")
Back to Journals » Therapeutics and Clinical Risk Management » Volume 11
Neurogenic orthostatic hypotension – management update and role of droxidopa
Received 2 April 2015
Accepted for publication 18 May 2015
Published 8 June 2015 Volume 2015:11 Pages 915—923
DOI https://doi.org/10.2147/TCRM.S68439
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Garry Walsh
Joy Vijayan,1 Vijay K Sharma1,2
1Department of Medicine, Division of Neurology, National University Health System, 2Yong Loo Lin School of Medicine, National University of Singapore, Singapore
Abstract: Orthostatic hypotension (OH) is defined as a significant decrease in blood pressure (BP) during the first 3 minutes of standing or a head up on a tilt table. Symptoms of OH are highly variable, ranging from mild light-headedness to recurrent syncope. OH occurs due to dysfunction of one or more components of various complex mechanisms that interplay closely to maintain BP in a normal range during various physiological and associated disease states. Various biochemical and electrophysiological studies are often undertaken to assess the severity and etiology of OH. In addition to the lifestyle modifications, various medications that stimulate the adrenergic receptors or increase central blood volume are used in patients with OH. Droxidopa is a newer agent that increases the levels of norepinephrine in postganglionic sympathetic neurons. Management strategies for OH are presented, including the mechanism of action of droxidopa and various studies performed to assess its efficacy.
Keywords: orthostatic hypotension, systemic blood pressure, midodrine, fludrocortisone, droxidopa
Introduction
Orthostatic hypotension (OH) is defined as a decrease in systolic blood pressure (BP) >20 mmHg or a diastolic BP >10 mmHg within 3 minutes of standing. An acceptable alternative is the demonstration of a similar fall in BP within 3 minutes, using a tilt table in the head up position, at an angle of at least 60°.1 OH can present with a wide variety of symptoms ranging from dizziness, light-headedness, cognitive slowing, and fatigue to syncope. The latter is by far the most common symptom for which the patient seeks medical attention. It is the third most frequent cause of syncope after vasovagal and cardiac syncopes.2 The incidence of OH and syncope increases with age, being more common in elderly residents of health care facilities (54%–68%) than in those in the community (6%).3–5 Patients with OH also carry an increased risk of falls and its attendant complications like fracture, predisposition to infections, and immobility.6 On a different note, in ~16%–18% of patients with documented OH, no symptoms are reported.7
There are several studies that have reported an increased rate of mortality in patients with OH. This increased risk in mortality occurs independent of the underlying vascular risk factors and cardiac dysfunction.8,9
Anatomical and physiological basis of orthostatic normotension
BP is maintained within a normal range by complex mechanisms and close interplay of several anatomical networks and loops that work across multiple systems. This is best exemplified by rapid regulation of BP upon assumption of the upright posture. These regulatory systems work efficiently within relatively narrow ranges of cardiac resistance, capacitance status, and fluid volume status (Figure 1). A clear understanding of these mechanisms is important in understanding the pathophysiological basis of OH and its associated clinical manifestations.10,11
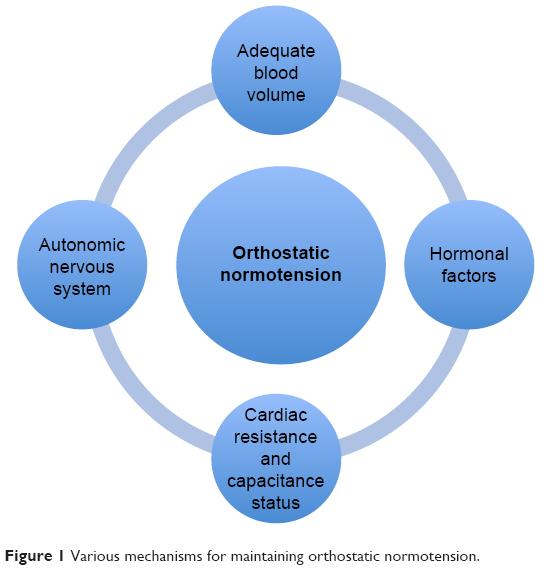 | Figure 1 Various mechanisms for maintaining orthostatic normotension. |
Attaining an upright posture initiates a complex sequence of physiological events in response to the gravity-mediated displacement of approximately 300–800 mL of blood into the splanchnic and lower limb vasculature.12,13 The ensuing reduced venous return to the heart results in decreased ventricular filling pressure, diminished cardiac output, and drop in BP. These hemodynamic changes activate several regulatory mechanisms to help stabilize the BP. The orthostatic response is mediated through three main phases, which consist of 1) the initial response during the first 30 seconds, 2) the early steady-state alteration (at 1–2 minutes), and 3) the prolonged orthostatic period (after at least 5 minutes upright). Table 1 lists various physiological mechanisms that play important roles during various phases of orthostatic response. Immediately after standing, the cardiac stroke volume remains normal due to the residual blood in the pulmonary circulation despite a decline in venous return. This is followed by a gradual fall in cardiac filling and arterial pressure, triggering the modulation of autonomic outflow to heart and blood vessels by a variety of reflexes.
 | Table 1 Physiological mechanisms involved in the maintenance of orthostatic normotension |
Heart is innervated by the sympathetic as well as the parasympathetic autonomic nervous systems. Sympathetic innervation is drawn from the neurons on the intermediolateral columns of the spinal cord at the first four thoracic cord levels. First-order neurons synapse in the superior, middle, and inferior cervical ganglia from where the second-order postganglionic neurons originate and innervate the heart through the mixed neural plexus. These neurons innervate the sinoatrial node, atrioventricular node, conduction system, and myocardial muscle fibers. The arteries and veins of the systemic circulation are also innervated primarily by the sympathetic nervous system.
First-order parasympathetic preganglionic fibers to the heart originate from the nucleus ambiguus and the dorsal motor nucleus of vagus nerve located in the medulla oblongata. These neurons, as part of the vagus nerve, join the cardiac mixed neural plexus and synapse with intracardiac ganglia to give rise to short postganglionic parasympathetic neurons. These second-order neurons innervate myocardial tissue, nodes, and the atrioventricular conduction system. Various reflexes that modulate the BP are controlled by a complex interplay of the autonomic nervous system, arterial baroreceptors, chemoreceptors, and several other cardiac receptors.
Arterial baroreflex plays the most important role in early buffering of BP fluctuations. This reflex is initiated by various pressure sensitive receptors and nerve endings located in the enlarged area of internal carotid artery just distal to the carotid bifurcation and within the aortic arch. These mechanoreceptors stimulate afferent fibers within the vagus and glossopharyngeal nerves, which subsequently synapse with neurons in the nucleus tractus solitarius (NTS). Impulses from the NTS modulate parasympathetic vagal motor activity in nucleus ambiguus and neurons in the ventrolateral medulla to control sympathetic outflow. With rising arterial pressure, afferent baroreceptor discharges in the NTS increase, resulting in an increased parasympathetic efferent activity, slowing of heart rate, and inhibition of the sympathetic activity and subsequent vasodilatation. On the contrary, a decrease in BP unloads the baroreceptors with reduced afferent impulses to NTS. This results in decreased parasympathetic vagal efferent activity to sinus node and an increase in sympathetic efferent activity. These effects increase the heart rate and induce systemic vasoconstriction to increase the BP.
Equally important physiological measures involved in the maintenance of orthostatic normotension include the contraction of muscles of the legs and abdomen to compress the capacitance and resistance vessels, leading to an elevation in the peripheral vascular resistance. This is further complemented by the activation of the venoarteriolar reflex and myogenic responses. With continued upright posture, various neurohumoral mechanisms and fluid volume status of the individual maintain the BP. Some of the important mechanisms are activation of the renin–angiotensin–aldosterone system and production of vasopressin, endothelin, and nitric oxide. In a simplistic manner, the autonomic nervous system is the major regulator of BP during the short- and medium-term periods after an orthostatic challenge as compared to various hormones of the renin–angiotensin–aldosterone system that maintain orthostatic normotension over prolonged periods.14
Classification and pathophysiology of OH
Various causes of OH can be broadly divided into neurogenic and nonneurogenic. A detailed clinical assessment is required to differentiate the two entities. Evaluation starts with the assessment of the volume and cardiac status, looking for historical and clinical clues of possible underlying endocrinological diseases and asking a detailed medication history. Other less important causes of OH include excessive peripheral venous pooling or vasodilatation secondary to alcohol, certain neuroendocrine tumors, and pregnancy.
Neurogenic OH results from the dysfunction of various reflexive regulatory systems that maintain orthostatic BP. The predominant mechanism responsible for neurogenic OH is the impairment of the sympathetic (vasoconstrictor) adrenergic system. From a pathophysiological standpoint, neurogenic hypotension can occur due to the dysfunction or loss of preganglionic neurons in the brainstem and spinal cord, dysfunction of the postganglionic fibers, and dysfunction of both pre- and postganglionic fibers (Table 2). Alternatively, neurogenic hypotension may result from either primary or secondary autonomic disorder where the site of pathology is within the central or peripheral nervous system.15
 | Table 2 Diseases commonly associated with autonomic dysfunction |
The primary autonomic degenerative disorders include multiple system atrophy, Parkinson’s disease, dementia with Lewy bodies, and pure autonomic failure. Autonomic dysfunctions in these disorders are due to the loss of preganglionic sympathetic neurons in the lateral intermediolateral column within the spinal cord and loss of neurons within the ventrolateral medulla or dorsal motor nucleus of the vagus nerve. These result in a functional disengagement of the peripheral sympathetic and parasympathetic postganglionic neurons. In Parkinson’s disease, the additional loss of postganglionic fibers can result in cardiac noradrenergic denervation. The pathological hallmark of primary neurodegenerative autonomic disorders is the deposition of intracellular inclusions called alpha-synuclein. In multiple system atrophy, these inclusions are seen within the oligodendrocytes, whereas in Parkinson’s disease and Lewy body disease, these intracellular inclusions are predominantly seen within neuronal population across various parts of the central nervous system and occasionally within the peripheral autonomic nervous system.
Various diseases associated with secondary autonomic disorders are mentioned in Table 2. Most of these diseases cause autonomic dysfunction as a result of involvement of the small fibers that subserve autonomic functions. The most common cause of secondary autonomic dysfunction is diabetes mellitus. Other less common causes include amyloidosis, malignancy, human immunodeficiency virus infection, and vitamin B12 deficiency.
Clinical features of OH
Wide variations are observed in the severity of symptoms of OH, ranging from minimal symptoms to severe disability. Orthostatic intolerance refers to symptoms and signs in the upright position due to circulatory abnormality. Most of the patients present with very nonspecific symptoms in the form of nonvertiginous giddiness, nausea, fatigue, light- or heavy-headedness, vaguely defined weakness of lower extremities, or cognitive slowing.16 More specific symptoms include posturally related visual blurring, secondary to decreased blood flow to the retina and occipital lobes; pain over the posterior nuchal and cervical regions due to ischemia to the paracervical and periscapular muscles (coat-hanger headache); or orthostatic dyspnea or chest pain due to ventilation–perfusion mismatch. Syncope can occur in some patients as a result of transient loss of cerebral perfusion.
Some aspects of OH that are clinically relevant but not well appreciated in the clinical context are described as follows:
- Patients with long-standing autonomic dysfunction may adapt to the orthostatic fall in BP and may show no associated clinical manifestations. This is possibly related to the leftward shift of the lower limit of cerebral autoregulation. Some of these patients may tolerate a fall in mean arterial BP to as low as 40 mmHg.
- Syncope due to OH tends to be more common in the early morning and may not be associated with diaphoresis or change in heart rate. Orthostatic intolerance is often exacerbated by heavy meals, alcohol intake, increase in ambient temperature, dehydration, or exercise.
- OH is commonly seen in elderly patients who have multiple comorbidities and who are on several types of medications. These patients may not have the full spectrum of autonomic dysfunction. There are several classes of medicines commonly associated with OH (Table 3).
- There are several variants of OH: i) initial OH characterized by a fall in BP by more than 40 mmHg immediately on standing is due to a transient mismatch between the cardiac output and the systemic vascular resistance; ii) classical OH that occurs between 30 seconds and 3 minutes after standing. This occurs due to impaired increase in systemic vascular resistance as a result of impaired autonomic reflexes; and iii) delayed or progressive OH that occurs beyond 3 minutes of standing. This tends to occur more commonly in the elderly with multiple comorbidities and is a sign of a mild or early sympathetic adrenergic failure.17–19
- Although OH can occur in isolation, it is mostly a part of some larger disease process. Therefore, in the latter, symptoms of generalized autonomic dysfunction in the form of thermoregulatory, pupillary, genitourinary, or gastrointestinal dysfunction are observed. A detailed clinical evaluation is often successful in elicitation of features suggestive of a primary or secondary autonomic disorder.
 | Table 3 Common drugs commonly associated with OH |
Evaluation and investigations
The evaluation and investigations of OH ranges from simple bedside clinical assessment to complex biochemical and advanced electrophysiological studies.
A comprehensive assessment should include studies to evaluate the severity of autonomic dysfunction (Table 4). Neurophysiological and imaging studies are designed to differentiate between primary and secondary diseases responsible for various autonomic dysfunctions (Table 5). A more detailed evaluation of underlying cardiac causes of syncope is required if there are no obvious abnormalities seen on autonomic tests.20
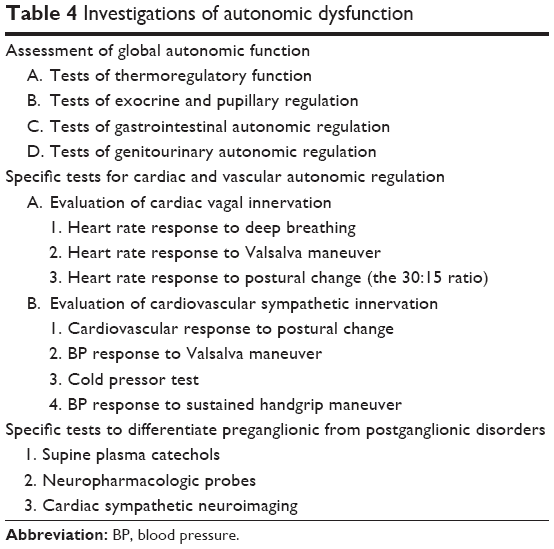 | Table 4 Investigations of autonomic dysfunction |
 | Table 5 General investigations for autonomic disorders |
The measurement of BP in patients with OH should include the measurement in the supine position after a period of adequate rest and measurements in the erect posture at 1 minute and 3 minutes. More specialized measurement techniques include the use of oscillometric devices and the measurement of beat-to-beat phasic BP recordings using Finometry.
Specific investigations for OH should focus on the evaluation of cardiovascular autonomic functions. This includes assessment of the cardiovagal (parasympathetic) and cardiovascular adrenergic (sympathetic) innervations.21–22
Tests of cardiovagal functions include heart rate responses to various challenges like deep breathing, Valsalva maneuver, and postural change. Heart rate variability is an important physiological parameter, with an increased heart rate during inspiration and decreased heart rate during expiration. Heart rate variability is most evident during deep breathing, at ~six breaths per minute. The difference in the heart rate during inspiration and expiration often parallels the cardiovagal function. Valsalva maneuver is performed by blowing for 15 seconds into a mouthpiece, connected to a mercury manometer to maintain a pressure of 40 mmHg. The Valsalva ratio (ratio between heart rate during the maneuver and the postmaneuver bradycardia) provides an index of cardiac vagal function. The heart rate response to postural change (30:15 ratio) also indicates the integrity of the cardiovagal function. This is calculated by dividing the longest R–R interval on the electrocardiogram at around the 13th beat by the shortest R–R interval around the 15th beat. Other tests of cardiovagal function include carotid sinus massage, cold face test, and the atropine test.
Tests of cardiovascular adrenergic (sympathetic) function evaluate the cardiovascular response to standing or tilt table testing and BP response to Valsalva maneuver. Patients with sympathetic failure are unable to maintain their BP or mount a tachycardic response on assuming an upright position on tilt table testing. The response to Valsalva maneuver assesses both the cardiovagal and adrenergic functions. In patients with adrenergic failure, BP shows a decrease during Phase II and an absence of overshoot during Phase IV. Other tests of cardiovascular adrenergic function include cold pressor test, sustained handgrip maneuver, and mental arithmetic test.
Management
Treatment of OH includes both pharmacological and nonpharmacological measures.23 Educating the patient regarding various underlying mechanisms of postural hypotension and strategies to circumvent or prevent a sudden fall in BP is often extremely helpful. Measures aimed at expanding the blood volume should also be emphasized. Table 6 lists the various measures used in the treatment of OH.
 | Table 6 Treatment of OH |
Nonpharmacological measures
A gradual stepwise process going through stages of sitting up for short periods of time and standing steady is important before attempting to walk. This is particularly important in the morning when the orthostatic stress is at its maximum. The orthostatic stress can also be ameliorated by keeping the headend of the bed elevated by 10°–20° during sleep. Adopting this method helps by activating the renin–angiotensin–aldosterone system and reducing nocturia. It has also been found to be effective in reducing supine hypertension. Patients should also be warned about exacerbating factors, including heavy meals, increased ambient temperature, alcohol intake, and avoiding certain medications. Patients should be educated about adopting certain maneuvers to counter a fall in BP such as tip toeing, leg crossing, and squatting. These acquired habits may be especially useful in patients who develop warning signs of an impending syncope. Small frequent meals prevent splanchnic pooling of blood and prevent postprandial hypotension. Use of abdominal binders and stockings are also helpful in ameliorating an orthostatic fall in BP.
Measures to expand the blood volume include a generous intake of fluid and salt supplementation. Salt supplementation can be achieved by adding extra salt to the meals or by taking salt tablets. Adequate salt intake can be verified by measuring the 24-hour urinary sodium, value below 170 mmol/24 hours suggests inadequate salt consumption and the need of augmenting additional salt intake. In patients with significant postural symptoms early in the morning, fluid or water loading can be very helpful. Ingestion of approximately 500 mL of water elicits a marked pressor response and increases the systolic BP by 30 mmHg, with peak effect approximately 2 hours after water intake.
Pharmacological measures
Several types of medications are used in the management of OH. Most of these agents primarily act by stimulating the adrenergic receptors or increasing the central blood volume.
Midodrine
Midodrine is the first of the two drugs approved by the Food and Drug Administration (FDA) for the treatment of symptomatic OH.24 It acts by selectively and directly stimulating alpha 1-adrenoreceptors located on the arteriolar and venous vasculature. It has also been used in the treatment of recurrent reflex syncope.25–27 Midodrine increases the standing BP and reduces the symptoms of orthostatic intolerance. The usual starting dose is 2.5 mg and titrated up to a maximum of 10 mg three times daily. The last dose of the day should be taken no later than 6 pm, and close monitoring for supine hypertension is required on starting the medication and subsequently on dose titration. Contraindications or extreme caution should be exercised in patients with organic heart disease, obstructive urinary symptoms, or those with endocrine diseases like pheochromocytoma and thyrotoxicosis. Appropriate dose modifications are needed in patients with renal failure.
Droxidopa
Droxidopa, also known as l-DOPS (l-threo-dihydroxyphenylserine), is the second of the two drugs approved by the FDA for the treatment of OH. It is a synthetic amino acid precursor that is converted by aromatic l-amino acid decarboxylase into norepinephrine. Droxidopa acts by several mechanisms28 to increase the levels of norepinephrine in postganglionic sympathetic neurons, leading to increased stimulation of adrenergic receptors. Unlike midodrine, it can cross the blood–brain barrier and enhance production of norepinephrine in the central nervous system. This can lead to increased activation of sympathetic preganglionic neurons in the spinal cord. Finally, norepinephrine synthesized from droxidopa can act as a circulating hormone and exert a pressor effect.
Following oral administration, droxidopa plasma levels reach their peak at approximately 3 hours. Its half-life is 2–3 hours.29 Plasma levels of norepinephrine and its metabolites also peak at approximately 3 hours. However, unlike l-DOPS, levels of norepinephrine decline at a much slower rate. This is probably related to the ongoing production of norepinephrine from l-DOPS within cellular storage sites and its subsequent entry into the blood stream.
Droxidopa was first used in the treatment of congenital dopamine beta hydroxylase deficiency.30 It was subsequently found to be useful in the treatment of OH associated with various disorders, including primary as well as secondary dysfunctions of the autonomic nervous system. Several studies have shown the efficacy of droxidopa in the symptomatic management of OH. In most of these trials, efficacy was assessed using the two-part OH Questionnaire (OHQ): OH Symptom Assessment (Item 1: dizziness/light-headedness), and OH Daily Activity Scale.31
Kaufmann et al provided Class I evidence that droxidopa was significantly superior to placebo in the treatment of neurogenic OH due to Parkinson’s disease, primary autonomic failure, multisystem atrophy, nondiabetic autonomic neuropathy, or dopamine beta-hydroxylase deficiency. In this study, open-label dose optimization of droxidopa lasting up to 14 days was followed by a 7-day washout and then a 7-day double-blind treatment. Improvement was assessed by the OHQ and an increase in standing systolic BP. Improvement was observed in the cardinal symptoms of neurogenic OH and on the impact of symptoms on daily activities like walking and standing in patients receiving droxidopa.32 In a randomized withdrawal study trial by Biaggioni et al patients underwent open-label droxidopa dose titration followed by 1 week of open-label treatment at individualized treatment and subsequently randomized to continue droxidopa or placebo. This study failed to demonstrate an improvement in the OHQ dizziness/light-headedness score, which was the primary outcome.33 However, patients receiving droxidopa reported a significant improvement in four of the other five OHQ symptom scores and all of the four symptom-impact scores as well as statistically significant self-reported ability to perform activities requiring standing for short or a long time. Many other studies have also demonstrated the usefulness of droxidopa in the management of OH.34–36
Other potential uses of droxidopa include improvement of orthostatic hypotensive symptoms in hemodialyzed patients37 and frozen gait in patients with Parkinson’s disease.38
The recommended starting dose of droxidopa is 100 mg, which can be escalated to a maximum of 600 mg three times a day, with the last dose being at least 3 hours prior to bedtime. The dose is increased on a weekly to two-weekly basis to achieve the intended symptomatic relief. The most common adverse effect is the development of worsening supine hypertension. Therefore, close monitoring of BP is required prior to and during the treatment with droxidopa, especially during dose increments. Elevating the head of the bed during sleep is believed to reduce the risk of supine hypertension. Droxidopa can exacerbate the symptoms in patients with ischemic heart disease, arrhythmias, and congestive heart failure. Other nonspecific adverse reactions like headache, dizziness, nausea, and fatigue may be seen in some patients. Occasionally, symptom complex resembling neuroleptic malignant syndrome may also occur, especially in patients taking concomitant levodopa and neuroleptics.
Fludrocortisone
Unlike midodrine and droxidopa, fludrocortisone acts primarily by increasing the central blood volume. It is a potent synthetic mineralocorticoid that promotes renal sodium reabsorption with resultant increase in central blood volume. Other potential mechanisms include sensitization of the vasculature to norepinephrine and angiotensin II.39 The usual starting dose is 0.1 mg/day that can be titrated to a maximum of 0.3 mg/day. Full pressor effect of fludrocortisone may take up to 2 weeks from its initiation. Patients usually gain approximately 2–3 kg when its effects are maximal, which with time tends to normalize back to their baseline with maintenance of BP. Adverse effects of fludrocortisone include hypokalemia, hypomagnesemia, and supine hypertension. Hypokalemia is usually dose dependent, appears within 1–2 weeks of treatment, and can occur in ~25% of patients taking fludrocortisone. Fludrocortisone has to be used with extreme caution in patients with congestive cardiac failure as it is known to increase the central blood volume. Chronic mineralocorticoid receptor stimulation can cause cardiac hypertrophy, and thus adversely affects the cardiovascular structure and function.40 Caution should be taken when patients are on warfarin as it is known to interact with warfarin and often a dose escalation is required.
Erythropoietin
Patients with OH frequently have a decreased red blood cell mass due to inadequate erythropoietin levels. This is possibly related to an inadequate circulating plasma norepinephrine level, which stimulates the secretion of erythropoietin. This decrease in effective blood volume exacerbates the preexisting hypotension. Administration of erythropoietin is known to increase the BP by an average of 10 mmHg. The postulated mechanisms for this increase in BP include an increase in blood volume, increase in blood viscosity, or due to vasoconstriction as a result of reduced levels of nitric oxide. Erythropoietin is administered subcutaneously at a dose of 25–75 U/kg three times a week to achieve a hematocrit value that reaches sex-specific normal values.41
Other medications
Several other classes of medication are also used in the treatment of OH and include sympathomimetic medications like ephedrine, acetylcholinesterase inhibitors (pyridostigmine), and cyclooxygenase inhibitors (indomethacin and flurbiprofen).
Conclusion
Neurogenic OH is a commonly encountered clinical condition and often seen as a manifestation of an underlying disease process. It occurs as a result of an impaired sympathetic tone of peripheral vasculature and can even be part of a broader autonomic disorder. Evaluation is two-pronged and includes assessment of the severity of autonomic dysfunction, while clinical and investigative studies aim at diagnosing the underlying disease process. Treatment approaches are aimed at improving the central blood volume and alpha-adrenoreceptor agonists like midodrine and droxidopa.
Disclosure
None of the authors declare any conflicts of interest related to this paper.
References
Freeman R, Wieling W, Axelrod FB, et al. Consensus statement on the definition of orthostatic hypotension, neutrally mediated syncope and the postural tachycardia syndrome. Clin Auton Res. 2011;21(2): 69–71. | ||
Soteriades ES, Evans JC, Larson MG, et al. Incidence and prognosis of syncope. N Engl J Med. 2002;347(12):878–885. | ||
Freeman R. Neurogenic Orthostatic Hypotension. N Engl J Med. 2008; 358(6):615–624. | ||
Shibao C, Grijalva CG, Raj SR, Biaggioni I, Griffin MR. Orthostatic hypotension-related hospitalizations in the United Sates. Am J Med. 2007;120(11):975–980. | ||
Ooi Wl, Hossain M, Lipsitz LA. The association between orthostatic hypotension and recurrent falls in nursing home residents. Am J Med. 2000;108(2):106–111. | ||
Berry SD, Miller RR. Falls: Epidemiology, pathophysiology and relationship to fracture. Curr Osteoporos Rep. 2008;6(4):149–154. | ||
Rose KM, Eigenbrodt ML, Biga RL, et al. Orthostatic hypotension predicts mortality in middle-aged adults: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2006;114(7):630–636. | ||
Rutan GH, Hermanson B, Bild DE, Kittner SJ, et al. Orthostatic hypotension in older adults. The Cardiovascular Health Study. CHS Collaborative Research Group. Hypertension. 1992;19(6 Pt 1): 508–519. | ||
Veronese N, De Rui M, Bolzetta F, et al. Orthostatic changes in blood pressure and mortality in the elderly: the Pro.V.A Study. Am J Hypertens. 2015 Mar 11. pii: hpv022. [Epub ahead of print]. | ||
Berry SD, Miller RR. Falls: Epidemiology, pathophysiology, and relationship to Fracture. Curr Osteoporos Rep. 2008;6(4):149–154. | ||
Joyner M, Sheppard T. Autonomic regulation of the circulation. In: Low P, ed. Clinical Autonomic disorders. 2nd ed. Philadelphia, Pa: Lippincott-Raven; 1997:61–71. | ||
Wieling W, VanLieshout JJ. Maintenance of postural normotension in humans. In Low, ed. Clinical Autonomic disorders. Philadelphia, Pa: Lippincott-Raven; 1997:73–82. | ||
Grubb BP. Neurocardiogenic syncope and related disorders of orthostatic intolerance. Circulation. 2005;111(22):2997–3006. | ||
Benarroch E, Freeman R, Kaufmann H. Autonomic nervous system. Textbook of Clinical Neurology. 3rd ed; Philadelphia, PA, Elsevier. 2007:383–404. | ||
Bennaroch E. The central autonomic network: functional organization and clinical correlations. Armonk, NY: Futura press; 1997:31–78. | ||
Golstein DS, Sharabi Y. Neurogenic orthostatic hypotension: a pathophysiological approach. Circulation. 2009;119(1):139–146. | ||
Freeman R, Kaufmann H. Disorders of Orthostatic tolerance-Orthostatic hypotension, postural tachycardia syndrome, and syncope. Continuum. 2007;13(6):50–88. | ||
Wieling W, Krediet CT, van Dijk N, Linzer M, Tschakovsky ME. Initial orthostatic hypotension: review of a forgotten condition. Clin Sci (Lond). 2007;112(3):157–165. | ||
Gibbons CH, Freeman R. Delayed orthostatic hypotension: a frequent cause of orthostatic intolerance. Neurology. 2006;67(1):28–32. | ||
The task force for the management diagnosis and management of syncope of the European Society of Cardiology (ESC). Guidelines for the diagnosis and management of syncope (version 2009). Eur Heart J. 2009;30:2631–2671. | ||
Ravits JM. AAEM minimonograph #48. Autonomic nervous testing. Muscle Nerve. 1997;20(8):919–937. | ||
Low PA, Tomalia VA, Park KJ. Autonomic function tests: some clinical applications. J Clin Neurol. 2013;9(1):1–8. | ||
Low PA, Singer W. Update on management of neurogenic orthostatic hypotension. Lancet Neurol. 2008;7(5):451–458. | ||
Kaufmann H, Brannan T, Krakoff L, Yahr MD, Mandeli J. Treatment of orthostatic hyotension due to autonomic failure with a peripheral alpha-adrenergic agonist (midodrine). Neurology. 1988;38(6):951–956. | ||
Low PA, Gilden Jl, Freeman R, Sheng KN, McElligott MA. Efficacy of midodrine vs placebo in neurogenic orthostatic hypotension. A randomized, double-blind multicenter study. Midodrine Study Group. JAMA. 1997;277(13):1046–1051. | ||
Wright RA, Kaufmann HC, Perera R, et al. A double-blind, dose-response study of midodrine in neurogenic orthostatic hypotension. Neurology. 1998;51(1):120–124. | ||
Izcovich A, Gonzalez Malla C, Manzotti M, Catalano HN, Guyatt G. Midodrine for orthostatic hypotension ad recurrent reflex syncopre: A systematic review. Neurology. 2014;83(13):1170–1177. | ||
Kaufmann H. L-dihydoxyphenylserine (droxidopa): a new therapy for neurogenic orthostatic hypotension: US expereince. Clin Auton Res. 2008;18(Suppl 1):19–24. | ||
Golstein DS, Homes C, Kaufmann H, Freeman R. Clinical pharmacokinetics of the norepinephrine precursor L-threo-DOPS in primary chronic autonomic failure. Clin Auton Res. 2004;14(6):363–368. | ||
Man in ‘t Veld AJ, Boomsma F, van den Meiracker AH, Schalekamp MA. Effect of unnatural noradrenaline precurosor on sympathetic control and orthostatic hypotension in dopamine-beta-hydroxylase deficiency. Lancet. 1987;2(8569):1172–1175. | ||
Kaufmann H, Malamut R, Norcliffe-Kaufmann L, Rosa K, Freeman R. The Orthostatic Hypotension Questionnaire (OHQ): validation of a novel symptom assessment scale. Clin Auton Res. 2012;22(2):79–90. | ||
Kaufman H, Freeman R, Biaggioni I, et al. Droxidopa for neurogenic hypotension: a randomized, placebo-controlled, phase 3 trial. Neurology. 2014;83(4):328–335. | ||
Biaggioni I, Freeman R, Mathias CJ, et al. Randomized withdrawal study of patients with symptomatic neurogenic orthostatic hypotension responsive to droxidopa. Hypertension. 2015;65(1):1001–1007. | ||
Freeman R, landsberg L, Young J. The treatment of neurogenic orthostatic hypotension with 3,4-DL-Threo-dihydroxyphenylserine: a randomized, placebo-controlled, crossover trial. Neurology. 1999;53(9): 2151–2157. | ||
Mathias CJ, Senard JM, Braune S, et al. L-threo-dihydroxyphenylserine (L-threo-DOPS; Droxidopa) in the management of neurogenic orthostatic hypotension: a multi-national, multi-center, dose-ranging study in multiple system atrophy and pure autonomic failure. Clin Auton Res. 2001;11(4):235–242. | ||
Kaaufmann H, Saadia D, Voustianiouk A, et al. Norepinephrine precursor therapy in neurogenic orthostatic hypotension. Circulation. 2003;108(6):724–728. | ||
Vannorsdall MD, Hariachar S, Hewitt LA. A randomized, placebo-controlled, phase 2 study of the efficacy and safety of droxidopa in patients with intradialytic hypotension. Postgrad Med. 2015;127(2): 133–143. | ||
Fukada K, Endo T, Yokoe M, et al. L-threo-3,4-dihydroxyphenylserine (L-DOPS) co-administered with entacapone improves freezing of gait in Parkinson’s disease. Med Hypotheses. 2013;80(2):209–212. | ||
Robertson D, Davis TJ. Recent advances in the treatment of orthostatic hypotension. Neurology. 1995;45:S26–S32. | ||
Lim PO, Farquharson CA, Shiels P, et al. Adverse cardiac effects of salt with fludrocortisone in hyperension. Hypertension. 2001;37: 856–861. | ||
Hoeldtke RD, Streeten DH. Treatment of orthostatic hypotension with erythropoietin. New Eng J Med. 1999;329:611–615. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms.php
and incorporate the Creative Commons Attribution
- Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.