Back to Journals » Drug Design, Development and Therapy » Volume 14
Neuroprotective Effect of Mesenchymal Stromal Cell-Derived Extracellular Vesicles Against Cerebral Ischemia-Reperfusion-Induced Neural Functional Injury: A Pivotal Role for AMPK and JAK2/STAT3/NF-κB Signaling Pathway Modulation
Authors Han M, Cao Y, Xue H, Chu X, Li T, Xin D, Yuan L, Ke H, Li G, Wang Z
Received 8 February 2020
Accepted for publication 3 June 2020
Published 20 July 2020 Volume 2020:14 Pages 2865—2876
DOI https://doi.org/10.2147/DDDT.S248892
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Jianbo Sun
Min Han,1– 3 Ying Cao,2 Hao Xue,1 Xili Chu,2 Tingting Li,2 Danqing Xin,2 Lin Yuan,2 Hongfei Ke,2 Gang Li,1 Zhen Wang2
1Department of Neurosurgery, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, People’s Republic of China; 2Department of Physiology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, People’s Republic of China; 3Department of Neurosurgery, The Fifth People’s Hospital of Jinan, Jinan, Shandong Province 250022, People’s Republic of China
Correspondence: Zhen Wang
Department of Physiology, School of Basic Medical Sciences, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, People’s Republic of China
Email [email protected]
Gang Li
Department of Neurosurgery, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan, Shandong 250012, People’s Republic of China
Email [email protected]
Introduction: Cerebral ischemia-reperfusion injury (CIRI) is the main factor that leads to poor prognosis of cerebral ischemia. Apoptosis has been shown to occur during the process of CIRI. Extracellular vesicles derived from mesenchymal stromal cells (MSCs-EVs) have shown broad potential for treating brain dysfunction and eliciting neuroprotective effects after stroke through neurogenesis and angiogenesis. However, the mechanism of action of extracellular vesicles during CIRI is not well known.
Methods: A middle cerebral artery occlusion (MCAO) model was induced by the modified Longa method, and MSCs-EVs were injected via the tail vein.
Results: Our results showed that MSCs-EVs significantly alleviated neurological deficits, reduced the volume of cerebral infarction and brain water content, improved pathological lesions in cortical brain tissue, and attenuated neuronal apoptosis in the cortex at 24 h and 48 h after MCAO in rats. Western blotting analysis showed that MSCs-EVs significantly upregulated p-AMPK and downregulated p-JAK2, p-STAT3 and p-NF-κB. In addition, an AMPK pathway blocker reversed the effect of MSCs-EVs on brain damage.
Conclusion: These results indicate that MSCs-EVs protected MCAO-injured rats, possibly by regulating the AMPK and JAK2/STAT3/NF-κB signaling pathways. This study supports the use of MSCs-EVs as a potential treatment strategy for MCAO in the future.
Keywords: extracellular vesicles, cerebral ischemia reperfusion, neuroprotection, AMPK, nuclear factor κB, JAK2/STAT3 signaling pathway
Introduction
Cerebrovascular disease ranks as the third leading cause of death in humans worldwide and is the leading cause of death among Chinese nationals.1 The prevalence, morbidity and mortality of stroke increase year to year and thus a huge social and economic burden. The incidence of cerebral ischemic stroke is significantly higher than that of hemorrhagic stroke. Ischemic stroke accounts for 75% of all stroke cases.2 The pathological type and pathogenesis of ischemic stroke are complicated and are often accompanied by cell apoptosis, the release of various inflammatory mediators, the infiltration and accumulation of inflammatory cells, the destruction of the blood-brain barrier, the secretion of inflammatory factors and the upregulation of adhesion molecules.3 Studies have shown that apoptosis after cerebral ischemia is one of the causes of secondary brain injury, indicating that apoptosis plays an important role in cerebral ischemia and cerebral ischemia-reperfusion injury (CIRI).4 Although the time window for endovascular mechanical thrombectomy has been extended, hemorrhagic transformation after thrombolytic or intravascular mechanical thrombectomy and rehabilitation after stroke are still the biggest challenges that plague neurologists. However, due to the complexity of the pathophysiological process of ischemic stroke, no thorough and effective treatment has been identified. Therefore, the molecular mechanism related to CIRI has been explored. Early intervention after cerebral ischemia has important clinical application value.5
Extracellular vesicles (EVs) are membranous vesicles with a diameter of 30 ~ 150 nm that can be secreted by cells and mediate the transmission of information between cells.6 Due to their circulatory stability and lower immunogenicity than that of stem cells, EVs have great potential in the field of biotherapy. EVs secreted by mesenchymal stromal cells (MSCs) to the outside are called MSCs-EVs and have shown beneficial effects in studies of myocardial infarction,7 liver fibrosis,8 wound healing,9 Alzheimer’s disease10 and spinal cord injury.11 Studies have also shown that MSCs-EVs have neuroprotective effects in cerebral ischemia-reperfusion rats.12
Adenosine 5ʹ-monophosphate (AMP)-activated protein kinase (AMPK), an important serine/threonine protein kinase, has frequently been referred to as the “metabolic master switch”.13 When the cell energy supply is reduced, AMPK can be activated to enhance energy production.14 The activation of AMPK can maintain cellular metabolic homeostasis by reducing energy expenditure and increasing energy utilization, thereby promoting poststroke repair.15 The activation of AMPK exerts a neuro-protective effect by reducing oxidative stress and inflammation after MCAO in rats.16
Apoptosis is the main causative factor in the pathogenesis of ischemic stroke, which involves several important signaling pathways. The Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway family is an important pathway through which most cytokines exert their biological functions, is normally expressed in the brain and plays a vital role in neuronal apoptosis.17,18 Among the JAK/STAT family, the JAK2/STAT3 pathway is the most conserved pathway and is most closely related to the pathophysiology of oxidative stress in the central nervous system.19,20 The JAK2/STAT3 signaling pathway has been found to have an important effect on the prognosis of neurological function in traumatic brain injury21 and cerebral ischemia.22 The activation of the JAK2 protein triggers the phosphorylation of STAT3. In addition, nuclear factor-kappa B (NF-κB), as a prototypical proinflammatory signaling molecule, is a critical transcription factor for inflammatory mediator induction and reduces the infiltration of inflammatory cells and apoptosis by targeting this pathway.23,24 However, in a model of ischemia-reperfusion, the mechanism by which MSCs-EVs play a neuroprotective role through the AMPK and JAK2/STAT3/NF-κB signaling pathways has not been fully elucidated. Therefore, this study aims to investigate the mechanism by which MSCs-EVs reduce brain injury induced by CIRI in rats by regulating the AMPK and the JAK2/STAT3/NF-κB pathway in a middle cerebral artery occlusion (MCAO) model in an attempt to provide a better option for the treatment of cerebral infarction in the future.
Materials and Methods
Materials
Details of the materials used in this study are shown in Supplementary Table S1.
Animals and Ethics Statement
All animal experimental procedures were performed according to the guidelines of the Chinese Care and Use legislation and were approved by the Animal Ethics Committee of Medical Department, Shandong University.
Adult male Sprague-Dawley (SD) rats of SPF grade aged 7–8 weeks and weighing 280–320 g were purchased from the Jinan Pengyue Experimental Animal Breeding Co., Ltd. (license number: SCXK (LU) 2019–0003). The animals were housed in standard rat cages at a constant temperature (24±1 °C) with a humidity of 45%-55% under a 12-h dark/light cycle 12 h, and all rats had free access to food and water. The experiment was started after 7 days of adaptive feeding.
Rat Middle Cerebral Artery Occlusion (MCAO)
MCAO was used to establish a cerebral ischemia-reperfusion model in rats based on the modified Longa method as previously described.25 In brief, rats were fasted for 6 h before the operation to reduce the complications of reflux and aspiration. Then, the animals were anesthetized with isoflurane. The neck skin and fascia were cut open, and the submandibular gland and sternocleidomastoid and scapular scapulohumeral muscles were exposed in turn and then pulled with hooks for fixation. The right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were separated, and then the ICA and CCA were clamped with micro-artery clips. Then, the ECA was ligated at the proximal end with a 5–0 polyester suture and cut 3.0 mm from the bifurcation of the CCA. Next, the ICA was fully dissociated, and microsurgical scissors were used to cut a small hole in the arterial wall 3 mm from the arterial bifurcation at the proximal end of the ECA. A prepared thread embolus (Guangzhou Jialing Biotechnology Co., Ltd.; product model: L3800) was inserted into the ECA and made parallel with the ICA, and the clip on the ICA was removed. After the micro-resistance was achieved, advancement of the embolus was stopped. Then, the ECA was tightened with a 5–0 polyester suture. The rats were anesthetized again after 2 h of ischemia, and then reperfusion was initiated by removing the thread embolus to the ECA trunk. The Sham group animals subsequently underwent sham surgical procedures without occlusion. Brain tissue samples were obtained 24 h or 48 h after reperfusion for assays. All animal models were established by the same researcher to reduce infarct variability. After the operation, each rat was house singly in a cage and provided free access to food and water.
Experimental Design and Animal Groups
The rats were randomly divided into eight groups: (1) the Sham 24 h + vehicle group, (2) the MCAO 24 h + vehicle group, (3) the MCAO 24 h + MSCs-EVs group, (4) the inhibitor (compound C, CC)-treated (MCAO 24 h + MSCs-EVs + CC) group, (5) the Sham 48 h + vehicle group, (6) the MCAO 48 h + vehicle group, (7) the MCAO 48 h + MSCs-EVs group and (8) the inhibitor-treated (MCAO 48 h + MSCs-EVs + CC) group. Each rat in the MSCs-EVs group was injected with 100 µg of MSCs-EVs suspended in 0.5 mL PBS through the caudal vein within 10 min after successful occlusion of the right MCA. The rats in the CC group were slowly stereotaxically injected with CC into the right ventricle 30 min before surgery to block the function of AMPK. The Sham groups were injected with the same volume of PBS as a control. The experiments were repeated at least three times.
Intracerebroventricular Injection
To further clarify the role of the AMPK pathway following MCAO, the rats in the AMPK + CC group were pretreated with CC (an effective inhibitor of AMPK). Dimethyl sulfoxide (DMSO) was used the solvent for CC, and prior to surgery, CC was dissolved at a concentration of 0.1 μM. Thirty minutes before MCAO surgery, CC (0.1 μM, 10 μL) was intracerebroventricularly injected to block the function of AMPK as described previously.26 Briefly, the animals were anesthetized with isoflurane and fixed on a stereotaxic apparatus. After the skull was exposed, 10 μL of the CC solution was drawn with a 25 μL Hamilton syringe, the syringe was fixed on the positioner, and the needle tip of the syringe was aligned with the front palate. The injection coordinates were determined based on ventricular anatomy: from bregma, 0.5 mm posterior along the midline, 1.5 mm to the right, and 4.0 mm deep. The solution was injected slowly and stably at a rate of approximately 0.5 μL/min. The needle was left in place for 10 min to prevent reflux after injection. The needle was slowly withdrawn, bone wax was used to close the hole, and the tissue was sutured layer by layer. The sham groups were injected with the same volume of sterile PBS as a control.
Neurological Deficit Scores
Before the animals were sacrificed, behavior was scored in a single-blinded manner using the modified Longa method; behavior was rated on a scale of 0–4:25 0 points: no neurological deficit; 1 point: unable to extend the contralateral forelimb and fails to go straight; 2 points: contralateral forelimb flexion and walks in a circle; 3 points: leans slightly to the contralateral side and walks in a circle toward the contralateral side; 4 points: walks in a circle toward the contralateral side. Animals with scores of 1, 2, or 3 points were selected for the experiment.
Brain Tissue Water Content Determination
Brain tissue was divided into two parts (the ipsilateral and contralateral cerebral hemispheres) after the animals were euthanized at the corresponding time points. The brain tissues were quickly weighed on an analytical balance with an accuracy of 0.01 mg (wet weight). Next, the hemispheres were dried in an oven at 105 °C for 24 h to obtain the dry weight content.27 The formula for brain water content was:
Brain water content (%) = [(wet weight-dry weight)/wet weight] ×100%.
2,3,5-Triphenyltetrazolium Chloride (TTC) Staining and Infarct Volume Measurement
The rats were sacrificed at the corresponding time points after cerebral ischemia-reperfusion with chloral hydrate. Whole brains were frozen at −20 °C for 20 min, and the brains were cut from the frontal pole with a blade. Each slice was approximately 2 mm thick. Sets of six serial slices from each brain were immersed in 2.0% TTC solution and incubated in a 37 °C incubator for 30 min in the dark. Brain tissues stained red were normal, while unstained white areas were regarded as infarcted regions. After fixation in 4% formalin for 24 h, the hemisphere infarct volume was measured by ImageJ software. The hemisphere lesion volume was calculated according to the following formula:28
Infarct volume = {[the intact contralateral hemisphere volume-(the intact ipsilateral hemisphere volume-the infarct volume)]/the intact contralateral hemisphere volume} × 100%.
Preparation of Paraffin Sections and Hematoxylin and Eosin (HE) Staining
At the corresponding time point, the chest of each rat was cut open to expose the chest cavity after deep anesthesia. The right atrial appendage was incised, PBS solution was perfused through the left ventricle until the effluent flowed out, and the entire brain tissue was completely removed after fixation with neutral formaldehyde. Then, paraffin sections were prepared. The sections were baked at 60 °C for 1 h and then dewaxed with xylene and gradient ethanol. Morphological changes in the brain tissue were observed under a microscope after staining with hematoxylin and differentiation with 10% hydrochloric acid.
Nissl Staining
Paraffin sections of brain tissues were baked at 55 °C for 30 min and then dewaxed with xylene and immersed in gradient ethanol. The slices were placed in Nissl staining solution and then immersed in differentiation solution in a 58 °C water bath for 40 min for differentiation. The sections were cleared with xylene after dehydration with ethanol (100%) and observed and photographed under a microscope.
TdT-Mediated dUTP Nick-End Labeling (TUNEL)
TUNEL staining was performed to detect the apoptosis rate as previously described.29 Briefly, paraffin sections were dewaxed in xylene for 10 min, treated with 3% H2O2 and incubated with sodium citrate (0.1 M). The sections were stained with DAB after treatment with the TUNEL reaction mixture. Five fields of view were randomly selected, observed and photographed under a fluorescence microscope. ImageJ software was used to analyze the ratio of positive cells. The neuronal death index was calculated as TUNEL-positive cells/total cells × 100%. The average apoptotic index in five fields was used as the neuronal apoptosis index of each rat.
Mesenchymal Stem Cell Culture and Reagents
MSCs were obtained from male SPF grade SD rats weighing approximately 80 g using previously described methods.30 Briefly, after sacrifice, the bilateral femurs and tibias were separated and disconnected at the ends. Then, the bone marrow in the medullary cavity was washed out with PBS. Next, the bone marrow suspension was filtered through a 70-μm cell strainer and centrifuged. The pellet was resuspended in DMEM containing 10% fetal bovine serum and 1% penicillin-streptomycin and then inoculated in a cell culture flask. After 72 h, the medium was exchanged to remove the unattached cells. When the cells were grown to a confluency of 80–90%, they were trypsinized and subcultured at a ratio of 1:3. Third-generation MSCs were used for the next experimental study. All of the above operations were performed under sterile conditions on a clean bench.
MSCs-EV Isolation
MSCs at 80% confluency (after passage three) were washed with PBS three times. Then, the culture medium was replaced with EV-free medium, which was generated by centrifugation at 120,000 × g for 2 h. After the culturing for 48 h, MSCs-EVs were extracted from the medium. EV isolation was performed as previously described.31 Briefly, the supernatant from MSC cultures was collected by centrifugation at 200 × g for 10 min and 20,000 × g for 20 min at 4 °C to remove any intact cells and cell debris. The supernatant was filtered through a 0.2 μm-filter and centrifuged at 100,000 × g for 75 min at 4 °C. The pellets were washed with PBS and subjected to an additional centrifugation at 100,000 × g for 75 min at 4 °C. The EV pellets were resuspended in PBS before storage at −80 °C. All the above steps were performed in an ultraclean platform.
Identification of MSCs-EVs
MSCs-EVs were prepared for transmission electron microscopy (TEM) examination (Hitachi H7700, Tokyo, Japan) as previously described.32 In brief, 5 μL of extracted MSCs-EVs in PBS was deposited onto carbon/formvar-coated Cu TEM grids and then dried for 10 min at 40 °C before staining with 1% uranyl acetate for 20 s. The grids were washed twice in deionized water and dried on Whatman filter paper. Then, MSCs-EVs were imaged by TEM at 80 kV and then air-dried for 20 min. The ultrastructure and size distribution of EVs were analyzed by NanoSight (Malvern, Malvern, UK), and EV markers (CD9 and TSG101) were measured by Western blotting.
Western Blotting Analysis
The procedures and protocols used for Western blotting analysis were described in previous reports.33,34 Briefly, ischemic tissue from the right cerebrum was collected for total protein extraction, and the protein concentration was measured with BCA kits. Denatured protein samples were diluted with 5× loading buffer. Then, equal amounts of protein extract were loaded in and separated on SDS-PAGE gels using acrylamide gradients. After electrophoresis, the separated proteins were transferred onto PVDF membranes via the wet transfer method. Then, the membranes were incubated at 4 °C overnight (while shaking) with the following diluted primary antibodies: β-actin (1:1000), p-AMPK (1:1000), AMPK (1:1000), p-JAK2 (1:500), JAK2 (1:500), p-STAT3 (1:1000), STAT3 (1:1000), p-NF-κB (1:1000), and NF-κB (1:1000), CD9 (1:500) and TSG101 (1:1000). Horseradish peroxidase-conjugated anti-rabbit or anti-mouse immunoglobulin IgG (1:10,000) was used as a secondary antibody for 1 h incubation at room temperature. The PVDF membranes were washed with TBST solution (3 times for 5 min each). The immunoreactive bands were visualized by enhanced chemiluminescence (ECL) reagents for 3–5 min and exposed to the Tanon Imaging System (Tanon-4600). The relative quantity of proteins was analyzed based on densitometry analysis performed with ImageJ software. The ratio of the gray value of each protein to the gray value of the internal reference (β-actin) was used as the relative protein expression level.
Quantification and Statistical Analysis
The results were analyzed using IBM SPSS software (version 20, IBM, New York, NY, United States) and are expressed as the mean ± standard deviation (SD). Multiple groups were compared using one-way analysis of variance followed by the Bonferroni test for post hoc comparisons. p<0.05 was considered significant.
Results
Characterization of MSCs-EVs
Western blotting analysis of MSCs-EVs showed expression of specific markers of EVs, CD9 and TSG101. Calnexin is an endoplasmic reticulum marker protein and was only rarely present in these EVs (Figure 1A). TEM showed that MSCs-EVs were saucer-shaped double-layer membrane structures with diameters in the range of 50–200 nm (Figure 1B). NanoSight analysis demonstrated that the average diameter of the particles was approximately 120 nm, which is consistent with the basic characteristics of EVs (Figure 1C).
 |
Figure 1 Characterization of MSCs-EVs. (A) Observations of the morphology of MSCs-EVs by transmission electron microscopy (scale bar=100 nm). (B) The expression of EV surface markers (TSG101 and CD9) was analyzed using Western blotting. (C) The size of the MSCs-EVs was analyzed using NanoSight. The experiments were repeated three times. |
MSC-EVs Alleviated Neurologic Deficits and Brain Water Content
Neurological deficit scores and brain water content are shown in Figure 2. The neurological deficit score of the MCAO group was lower than that of the Sham group at 24 h and 48 h (all p<0.001), and MSCs-EVs improved the neurological deficit score compared with that of the MCAO group at 24 h and 48 h (24 h: p<0.05, 48 h: p<0.01; Figure 2A). The brain water content was significantly increased at 24 h after embolization (p<0.001) compared to that in the Sham group, and MSCs-EVs reduced the brain water content at 24 h after embolization compared with that in the MCAO insult group (p<0.01; Figure 2B). These results suggest that MSCs-EVs exert a neuroprotective effect after CIRI.
 |
Figure 2 Neurological deficit scores and brain water content in different rat brain regions at 24 h and 48 h. (A) Neurological function of the MCAO and Sham groups was assessed 24 h and 48 h after MCAO (n = 8). (B) Brain water content in the brains of rats from the MCAO and sham groups at 24 h and 48 h. The data are expressed as the mean ± SD (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001 by ANOVA with Bonferroni test for post hoc comparisons. |
MSCs-EVs Reduced the Volume of Cerebral Infarction
The results of TTC staining showed that normal brain tissue was red, while brain tissue in the ischemic reperfusion area was white. A typical TTC image of cerebral infarction is shown in Figure 3A. The volume of cerebral infarction was significantly increased at 24 h and 48 h after embolization (all p<0.001) compared to that in the Sham group, and MSCs-EVs significantly reduced the volume of cerebral infarction at 24 h and 48 h (all p<0.001) compared with that in the MCAO insult group (Figure 3B). These results indicate that MSCs-EVs reduced the volume of cerebral infarction induced by CIRI in MCAO rats and exerted a neuroprotective effect.
 |
Figure 3 Effects of MSCs-EVs on MCAO-induced cerebral infarction. (A) TTC-stained coronal sections from representative rats from each group at 24 h and 48 h. (B) Quantitative analysis of the cerebral infarct volume. The data are expressed as the mean ± SD (n = 5). ***p < 0.001 by ANOVA with Bonferroni test for post hoc comparisons. |
MSCs-EVs Inhibited Neuropathological Changes
HE and Nissl staining were used to evaluate pathological changes in neurons after CIRI. As shown in Figure 4A, cerebral cortical neurons in the Sham group exhibited normal morphology, clear outlines and uniform cytoplasmic coloration. The nuclei were located in the center of the cells, and the brain tissue was clearly textured. In the ischemia-reperfusion group, the cortical neurons were damaged to varying degrees. The neurons were atrophied, the outlines were blurred, and the cytoplasm was deeply stained. The boundary between the nucleus and the cytoplasm was not clear, the texture of the brain tissue was disordered, and the number of Nissl bodies was obviously decreased. Few apoptotic cells (TUNEL-positive cells) were present in the Sham operation group, while the apoptosis index of the ischemia-reperfusion group was significantly increased. TUNEL-positive cells showed cell body atrophy, irregular shapes, and pyknosis of the nucleus, and were brown under a microscope. The results showed that the number of TUNEL-positive cells was significantly increased at 24 h and 48 h after embolization (all p<0.001) compared to that in the sham group and that MSCs-EVs significantly reduced the number of TUNEL-positive cells at 24 h and 48 h (all p<0.001) compared with that in the MCAO insult group (Figure 4A and B). These results indicate that MSCs-EVs decreased neuronal pathological injury and cell apoptosis in rats after CIRI.
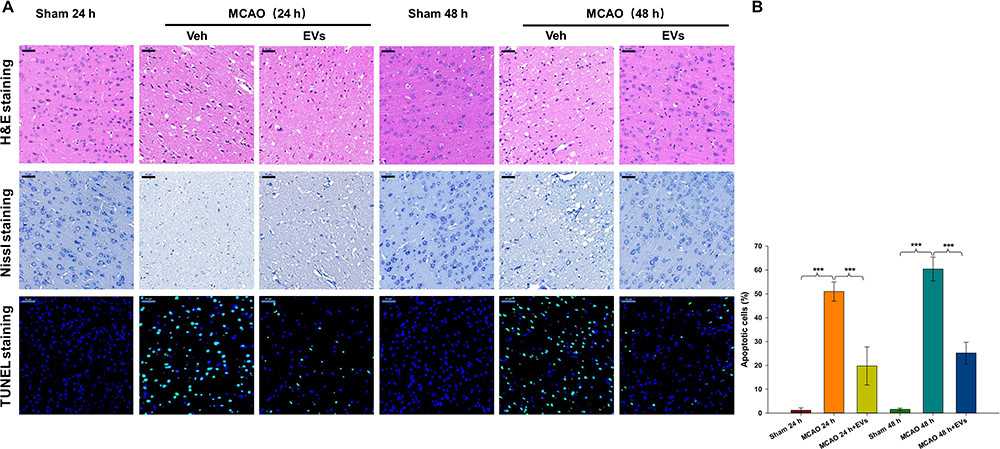 |
Figure 4 Effects of MSCs-EVs on MCAO-induced neuronal pathological changes. (A) Representative HE, Nissl and TUNEL staining in the cortex following MCAO insult. (B) MSCs-EVs effectively reduced apoptosis in cerebral cells after MCAO (scale bar=50 μm). The data are expressed as the mean ± SD (n = 3). ***p < 0.001 by ANOVA with Bonferroni test for post hoc comparisons. |
Changes in AMPK, p-AMPK, JAK2, p-JAK2, STAT3, p-STAT3, NF-κB and p-NF-κB Expression in the Brain Tissues of Rats After Cerebral Ischemia
The current study analyzed the potential MSC-EV-mediated mechanism of apoptosis and neuronal pathological injury in neuronal cells. The expression of p-AMPK in the lesioned cortex was increased in the MCAO group compared with the Sham group, and MSCs-EVs upregulated p-AMPK expression compared with that in the MCAO group at 24 h (p<0.001; Figure 5A and C). Compared with those in the Sham group, the expression levels of p-JAK2 in the cortex were increased at 24 h post-MCAO (p<0.001), and MSCs-EVs significantly decreased the expression of p-JAK2 in the cortex at 24 h (p<0.001; Figure 5A and C). Compared with those in the Sham group, the expression levels of p-STAT3 in the cortex were increased at 24 h and 48 h post-MCAO (all p<0.001). MSCs-EVs significantly decreased the expression of p-STAT3 in the cortex at 48 h (p<0.001; Figure 5B and D) compared with that observed in the MCAO group. Compared with those in the Sham group, the expression levels of p-NF-κB in the cortex were increased at 24 h and 48 h post-MCAO (all p<0.05), and MSCs-EVs significantly decreased the expression of p-NF-κB in the cortex at 48 h (p<0.01; Figure 5B and D) compared with that observed in the MCAO group. These results indicate that MSCs-EVs reduced neuropathological damage and decreased nerve cell apoptosis by upregulating the AMPK-mediated JAK2/STAT3/NF-κB signaling pathway.
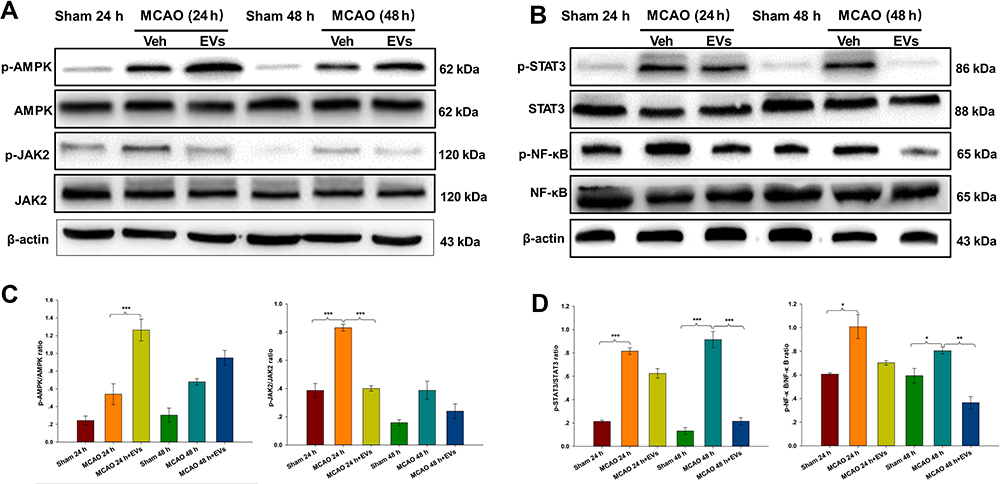 |
Figure 5 MSCs-EVs activated the phosphorylation of AMPK and downregulated the JAK2/STAT3/NF-κB signaling pathway. (A and C) Western blot analysis of p-AMPK, AMPK, p-JAK2, JAK2, and β-actin and the relative expression of the target proteins in brain tissue homogenates. (B and D) Western blot analysis of p-STAT3, STAT3, p-NF-κB, NF-κB, and β-actin and the relative expression of the target proteins in brain tissue homogenates. The data are expressed as the mean ± SD (n = 3). *p < 0.05, **p < 0.01, ***p < 0.001 by ANOVA with Bonferroni test for post hoc comparisons. |
An AMPK Signaling Pathway Blocker (Compound C, CC) Reversed the Neuroprotective Effect of MSCs-EVs on MCAO-Induced Brain Damage
Representative TTC staining images of the cortex following MCAO insult are shown in Figure 6A. MSCs-EVs significantly reduced the volume of cerebral infarction at 24 h and 48 h (all p<0.001) compared with that in the MCAO insult group (Figure 6B). TTC staining showed that the inhibition of the AMPK signaling pathway by CC reversed the neuroprotective effect of MSCs-EVs on the MCAO-induced brain and increased the cerebral infarct area. The volume of cerebral infarction was increased 48 h (p<0.01) after the administration of CC compared to that in the MSCs-EVs group (Figure 6B).
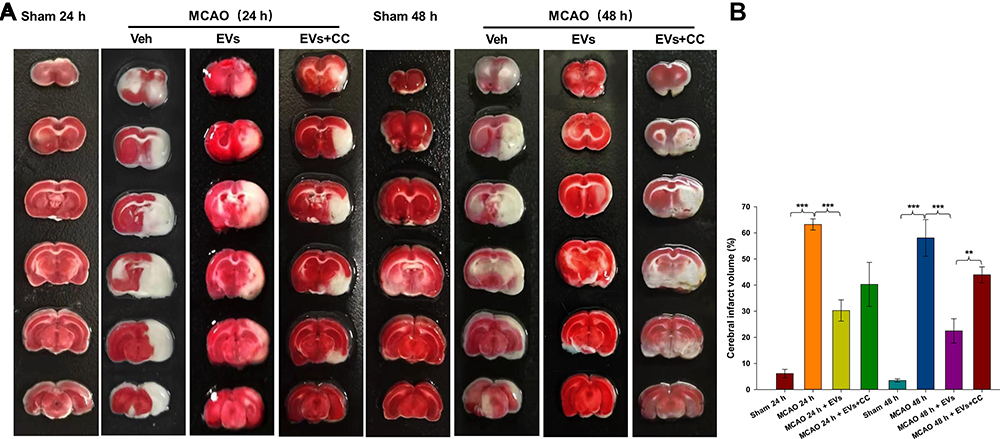 |
Figure 6 Effects of compound C (CC) on MCAO-induced cerebral infarction. (A) TTC-stained coronal sections from representative rats from each group at 24 h and 48 h. (B) Quantitative analysis of the cerebral infarct volume. The data are expressed as the mean ± SD (n = 5). **p < 0.01, ***p < 0.001 by ANOVA with Bonferroni test for post hoc comparisons. |
Effects of Compound C on the Expression of AMPK and p-AMPK
As indicated in Figure 7A, the expression of p-AMPK in the lesioned cortex was significantly increased in the MSCs-EVs group compared with the MCAO group at 24 h (p<0.05) and 48 h (p<0.001), and the expression of p-AMPK was increased 48 h (p<0.001) after the administration of CC compared to that in the MSCs-EVs group (Figure 7B). The results confirmed that MSCs-EVs reduced the release of inflammatory factors after cerebral ischemia by activating the AMPK signaling pathway.
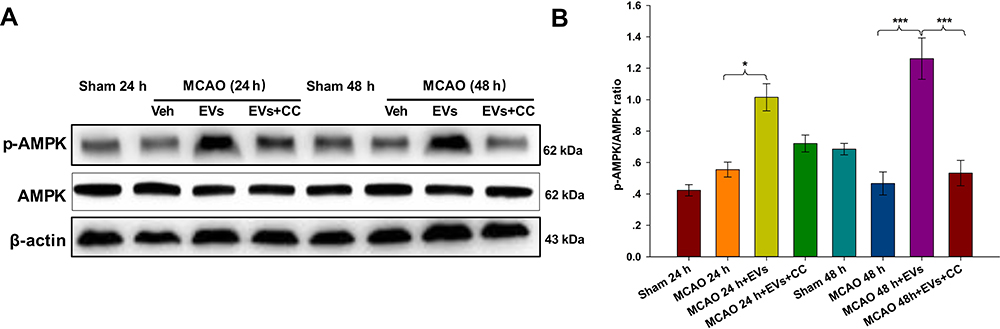 |
Figure 7 Effects of compound C (CC) on the expression of AMPK and p-AMPK. The non-pretreated MCAO group before operation; the MCAO + MSCs-EVs group (100 µg of MSCs-EVs suspended in 0.5 mL PBS was injected via the caudal vein within 10 min after MCAO); and the MCAO + MSCs-EVs + CC group (10 μL CC was injected intracerebroventricularly 30 min before the MCAO operation, and 100 µg of MSCs-EVs suspended in 0.5 mL PBS was injected via the caudal vein within 10 min after MCAO). There were 3 rats in each group. (A) Western blot analysis indicated significantly higher p-AMPK expression in the MSCs-EVs group than in the vehicle group. These effects were partially reversed by CC. (B) Quantitative analysis of the cerebral infarct volume. The data are expressed as the mean ± SD (n = 3). *p < 0.05, ***p < 0.001 by ANOVA with Bonferroni test for post hoc comparisons. |
Discussion
Apoptosis is the main causative factor in the pathogenesis of ischemic stroke. Reducing neuronal apoptosis is essential for suppressing stroke-related injuries. In this study, we investigated MSCs-EV-induced neuroprotection, which attenuates neuronal apoptosis and CIRI as well as its mechanisms and signaling pathways. We also investigated the relationship between AMPK and JAK2/STAT3/NF-κB signaling mediated by MSCs-EVs. The results showed that during CIRI, MSCs-EVs activated p-AMPK, inhibited JAK2/STAT3/NF-κB, increased MCAO-induced neurological impairment scores, decreased brain water content and cerebral infarction volume, attenuated pathological damage, and reduced apoptosis. We believe that MSCs-EV-induced neuroprotection may be achieved by activating AMPK and reducing the JAK2/STAT3/NF-κB pathway, thereby improving the damage caused by cerebral ischemia.
AMPK is mainly responsible for peripheral energy balance, fat metabolism and ATP conservation and synthesis.35 AMPK has frequently been referred to as the “metabolic master switch”, and it has the ability to be rapidly activated by anti-inflammatory stimuli.13 When the cell energy supply is reduced, AMPK can be activated to enhance energy production14 and represents a potential protective mechanism in the early stages of cerebral ischemia.36
The JAK/STAT family is an important intracellular signal transduction pathway mediated by cytokines and oxidative stress.37 As the most conserved members of the family, JAK2/STAT3 are closely related to CIRI and can be activated in the early stage of cerebral infarction to induce the expression of proinflammatory factors to aggravate tissue damage.38–40 Studies have shown that the aberrant activation of the JAK2/STAT3 pathway is involved in neuroinflammatory injury after ischemic stroke41 and that MCAO injury increases STAT3 protein levels in cortex microglia.42
Among the many measures for stroke treatment, MSCs-EVs have great potential in the field of biotherapy due to their advantages, such as good circulatory stability, good biocompatibility, low toxicity and low immunogenicity. Meanwhile, there are no ethical restrictions on the use of EVs for noncell therapies, and the dosage and starting and ending times of EVs are more controllable. Additionally, they have lower risks of vessel blockage and microvascular thrombosis, and are easy to preserve, produce on a large scale and genetically modify.43 MSCs-EVs maintain biological activity similar to that of their parental cells, allowing them to home to the site of inflammation to regulate immune responses and participate in tissue repair.44,45 A key characteristic of EVs is their ability to penetrate the blood-brain barrier and release related RNAs and proteins into the central nervous system.46 Studies have shown that MSCs-EVs can exert neuroprotective effects by promoting the growth and repair of blood vessels, repairing and regenerating nerve cells, and inhibiting apoptosis of nerve cells after stroke.47 Studies have shown that EVs can stimulate the growth of nerve cells,48 induce neuronal proliferation,49 increase the number of axons,50 regulate the peripheral immune response,51 reduce the area of infarction after cerebral ischemia, improve brain function,12 and promote axonal growth and white matter recovery.52 At present, encouraging results have been obtained in preclinical studies on EVs. Therefore, from a clinical perspective, the application of EVs for the treatment of cerebral ischemia has attracted much attention. To date, although there are no clinical trials on EV transplantation for ischemic stroke patients, there is growing interest in EV-based therapies. This study serves as a platform for the development of protocols for the use of EVs in clinical trials and provides new ideas for the treatment of cerebral ischemia reperfusion.
In conclusion, MSCs-EVs can reduce neuronal apoptosis induced by CIRI. The administration of CC abolishes the protective effects of MSCs-EVs, suggesting that the phosphorylation of AMPK and JAK2/STAT3/NF-κB is closely associated with the neuroprotective signaling pathway activated by MSCs-EVs. Thus, the regulation of the AMPK and JAK2/STAT3/NF-κB pathways may also be a potential approach for subsequently reducing apoptosis and providing neuroprotection.
Conclusions
In summary, we found that MSCs-EVs reduced apoptosis and alleviated tissue damage caused by MCAO in rats. This protective mechanism of MSCs-EVs may occur via the regulation of the AMPK and JAK2/STAT3/NF-κB signaling pathways. Our study indicated that MSCs-EVs have a neuroprotective effect in MCAO.
Abbreviations
AMPK, AMP-activated protein kinase; ECA, external carotid artery; EVs, extracellular vesicles; JAK2/STAT3, Janus kinase2/signal transducer and activator of transcription3; HE, hematoxylin and eosin staining; ICA, internal carotid artery; MSCs-EVs, extracellular vesicles derived from mesenchymal stromal cells; MCAO, middle cerebral artery occlusion; NF-κB, nuclear factor kappa B; RT-PCR, reverse transcription-polymerase chain reaction; TEM, transmission electron microscopy; TTC, 2,3,5-triphenyltetrazolium chloride; TUNEL, terminal-deoxynucleotidyl transferase mediated nick end labeling.
Disclosure
No potential conflict of interest was reported by the authors.
References
1. Zhou M, Wang H, Zhu J, et al. Cause-specific mortality for 240 causes in China during 1990–2013: a systematic subnational analysis for the global burden of disease study 2013. Lancet. 2016;387(10015):251–272.
2. Zevallos J, Santiago F, Gonzalez J, et al. Burden of stroke in puerto rico. Int J Stroke. 2015;10(1):117–119. doi:10.1111/ijs.12350
3. Pantoni L, Sarti C, Inzitari D. Cytokines and cell adhesion molecules in cerebral ischemia: experimental bases and therapeutic perspectives. Arterioscler Thromb Vasc Biol. 1998;18(4):503–513. doi:10.1161/01.ATV.18.4.503
4. Zhao J, Zhao X, Tian J, et al. Theanine attenuates hippocampus damage of rat cerebral ischemia-reperfusion injury by inhibiting HO-1 expression and activating ERK1/2 pathway. Life Sci. 2020;241:117160. doi:10.1016/j.lfs.2019.117160
5. Johann S, Beyer C. Neuroprotection by gonadal steroid hormones in acute brain damage requires cooperation with astroglia and microglia. J Steroid Biochem Mol Biol. 2013;137:71–81. doi:10.1016/j.jsbmb.2012.11.006
6. Thery C, Zitvogel L, Amigorena S. Exosomes: composition, biogenesis and function. Nat Rev Immunol. 2002;2(8):569–579. doi:10.1038/nri855
7. Wei Z, Qiao S, Zhao J, et al. miRNA-181a over-expression in mesenchymal stem cell-derived exosomes influenced inflammatory response after myocardial ischemia-reperfusion injury. Life Sci. 2019;232:116632. doi:10.1016/j.lfs.2019.116632
8. Mardpour S, Ghanian MH, Sadeghi-Abandansari H, et al. Hydrogel-mediated sustained systemic delivery of mesenchymal stem cell-derived extracellular vesicles improves hepatic regeneration in chronic liver failure. ACS Appl Mater Interfaces. 2019;11(41):37421–37433. doi:10.1021/acsami.9b10126
9. Carrasco E, Soto-Heredero G, Mittelbrunn M. The role of extracellular vesicles in cutaneous remodeling and hair follicle dynamics. Int J Mol Sci. 2019;20(11):E2758. doi:10.3390/ijms20112758
10. Liew LC, Katsuda T, Gailhouste L, Nakagama H, Ochiya T. Mesenchymal stem cell-derived extracellular vesicles: a glimmer of hope in treating Alzheimer’s disease. Int Immunol. 2017;29(1):11–19. doi:10.1093/intimm/dxx002
11. Zhou X, Chu X, Yuan H, et al. Mesenchymal stem cell derived EVs mediate neuroprotection after spinal cord injury in rats via the microRNA-21-5p/FasL gene axis. Biomed Pharmacother. 2019;115:108818. doi:10.1016/j.biopha.2019.108818
12. Ophelders DR, Wolfs TG, Jellema RK, et al. Mesenchymal stromal cell-derived extracellular vesicles protect the fetal brain after hypoxia-ischemia. Stem Cells Transl Med. 2016;5(6):754–763. doi:10.5966/sctm.2015-0197
13. Sag D, Carling D, Stout RD, Suttles J. Adenosine 5ʹ-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol. 2008;181(12):8633–8641. doi:10.4049/jimmunol.181.12.8633
14. Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–262. doi:10.1038/nrm3311
15. Qiu J, Wang M, Zhang J, et al. The neuroprotection of Sinomenine against ischemic stroke in mice by suppressing NLRP3 inflammasome via AMPK signaling. Int Immunopharmacol. 2016;40:492–500. doi:10.1016/j.intimp.2016.09.024
16. Yu J, Wang W-N, Matei N, et al. Ezetimibe attenuates oxidative stress and neuroinflammation via the AMPK/Nrf2/TXNIP pathway after MCAO in rats. Oxid Med Cell Longev. 2020;2020:4717258. doi:10.1155/2020/4717258
17. Song F, Zeng K, Liao L, Yu Q, Tu P, Wang X. Schizandrin a inhibits microglia-mediated neuroninflammation through inhibiting TRAF6-NF-kappaB and Jak2-Stat3 signaling pathways. PLoS One. 2016;11(2):e0149991. doi:10.1371/journal.pone.0149991
18. Li Y, Sun W, Han N, Zou Y, Yin D. Curcumin inhibits proliferation, migration, invasion and promotes apoptosis of retinoblastoma cell lines through modulation of miR-99a and JAK/STAT pathway. BMC Cancer. 2018;18(1):1230. doi:10.1186/s12885-018-5130-y
19. Zeng KW, Wang S, Dong X, Jiang Y, Tu PF. Sesquiterpene dimer (DSF-52) from Artemisia argyi inhibits microglia-mediated neuroinflammation via suppression of NF-kappaB, JNK/p38 MAPKs and Jak2/Stat3 signaling pathways. Phytomedicine. 2014;21(3):298–306. doi:10.1016/j.phymed.2013.08.016
20. Jere SW, Abrahamse H, Houreld NN. The JAK/STAT signaling pathway and photobiomodulation in chronic wound healing. Cytokine Growth Factor Rev. 2017;38:73–79. doi:10.1016/j.cytogfr.2017.10.001
21. Zhao J, Li G, Zhang Y, Su X, Hang C. The potential role of JAK2/STAT3 pathway on the anti-apoptotic effect of recombinant human erythropoietin (rhEPO) after experimental traumatic brain injury of rats. Cytokine. 2011;56(2):343–350. doi:10.1016/j.cyto.2011.07.018
22. Hu GQ, Du X, Li YJ, Gao XQ, Chen BQ, Yu L. Inhibition of cerebral ischemia/reperfusion injury-induced apoptosis: nicotiflorin and JAK2/STAT3 pathway. Neural Regen Res. 2017;12(1):96–102. doi:10.4103/1673-5374.198992
23. Lawrence T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb Perspect Biol. 2009;1(6):a001651. doi:10.1101/cshperspect.a001651
24. Lu M, Wang S, Han X, Lv D. Butein inhibits NF-kappaB activation and reduces infiltration of inflammatory cells and apoptosis after spinal cord injury in rats. Neurosci Lett. 2013;542:87–91. doi:10.1016/j.neulet.2013.03.004
25. Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20(1):84–91. doi:10.1161/01.STR.20.1.84
26. Dong H, Zhou W, Xin J, et al. Salvinorin A moderates postischemic brain injury by preserving endothelial mitochondrial function via AMPK/Mfn2 activation. Exp Neurol. 2019;322:113045. doi:10.1016/j.expneurol.2019.113045
27. Shao A, Guo S, Tu S, et al. Astragaloside IV alleviates early brain injury following experimental subarachnoid hemorrhage in rats. Int J Med Sci. 2014;11(10):1073–1081. doi:10.7150/ijms.9282
28. Zhang YM, Xu H, Sun H, Chen SH, Wang FM. Electroacupuncture treatment improves neurological function associated with regulation of tight junction proteins in rats with cerebral ischemia reperfusion injury. Evid Based Complement Alternat Med. 2014;(2014):989340. doi:10.1155/2014/105186
29. Liu L, Chen H, Jin J, et al. Melatonin ameliorates cerebral ischemia/reperfusion injury through SIRT3 activation. Life Sci. 2019;239:117036. doi:10.1016/j.lfs.2019.117036
30. Phinney DG. Isolation of mesenchymal stem cells from murine bone marrow by immunodepletion. Methods Mol Biol. 2008;449:171–186. doi:10.1007/978-1-60327-169-1_12
31. Dabrowska S, Andrzejewska A, Strzemecki D, Muraca M, Janowski M, Lukomska B. Human bone marrow mesenchymal stem cell-derived extracellular vesicles attenuate neuroinflammation evoked by focal brain injury in rats. J Neuroinflammation. 2019;16(1):216. doi:10.1186/s12974-019-1602-5
32. Wei F, Li Z, Crawford R, Xiao Y, Zhou Y. Immunoregulatory role of exosomes derived from differentiating mesenchymal stromal cells on inflammation and osteogenesis. J Tissue Eng Regen Med. 2019;13(11):1978–1991. doi:10.1002/term.2947
33. Chang CL, Sung PH, Chen KH, et al. Adipose-derived mesenchymal stem cell-derived exosomes alleviate overwhelming systemic inflammatory reaction and organ damage and improve outcome in rat sepsis syndrome. Am J Transl Res. 2018;10(4):1053–1070.
34. Day YJ, Chen KH, Chen YL, et al. Preactivated and disaggregated shape-changed platelets protected against acute respiratory distress syndrome complicated by sepsis through inflammation suppression. Shock. 2016;46(5):575–586. doi:10.1097/SHK.0000000000000617
35. Ma C, Wang X, Xu T, et al. Qingkailing injection ameliorates cerebral ischemia-reperfusion injury and modulates the AMPK/NLRP3 inflammasome signalling pathway. BMC Complement Altern Med. 2019;19(1):320. doi:10.1186/s12906-019-2703-5
36. Shen P, Hou S, Zhu M, Zhao M, Ouyang Y, Feng J. Cortical spreading depression preconditioning mediates neuroprotection against ischemic stroke by inducing AMP-activated protein kinase-dependent autophagy in a rat cerebral ischemic/reperfusion injury model. J Neurochem. 2017;140(5):799–813. doi:10.1111/jnc.13922
37. Wu Y, Xu J, Xu J, Zheng W, Chen Q, Jiao D. Study on the mechanism of JAK2/STAT3 signaling pathway-mediated inflammatory reaction after cerebral ischemia. Mol Med Rep. 2018;17(4):5007–5012. doi:10.3892/mmr.2018.8477
38. Satriotomo I, Bowen KK, Vemuganti R. JAK2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J Neurochem. 2006;98(5):1353–1368. doi:10.1111/j.1471-4159.2006.04051.x
39. Xie HF, Xu RX, Wei JP, Jiang XD, Liu ZH. P-JAK2 and P-STAT3 protein expression and cell apoptosis following focal cerebral ischemia-reperfusion injury in rats. Nan Fang Yi Ke Da Xue Xue Bao. 2007;27(2):208–11, 218.
40. Planas AM, Gorina R, Chamorro A. Signalling pathways mediating inflammatory responses in brain ischaemia. Biochem Soc Trans. 2006;34(Pt 6):1267–1270. doi:10.1042/BST0341267
41. Alhadidi Q, Shah ZA. Cofilin mediates LPS-induced microglial cell activation and associated neurotoxicity through activation of NF-kappaB and JAK-STAT pathway. Mol Neurobiol. 2018;55(2):1676–1691. doi:10.1007/s12035-017-0432-7
42. Qin C, Fan WH, Liu Q, et al. Fingolimod protects against ischemic white matter damage by modulating microglia toward M2 polarization via STAT3 pathway. Stroke. 2017;48(12):3336–3346. doi:10.1161/STROKEAHA.117.018505
43. Dabrowska S, Andrzejewska A, Lukomska B, Janowski M. Neuroinflammation as a target for treatment of stroke using mesenchymal stem cells and extracellular vesicles. J Neuroinflammation. 2019;16(1):178. doi:10.1186/s12974-019-1571-8
44. Tetta C, Bruno S, Fonsato V, Deregibus MC, Camussi G. The role of microvesicles in tissue repair. Organogenesis. 2011;7(2):105–115. doi:10.4161/org.7.2.15782
45. Bruno S, Grange C, Deregibus MC, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol. 2009;20(5):1053–1067. doi:10.1681/ASN.2008070798
46. Blandford SN, Galloway DA, Moore CS. The roles of extracellular vesicle microRNAs in the central nervous system. Glia. 2018;66(11):2267–2278. doi:10.1002/glia.23445
47. Long Q, Upadhya D, Hattiangady B, et al. Intranasal MSC-derived A1-exosomes ease inflammation, and prevent abnormal neurogenesis and memory dysfunction after status epilepticus. Proc Natl Acad Sci U S A. 2017;114(17):E3536–E3545. doi:10.1073/pnas.1703920114
48. Zhang Y, Chopp M, Liu XS, et al. Exosomes derived from mesenchymal stromal cells promote axonal growth of cortical neurons. Mol Neurobiol. 2017;54(4):2659–2673. doi:10.1007/s12035-016-9851-0
49. El Bassit G, Patel RS, Carter G, et al. MALAT1 in human adipose stem cells modulates survival and alternative splicing of PKCdeltaII in HT22 cells. Endocrinology. 2017;158(1):183–195. doi:10.1210/en.2016-1819
50. Xin H, Li Y, Liu Z, et al. MiR-133b promotes neural plasticity and functional recovery after treatment of stroke with multipotent mesenchymal stromal cells in rats via transfer of exosome-enriched extracellular particles. Stem Cells. 2013;31(12):2737–2746. doi:10.1002/stem.1409
51. Doeppner TR, Herz J, Gorgens A, et al. Extracellular vesicles improve post-stroke neuroregeneration and prevent postischemic immunosuppression. Stem Cells Transl Med. 2015;4(10):1131–1143. doi:10.5966/sctm.2015-0078
52. Otero-Ortega L, Laso-Garcia F, Gomez-de Frutos MD, et al. White matter repair after extracellular vesicles administration in an experimental animal model of subcortical stroke. Sci Rep. 2017;7:44433. doi:10.1038/srep44433
 © 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2020 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.