Back to Journals » International Medical Case Reports Journal » Volume 14
Neuromyelitis Optica Spectrum Disorder: A Case Report
Authors Yaregal S, Bekele N ![]() , Gebrewold Y
, Gebrewold Y ![]() , Tadesse A
, Tadesse A ![]()
Received 18 August 2021
Accepted for publication 14 September 2021
Published 21 September 2021 Volume 2021:14 Pages 643—648
DOI https://doi.org/10.2147/IMCRJ.S334362
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Samson Yaregal,1 Nebiyu Bekele,1 Yonathan Gebrewold,2 Abilo Tadesse1
1Department of Internal Medicine, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia; 2Department of Radiology, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia
Correspondence: Abilo Tadesse
Department of Internal Medicine, College of Medicine and Health Sciences, University of Gondar, Gondar, Ethiopia
Email [email protected]
Background: Neuromyelitis optica spectrum disorder is an autoimmune, astrocytopathic CNS disease, mainly involving the optic nerves, spinal cord, and brain stem regions. The “International Panel for NMOSD Diagnosis (IPND) Diagnostic Criteria” was implemented to define the disorder.
Case Presentation: A 38-year-old patient presented with visual loss of eight months’ duration and weakness of the lower extremities of one week’s duration. The patient had bilateral optic atrophy on fundoscopic examination, and flaccid paraplegia with sensory loss below T4 level. Serological tests for syphilis, HIV infection, and SLE were negative. Aquaporin-4 antibody test was not done due to limited clinical setup. T2-spine MRI revealed long central thoracic segment (T3 to T6) hyperintense lesion with mild cord expansion. Long segment central canal dilation (syrinx) was noted in the cord proximal to the lesion. Diagnosis of opticospinal variant, NMOSD was made using IPND diagnostic criteria. The patient was started on dexamethasone 50 mg, IV, four times daily (QID) for one week, and changed to prednisolone 1 mg/kg (40 mg) PO daily for one month, to be tapered over three-to-six months. The patient was scheduled to initiate azathioprine 50 mg PO twice daily.
Conclusion: The case emphasizes the existence of neuromyelitis optica spectrum disorder in clinical settings of the developing world. High index of suspicion of this rare disease is required to avoid delayed diagnosis and treatment.
Keywords: neuromyelitis optica spectrum disorder, Northwest Ethiopia
Background
Neuromyelitis optica spectrum disorder (NMOSD) is an autoimmune astrocytopathic disorder characterized by optic neuritis (ON), transverse myelitis (LETM), and brain stem lesions.1,2 Autopsied case reports of “neuromyelite optique aigue” were first reported by Eugene Devic in 1894. Since then, opticospinal variant, NMOSD has also been designated as Devic’s disease. Discovery of aquaporin-4 antibody, and distinct pathophysiological, clinical, and MRI findings of NMOSD have led to it being recognized as a separate disease from multiple sclerosis.1–4 Aquaporin-4 antibody is a specific biomarker for NMOSD and is considered as an additional criterion supporting the diagnosis.5 Immunosuppressant drugs and immunotherapies are the treatment of choice for NMOSD with variable outcomes.7 Here, we discuss this rare clinical disorder with available literature.
Case Presentation
A 38-year-old patient presented to the ophthalmology clinic, University of Gondar hospital with a complaint of progressive bilateral visual loss of four months' duration associated with throbbing, temporal headache. No history of reddening of eyes, vomiting or neck stiffness. On ophthalmologic examination, the patient was able to count fingers at three meters. A fundoscopic examination showed pale optic discs with no background changes on both eyes, which indicated bilateral optic atrophy. Brain MRI revealed no abnormality. Betaxolol eye drops (0.5%) were given but there was no improvement in vision. The patient was referred to an outpatient medical clinic and was evaluated by a neurologist after presenting with a complaint of headache and bilateral visual loss of five months' duration. Bilateral optic atrophy was detected on fundoscopic examination. There was no history of memory deficit, other cranial nerve palsies, sensory or motor deficit, or bladder or bowel dysfunction. There was normal CSF chemistry and cellularity. Repeated brain MR imaging revealed no abnormality. A diagnosis of idiopathic intracranial hypertension was made, and the patient was put on acetazolamide 500 mg PO twice daily and therapeutic lumbar puncture as required. The patient claimed to have a slight improvement in vision with this therapy. Three months later, the patient was admitted to a medical ward, University of Gondar hospital after presenting with weakness of lower extremities of a week's duration associated with numbness and tingling sensation, and bladder and bowel dysfunction. The patient’s vision in both eyes deteriorated during episodes of lower extremity weakness. On physical examination, the patient was acutely sick. Vital signs were: blood pressure (BP)=100/60 mmHg, pulse rate (PR)=90 beats per minute, respiratory rate (RR)=24 breaths per minute, and temperature (T0)=37.2°C. There were pertinent findings on neurologic examination. The patient was conscious, and oriented in time, place, and person. No objective memory deficit. The patient could count fingers at two meters. There was no other cranial nerve palsy. Flaccid paraplegia with sensory loss below T4 was detected. No meningeal irritation signs. Findings on other systems were unremarkable. On investigation, complete blood count, urinalysis, and serum liver biochemical and renal function tests were within normal limits. Serological tests for HIV, hepatitis B and hepatitis C viruses were negative. The serum VDRL test was negative. Lumbar puncture revealed elevated CSF protein (145 mg/dL), while CSF glucose was normal (55 mg/dL). CSF stains showed no cells or organisms. The CSF VDRL test was negative. Serum aquaporin-4 (AQP4)antibody test was not done due to limited clinical setup. The chestX-ray showed no abnormality. T2-spine MRI revealed long central thoracic segment (T3 to T6) hyperintense lesion with mild cord expansion. Long segment central canal dilation (syrinx) was noted in the cord proximal to the lesion (Figures 1–3). A diagnosis of neuromyelitis optica spectrum disorder (opticospinal variant) was made using the “International Panel for NMOSD Diagnosis (IPND) Diagnostic Criteria” (Table 1). The patient was started on dexamethasone 50 mg IV QID for a week and changed to prednisolone 1mg/kg (40mg) PO daily for one month, and tapered over three-to-six months. The patient was scheduled to initiate azathioprine, 50 mg, PO twice daily. Unfractionated heparin 5000 IU SC twice daily for two weeks and early physiotherapy were initiated. The patient was discharged after two weeks in the medical ward, and was referred to a neurology follow-up clinic. The patient never showed up at the follow-up clinic after discharge.
 |
Table 1 The 2015 International Panel for NMOSD Diagnostic Criteria for Adult Patients |
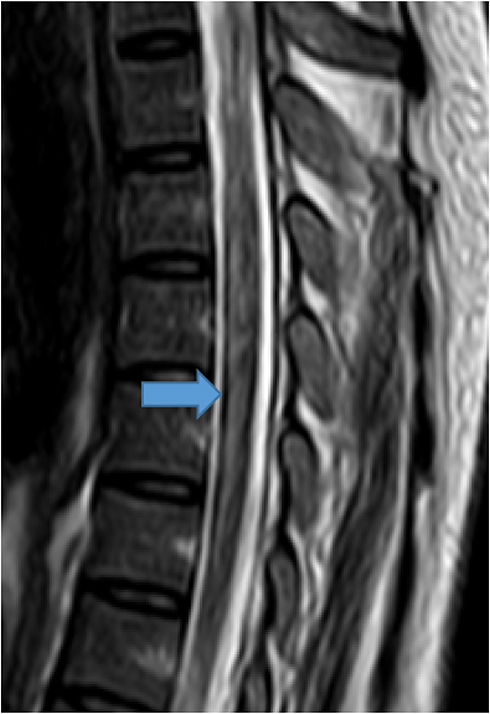 |
Figure 1 T2-weighted mid-sagittal image centered at T3. Long contiguous segment (T3 to T6) central T2 hyperintense lesion mildly expanding the cord. |
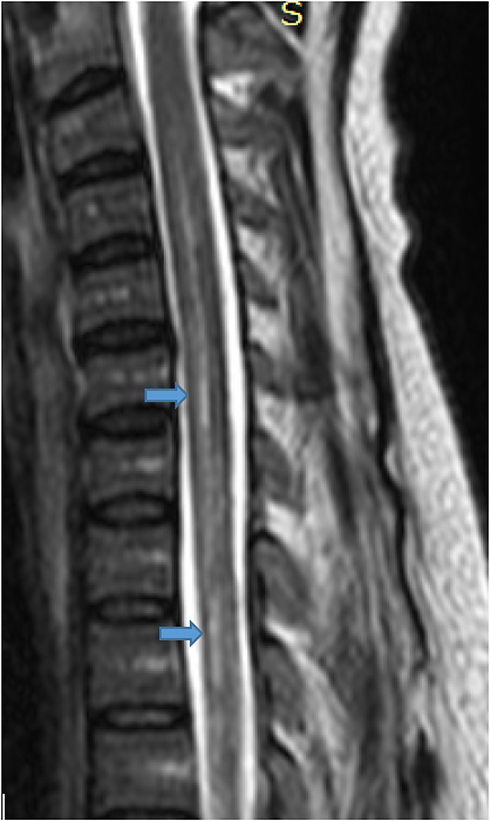 |
Figure 2 T2-weighted mid-sagittal image proximal to the level on previous image shows long segment expansion of the central canal (syrinx). |
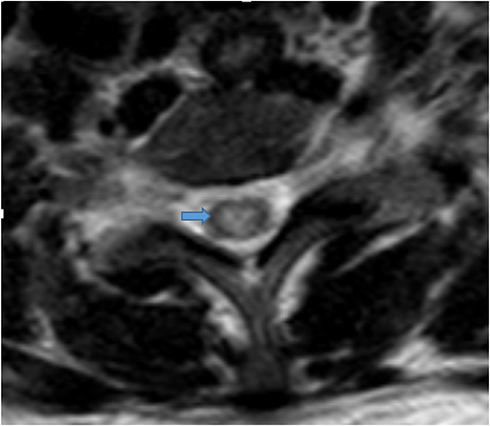 |
Figure 3 Axial T2-weighted image at the level of T3 revealed central cord hyperintensity. |
Discussion
Neuromyelitis optica spectrum disorder is an autoimmune disease of the central nervous system with a predilection for the optic nerves, spinal cord, and brain stem regions. The epicenter of autoimmunity is defined by IgG antibody to aquaporin-4, a water channel protein in astrocyte foot processes of CNS structures. Serum AQP4-IgG is detected in 80% of patients with NMOSD. Half of AQP4-IgG negative NMOSD are categorized under myelin oligodendrocyte glycoprotein (MOG)-antibody positive oligodendrocytopathic NMOSD. Details of double-negative NMOSD remain to be explained. Complement and antibody mediated cellular toxicities are the immunopathologic drivers of NMOSD.1,2 Epidemiological frequencies of NMOSD have shown peaks in middle-aged adults and among those of non-White descent.6 Clinical syndromes in NMOSD include bilateral optic neuritis, longitudinally extensive transverse myelitis, area postrema syndrome, acute brain stem syndrome, diencephalic syndrome, and symptomatic cerebral syndrome. Area postrema syndrome presents with intractable hiccups, nausea, and vomiting. Narcolepsy, insomnia, and inappropriate diuresis are features of diencephalic syndrome. Acute brain stem syndrome presents with cranial nerve palsy, long tract signs, and ataxia. Acute confusional state, delirium, and seizures are features of symptomatic cerebral syndrome.1–4 The patient fulfilled the diagnostic criteria for “NMOSD with unknown AQP4-IgG” as evidenced by the presence of two core clinical characteristics such as optic neuritis and acute myelitis. It has shown dissemination in space with two sites. The T2-spine MR image revealed LETM with spinal lesion extending ≥3 contiguous vertebral segments (Table 1).5 Differential diagnoses of the case include multiple sclerosis, systemic lupus erythematosus (SLE), neurosarcoidosis, and subacute combined neurodegeneration (SCND). Patchy, peripheral, short spinal segment lesions are a common finding in multiple sclerosis.3,4 The lack of other clinical features of SLE and negative serological test for ANA exclude opticospinal lupus. Normal chest X-ray finding does not favor sarcoidosis. Absence of risk factors for vitamin B12 deficiency like malabsorption of any cause makes SCND less likely. Acute attacks of NMOSD are usually treated with intravenous methylprednisolone. Therapeutic plasma exchange is considered in intravenous steroid refractory cases. Maintenance therapy with immunosuppressive drugs such as oral prednisone, azathioprine, mycophenolate mofetil and rituximab has shown benefit in reducing subsequent relapses. Inebilizumab, eculizumab, and satralizumab are approved immunotherapies to treat NMOSD and prevent relapses, and reduce burden of accrual neurological deficits. Sivelestat, ublituximab, ravulizumab, and aquaporumab are newer biologics under clinical evaluation.7 In Ethiopia, prednisolone, methotrexate, and azathioprine are readily available to treat NMOSD, while mycophenolate mofetil and rituximab are accessible on an “on and off” basis. The newer novel immunotherapies are not available.
Three cases of neuromyelitis optica have been reported in Ethiopian medical literature. Summary of case presentations, treatment regimens and their outcomes were mentioned as follows.8–10
Case 1
A 24-year-old female patient from Southern Ethiopia presented with left eye visual loss and recurrent quadriparesis of five years' duration. The patient was diagnosed with acute myelitis with longitudinally extensive transverse myelitis (LETM) and optic neuritis. A T2-brain MRI showed no abnormality. T2-spine MRI revealed central hyperintense lesions from C1 to C7 vertebral segments. Serological tests for syphilis (VDRL), HIV infection, and SLE (ANA) were negative. Serum aquaporin-4 antibody test was not done. The patient was initially treated with intravenous methylprednisolone for five days and maintained on oral prednisolone and azathioprine for six months. The patient regained remarkable strength in upper and lower extremities with left eye visual loss.8
Case 2
A 32-year-old female patient from central Ethiopia presented with right eye visual impairment with recurrent paraparesis of two years' duration. The patient was diagnosed with optic neuritis and acute myelitis with LETM. T2-brain MRI showed an atrophied right optic nerve. T2-spine MRI showed central hyperintense lesions from T4 to T7 vertebral segments. Serum aquaporin-4 antibody was positive. Serological tests for syphilis (VDRL), HIV infection, and SLE (ANA) were negative. The patient was treated with intravenous methylprednisolone for five days and maintained with oral prednisolone and azathioprine for four months. The patient was fully functional with residual right eye visual impairment.9
Case 3
A 41-year-old female patient from central Ethiopia presented with left eye visual impairment and recurrent paraparesis of seven years' duration. She was diagnosed with left eye visual impairment and acute myelitis with LETM. T2-brain MRI showed hyperintense lesions within the deep white matter of frontal and parietal lobes. T2-spine MRI revealed a central hyperintense lesion extending from C3 to T7 vertebral segments. Lumbar puncture revealed normal findings. Serological tests for aquaporin-4, syphilis, HIV infection, and SLE were not done.10
All these reported cases were opticospinal variants of NMOSD, which was easy to recognize without serological test (AQP-4 antibody). Other variants of NMOSD might have been underreported due to lack of serological test and limited clinical exposure.
There are few reported cases of NMOSD in sub-Saharan Africa. Sixteen cases of NMOSD were detected in the Senegalese cohort. Average age of patients was 30 years. Ten patients presented with opticospinal variant, the rest with isolated transverse myelitis. Ten patients had positive AQP-4 antibody tests. Favorable clinical outcome was documented after three months of systemic corticosteroid and azathioprine therapy.11 Another report from East Africa documented 11 cases of NMOSD. Average age at presentation was 30 years. Eight patients presented with opticospinal variant, while the rest had acute brainstem syndrome. Seven patients had positive AQP-4 antibody tests. All patients were treated with systemic corticosteroid and azathioprine/mycophenolate mofetil. Four of them required plasma exchange. All cases had fewer relapses and disability at the end of review period.12
This clinical literature emphasizes the existence of neuromyelitis optica spectrum disorder in clinical settings of the developing world. The low expectations and diagnostic challenges of this rare disease in limited clinical settings might contribute to delayed diagnosis and treatment.
Ethical Approval
Our institution does not require ethical approval for reporting individual case reports.
Consent for Publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Acknowledgments
We are grateful to the medical personnel who were caring for the patient.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work.
Funding
The authors received no financial support from any source.
Disclosure
The authors declare no potential conflict of interest with respect to the research, authorship or publication of the article.
References
1. Jacob A, Mc Keon A, Nakashima I, et al. Current concepts of neuromyelitis optica (NMO) and NMO spectrum disorders. J Neurol Neurosurg Psychiatry. 2013;84:922–930. doi:10.1136/jnnp-2012-302310
2. Fujihara K. Neuromyelitis optica spectrum disorders: Still evolving and broadening. Curr Opin Neurol. 2019;32(3):385–394. doi:10.1097/WCO.0000000000000694
3. Dutra BG, Rocha AF, Nunes RH, Funior ACMM. Neuromyelitis optica spectrum disorders: Spectrum of MR imaging findings and their differential diagnosis. RadioGraphics. 2018;38:169–193. doi:10.1148/rg.2018170141
4. Wang KY, Chetta J, Bains P, et al. Spectrum of MRI brain lesion patterns in neuromyelitis optica spectrum disorder. A pictorial review. Br J Radiol. 2018;91:20170690. doi:10.1259/bjr.20170690
5. Wingerchuk DM, Banwell B, Bennett J, et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurol. 2015;85:177–189. doi:10.1212/WNL.0000000000001729
6. Papp V, Magyari M, Aktas O, et al. Worldwide incidence and prevalence of neuromyelitis optica. Systematic Review Neurol. 2021;96(2):59–77.
7. Held F, Klein A-K, Berthele A. Drug treatment of neuromyelitis optica spectrum disorders: Out with the old, in with the new? ImmunoTargets Therapy. 2021;10:87–101. doi:10.2147/ITT.S287652
8. Jemal A, Bane A, Ali S. A 24-year-old female with neuromyelitis optica from Ethiopia. Ethiop Med J. 2017;55(4):320–323.
9. Melka D. Aquaporin-4-IgG positive neuromyelitis optica spectrum disorder from Ethiopia: A case report. Ethiop J Health Sci. 2020;30(5):847–852.
10. Asrat R, Sisay G, Sultan A, et al. Neuromyelitis optica (NMO, NMOSD). J Psych. 2020;23(9):1–2.
11. Gaye NM, Fall M, Diop AM, et al. Neuromyelitis optica spectrum disorders (NMO-SD) in a sub-Saharan Africa country: A preliminary study of sixteen cases. Multi Scler Relat Disord. 2019;27:179–183. doi:10.1016/j.msard.2018.10.108
12. Sokhi D, Suleiman A, Manji S, Hooker J, Mativo P. Cases of neuromyelitis optica spectrum disorder from the East Africa region, high lighting challenges in diagnostics and health care access. Neurological Sci. 2021;22:100320.
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.