane via the monocarboxylate transporter and then through the mitochondrial membrane via the pyruvate transporter, thus entering the mitochondrial matrix to exert its role.158 In vivo, DCA is metabolized mainly by the zeta-1 family isoform of glutathione transferase (GSTZ1).159 GSTZ1 is distributed in tissues such as the liver, kidney, testis, heart, and brain, but the amount in the liver is much higher than that in other organs, so the liver is the main metabolizing organ of DCA.160 Further studies showed that 86% of GSTZ1 in rat liver was in the cytoplasm and 14% in mitochondria.161
As early as the last century, it was discovered that DCA has nonlinear kinetics at single injection doses ≥ 35 mg/kg.162 This is because DCA limits its own metabolism by inhibiting GSTZ1, and the plasma clearance of the drug is reduced after multiple repeated administrations.163 More importantly, the activity of GSTZ1 and the time to recovery of activity were related to the duration of DCA administration and the dose.10 After a single intraperitoneal injection of DCA (45 mg/kg) in male Fischer-344 rats, there was a significant loss of GSTZ1 protein and activity within 12 hours, with a minimum value of less than 40% of the initial value, which was not restored until 10 to 12 days after the administration of the drug.163 Li et al found that GSTZ1 in rat mitochondria had a 2.5-fold higher apparent Km for glutathione than cytoplasmic GSTZ1, while DCA had the same apparent Km values, which suggests that there may be a difference between cytoplasmic GSTZ1 and mitochondrial GSTZ1 activity.164 However, Smeltz et al found that when adult male rats were orally administered DCA (100 mg/kg), with DCA acting as a substrate together with glutathione (GSH), the half-life of GSTZ1 in adult rat cytoplasm was 0.82 ± 0.02 hr and that in mitochondria was 0.54 ± 0.05 hr, which suggests that mitochondrial GSTZ1 is more susceptible to DCA inhibition.165 Zhong et al found that although the contribution of mitochondrial GSTZ1 to total GSTZ1 increased with age, the contribution of mitochondrial GSTZ1 was still inferior to that of cytoplasmic GSTZ1 when repeated DCA treatments were administered.166 Although there are differences in the kinetics of mitochondrial and cytoplasmic GSTZ1, cytoplasmic GSTZ1 remains a key area of interest for studying DCA metabolism, considering the long-term clinical use of the drug.167
In addition to inhibition by DCA, GSTZ1 activity is affected by age.155 After treatment with DCA (25 mg/kg/day) for 6 months, the t1/2 for DCA was 6.4±3.4 hours in the paediatric group (mean 5.2±1.8 years) and 21±5.8 hours in the adult group.158 The total activity of GSTZ1 in hepatocyte lysates of rats after DCA (100 mg/kg) administration for 8 days was 113 ± 37 and 115 ± 33 nmol glyoxylate/min/kg in young male and female rats, respectively, compared with 44.7 ± 13.4 nmol glyoxylate/min/kg in 52-week-old female rats.161 This may be because children have a higher liver/body weight ratio, or it may be the result of the effect of chloride ions.10 Jahn et al found that chloride ion concentrations in human liver from donors aged 1 day to 84 years decreased with age, consistent with age-related changes in GSTZ1, and the chloride ion concentrations of the cytoplasm (mean 105 mmol/L) were much larger than those of mitochondria (mean 4.2 mmol/L), which could also explain the greater susceptibility of mitochondrial GSTZ1 to inactivation compared to that in the cytoplasm.168 Zhong et al found that chloride, bromide, iodide, and sulfite attenuated the loss of GSTZ1 activity in human hepatocyte cytosol in a concentration-dependent manner after 2 h of DCA (0.5 mmol/L) incubation, but the mechanism by which chloride ions and other ions affect the loss of GSTZ1 activity is still unclear.169 In addition, Jahn et al showed that miR-376c-3p prevents the expression of GSTZ1 by repressing translation, and the expression of this miRNA is lower in adults than in children, which provides new ideas for studying the factors that influence GSTZ1.170
GSTZ1 genotypes are also important in influencing the metabolism of DCA.171 There are five common haplotypes based on the three common asynchronous single-nucleotide polymorphisms (rs7975, rs7972, rs1046428) in the GSTZ1 gene coding region, which, in order of population frequency, are EGT (GSTZ1C, 50%), KGT (GSTZ1B, 28%), EGM (GSTZ1D, 15%), KRT (GSTZ1A, 7%), and KGM (GSTZ1F, 0.4%).172,173 The residual activity of the variants was different for each variant in a single DCA-induced inactivation: 1A-1A (12%) > 1B-1B≈1C-1C ≈1D-1D (<5%).174 In vitro studies on human hepatocyte cytoplasm showed that GSTZ1A homozygous or heterozygous samples had 3 times higher DCA dechlorination activity than those carrying other alleles.175 However, with multiple administrations, after 5 or more days of DCA treatment (2.5 µg/kg/day, once or twice), those with one copy of the GSTZ1C variant had a lower reduction in DCA clearance than those without the GSTZ1C variant, implying that the GSTZ1 enzyme activity of those with a copy of the GSTZ1C variant is less affected by multiple DCA administrations.176 James and Stacpoole classified individuals into fast-metabolizing (at least one GSTZ1C copy) and slow-metabolizing (no GSTZ1C copy) phenotypes based on the level of GSTZ1 inactivation following multiple doses of DCA.167 Notably, the chloride concentrations that provided 50% inactivation protection (EC50) in the EGT and EGM homozygotes were similar; however, the EC50 concentration of chloride in the EGT/KRT haplotype was approximately 2.5-fold higher than that of the EGT/EGT and EGM/EGM haplotypes.169 This is consistent with the faster inactivation of GSTZ1 in individuals carrying the GSTZ1A variant and suggests that the mechanism of the protective effect of chloride ions may involve binding to a specific regulatory site on GSTZ1, such that different GSTZ1 variants have different affinities for chloride ions. Interestingly, in the study by Shroads et al, patients received oral DCA (12.5 mg/kg/12 h) or placebo for 6 months, after which subjects in the placebo group were switched to the DCA group (patients were treated for a total of 30 months), and the results showed that although the plasma trough concentration of DCA was lower in EGT carriers than in non-EGT carriers, none of the genotypes showed a gradual accumulation of plasma drug concentrations, and the differences in kinetic parameters stabilized.171 Therefore, it is theoretically possible to achieve controllable blood concentrations by administering different doses of DCA according to GSTZ1 haplotypes at the time of treatment, and it is also valuable to further develop models for the nonlinear pharmacokinetics of DCA according to age and GSTZ1 genotype.177
Adverse Reactions
The toxicity of DCA, as a byproduct of water chlorination disinfection, has been an area of concern. In animal experiments, it was found that long-term use at high doses could induce liver cancer. The addition of DCA to drinking water (3.5 g/L for 93 weeks) increased the incidence of liver tumours in mice.178,179 However, epidemiological evidence does not demonstrate a direct carcinogenic effect of DCA in humans, and long-term use of DCA has been found to have no haematological, hepatic, or renal toxicity.10,180 For example, Abdelmalak et al followed eight patients with congenital lactic acidosis who received oral DCA (5.12 mg/kg/9 h) for 7.16 to 5.1 years, and their renal, liver, electrolyte, and hepatic status remained stable.181
The biggest limiting issue found with DCA in clinical trials is reversible peripheral neuropathy, with dose dependence and individual variability.182 DCA is catalysed by GSTZ1 and converted to glyoxalate in a reaction that needs but does not consume GSH.183 The intermediates of tyrosine catabolism, maleoylacetoacetate and maleoylacetone, are physiological substrates of GSTZ1, which converts them to fumarylacetoacetate and fumarylacetone.184 Fumarylacetoacetate hydrolase (FAH) converts fumarylacetoacetate to two nontoxic substances, fumarate and acetoacetate.185 Maleoylacetoacetate, maleoylacetone, fumarylacetoacetate, and fumarylacetone are considered to be toxic.174 The accumulation of maleoylacetone and fumarylacetone inhibits the conversion of δ-aminolaevulinic acid (δ-ALA) to porphobilinogen by ALA dehydratase, leading to the accumulation of δ-ALA, a neurotoxic substance.186,187 Patients with hereditary tyrosinemia caused by abnormal FAH function develop symptoms of hepatic, neurologic, and renal system damage, including hepatocellular carcinoma and peripheral neuropathy, as a result of the accumulation of these toxic substances.188 Toxic metabolites as well as δ-ALA have been found in the urine of adults and children treated with DCA, and the accumulation of toxic products has been correlated with the GSTZ1 genotype, suggesting that DCA produces many neurotoxic metabolites due to the inhibition of GSTZ1 (Figure 5).176 The metabolite of DCA, glyoxalate, is further metabolized by lactate dehydrogenase (LDH) to glycine, carbon dioxide, and oxalate.189,190 Oxalate is considered relevant to peripheral neuropathy, and its metabolism consumes thiamine.191,192 In addition, thiamine is a cofactor of pyruvate dehydrogenase.193 After 7 weeks of DCA (1.1 g/kg) treatment in rats, oxalate in the urine was 86% higher in rats treated with DCA than in controls but was only 28% higher in those treated with DCA plus thiamine than in controls; thus, DCA may lead to peripheral neuropathy due to thiamine deficiency and oxalate accumulation.182 A trial of DCA in combination with thiamine in a 13-year-old girl with complex I deficiency found that the patient still had peripheral neuropathy; however, the study had an inadequate sample size and did not consider the effect of the GSTZ1 genotype.194 More experimental validation is needed to verify whether thiamine can alleviate DCA-induced peripheral neuropathy. Thiamine is used to treat mitochondrial diseases as a drug that enhances pyruvate dehydrogenase, similar to DCA, and the combination may result in better efficacy.195 Broxton found that DCA (25 mmol/L) with thiamine (25 mmol/L) showed isolated or synergistic therapeutic effects in pyruvate dehydrogenase complex-deficient Cryptobacterium hidradii nematodes.196 In a randomized controlled trial of patients with MELAS and A3243G mutations, DCA (25 mg/kg/day) was discontinued due to strong peripheral neurotoxicity.197 Several other clinical trials in solid tumours and congenital lactic acidosis have shown that DCA-induced peripheral neurotoxicity at therapeutic doses is tolerable.198–200 For example, a study of patients with advanced solid malignancies found that DCA (6.25 mg/kg/12 h) was associated with fatigue, neuropathy, and nausea in only a subset of patients and that the response to DCA varied among patients.201 More importantly, adults and children do not respond consistently to DCA, children tolerate DCA better than adults, and the use of DCA for several years in children and adolescents to treat primary mitochondrial disease is considered to have an excellent long-term safety record.181,202 Therefore, the clinical side effects of DCA may be due to the different abilities of different patients to metabolize DCA, which is also affected by age and GSTZ1 genotype, as mentioned above. Tian et al found in six adult patients treated with DCA (mean 25 mg/kg/day) that a patient without the EGT variant had a higher neuropathy score on Day 84 of treatment compared with other patients carrying the EGT variant and that this patient’s plasma trough concentrations of DCA on Day 56 and Day 84 were 2- to 3-fold higher than the mean values of the other patients, suggesting that peripheral neuropathy caused by DCA is related to the accumulation of DCA resulting from GSTZ1 suppression.203 Therefore, individualized dose design is one way to address the peripheral neurotoxicity of DCA.167 In a study of 15 adults with recurrent gliomas or intracranial metastases, Dunbar et al stratified the administration of DCA according to whether the patient carried the EGT variant: patients carrying the EGT variant were administered DCA (8.0 mg/kg/12 h, 12.5 mg/kg/12 h, 5.0 mg/kg/12 h), and patients without the EGT variant were administered a dose of 4.0 mg/kg/12 h; no patients discontinued the drug due to adverse effects of DCA.200 This suggests that it is feasible to group patients by whether they carry the EGT variant and administer different concentrations of DCA individually. Dunbar et al concluded that the starting oral dose for patients who do not carry the variant should be 5 mg/kg/12 h, which can be increased appropriately based on the absence of peripheral neuropathy, and that subjects carrying the EGT variant should be able to tolerate at least 6.25 mg/kg/h.200 However, because of the differences in the metabolism of DCA between children and adults, this finding may apply only to adults, and there are currently no clinical trials in children designed to administer DCA based on the GSTZ1 genotype.181,202 Mangal et al developed a population pharmacokinetic model for DCA based on data from a randomized controlled trial of DCA in children with congenital lactic acidosis and recommended optimal DCA dosages of 12.5 and 10.6 mg/kg twice daily for children who are EGT carriers and non-EGT carriers, respectively.23,204 More data based on GSTZ1 genotypic dosing are needed in all age groups to determine the dose of DCA in clinical therapy.
 |
Figure 5 Metabolism of DCA and related neurotoxic products. Abbreviations: DCA, dichloroacetate; GSTZ1, glutathione transferase zeta 1; GSH, glutathione; δ-ALA, δ-aminolevulinic acid; FAH, fumarylacetoacetate hydrolase; LDH, lactate dehydrogenase. Note: Created with BioRender.com. |
Various synthetic nanocarriers and cell-derived nanovesicles now offer new possibilities for drug delivery.205,206 They can improve drug absorption, enable precise drug delivery and reduce side effects and have been used as a potential therapeutic option for cancer, diabetes and other diseases.207–210 In tumour therapy, to enhance drug delivery targeting and reduce side effects, DCA is commonly delivered in metal-organic frameworks.211–215 For example, Lázaro et al functionalized DCA using zirconium (Zr) terephthalate (UiO-66) nanoparticles to achieve cytotoxicity and selectivity against different cancer cell lines.216 For drug delivery in the nervous system, cell-derived exosomes can cross the BBB and exhibit high biocompatibility, good stability, low accumulation, and ease of modification, which show more advantages than traditional synthetic delivery carriers and have been widely used in therapeutic research on neurological diseases.217,218 There have been no studies using nanovesicles to deliver DCA for the treatment of brain diseases. DCA, as a small-molecule lipid-soluble compound, binds itself passively to the lipid bilayer of exosomes; therefore, utilizing exosomes and delivering DCA to the brain through passive or active targeting strategies may allow for the administration of controlled concentrations of DCA to avoid its side effects.218,219 In addition, multitarget drug delivery utilizing specific markers of disease pathophysiological processes, such as ROS, inflammatory factors, and pH. is another method to enhance drug targeting.220 In a study on AD, Hu et al designed nanoparticles based on the specific response of Congo red to amyloid plaques and the redox sensitivity of boronate ester bonds, using Congo red/boronate ester bonds as the ligands, and realized the targeted delivery of AD therapeutic agents and controlled release by H2O2.221 Qiao et al designed nanoparticles based on poly[(2-angiopep)ethyl(p-boronic acid benzyl)diethylammonium bromide] (BAP)’s ability to respond to ROS, using angiopep-2/zwitterionic lipid distearoyl phosphoethanol-aminepolycarboxybetaine/BAP as a ligand to design the nanoparticle, thereby enabling the drug to overcome the limitations of the BBB, specifically target glioma cells, escape endosomes/lysosomes, and release the drug in response to ROS.222 Goyal et al exploited the characteristics of pH and redox sensitivity of Lf (part of the transferrin family)-conjugated aggregates in the treatment of amnesia to design Lf/disulfide linkage nanoparticles, thus enabling drug-targeted delivery by detecting the pH microenvironment.223 Modulation of metabolism is the commonality of DCA’s pharmacological effects in the aforementioned neurological disorders; metabolic disorders in the brain are associated with decreased pH and increased oxidative stress, and in some diseases, there is an accumulation of specific markers such as Aβ. These existing studies may provide ideas for the future targeting of ROS, pH, and disease-specific markers for the delivery of DCA.
Conclusion and Prospects
In this review, we first summarized the current studies on the treatment and mechanisms of DCA in some neurological disorders. Second, we analysed the limitations of the clinical application of DCA. Finally, possible coping strategies were proposed based on its limitations.
Current studies on the neuroprotective effects of DCA involve a variety of diseases (Table 1). The neuroprotective mechanisms of DCA include regulating metabolism, improving oxidative stress, reducing neuroinflammation, reducing apoptosis, reducing autophagy in the brain, protecting the BBB, improving endothelial progenitor cell function, improving mitochondrial dynamics, and reducing Aβ protein production. Studies have focused on cerebral I/RI-related disorders, and these studies have shown that DCA reduces excess ROS production primarily by improving metabolism, thereby reversing the subsequent cascade of damage caused by reperfusion. In neurodegenerative diseases, major studies have found that DCA attenuates motor neuron death in ALS model animals and the production of Aβ protein in AD model cells by modulating metabolism. In ALS, DCA reduced the toxicity of AbGCs to neurons, which may be related to the attenuation of lactic acid toxicity and the inhibition of AbGC growth and proliferation. On the other hand, DCA favours motor neuron survival by regulating glycolytic flux in glycolytic muscles. Additionally, a study in VD animals found that DCA improves brain function by enhancing the function of endothelial progenitor cells. These studies suggest that DCA has the potential to be beneficial when it acts on different cells, but these studies are limited, and further exploration of the effects of DCA on different cells is needed. Notably, the exertion of all these pharmacological effects seems to be related to the regulation of PGC-1α, which senses changes in cellular energy metabolism and is the connection point for multiple pathways of metabolism, oxidation, inflammation, the production of Aβ and mitochondrial dynamics. However, due to the complex metabolic profile of cells in vivo, the specific mechanisms by which DCA regulates PGC-1α remain to be explored, and the changes in the AMP/ADP and NAD/NADH ratios induced by DCA may be different in different situations. It is now well established that DCA enhances mitochondrial respiration and enhances ROS production in all cells, but this is not sufficient to affect normal cells under normal conditions.65 Moreover, PDH in normal neurons maintains a low level of phosphorylation, close to maximal activity.224 The role of DCA needs to be specifically analysed in different pathological situations. In cells with a severely impaired or inhibited ETC, for example, the application of DCA to tumour cells, AbGCs in ALS, and neurons in hypoxia may disrupt the protective process of cellular switching to glycolysis, resulting in accelerated death due to cellular energy depletion. In other cases, such as cerebral I/RI, the enhancement of mitochondrial metabolism by DCA may be accompanied by some ROS damage to the cell, but it attenuates the more serious consequences of insufficient energy metabolism and protects neurons overall. In addition, current studies on the neuroprotective effects of DCA are only at the cellular or animal level. DCA is metabolized primarily in the cytoplasm of hepatocytes by the enzyme GSTZ1, which can be inhibited by DCA itself. The activity of GSTZ1 is affected by age, and in general, the older the person is, the lower the GSTZ1 activity. Furthermore, the activity of GSTZ1 and the extent to which DCA inhibits it after multiple administrations varies between different GSTZ1 genotypes. Therefore, during clinical use, peripheral neurotoxicity often results from inhibition of GSTZ1 activity. Accumulation of the toxic product δ-ALA due to GSTZ1 inhibition and accumulation of oxalate, DCA’s own metabolite, may be responsible for its peripheral neurotoxicity. Coadministration of thiamine with DCA may alleviate oxalate accumulation and reduce peripheral neurotoxicity. Alternatively, individualized dosing by dividing patients into populations with different metabolic rates based on their age and GSTZ1 genotype or targeted delivery of DCA using nanovesicle-targeted dosing may be possible strategies to enhance its therapeutic efficacy and address its accumulation and peripheral neurotoxicity.
In conclusion, current animal and cellular experiments suggest that DCA is a promising neuroprotective drug with multiple pharmacological activities. Since energy metabolism disorders and mitochondrial dysfunction are important pathologic mechanisms in a variety of neurological disorders, further preclinical and clinical studies of this drug are warranted.
Funding
This work was supported by the Natural Science Foundation of Shandong Province [ZR2016HP06]; the Science and Technology Planning Project of Traditional Chinese Medicine of Shandong Province [M-2023094]; the Shandong Medical and Health Science and Technology Development Project [2019WS605, 202104110334, 202104010549]; the Weifang Medical University Students Innovation and Entrepreneurship Training Program Project [X2021514, X2021517]; and the Weifang Science and Technology Development Project [2021YX028].
Disclosure
The authors report no conflicts of interest in this work.
References
1. Tovar-y-Romo LB, Penagos-Puig A, Ramirez-Jarquin JO. Endogenous recovery after brain damage: molecular mechanisms that balance neuronal life/death fate. J Neurochem. 2016;136(1):13–27. doi:10.1111/jnc.13362
2. Campos AC, Fogaca MV, Sonego AB, Guimaraes FS. Cannabidiol, neuroprotection and neuropsychiatric disorders. Pharmacol Res. 2016;112:119–127. doi:10.1016/j.phrs.2016.01.033
3. Perez-Olives C, Rivas-Santisteban R, Lillo J, Navarro G, Franco R. Recent Advances in the Potential of Cannabinoids for Neuroprotection in Alzheimer’s, Parkinson’s, and Huntington’s Diseases. Adv Exp Med Biol. 2021;1264:81–92.
4. Betjemann JP, Lowenstein DH. Status epilepticus in adults. Lancet Neurol. 2015;14(6):615–624. doi:10.1016/S1474-4422(15)00042-3
5. Zhu T, Wang L, Wang LP, Wan Q. Therapeutic targets of neuroprotection and neurorestoration in ischemic stroke: applications for natural compounds from medicinal herbs. Biomed Pharmacother. 2022;148:112719. doi:10.1016/j.biopha.2022.112719
6. Tao T, Liu M, Chen M, et al. Natural medicine in neuroprotection for ischemic stroke: challenges and prospective. Pharmacol Ther. 2020;216:107695. doi:10.1016/j.pharmthera.2020.107695
7. Carrera I, Cacabelos R. Current drugs and potential future neuroprotective compounds for Parkinson’s disease. Curr Neuropharmacol. 2019;17(3):295–306. doi:10.2174/1570159X17666181127125704
8. Pottoo FH, Salahuddin M, Khan FA, et al. Combinatorial Regimen of Carbamazepine and Imipramine Exhibits Synergism against Grandmal Epilepsy in Rats: Inhibition of Pro-Inflammatory Cytokines and PI3K/Akt/mTOR Signaling Pathway. Pharmaceuticals (Basel). 2021;14(11):1204. doi:10.3390/ph14111204
9. Stacpoole PW. The dichloroacetate dilemma: environmental hazard versus therapeutic goldmine--both or neither?. Environ Health Perspect. 2011;119(2):155–158. doi:10.1289/ehp.1002554
10. James MO, Jahn SC, Zhong G, Smeltz MG, Hu Z, Stacpoole PW. Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1. Pharmacology & Therapeutics (Oxford). 2017;170:166–180. doi:10.1016/j.pharmthera.2016.10.018
11. Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329 (Pt 1)(1):191–196. doi:10.1042/bj3290191
12. Anwar S, Shamsi A, Mohammad T, Islam A, Hassan MI. Targeting pyruvate dehydrogenase kinase signaling in the development of effective cancer therapy. Biochim Biophys Acta Rev Cancer. 2021;1876(1):188568. doi:10.1016/j.bbcan.2021.188568
13. Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci. 2014;71(14):2577–2604. doi:10.1007/s00018-013-1539-2
14. Abemayor E, Kovachich GB, Haugaard N. Effects of dichloroacetate on brain pyruvate dehydrogenase. J Neurochem. 1984;42(1):38–42. doi:10.1111/j.1471-4159.1984.tb09694.x
15. Kankotia S, Stacpoole PW. Dichloroacetate and cancer: new home for an orphan drug?. Biochim Biophys Acta. 2014;1846(2):617–629. doi:10.1016/j.bbcan.2014.08.005
16. Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008;99(7):989–994. doi:10.1038/sj.bjc.6604554
17. Powell SF, Mazurczak M, Dib EG, et al. Phase II study of dichloroacetate, an inhibitor of pyruvate dehydrogenase, in combination with chemoradiotherapy for unresected, locally advanced head and neck squamous cell carcinoma. Invest New Drugs. 2022;40(3):622–633. doi:10.1007/s10637-022-01235-5
18. Tataranni T, Piccoli C. Dichloroacetate (DCA) and cancer: an overview towards clinical applications. Oxid Med Cell Longev. 2019;2019:8201079. doi:10.1155/2019/8201079
19. Jin J, Yuan P, Yu W, et al. Mitochondria-targeting polymer micelle of Dichloroacetate induced pyroptosis to enhance osteosarcoma immunotherapy. ACS Nano. 2022;16(7):10327–10340. doi:10.1021/acsnano.2c00192
20. Shen H, Yu M, Tsoli M, et al. Targeting reduced mitochondrial DNA quantity as a therapeutic approach in pediatric high-grade gliomas. Neuro Oncol. 2020;22(1):139–151. doi:10.1093/neuonc/noz140
21. Sun H, Zhu A, Zhou X, Wang F. Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel. Oncotarget. 2017;8(32):52642–52650. doi:10.18632/oncotarget.16991
22. Lu X, Zhou D, Hou B, et al. Dichloroacetate enhances the antitumor efficacy of chemotherapeutic agents via inhibiting autophagy in non-small-cell lung cancer. Cancer Manag Res. 2018;10:1231–1241. doi:10.2147/CMAR.S156530
23. Mangal N, James MO, Stacpoole PW, Schmidt S. Model Informed Dose Optimization of Dichloroacetate for the Treatment of Congenital Lactic Acidosis in Children. J Clin Pharmacol. 2018;58(2):212–220. doi:10.1002/jcph.1009
24. Mainali R, Zabalawi M, Long D, et al. Dichloroacetate reverses sepsis-induced hepatic metabolic dysfunction. Elife. 2021;10:e64611.
25. Li X, Liu J, Hu H, et al. Dichloroacetate ameliorates cardiac dysfunction caused by ischemic insults through AMPK signal pathway-not only shifts metabolism. Toxicol Sci. 2019;167(2):604–617. doi:10.1093/toxsci/kfy272
26. Tian L, Wu D, Dasgupta A, et al. Epigenetic metabolic reprogramming of right ventricular fibroblasts in pulmonary arterial hypertension: a pyruvate dehydrogenase kinase-dependent shift in mitochondrial metabolism promotes right ventricular fibrosis. Circ Res. 2020;126(12):1723–1745. doi:10.1161/CIRCRESAHA.120.316443
27. Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80(1):315–360. doi:10.1152/physrev.2000.80.1.315
28. Engl E, Attwell D. Non-signalling energy use in the brain. J Physiol. 2015;593(16):3417–3429. doi:10.1113/jphysiol.2014.282517
29. Collier JJ, Olahova M, McWilliams TG, Taylor RW. Mitochondrial signalling and homeostasis: from cell biology to neurological disease. Trends Neurosci. 2023;46(2):137–152. doi:10.1016/j.tins.2022.12.001
30. Garabadu D, Agrawal N, Sharma A, Sharma S. Mitochondrial metabolism: a common link between neuroinflammation and neurodegeneration. Behav Pharmacol. 2019;30(8):642–652. doi:10.1097/FBP.0000000000000505
31. Tian H, Chen X, Liao J, et al. Mitochondrial quality control in stroke: from the mechanisms to therapeutic potentials. J Cell Mol Med. 2022;26(4):1000–1012. doi:10.1111/jcmm.17189
32. Chang LH, Shimizu H, Abiko H, et al. Effect of dichloroacetate on recovery of brain lactate, phosphorus energy metabolites, and glutamate during reperfusion after complete cerebral ischemia in rats. J Cereb Blood Flow Metab. 1992;12(6):1030–1038. doi:10.1038/jcbfm.1992.140
33. Katayama Y, Welsh FA. Effect of dichloroacetate on regional energy metabolites and pyruvate dehydrogenase activity during ischemia and reperfusion in gerbil brain. J Neurochem. 1989;52(6):1817–1822. doi:10.1111/j.1471-4159.1989.tb07262.x
34. Peng J, Huang H, Wang F. 二氯醋酸钠对缺血再灌注大鼠脑保护作用的观察 [The protective effects of dichloroacetate on cerebral ischemia after reperfusion of fed rats]. Zhonghua Yi Xue Za Zhi. 1996;76(7):509–511. Chinese.
35. Hu CL. Treatment of experimental ischemic cerebral lactic acidosis in rats with dichloroacetate. Chin J Neurol Psychiatry. 1992;25(6):355.
36. Peeling J, Sutherland G, Brown RA, Curry S. Protective effect of dichloroacetate in a rat model of forebrain ischemia. Neurosci Lett. 1996;208(1):21–24. doi:10.1016/0304-3940(96)12542-8
37. Zhao X, Li S, Mo Y, et al. DCA Protects against Oxidation Injury Attributed to Cerebral Ischemia-Reperfusion by Regulating Glycolysis through PDK2-PDH-Nrf2 Axis. Oxid Med Cell Longev. 2021;2021:5173035. doi:10.1155/2021/5173035
38. Hong DK, Kho AR, Choi BY, et al. Combined treatment with Dichloroacetic acid and pyruvate reduces hippocampal neuronal death after transient cerebral ischemia. Front Neurol. 2018;9:137.
39. Sun Y, Li T, Xie C, et al. Dichloroacetate treatment improves mitochondrial metabolism and reduces brain injury in neonatal mice. Oncotarget. 2016;7(22):31708–31722. doi:10.18632/oncotarget.9150
40. Guan X, Wei D, Liang Z, et al. FDCA Attenuates Neuroinflammation and Brain Injury after Cerebral Ischemic Stroke. ACS Chem Neurosci. 2023;14(20):3839–3854. doi:10.1021/acschemneuro.3c00456
41. Gao X, Gao YY, Yan HY, et al. PDK4 decrease neuronal apoptosis via inhibiting ROS-ASK1/P38 pathway in early brain injury after subarachnoid hemorrhage. Antioxid Redox Signal. 2022;36(7–9):505–524. doi:10.1089/ars.2021.0083
42. Zhao H, Mao J, Yuan Y, et al. Sodium Dichloroacetate stimulates angiogenesis by improving endothelial precursor cell function in an AKT/GSK-3beta/Nrf2 dependent pathway in vascular dementia rats. Front Pharmacol. 2019;10:523. doi:10.3389/fphar.2019.00523
43. Kho AR, Choi BY, Lee SH, et al. The Effects of Sodium Dichloroacetate on Mitochondrial Dysfunction and Neuronal Death Following Hypoglycemia-Induced Injury. Cells. 2019;8(5):405. doi:10.3390/cells8050405
44. DeVience SJ, Lu X, Proctor JL, et al. Enhancing Metabolic Imaging of Energy Metabolism in Traumatic Brain Injury Using Hyperpolarized [1-(13)C]Pyruvate and Dichloroacetate. Metabolites. 2021;11(6):335. doi:10.3390/metabo11060335
45. Durie D, McDonald TS, Borges K. The effect of dichloroacetate in mouse models of epilepsy. Epilepsy Res. 2018;145:77–81. doi:10.1016/j.eplepsyres.2018.06.004
46. Lee SH, Choi BY, Kho AR, et al. Combined treatment of dichloroacetic acid and pyruvate increased neuronal survival after seizure. Nutrients. 2022;14(22):4804. doi:10.3390/nu14224804
47. Miquel E, Cassina A, Martinez-Palma L, et al. Modulation of astrocytic mitochondrial function by dichloroacetate improves survival and motor performance in inherited amyotrophic lateral sclerosis. PLoS One. 2012;7(4):e34776. doi:10.1371/journal.pone.0034776
48. Martinez-Palma L, Miquel E, Lagos-Rodriguez V, Barbeito L, Cassina A, Cassina P. Mitochondrial Modulation by Dichloroacetate Reduces Toxicity of Aberrant Glial Cells and Gliosis in the SOD1G93A Rat Model of Amyotrophic Lateral Sclerosis. Neurotherapeutics. 2019;16(1):203–215. doi:10.1007/s13311-018-0659-7
49. Palamiuc L, Schlagowski A, Ngo ST, et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol Med. 2015;7(5):526–546. doi:10.15252/emmm.201404433
50. Parkin ET, Hammond JE, Owens L, Hodges MD. The orphan drug dichloroacetate reduces amyloid beta-peptide production whilst promoting non-amyloidogenic proteolysis of the amyloid precursor protein. PLoS One. 2022;17(1):e0255715. doi:10.1371/journal.pone.0255715
51. Andreassen OA, Ferrante RJ, Huang HM, et al. Dichloroacetate exerts therapeutic effects in transgenic mouse models of Huntington’s disease. Ann Neurol. 2001;50(1):112–117. doi:10.1002/ana.1085
52. O’Hara D, Davis GM, Adlesic NA, Hayes JM, Davey GP. Dichloroacetate Stabilizes Mitochondrial Fusion Dynamics in Models of Neurodegeneration. Front Mol Neurosci. 2019;12:219. doi:10.3389/fnmol.2019.00219
53. Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nat Med. 2011;17(11):1391–1401. doi:10.1038/nm.2507
54. Kalogeris T, Baines CP, Krenz M, Korthuis RJ. Cell biology of ischemia/reperfusion injury. Int Rev Cell Mol Biol. 2012;298:229–317. doi:10.1016/B978-0-12-394309-5.00006-7
55. Pradeep H, Diya JB, Shashikumar S, Rajanikant GK. Oxidative stress--assassin behind the ischemic stroke. Folia Neuropathol. 2012;50(3):219–230. doi:10.5114/fn.2012.30522
56. Bramlett HM, Dietrich WD. Pathophysiology of cerebral ischemia and brain trauma: Similarities and differences. J Cereb Blood Flow Metab. 2004;24(2):133–150. doi:10.1097/01.WCB.0000111614.19196.04
57. Martin E, Rosenthal RE, Fiskum G. Pyruvate dehydrogenase complex: metabolic link to ischemic brain injury and target of oxidative stress. J Neurosci Res. 2005;79(1–2):240–247. doi:10.1002/jnr.20293
58. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018;122(6):877–902. doi:10.1161/CIRCRESAHA.117.311401
59. Zucker SN, Fink EE, Bagati A, et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol Cell. 2014;53(6):916–928. doi:10.1016/j.molcel.2014.01.033
60. Chhunchha B, Kubo E, Singh DP. Sulforaphane-Induced Klf9/Prdx6 Axis Acts as a Molecular Switch to Control Redox Signaling and Determines Fate of Cells. Cells. 2019;8(10):1159. doi:10.3390/cells8101159
61. Cahill J, Zhang JH. Subarachnoid hemorrhage: is it time for a new direction?. Stroke. 2009;40(3 Suppl):S86–S87. doi:10.1161/STROKEAHA.108.533315
62. Lilla N, Fullgraf H, Stetter C, Kohler S, Ernestus RI, Westermaier T. First Description of Reduced Pyruvate Dehydrogenase Enzyme Activity Following Subarachnoid Hemorrhage (SAH). Front Neurosci. 2017;11:37. doi:10.3389/fnins.2017.00037
63. Westermaier T, Jauss A, Eriskat J, Kunze E, Roosen K. Time-course of cerebral perfusion and tissue oxygenation in the first 6 h after experimental subarachnoid hemorrhage in rats. J Cereb Blood Flow Metab. 2009;29(4):771–779. doi:10.1038/jcbfm.2008.169
64. Chen Y, Galea I, Macdonald RL, Wong G, Zhang JH. Rethinking the initial changes in subarachnoid haemorrhage: Focusing on real-time metabolism during early brain injury. EBioMedicine. 2022;83:104223. doi:10.1016/j.ebiom.2022.104223
65. Stockwin LH, Yu SX, Borgel S, et al. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer. 2010;127(11):2510–2519. doi:10.1002/ijc.25499
66. Adam-Vizi V. Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources. Antioxid Redox Signal. 2005;7(9–10):1140–1149. doi:10.1089/ars.2005.7.1140
67. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–185. doi:10.1016/j.cmet.2006.02.002
68. Chouchani ET, Pell VR, James AM, et al. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab. 2016;23(2):254–263.
69. Serrander L, Cartier L, Bedard K, et al. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J. 2007;406(1):105–114. doi:10.1042/BJ20061903
70. Bugger H, Pfeil K. Mitochondrial ROS in myocardial ischemia reperfusion and remodeling. Biochim Biophys Acta Mol Basis Dis. 2020;1866(7):165768. doi:10.1016/j.bbadis.2020.165768
71. Kinugasa Y, Ogino K, Furuse Y, et al. Allopurinol improves cardiac dysfunction after ischemia-reperfusion via reduction of oxidative stress in isolated perfused rat hearts. Circ J. 2003;67(9):781–787. doi:10.1253/circj.67.781
72. Ricci L, Stanley FU, Eberhart T, Mainini F, Sumpton D, Cardaci S. Pyruvate transamination and NAD biosynthesis enable proliferation of succinate dehydrogenase-deficient cells by supporting aerobic glycolysis. Cell Death Dis. 2023;14(7):403. doi:10.1038/s41419-023-05927-5
73. Chouchani ET, Pell VR, Gaude E, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515(7527):431–435.
74. Oh CJ, Ha CM, Choi YK, et al. Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury. Kidney Int. 2017;91(4):880–895. doi:10.1016/j.kint.2016.10.011
75. Brand MD. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med. 2016;100:14–31. doi:10.1016/j.freeradbiomed.2016.04.001
76. Lin G, Hill DK, Andrejeva G, et al. Dichloroacetate induces autophagy in colorectal cancer cells and tumours. Br J Cancer. 2014;111(2):375–385. doi:10.1038/bjc.2014.281
77. Fisher M, Savitz SI. Pharmacological brain cytoprotection in acute ischaemic stroke - renewed hope in the reperfusion era. Nat Rev Neurol. 2022;18(4):193–202. doi:10.1038/s41582-021-00605-6
78. Mei ZG, Huang YG, Feng ZT, et al. Electroacupuncture ameliorates cerebral ischemia/reperfusion injury by suppressing autophagy via the SIRT1-FOXO1 signaling pathway. Aging (Albany NY). 2020;12(13):13187–13205. doi:10.18632/aging.103420
79. Wang P, Shao BZ, Deng Z, Chen S, Yue Z, Miao CY. Autophagy in ischemic stroke. Prog Neurobiol. 2018;163–164:98–117. doi:10.1016/j.pneurobio.2018.01.001
80. Mo Y, Sun YY, Liu KY. Autophagy and inflammation in ischemic stroke. Neural Regen Res. 2020;15(8):1388–1396. doi:10.4103/1673-5374.274331
81. Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015;22(3):377–388. doi:10.1038/cdd.2014.150
82. Zou Y, Luo X, Feng Y, et al. Luteolin prevents THP-1 macrophage pyroptosis by suppressing ROS production via Nrf2 activation. Chem Biol Interact. 2021;345:109573. doi:10.1016/j.cbi.2021.109573
83. Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281(5381):1309–1312. doi:10.1126/science.281.5381.1309
84. Kaur I, Behl T, Sachdeva M, Bungau S, Venkatachalam T. Exploring the Mitochondrial Apoptotic Cell Death Landscape and Associated Components Serving as Molecular Targets, Primarily for Synthetic and Natural Drugs Targeting Oncology Therapeutics. Curr Mol Pharmacol. 2021;14(6):1066–1082. doi:10.2174/1874467214666210120145537
85. Ding L, Li J, Li W, et al. p53- and ROS-mediated AIF pathway involved in TGEV-induced apoptosis. J Vet Med Sci. 2018;80(11):1775–1781. doi:10.1292/jvms.18-0104
86. Liberale L, Ministrini S, Carbone F, Camici GG, Montecucco F. Cytokines as therapeutic targets for cardio- and cerebrovascular diseases. Basic Res Cardiol. 2021;116(1):23. doi:10.1007/s00395-021-00863-x
87. Wang P, Chen M, Yang Z, et al. Activation of pyruvate dehydrogenase activity by dichloroacetate improves survival and neurologic outcomes after cardiac arrest in rats. Shock. 2018;49(6):704–711. doi:10.1097/SHK.0000000000000971
88. Kim Y, Cho AY, Kim HC, Ryu D, Jo SA, Jung YS. Effects of Natural Polyphenols on Oxidative Stress-Mediated Blood-Brain Barrier Dysfunction. Antioxidants (Basel). 2022;11(2):197.
89. Pun PB, Lu J, Moochhala S. Involvement of ROS in BBB dysfunction. Free Radic Res. 2009;43(4):348–364. doi:10.1080/10715760902751902
90. Nitta T, Hata M, Gotoh S, et al. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J Cell Biol. 2003;161(3):653–660. doi:10.1083/jcb.200302070
91. Lee JY, Kim YH, Koh JY. Protection by pyruvate against transient forebrain ischemia in rats. J Neurosci. 2001;21(20):RC171. doi:10.1523/JNEUROSCI.21-20-j0002.2001
92. Vander HM, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029–1033. doi:10.1126/science.1160809
93. Bir SC, Khan MW, Javalkar V, Toledo EG, Kelley RE. Emerging Concepts in Vascular Dementia: A Review. J Stroke Cerebrovasc Dis. 2021;30(8):105864. doi:10.1016/j.jstrokecerebrovasdis.2021.105864
94. Williamson K, Stringer SE, Alexander MY. Endothelial progenitor cells enter the aging arena. Front Physiol. 2012;3:30. doi:10.3389/fphys.2012.00030
95. Jian KT, Shi Y, Zhang Y, Mao YM, Liu JS, Xue FL. Time course effect of hypoxia on bone marrow-derived endothelial progenitor cells and their effects on left ventricular function after transplanted into acute myocardial ischemia rat. Eur Rev Med Pharmacol Sci. 2015;19(6):1043–1054.
96. Brown RH, Al-Chalabi A. Amyotrophic Lateral Sclerosis. N Engl J Med. 2017;377(2):162–172. doi:10.1056/NEJMra1603471
97. Philips T, Rothstein JD. Glial cells in amyotrophic lateral sclerosis. Exp Neurol. 2014;262 Pt B:111–120. doi:10.1016/j.expneurol.2014.05.015
98. Kang SH, Li Y, Fukaya M, et al. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16(5):571–579. doi:10.1038/nn.3357
99. Smith EF, Shaw PJ, De Vos KJ. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci Lett. 2019;710:132933. doi:10.1016/j.neulet.2017.06.052
100. Feldman EL, Goutman SA, Petri S, et al. Amyotrophic lateral sclerosis. Lancet. 2022;400(10360):1363–1380. doi:10.1016/S0140-6736(22)01272-7
101. Valbuena GN, Tortarolo M, Bendotti C, Cantoni L, Keun HC. Altered Metabolic Profiles Associate with Toxicity in SOD1(G93A) Astrocyte-Neuron Co-Cultures. Sci Rep. 2017;7(1):50. doi:10.1038/s41598-017-00072-4
102. Pehar M, Cassina P, Vargas MR, et al. Astrocytic production of nerve growth factor in motor neuron apoptosis: implications for amyotrophic lateral sclerosis. J Neurochem. 2004;89(2):464–473. doi:10.1111/j.1471-4159.2004.02357.x
103. Izrael M, Slutsky SG, Revel M. Rising Stars: Astrocytes as a Therapeutic Target for ALS Disease. Front Neurosci. 2020;14:824. doi:10.3389/fnins.2020.00824
104. Fan YY, Huo J. A1/A2 astrocytes in central nervous system injuries and diseases: Angels or devils?. Neurochem Int. 2021;148:105080. doi:10.1016/j.neuint.2021.105080
105. Diaz-Garcia CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G. Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab. 2017;26(2):361–374. doi:10.1016/j.cmet.2017.06.021
106. Jia L, Liao M, Mou A, et al. Rheb-regulated mitochondrial pyruvate metabolism of Schwann cells linked to axon stability. Dev Cell. 2021;56(21):2980–2994. doi:10.1016/j.devcel.2021.09.013
107. Staub F, Mackert B, Kempski O, Peters J, Baethmann A. Swelling and death of neuronal cells by lactic acid. J Neurol Sci. 1993;119(1):79–84. doi:10.1016/0022-510X(93)90194-4
108. Zimmerman MC, Oberley LW, Flanagan SW. Mutant SOD1-induced neuronal toxicity is mediated by increased mitochondrial superoxide levels. J Neurochem. 2007;102(3):609–618. doi:10.1111/j.1471-4159.2007.04502.x
109. Bartosz G. Peroxynitrite: mediator of the toxic action of nitric oxide. Acta Biochim Pol. 1996;43(4):645–659. doi:10.18388/abp.1996_4461
110. Kirkinezos IG, Bacman SR, Hernandez D, et al. Cytochrome c association with the inner mitochondrial membrane is impaired in the CNS of G93A-SOD1 mice. J Neurosci. 2005;25(1):164–172. doi:10.1523/JNEUROSCI.3829-04.2005
111. Cassina P, Cassina A, Pehar M, et al. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci. 2008;28(16):4115–4122. doi:10.1523/JNEUROSCI.5308-07.2008
112. Delaney LM, Ho N, Morrison J, Farias NR, Mosser DD, Coomber BL. Dichloroacetate affects proliferation but not survival of human colorectal cancer cells. Apoptosis. 2015;20(1):63–74. doi:10.1007/s10495-014-1046-4
113. Fischer LR, Culver DG, Tennant P, et al. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185(2):232–240. doi:10.1016/j.expneurol.2003.10.004
114. Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20(7):2534–2542. doi:10.1523/JNEUROSCI.20-07-02534.2000
115. Hegedus J, Putman CT, Tyreman N, Gordon T. Preferential motor unit loss in the SOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. The Journal of Physiology. 2008;586(14):3337–3351. doi:10.1113/jphysiol.2007.149286
116. Baker JS, McCormick MC, Robergs RA. Interaction among skeletal muscle metabolic energy systems during intense exercise. J Nutr Metab. 2010;2010:905612. doi:10.1155/2010/905612
117. Bassel-Duby R, Olson EN. Signaling Pathways in Skeletal Muscle Remodeling. Annu Rev Biochem. 2006;75(1):19–37. doi:10.1146/annurev.biochem.75.103004.142622
118. Malgoyre A, Chabert C, Tonini J, Koulmann N, Bigard X, Sanchez H. Alterations to mitochondrial fatty-acid use in skeletal muscle after chronic exposure to hypoxia depend on metabolic phenotype. J Appl Physiol. 2017;122(3):666–674. doi:10.1152/japplphysiol.00090.2016
119. Aon MA, Bhatt N, Cortassa SC. Mitochondrial and cellular mechanisms for managing lipid excess. Front Physiol. 2014;5:282. doi:10.3389/fphys.2014.00282
120. Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J. 2003;375(Pt 2):365–371. doi:10.1042/bj20030022
121. Kim MJ, Sinam IS, Siddique Z, Jeon JH, Lee IK. The link between mitochondrial dysfunction and sarcopenia: an update focusing on the role of pyruvate dehydrogenase kinase 4. Diabetes Metab J. 2023;47(2):153–163. doi:10.4093/dmj.2022.0305
122. Dupuis L, Gonzalez DAJ, Echaniz-Laguna A, et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS One. 2009;4(4):e5390. doi:10.1371/journal.pone.0005390
123. Massao HS, de Oliveira CC, Mendonca JR, Piltcher HE, Fernandes LC, Curi R. Palmitate acutely raises glycogen synthesis in rat soleus muscle by a mechanism that requires its metabolization (Randle cycle). FEBS Lett. 2003;541(1–3):109–114.
124. Zhao Y, Zheng Q, Hong Y, et al. beta(2)-Microglobulin coaggregates with Abeta and contributes to amyloid pathology and cognitive deficits in Alzheimer’s disease model mice. Nat Neurosci. 2023;26(7):1170–1184. doi:10.1038/s41593-023-01352-1
125. Musiek ES, Holtzman DM. Three dimensions of the amyloid hypothesis: time, space and ‘wingmen’. Nat Neurosci. 2015;18(6):800–806. doi:10.1038/nn.4018
126. Hitschler L, Lang T. The transmembrane domain of the amyloid precursor protein is required for antiamyloidogenic processing by alpha-secretase ADAM10. J Biol Chem. 2022;298(6):101911. doi:10.1016/j.jbc.2022.101911
127. Velliquette RA, O’Connor T, Vassar R. Energy inhibition elevates beta-secretase levels and activity and is potentially amyloidogenic in APP transgenic mice: possible early events in Alzheimer’s disease pathogenesis. J Neurosci. 2005;25(47):10874–10883. doi:10.1523/JNEUROSCI.2350-05.2005
128. O’Connor T, Sadleir KR, Maus E, et al. Phosphorylation of the translation initiation factor eIF2alpha increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60(6):988–1009. doi:10.1016/j.neuron.2008.10.047
129. Walia V, Kaushik D, Mittal V, et al. Delineation of neuroprotective effects and possible benefits of antioxidantstherapy for the treatment of Alzheimer’s diseases by targeting mitochondrial-derived reactive oxygen species: bench to bedside. Mol Neurobiol. 2022;59(1):657–680. doi:10.1007/s12035-021-02617-1
130. Song T, Song X, Zhu C, et al. Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimer’s disease: a meta-analysis of in vivo magnetic resonance spectroscopy studies. Ageing Res Rev. 2021;72:101503. doi:10.1016/j.arr.2021.101503
131. Yulug B, Altay O, Li X, et al. Combined metabolic activators improve cognitive functions in Alzheimer’s disease patients: a randomised, double-blinded, placebo-controlled phase-II trial. Transl Neurodegener. 2023;12(1):4. doi:10.1186/s40035-023-00336-2
132. Sajan MP, Leitges M, Park C, et al. Control of beta-site amyloid precursor protein-cleaving Enzyme-1 expression by protein kinase C-lambda/iota and nuclear factor kappa-B. Curr Alzheimer Res. 2021;18(12):941–955. doi:10.2174/1567205019666211222120448
133. Bahn G, Park JS, Yun UJ, et al. NRF2/ARE pathway negatively regulates BACE1 expression and ameliorates cognitive deficits in mouse Alzheimer’s models. Proc Natl Acad Sci U S A. 2019;116(25):12516–12523. doi:10.1073/pnas.1819541116
134. Ho CL, Kao NJ, Lin CI, Cross TL, Lin SH. Quercetin Increases Mitochondrial Biogenesis and Reduces Free Radicals in Neuronal SH-SY5Y Cells. Nutrients. 2022;14(16):3310. doi:10.3390/nu14163310
135. Zhao H, Sun M, Zhang Y, et al. Connecting the dots: the cerebral lymphatic system as a bridge between the central nervous system and peripheral system in health and disease. Aging Dis. 2023. doi:10.14336/AD.2023.05162023
136. Perez OJ, Swerdlow RH. Mitochondrial dysfunction in Alzheimer’s disease: role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol. 2019;176(18):3489–3507. doi:10.1111/bph.14585
137. Borsche M, Pereira SL, Klein C, Grunewald A. Mitochondria and Parkinson’s disease: clinical, molecular, and translational aspects. J Parkinsons Dis. 2021;11(1):45–60. doi:10.3233/JPD-201981
138. Illarioshkin SN, Klyushnikov SA, Vigont VA, Seliverstov YA, Kaznacheyeva EV. Molecular Pathogenesis in Huntington’s Disease. Biochemistry (Mosc). 2018;83(9):1030–1039. doi:10.1134/S0006297918090043
139. Zhuang L, Jia K, Chen C, et al. DYRK1B-STAT3 drives cardiac hypertrophy and heart failure by impairing mitochondrial bioenergetics. Circulation. 2022;145(11):829–846. doi:10.1161/CIRCULATIONAHA.121.055727
140. Rius-Perez S, Torres-Cuevas I, Millan I, Ortega AL, Perez S. PGC-1alpha, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid Med Cell Longev. 2020;2020:1452696. doi:10.1155/2020/1452696
141. Jamwal S, Blackburn JK, Elsworth JD. PPARgamma/PGC1alpha signaling as a potential therapeutic target for mitochondrial biogenesis in neurodegenerative disorders. Pharmacol Ther. 2021;219:107705. doi:10.1016/j.pharmthera.2020.107705
142. Yadav H, Quijano C, Kamaraju AK, et al. Protection from obesity and diabetes by blockade of TGF-beta/Smad3 signaling. Cell Metab. 2011;14(1):67–79. doi:10.1016/j.cmet.2011.04.013
143. Villena JA. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015;282(4):647–672. doi:10.1111/febs.13175
144. Wu S, Zou MH. AMPK, Mitochondrial function, and cardiovascular disease. Int J Mol Sci. 2020;21(14):4987.
145. Tang BL. Sirt1 and the Mitochondria. Mol Cells. 2016;39(2):87–95.
146. Canto C, Gerhart-Hines Z, Feige JN, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458(7241):1056–1060. doi:10.1038/nature07813
147. El SS. Biochemical origin of the Warburg effect in light of 15 years of research experience: a novel evidence-based view (An Expert Opinion Article). Onco Targets Ther. 2023;16:143–155. doi:10.2147/OTT.S397593
148. Titov DV, Cracan V, Goodman RP, Peng J, Grabarek Z, Mootha VK. Complementation of mitochondrial electron transport chain by manipulation of the NAD + /NADH ratio. Science. 2016;352(6282):231–235. doi:10.1126/science.aad4017
149. Lee JJ, Ng SC, Hsu JY, et al. Galangin Reverses H(2)O(2)-Induced Dermal Fibroblast Senescence via SIRT1-PGC-1alpha/Nrf2 Signaling. Int J Mol Sci. 2022;23(3):1387.
150. Perez S, Rius-Perez S, Finamor I, et al. Obesity causes PGC-1alpha deficiency in the pancreas leading to marked IL-6 upregulation via NF-kappaB in acute pancreatitis. J Pathol. 2019;247(1):48–59. doi:10.1002/path.5166
151. Niel R, Le Moyec L, Launay T, et al. Physical performance level in sarcomeric mitochondria creatine kinase knockout mouse model throughout ageing. Exp Gerontol. 2021;146:111246. doi:10.1016/j.exger.2021.111246
152. Li Y, Feng YF, Liu XT, et al. Songorine promotes cardiac mitochondrial biogenesis via Nrf2 induction during sepsis. Redox Biol. 2021;38:101771. doi:10.1016/j.redox.2020.101771
153. Liu J, Du S, Kong Q, et al. HSPA12A attenuates lipopolysaccharide-induced liver injury through inhibiting caspase-11-mediated hepatocyte pyroptosis via PGC-1alpha-dependent acyloxyacyl hydrolase expression. Cell Death Differ. 2020;27(9):2651–2667. doi:10.1038/s41418-020-0536-x
154. Wang R, Li JJ, Diao S, et al. Metabolic stress modulates Alzheimer’s beta-secretase gene transcription via SIRT1-PPARgamma-PGC-1 in neurons. Cell Metab. 2013;17(5):685–694. doi:10.1016/j.cmet.2013.03.016
155. James MO, Yan Z, Cornett R, et al. Pharmacokinetics and metabolism of [14C]dichloroacetate in male Sprague-Dawley rats. Identification of glycine conjugates, including hippurate, as urinary metabolites of dichloroacetate. Drug Metab Dispos. 1998;26(11):1134–1143.
156. Kuroda Y, Toshima K, Watanabe T, et al. Effects of dichloroacetate on pyruvate metabolism in rat brain in vivo. Pediatr Res. 1984;18(10):936–938. doi:10.1203/00006450-198410000-00005
157. Lin EL, Mattox JK, Daniel FB. Tissue distribution, excretion, and urinary metabolites of dichloroacetic acid in the male Fischer 344 rat. J Toxicol Environ Health. 1993;38(1):19–32. doi:10.1080/15287399309531697
158. Shroads AL, Guo X, Dixit V, Liu HP, James MO, Stacpoole PW. Age-dependent kinetics and metabolism of dichloroacetate: possible relevance to toxicity. J Pharmacol Exp Ther. 2008;324(3):1163–1171. doi:10.1124/jpet.107.134593
159. Saghir SA, Schultz IR. Low-dose pharmacokinetics and oral bioavailability of dichloroacetate in naive and GST-zeta-depleted rats. Environ Health Perspect. 2002;110(8):757–763. doi:10.1289/ehp.02110757
160. Lantum HB, Baggs RB, Krenitsky DM, Board PG, Anders MW. Immunohistochemical localization and activity of glutathione transferase zeta (GSTZ1-1) in rat tissues. Drug Metab Dispos. 2002;30(6):616–625. doi:10.1124/dmd.30.6.616
161. Squirewell EJ, Smeltz MG, Rowland-Faux L, Horne LP, Stacpoole PW, James MO. Effects of Multiple Doses of Dichloroacetate on GSTZ1 Expression and Activity in Liver and Extrahepatic Tissues of Young and Adult Rats. Drug Metab Dispos. 2020;48(11):1217–1223. doi:10.1124/dmd.120.000142
162. Wells PG, Moore GW, Rabin D, Wilkinson GR, Oates JA, Stacpoole PW. Metabolic effects and pharmacokinetics of intravenously administered dichloroacetate in humans. Diabetologia. 1980;19(2):109–113. doi:10.1007/BF00421855
163. Anderson WB, Board PG, Gargano B, Anders MW. Inactivation of glutathione transferase zeta by dichloroacetic acid and other fluorine-lacking alpha-haloalkanoic acids. Chem Res Toxicol. 1999;12(12):1144–1149. doi:10.1021/tx990085l
164. Li W, James MO, McKenzie SC, Calcutt NA, Liu C, Stacpoole PW. Mitochondrion as a novel site of dichloroacetate biotransformation by glutathione transferase zeta 1. J Pharmacol Exp Ther. 2011;336(1):87–94. doi:10.1124/jpet.110.173195
165. Smeltz MG, Hu Z, Zhong G, et al. Mitochondrial Glutathione Transferase Zeta 1 Is Inactivated More Rapidly by Dichloroacetate than the Cytosolic Enzyme in Adult and Juvenile Rat Liver. Chem Res Toxicol. 2019;32(10):2042–2052. doi:10.1021/acs.chemrestox.9b00207
166. Zhong G, James MO, Smeltz MG, et al. Age-Related Changes in Expression and Activity of Human Hepatic Mitochondrial Glutathione Transferase Zeta1. Drug Metab Dispos. 2018;46(8):1118–1128. doi:10.1124/dmd.118.081810
167. James MO, Stacpoole PW. Pharmacogenetic considerations with dichloroacetate dosing. Pharmacogenomics. 2016;17(7):743–753. doi:10.2217/pgs-2015-0012
168. Jahn SC, Rowland-Faux L, Stacpoole PW, James MO. Chloride concentrations in human hepatic cytosol and mitochondria are a function of age. Biochem Biophys Res Commun. 2015;459(3):463–468. doi:10.1016/j.bbrc.2015.02.128
169. Zhong G, Li W, Gu Y, Langaee T, Stacpoole PW, James MO. Chloride and other anions inhibit dichloroacetate-induced inactivation of human liver GSTZ1 in a haplotype-dependent manner. Chem Biol Interact. 2014;215:33–39. doi:10.1016/j.cbi.2014.02.015
170. Jahn SC, Gay LA, Weaver CJ, et al. Age-related changes in miRNA expression influence GSTZ1 and other drug metabolizing enzymes. Drug Metab Dispos. 2020;48(7):563–569. doi:10.1124/dmd.120.090639
171. Shroads AL, Coats BS, McDonough CW, Langaee T, Stacpoole PW. Haplotype variations in glutathione transferase zeta 1 influence the kinetics and dynamics of chronic dichloroacetate in children. J Clin Pharmacol. 2015;55(1):50–55. doi:10.1002/jcph.371
172. Langaee T, Wagner R, Horne LP, et al. Personalized dosing of Dichloroacetate using GSTZ1 clinical genotyping assay. Genet Test Mol Biomarkers. 2018;22(4):266–269. doi:10.1089/gtmb.2017.0261
173. Langaee TY, Zhong G, Li W, et al. The influence of human GSTZ1 gene haplotype variations on GSTZ1 expression. Pharmacogenet Genomics. 2015;25(5):239–245. doi:10.1097/FPC.0000000000000129
174. Lantum HB, Board PG, Anders MW. Kinetics of the biotransformation of maleylacetone and chlorofluoroacetic acid by polymorphic variants of human glutathione transferase zeta (hGSTZ1-1). Chem Res Toxicol. 2002;15(7):957–963. doi:10.1021/tx010095y
175. Li W, Gu Y, James MO, et al. Prenatal and postnatal expression of glutathione transferase zeta 1 in human liver and the roles of haplotype and subject age in determining activity with dichloroacetate. Drug Metab Dispos. 2012;40(2):232–239. doi:10.1124/dmd.111.041533
176. Shroads AL, Langaee T, Coats BS, et al. Human polymorphisms in the glutathione transferase zeta 1/maleylacetoacetate isomerase gene influence the toxicokinetics of dichloroacetate. J Clin Pharmacol. 2012;52(6):837–849. doi:10.1177/0091270011405664
177. Jiang Y, Milavetz G, James MO, An G. A mechanism-based pharmacokinetic enzyme turnover model for dichloroacetic acid autoinhibition in rats. J Pharm Sci. 2017;106(5):1396–1404. doi:10.1016/j.xphs.2017.01.032
178. Wehmas LC, DeAngelo AB, Hester SD, et al. Metabolic disruption early in life is associated with latent carcinogenic activity of dichloroacetic acid in mice. Toxicol Sci. 2017;159(2):354–365. doi:10.1093/toxsci/kfx146
179. Wood CE, Hester SD, Chorley BN, et al. Latent carcinogenicity of early-life exposure to dichloroacetic acid in mice. Carcinogenesis. 2015;36(7):782–791. doi:10.1093/carcin/bgv057
180. Program NT Report on carcinogens monograph on haloacetic acids found as water disinfection by-products: RoC monograph 12. Research Triangle Park (NC): National Toxicology Program; 2018.
181. Abdelmalak M, Lew A, Ramezani R, et al. Long-term safety of dichloroacetate in congenital lactic acidosis. Mol Genet Metab. 2013;109(2):139–143. doi:10.1016/j.ymgme.2013.03.019
182. Stacpoole PW, Harwood HJ, Cameron DF, et al. Chronic toxicity of dichloroacetate: possible relation to thiamine deficiency in rats. Fundam Appl Toxicol. 1990;14(2):327–337. doi:10.1016/0272-0590(90)90212-3
183. Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Pharmacokinetics, metabolism and toxicology of dichloroacetate. Drug Metab Rev. 1998;30(3):499–539. doi:10.3109/03602539808996323
184. Board PG, Anders MW. Human glutathione transferase zeta. Methods Enzymol. 2005;401:61–77.
185. St-Louis M, Tanguay RM. Mutations in the fumarylacetoacetate hydrolase gene causing hereditary tyrosinemia type I: overview. Hum Mutat. 1997;9(4):291–299. doi:10.1002/(SICI)1098-1004(1997)9:4<291::AID-HUMU1>3.0.CO;2-9
186. Lantum HB, Cornejo J, Pierce RH, Anders MW. Perturbation of maleylacetoacetic acid metabolism in rats with dichloroacetic Acid-induced glutathione transferase zeta deficiency. Toxicol Sci. 2003;74(1):192–202. doi:10.1093/toxsci/kfg104
187. Andrade V, Mateus ML, Batoreu MC, Aschner M, Dos SA. Urinary delta-ALA: a potential biomarker of exposure and neurotoxic effect in rats co-treated with a mixture of lead, arsenic and manganese. Neurotoxicology. 2013;38:33–41. doi:10.1016/j.neuro.2013.06.003
188. Morrow G, Tanguay RM. Biochemical and clinical aspects of hereditary tyrosinemia type 1. Adv Exp Med Biol. 2017;959:9–21.
189. Baker PR, Cramer SD, Kennedy M, Assimos DG, Holmes RP. Glycolate and glyoxylate metabolism in HepG2 cells. Am J Physiol Cell Physiol. 2004;287(5):C1359–C1365. doi:10.1152/ajpcell.00238.2004
190. Salido E, Pey AL, Rodriguez R, Lorenzo V. Primary hyperoxalurias: disorders of glyoxylate detoxification. Biochim Biophys Acta. 2012;1822(9):1453–1464. doi:10.1016/j.bbadis.2012.03.004
191. Sidhu H, Gupta R, Thind SK, Nath R. Oxalate metabolism in thiamine-deficient rats. Ann Nutr Metab. 1987;31(6):354–361. doi:10.1159/000177294
192. Berini SE, Tracy JA, Engelstad JK, Lorenz EC, Milliner DS, Dyck PJ. Progressive polyradiculoneuropathy due to intraneural oxalate deposition in type 1 primary hyperoxaluria. Muscle Nerve. 2015;51(3):449–454. doi:10.1002/mus.24495
193. Evans OB, Stacpoole PW. Prolonged hypolactatemia and increased total pyruvate dehydrogenase activity by dichloroacetate. Biochem Pharmacol. 1982;31(7):1295–1300. doi:10.1016/0006-2952(82)90019-3
194. Kurlemann G, Paetzke I, Moller H, et al. Therapy of complex I deficiency: peripheral neuropathy during dichloroacetate therapy. Eur J Pediatr. 1995;154(11):928–932. doi:10.1007/BF01957508
195. El-Hattab AW, Zarante AM, Almannai M, Scaglia F. Therapies for mitochondrial diseases and current clinical trials. Mol Genet Metab. 2017;122(3):1–9. doi:10.1016/j.ymgme.2017.09.009
196. Broxton CN, Kaur P, Lavorato M, et al. Dichloroacetate and thiamine improve survival and mitochondrial stress in a C. elegans model of dihydrolipoamide dehydrogenase deficiency. JCI Insight. 2022;7(20). doi:10.1172/jci.insight.156222
197. Kaufmann P, Engelstad K, Wei Y, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology. 2006;66(3):324–330. doi:10.1212/01.wnl.0000196641.05913.27
198. Garon EB, Christofk HR, Hosmer W, et al. Dichloroacetate should be considered with platinum-based chemotherapy in hypoxic tumors rather than as a single agent in advanced non-small cell lung cancer. J Cancer Res Clin Oncol. 2014;140(3):443–452. doi:10.1007/s00432-014-1583-9
199. Michelakis ED, Sutendra G, Dromparis P, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2(31):31ra34. doi:10.1126/scitranslmed.3000677
200. Dunbar EM, Coats BS, Shroads AL, et al. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs. 2014;32(3):452–464. doi:10.1007/s10637-013-0047-4
201. Chu QS, Sangha R, Spratlin J, et al. A phase I open-labeled, single-arm, dose-escalation, study of dichloroacetate (DCA) in patients with advanced solid tumors. Invest New Drugs. 2015;33(3):603–610. doi:10.1007/s10637-015-0221-y
202. Stacpoole PW, Gilbert LR, Neiberger RE, et al. Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics. 2008;121(5):e1223–e1228. doi:10.1542/peds.2007-2062
203. Tian DD, Bennett SK, Coupland LA, et al. GSTZ1 genotypes correlate with dichloroacetate pharmacokinetics and chronic side effects in multiple myeloma patients in a pilot phase 2 clinical trial. Pharmacol Res Perspect. 2019;7(6):e526. doi:10.1002/prp2.526
204. Stacpoole PW, Kerr DS, Barnes C, et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics. 2006;117(5):1519–1531. doi:10.1542/peds.2005-1226
205. Liang Y, Duan L, Lu J, Xia J. Engineering exosomes for targeted drug delivery. Theranostics. 2021;11(7):3183–3195. doi:10.7150/thno.52570
206. Wong KH, Riaz MK, Xie Y, et al. Review of Current Strategies for Delivering Alzheimer’s Disease Drugs across the Blood-Brain Barrier. Int J Mol Sci. 2019;20(2):381. doi:10.3390/ijms20020381
207. Verma R, Kaushik A, Almeer R, Rahman MH, Abdel-Daim MM, Kaushik D. Improved Pharmacodynamic Potential of Rosuvastatin by Self-Nanoemulsifying Drug Delivery System: an in vitro and in vivo Evaluation. Int J Nanomedicine. 2021;16:905–924. doi:10.2147/IJN.S287665
208. Verma R, Mittal V, Pandey P, et al. Exploring the role of self-nanoemulsifying systems in drug delivery: challenges, issues, applications and recent advances. Curr Drug Deliv. 2023;20(9):1241–1261. doi:10.2174/1567201819666220519125003
209. Verma R, Kaushik D. In vitro Lipolysis as a Tool for the Establishment of IVIVC for Lipid-Based Drug Delivery Systems. Curr Drug Deliv. 2019;16(8):688–697. doi:10.2174/1567201816666190620115716
210. Liang Y, Iqbal Z, Lu J, et al. Cell-derived nanovesicle-mediated drug delivery to the brain: Principles and strategies for vesicle engineering. Mol Ther. 2023;31(5):1207–1224. doi:10.1016/j.ymthe.2022.10.008
211. Pracharova J, Novohradsky V, Kostrhunova H, et al. Half-sandwich Os(ii) and Ru(ii) bathophenanthroline complexes: anticancer drug candidates with unusual potency and a cellular activity profile in highly invasive triple-negative breast cancer cells. Dalton Trans. 2018;47(35):12197–12208. doi:10.1039/C8DT02236D
212. Starha P, Travnicek Z, Vanco J, Dvorak Z. Half-Sandwich Ru(II) and Os(II) Bathophenanthroline Complexes Containing a Releasable Dichloroacetato Ligand. Molecules. 2018;23(2):420. doi:10.3390/molecules23020420
213. Abanades LI, Haddad S, Rodrigo-Munoz JM, et al. Mechanistic Investigation into the Selective Anticancer Cytotoxicity and Immune System Response of Surface-Functionalized, Dichloroacetate-Loaded, UiO-66 Nanoparticles. ACS Appl Mater Interfaces. 2018;10(6):5255–5268. doi:10.1021/acsami.7b17756
214. Abanades LI, Haddad S, Rodrigo-Munoz JM, et al. Surface-Functionalization of Zr-Fumarate MOF for Selective Cytotoxicity and Immune System Compatibility in Nanoscale Drug Delivery. ACS Appl Mater Interfaces. 2018;10(37):31146–31157. doi:10.1021/acsami.8b11652
215. Yang J, Cao Q, Zhang H, et al. Targeted reversal and phosphorescence lifetime imaging of cancer cell metabolism via a theranostic rhenium(I)-DCA conjugate. Biomaterials. 2018;176:94–105. doi:10.1016/j.biomaterials.2018.05.040
216. Lázaro AI, Abanades LS, Forgan RS. Enhancing anticancer cytotoxicity through bimodal drug delivery from ultrasmall Zr MOF nanoparticles. Chem Commun (Camb). 2018;54(22):2792–2795. doi:10.1039/C7CC09739E
217. Choi H, Choi Y, Yim HY, Mirzaaghasi A, Yoo JK, Choi C. Biodistribution of Exosomes and Engineering Strategies for Targeted Delivery of Therapeutic Exosomes. Tissue Eng Regen Med. 2021;18(4):499–511. doi:10.1007/s13770-021-00361-0
218. Meng W, He C, Hao Y, Wang L, Li L, Zhu G. Prospects and challenges of extracellular vesicle-based drug delivery system: considering cell source. Drug Deliv. 2020;27(1):585–598. doi:10.1080/10717544.2020.1748758
219. Do AD, Kurniawati I, Hsieh CL, Wong TT, Lin YL, Sung SY. Application of Mesenchymal Stem Cells in Targeted Delivery to the Brain: Potential and Challenges of the Extracellular Vesicle-Based Approach for Brain Tumor Treatment. Int J Mol Sci. 2021;22(20):11187. doi:10.3390/ijms222011187
220. Luo Y, Yang H, Zhou YF, Hu B. Dual and multi-targeted nanoparticles for site-specific brain drug delivery. J Control Release. 2020;317:195–215. doi:10.1016/j.jconrel.2019.11.037
221. Hu B, Dai F, Fan Z, Ma G, Tang Q, Zhang X. Nanotheranostics: Congo Red/Rutin-MNPs with Enhanced Magnetic Resonance Imaging and H2O2-Responsive Therapy of Alzheimer’s Disease in APPswe/PS1dE9 Transgenic Mice. Adv Mater. 2015;27(37):5499–5505. doi:10.1002/adma.201502227
222. Qiao C, Yang J, Shen Q, et al. Traceable Nanoparticles with Dual Targeting and ROS Response for RNAi-Based Immunochemotherapy of Intracranial Glioblastoma Treatment. Adv Mater. 2018;30(18):e1705054. doi:10.1002/adma.201705054
223. Goyal K, Konar A, Kumar B, Koul V. Lactoferrin-conjugated pH and redox-sensitive polymersomes based on PEG-S-S-PLA-PCL-OH boost delivery of bacosides to the brain. Nanoscale. 2018;10(37):17781–17798. doi:10.1039/C8NR03828G
224. Itoh Y, Esaki T, Shimoji K, et al. Dichloroacetate effects on glucose and lactate oxidation by neurons and astroglia in vitro and on glucose utilization by brain in vivo. Proc Natl Acad Sci U S A. 2003;100(8):4879–4884. doi:10.1073/pnas.0831078100
 © 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2023 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.