Back to Archived Journals » Reports in Medical Imaging » Volume 14
Neurofibromatosis Type 1 with Neck and Thoraco-Abdominal Involvement: A Case Series Showing Different Localization and MRI Features
Authors Tortora S, Esposito A ![]() , Della Pepa G
, Della Pepa G ![]() , Paternò M
, Paternò M ![]() , Cagnoli GA, Cesaretti C
, Cagnoli GA, Cesaretti C ![]() , Natacci F, Carrafiello G
, Natacci F, Carrafiello G
Received 22 January 2021
Accepted for publication 18 March 2021
Published 6 April 2021 Volume 2021:14 Pages 41—51
DOI https://doi.org/10.2147/RMI.S300065
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Tarik Massoud
Silvia Tortora,1 Andrea Esposito,2 Gianmarco Della Pepa,1 Michele Paternò,1 Giulia Anna Cagnoli,3 Claudia Cesaretti,3 Federica Natacci,3 Gianpaolo Carrafiello2
1School of Diagnostic and Interventional Radiology, University of Milan, Milan, 20122, Italy; 2Radiology Department, Foundation IRCCS Ca’ Granda Maggiore Policlinico Hospital, Milan, 20122, Italy; 3Medical Genetics Unit, Woman-Child-Newborn Department, Foundation IRCCS Ca’ Granda Maggiore Policlinico Hospital, Milan, 20122, Italy
Correspondence: Silvia Tortora
School of Diagnostic and Interventional Radiology, University of Milan, Via Festa del Perdono 7, Milan, 20122, Italy
Email [email protected]
Andrea Esposito
Radiology Department, Foundation IRCCS Ca’ Granda Maggiore Policlinico Hospital, Via F. Sforza 35, Milan, 20122, Italy
Email [email protected]
Abstract: Neurofibromatosis type 1 (NF1) is an autosomal dominant disease caused by the mutation of the tumour suppressor gene NF1 located on chromosome 17q11.2, occurring in approximately 1 in 2000– 2500 people. The diagnosis of NF1 is made according to the presence of two or more diagnostic criteria of the National Institute of Health Consensus Development Conference. We present six cases of NF1 with neck and thoraco-abdominal involvement that we studied with MRI. The patients we present in this case series are asymptomatic and have in common the presence of multiple neurofibromas ubiquitously distributed in various body districts, including nerve roots, mediastinum, abdominal cavity, skin and subcutaneous tissue. Currently, there are no clear indications on the use of imaging in the diagnosis and follow-up of patients with NF1, although there is increasing evidence of the usefulness of imaging techniques and in particular of MRI. MRI with DWI and ADC mapping is a technique that should be proposed as a new standard of care for patients with NF1 since it can be used to distinguish a benign from a malignant tumour in relation to tumour size and ADC map values.
Keywords: neurofibromatosis 1, neurofibroma, plexiform neurofibroma, nerve sheath tumour, magnetic resonance imaging
Introduction
Neurofibromatosis type 1 (NF1) is the most common of the phacomatoses,1,2 occurring in approximately 1 in 2000–2500 people.3–8 It is an autosomal dominant disease caused by the mutation of the tumour suppressor gene NF1 located on chromosome 17q11.2.3 In about 50% of cases the disease arises as a de novo mutation.1,5 It has complete penetrance and variable expression.9
NF1 is a multisystem disease characterized by involvement of the central and peripheral nervous system (optic gliomas, spinal and peripheral nerve neurofibromas, neurological or cognitive impairment, malignant tumours of the nerve sheath) and extra-neurological manifestations (café-au-lait spots, skinfold freckling, Lisch nodules, bone lesions, pheochromocytoma, etc.).5,9,10 Neurofibromas can be divided into three types: localized, diffuse and plexiform. Localized neurofibromas usually present as a focal mass with well-defined margins that involve a single nerve fascicle.9–15 Diffuse neurofibromas have a plaque-like appearance associated with thickened skin, with poorly defined margins that develops in the subcutaneous fat and infiltrates the connective septa.14 Plexiform neurofibromas are composed of the same cell type as other neurofibromas, with a more represented extracellular matrix, greater vascularity and they develop along a nerve and may involve multiple nerve fascicles.3 About 10% of patients with multiple neurofibromatosis develop malignant peripheral nerve sheath tumours (MPNSTs).10
MPNST may originate de novo or more frequently from a pre-existing plexiform neurofibroma that undergoes sarcomatous transformation.1
The diagnosis of NF1 is made according to the presence of two or more diagnostic criteria of the National Institute of Health Consensus Development Conference.9,11
We present six cases of NF1, studied with MRI, with different sites of neck and thoraco-abdominal involvement. MRI with DWI and ADC mapping is a technique that should be proposed as a new standard of care for patients with NF1 since it can help to distinguish a benign from a malignant tumour in relation to tumour size and ADC map values.16,17
Ethics Statement
The patients provided written informed consent for publication of their case reports and accompanying images. This study was conducted in accordance with the Declaration of Helsinki and was accepted by the Ethics Committee of our Institution, Foundation IRCCS Ca’ Granda Maggiore Policlinico Hospital.
Case Series
Case 1
We present the case of an asymptomatic 30-year-old man with diagnosis of Neurofibromatosis type 1 (NF1), suspected in early childhood due to the presence of café-au-lait spots and confirmed after multiple cutaneous tumours appeared.
He underwent different imaging diagnostic exams that demonstrated multiple localizations of neurofibromatosis at basicranium, neck, in the nerve root canal along the entire extension of the spinal column and in abdominal and pelvic cavities.
The periodic annual follow-up MRI showed the presence of numerous, diffuse subcutaneous, intramuscular neurofibromas with neurofibromas that also involved the lumbar, celiac and superior mesenteric ganglia, the iliac plexuses and the obturator nerves bilaterally and the rectal plexuses.
In axial T2-weighted sequences, neurofibromas show inhomogeneous hyperintense appearance (Figure 1A and B).
 |
Figure 1 A 30-year old man affected by Neurofibromatosis 1 with multiple diffuse neurofibromas in the upper and lower abdomen (yellow arrows) and a malignant peripheral nerve sheath tumour (MPSNT) in the left para-aortic retroperitoneum (asterisks). Neurofibromas show inhomogeneous hyperintense appearance in axial T2-weighted sequences (A, B), low signal intensity in axial T1-weighted images (C, D), with faint enhancement in post-contrast T1-weighted sequences (E, F). The retroperitoneal MPNST shows inhomogeneous appearance in T2 and pre-contrast T1 sequences (A, C) and a faint and predominantly peripheral enhancement in post-contrast T1-weighted image (E). Some portions of this lesion show hyperintense signal in DWI sequence (G) with an ADC value <1.0 × 10−3 mm2/s on ADC map (H). |
Axial T1-weighted images show low signal intensity (Figure 1C and D) of the masses, which have faint enhancement in post-contrast T1-weighted sequences (Figure 1E and F).
In this context, in the left para-aortic retroperitoneal space there is a solid lesion which is referable to a retroperitoneal malignant peripheral nerve sheath tumour (MPNST) with inhomogeneous T1 pre-contrast appearance with extensive spots of hyperintensity of likely haemorrhagic nature (Figure 1C) and inhomogeneous appearance also in T2 sequence (Figure 1A); some portions of this lesion show hyperintense signal in DWI sequence (Figure 1G) with an ADC value <1.0 × 10−3 mm2/s on ADC map (Figure 1H).
This lesion shows a faint and predominantly peripheral enhancement in post-contrast T1-weighted image (Figure 1E).
About 10% of patients with multiple neurofibromatosis develop MPNSTs, which may originate de novo or more frequently from a pre-existing plexiform neurofibroma that undergoes sarcomatous transformation.
Case 2
The second case is represented by a 29-year-old man with NF1, diagnosed at the age of 2 due to the presence of diffuse cafe-au-lait spots and a sacral neurofibroma, associated with right convex thoracic scoliosis.
Over the years, there was evidence of progression of the disease with the appearance of multiple subcutaneous neurofibromas, a plexiform neurofibroma in the gluteal region and in the auricle of the right ear and other similar masses at the conjugate foramina of the dorsal and lumbar metameres, without significant spinal canal involvement or spinal cord compression.
In particular, the man underwent an MR of the thorax to assess an already known voluminous expansive neurofibroma localized in the left paravertebral dorsal site, from the fourth thoracic vertebral body to the ninth, closed to the descending thoracic aorta.
In axial pre-contrast T1-weighted image, the left paravertebral lesion is iso-hypointense (Figure 2A), with faint enhancement in post-contrast T1 sequence (Figure 2B). In the axial fat-saturated T2-weighted image (Figure 2C) and True Fast Imaging with steady-state free precession (TRUFI) sequence (Figure 2D), it has an inhomogeneous high signal intensity; the coronal TRUFI sequence shows the cranio-caudal extension of the lesion, from a plane passing at the level of the posterior arch of the second rib to the subcarinal region. The lesion is very near to the descending thoracic aorta (Figure 2E).
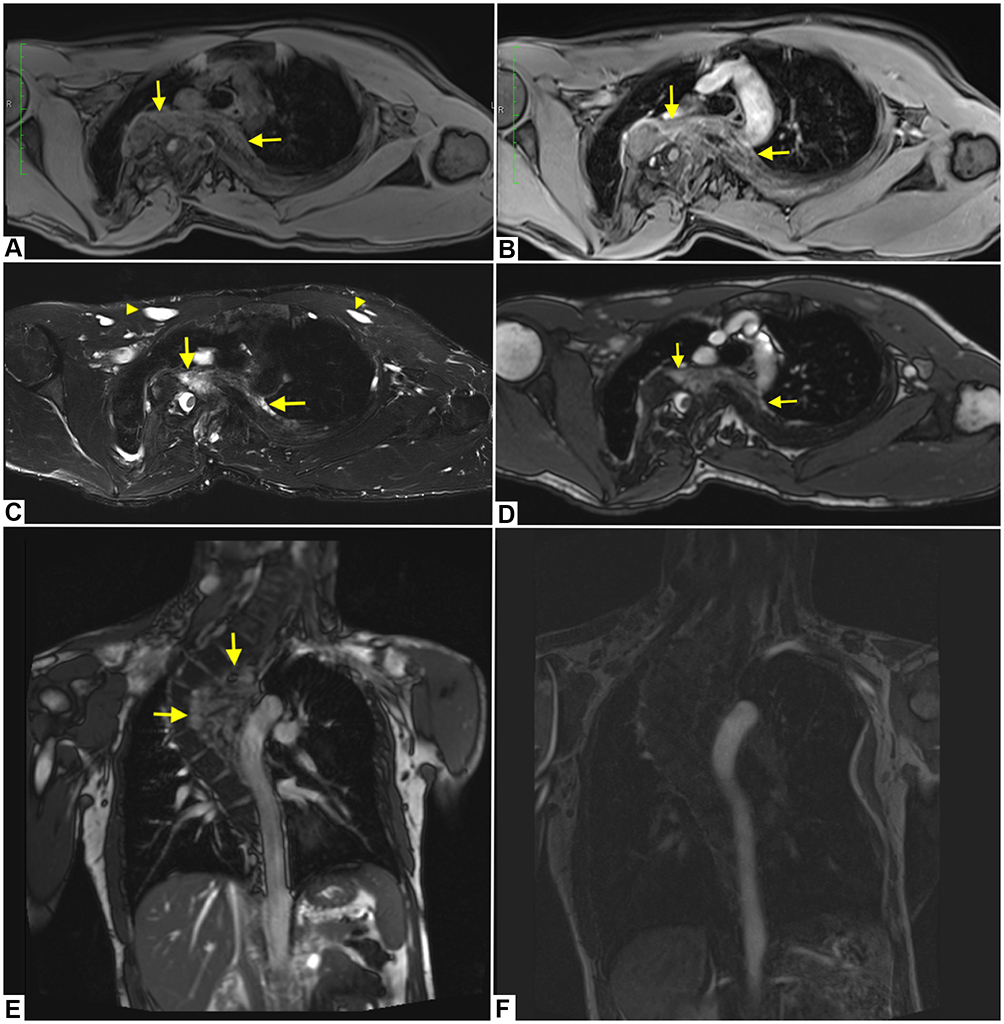 |
Figure 2 A 29-year old man with Neurofibromatosis 1 with multiple thoracic neurofibromas. Images show a voluminous left paravertebral lesion (yellow arrows) which is iso-hypointense in pre-contrast T1 image (A), with faint enhancement in post-contrast T1 sequence (B). In axial fat-saturated T2-weighted sequence (C) and True Fast Imaging with steady-state free precession (TRUFI) image (D), it has an inhomogeneous high signal intensity. Coronal TRUFI sequence shows the cranio-caudal extension of the lesion (E). In post-contrast coronal fat-saturated T1-weighted image the aorta is pervious and of regular calibre (F). Axial fat-saturated T2-weighted image shows multiple neurofibromas of the chest wall (yellow arrowheads) (C). |
In coronal post-contrast T1-weighted image, the aorta is pervious and of regular calibre (Figure 2F).
Additional neurofibromas were observed in the bilateral paravertebral regions, along the costovertebral joints, in the context of the intercostal and pectoral muscles (Figure 2C).
Case 3
This case deals with a 44-year-old man with known NF1, with a family history of NF1 and multiple manifestations of the disease, including cafe-au-lait spots, diffuse freckles, multiple neurofibromas on the trunk, face and in the neck region.
The patient underwent MRI of the neck which showed multiple lobulated, solid formations.
In particular, a lesion is observed in the right postero-lateral pharynx (Figure 3A-C) with compression on the right posterior-lateral wall of the oropharynx, larynx and on the hypopharynx.
 |
Figure 3 A 44-year-old man with Neurofibromatosis 1 (NF1) and family history of NF1. Images show a lesion in the right postero-lateral pharynx (yellow arrows), isointense in T1-weighted sequence (A), hyperintense in T2-weighted sequence (B) and in diffusion-weighted (DW) sequence (C) with ADC value greater than 1.0 × 10−3 mm2/s. In the right lateral-cervical region, a plexiform neurofibroma with “bag of worms” appearance is present (yellow arrowheads) (F), with isointense signal in T1-weighted sequence (D), hyperintense in T2-weighted sequence (E) and with faint enhancement in post-contrast axial fat-saturated T1-weighted sequence (G). |
Another similar voluminous lesion is observed in the right latero-cervical region (Figure 3D-G); it is polylobate and apparently formed by the confluence of several round foci, which extend into the upper mediastinum (Figure 3D) and compress the right internal jugular vein which is regularly opacified (Figure 3G).
A further lesion is found in the left anterolateral subcutaneous adipose tissue, at the middle third of the neck, anterior to the sternocleidomastoid muscle.
All of the above formations are characterized by isointense signal in T1-weighted sequences (Figure 3A and D), marked hyperintensity in T2-weighted sequences (Figure 3B, E, F); in axial post-contrast T1 image the abovementioned lesions show faint enhancement (Figure 3G). The lesion in the right postero-lateral pharynx shows hyperintense signal in diffusion-weighted (DW) sequences (Figure 3C), with ADC value greater than 1.0 ×1.0 × 10−3 mm2/s.
All the abovementioned lesions are referable to neurofibromas.
Case 4
We present the case of a 50-year-old, osteopenic woman, with a diagnosis of NF1 in adolescence, due to the presence of diffuse cutaneous neurofibromas on the trunk and limbs.
The patient underwent previous surgery for the removal of neurofibromas in the right forearm, lip, and gluteus.
At the age of 41, she was diagnosed with relapsing-remitting multiple sclerosis after showing persistent paresthesia of the hands.
The periodic follow-up MRI demonstrated the appearance of two solid, polylobate masses, with well-defined margins, in the right hypochondrium.
Axial T2-weighted image shows an inhomogeneous high signal intensity of the lesions (Figure 4A), which demonstrate hyperintense signal in axial diffusion-weighted (DW) sequence (Figure 4B), with ADC value greater than 1.0 × 10−3 mm2/s.
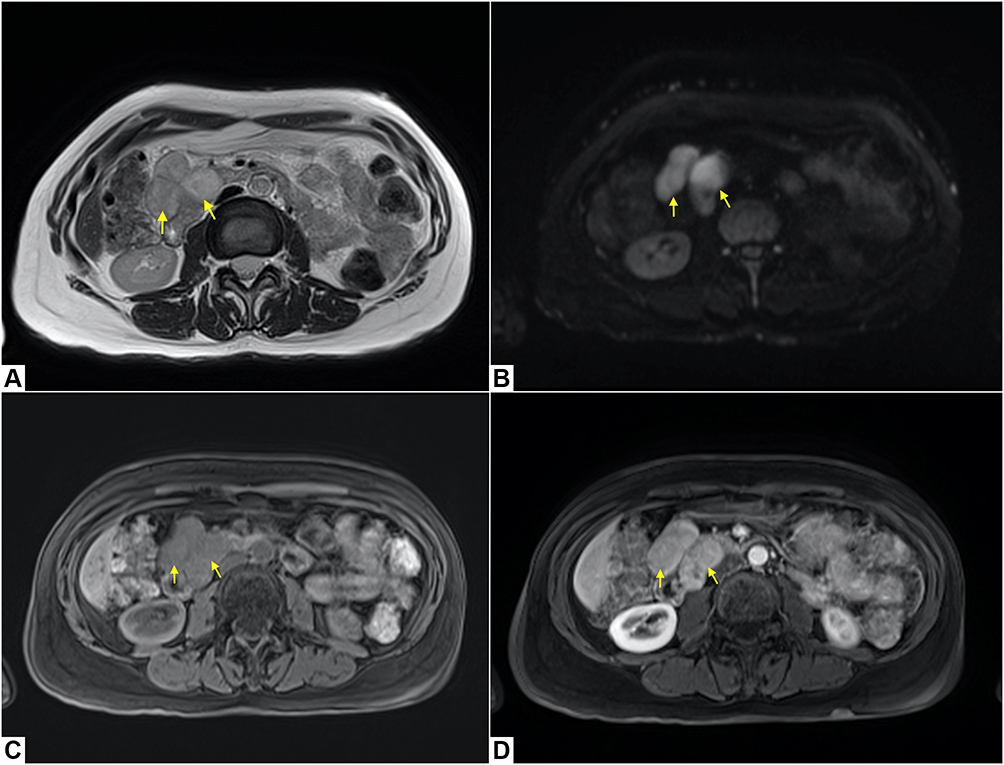 |
Figure 4 A 50-year-old woman affected by Neurofibromatosis 1 with two solid polylobate neurofibromas with well-defined margins in the right hypochondrium (yellow arrows). Axial T2-weighted image shows an inhomogeneous high signal intensity of the lesions (A), with hyperintense signal in axial diffusion-weighted (DW) sequence (B) and an ADC value greater than 1.0 × 10−3 mm2/s. In axial T1 pre-contrast image, the lesions appear isointense to the muscle (C) and show early enhancement in axial T1-weighted post-contrast image (D). |
In axial pre-contrast T1-weighted image, the lesions appear isointense to the muscle (Figure 4C) and show early enhancement in axial T1-weighted post-contrast image (Figure 4D).
Case 5
This is the case of a 20-year-old woman affected by NF1, diagnosed eight years earlier, due to the presence of cafe-au-lait spots. No relevant clinical conditions or hospitalizations occurred.
She underwent an MR, after the detection of solid lesions at ultrasound examination, in the axillary and lateral cervical region.
MRI shows several bilateral, ovoid, contiguous solid masses along the course of the vagus nerve in the neurovascular bundle of the neck up to a plane passing through the upper mediastinum. Coronal T2-weighted MR images demonstrate a homogeneous high signal intensity of the masses, compatible with neurofibromas due to signal intensity, the location and course (Figure 5A and B). The lesions appear isointense to the muscle in axial T1-weighted images.
 |
Figure 5 A 20-year old woman affected by Neurofibromatosis 1 with several ovoid bilateral contiguous neurofibromas along the course of the vagus nerve, rope-like in appearance (yellow arrows). Coronal T2-weighted images demonstrate a homogeneous high signal intensity of the masses (A, B). A plexiform neurofibroma is present along the course of the left accessory nerve (yellow arrowheads) with high signal intensity in axial T2-weighted image (C), low signal intensity in axial fat-saturated pre-contrast T1 image (D). Axial ADC map (E) shows ADC values greater than 1.0 × 10−3 mm2/s. |
Another separate ovoid polylobate formation is detected along the course of the left accessory nerve, with high signal intensity in axial T2-weighted image (Figure 5C), low signal intensity to the muscle in fat-saturated axial pre-contrast T1-weighted image (Figure 5D). Axial ADC map (Figure 5E) shows ADC value greater than 1.0 × 10−3 mm2/s. This lesion is compatible with a plexiform neurofibroma.
Case 6
We present the case of a 36-year-old man with a family history of NF1, whose mother died at the age of 37 from a malignant peripheral nerve sheath tumour (MPNST). He presented with cafe-au-lait spots, freckles in the armpits and diffuse neurofibromas in the subcutaneous adipose tissue, in the right brachial plexus, in the paravertebral and lateral cervical regions.
The patient underwent previous surgery for the removal of several neurofibromas. An abdominal MR demonstrates the presence of multiple neurofibromas, diffusely localized both in the muscles and in the intra-abdominal cavity with an inhomogeneous high signal intensity of the masses in the axial T2-weighted images. In axial T1-weighted MR images, the masses appear isointense to the muscle and show heterogeneous enhancement in axial T1-weighted post-contrast images (Figure 6B and D).
 |
Figure 6 A 36-year-old man with a family history of Neurofibromatosis 1 with multiple neurofibromas. Scattered lesions can be seen in the subcutaneous soft tissue, in the muscles, and within the intra-abdominal cavity. One neurofibroma is located caudally to the right kidney (yellow arrows (A, B)); a second neurofibroma is localized above the sacral promontory (yellow arrowheads (C, D)); a third neurofibroma is located in the left psoas (white arrows (C, D)). The lesions show heterogeneous enhancement in axial post-contrast T1 images (B, D). |
In the upper abdominal cavity, the largest lesion is located caudally to the right kidney (Figure 6A and B) while in the pelvis, the biggest one is appreciable in the median line, just above the sacral promontory (Figure 6C and D). Of the intramuscular lesions, the greatest one is detected in the context of the left psoas (Figure 6C and D). The lesions show suspicious features and should be monitored closely.
Discussion
Neurofibromatosis type 1 (NF1) is an autosomal dominant disease caused by a mutation or microdeletion of the tumour suppressor gene NF1 located on chromosome 17q11.2.3
According to National Institutes of Health Consensus Development Conference statement, the diagnostic criteria are the presence of two or more of the following features:
- Six or more café-au-lait spots over 5 mm in greatest diameter in pre-pubertal individuals and over 15 mm in greatest diameter in post-pubertal individuals.
- Two or more neurofibromas of any type or one plexiform neurofibroma.
- Freckles in the axillary or inguinal regions.
- Optic glioma.
- Two or more Lisch nodules. (iris hamartomas)
- A distinctive osseous lesion such as sphenoid dysplasia or thinning of long bone cortex with or without pseudarthrosis.
- A first-degree relative (parent, sibling, or offspring) with NF1 by the abovementioned criteria.10–12
Genetic analysis to characterize NF1 mutation is feasible, and is used to confirm a clinical diagnosis or to set up prenatal diagnosis or preimplantation genetic diagnosis. Currently, few genotype–phenotype correlation are known.8
A hallmark of NF1 is represented by neurofibromas, benign tumours (WHO grade I) of peripheral nerve sheaths made up of different types of cells such as Schwann cells, fibroblasts, mast cells and macrophages.13 Neurofibromas can be located in any part of the body (viscera and superficial and deep soft tissues), along the course of the peripheral nerves.10,14
Neurofibromas can be divided into three types: localized, diffuse and plexiform.
- Localized neurofibromas usually present as a focal mass with well-defined margins that involve a single nerve fascicle.9–15 In MRI, the presence of a“tail sig” suggests its neurogenic origin whereas the presence of a“target sig”, characterized by a T2 hyperintense peripheral region of myxomatous tissue and a T2 hypointense central region of fibro-collagen tissue, suggests the diagnosis of neurofibroma.16,17
- Diffuse neurofibromas have a plaque-like appearance associated with thickened skin; it is a lesion with poorly defined margins that develops in the subcutaneous fat and infiltrates the connective septa.14
- Plexiform neurofibromas are composed of the same cell type as other neurofibromas, but they have a more represented extracellular matrix and greater vascularity.3 They develop along a nerve and may involve multiple nerve fascicles. Macroscopic appearance can be rope-like if it involves a main nerve or can resemble a bag of worms if it extends widely along nerve branches.18 Any compression on the surrounding structures can cause functional impairment.
About 10% of patients with multiple neurofibromatosis develop malignant peripheral nerve sheath tumours (MPNSTs).10
MPNST may originate de novo or more frequently from a pre-existing plexiform neurofibroma that undergoes sarcomatous transformation.1
Therefore, it would be advisable to increase the surveillance of patients with NF1 and plexiform neurofibromas to diagnose any malignant transformation as soon as possible.
The clinical suspicion of malignant transformation of a plexiform neurofibroma arises from rapid increase in growth, change in consistency, the onset of persistent pain and neurological deficits.3,19
The standard of care for the management and follow-up of patients with neurofibromatosis consists of an annual physical examination and the treatment of symptomatic manifestations.20
Currently, there are no clear indications on the use of imaging for diagnosis and follow-up of patient with neurofibromatosis, although there is increasing evidence of the usefulness of imaging techniques and in particular of MRI in these cases.16,19
MRI can be localized or whole body. The localized MRI (with or without intravenous contrast administration) can be used to identify neurofibromas in a symptomatic patient, to evaluate the characteristics of benignity or malignancy, its extension and the anatomical relationship with the surrounding structures for a possible surgical removal. Whole-body MRI (WB-MRI) is useful for assessing tumour burden in a single session.20 An additional advantage is that WB-MRI can be completed with a typical scan time of 45–60 minutes on both 1.5T and 3.0T systems and uses the same protocols for patients of all ages. Variable approaches have been used for WB-MRI acquisition and image analysis in NF.21
According to Fayad et al, the WB-MRI protocol consists of T1-weighted sequences volume interpolated breath-hold examination (VIBE) (TR/TE = 0.88/2/43 ms, field of view = 50 × 50 cm2, matrix = 256 × 256, slice thickness = 2 mm), short tau inversion recovery (STIR, TR/TE = 6640/84 ms, field of view = 50 × 50 cm2, matrix = 256 × 256, slice thickness = 2 mm with interpolation), and quantitative DWI (TR/TE = 4100/70 ms, b = 50, 400, 800 s/mm2, field of view = 50 × 50 cm2, averages = 4, acceleration factor = 2, slice thickness = 5 mm, bandwidth = 1900 kHz, phase encode lines = 24, echo spacing = 0.64–0.88) with ADC mapping. Subsequently, a postcontrast T1 weighted sequence (VIBE, TR/TE = 0.88/2/43 ms, FOV = 50 × 50 cm2, matrix = 256 × 256, slice thickness = 2 mm) after intravenous contrast agent administration.22
In our cases, the different body districts were analyzed separately, using the same protocol for all the districts. The following sequences were performed: a T2 motion insensitive multi-shot Turbo Spin Echo (TSE) (BLADE) with and without fat-saturation (TR/TE: 5000 ms/100 ms; field of view: 38 x 46.3 cm2; flip angle: 140; slice thickness = 7 mm), T1 in-phase and out-of-phase (TR/TE: 150 ms/2.38 ms; field of view: 42 x 48 cm2; flip angle: 140; slice thickness = 5 mm), T1 Turbo-Spin-Echo (TSE) (TR/TE: 610/20; field of view: 38 x 46.3 cm2; flip angle: 140; slice thickness = 5 mm), and quantitative DWI (TR/TE = 5400 ms/76ms, b = 50, 400, 800 s/mm2, field of view = 50 × 50 cm2, slice thickness = 7 mm) with ADC mapping. Finally, a T1 volume interpolated breath-hold examination (VIBE) was obtained before and after intravenous administration of contrast agent (TR/TE: 4 ms/1.5 ms; field of view: 40 x 45 cm2; flip angle: 10; slice thickness = 3 mm).
The specificity and sensitivity in identifying malignant and premalignant lesions can be increased by functional MRI and metabolic (18F-fluorodeoxyglucose positron emission tomography/computed tomography FDG-PET/CT) investigations.5
In localized MRI, an average diameter of the lesion >4.2 cm and an ADC < 1.0 × 10−3 mm2/s were useful cut-off values for diagnosing malignancy, with 100% of sensitivity as neurofibromas usually have an ADC value >1.0 × 10−3 mm2/s.17
Similarly, whole-body MRI with DWI and ADC mapping can be used to distinguish a benign from a malignant tumour (which requires biopsy and possible excision) in relation to tumour size and ADC map values.17
Although FDG-PET/CT is currently considered the most accurate method to identify a MPNST,5 whole-body MRI with DWI and ADC mapping is an alternative technique that should be proposed as a new standard of care for patients with NF type 1.20
Conclusion
We presented six cases of patients with NF1, who underwent MRI, showing different sites of neck and thoraco-abdominal involvement and their main MRI features.
Disclosure
The authors report no conflicts of interest for this work.
References
1. Becker B, Strowd RE
2. Fortman BJ, Kuszyk BS, Urban BA, Fishman EK. Neurofibromatosis type 1: a diagnostic mimicker at CT. Radiographics. 2001;21(3):601–612. doi:10.1148/radiographics.21.3.g01ma05601
3. Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62(5):1573–1577.
4. Huson SM, Norton A. Neurofibromatosis types 1 and 2. In.: Genetic Predisposition to Cancer.
5. Cimino PJ, Gutmann DH. Neurofibromatosis type 1. In: Handbook of Clinical Neurology,
6. Drouet A. Seizures in neurofibromatosis. What is the risk? Revue Neurologique. 2011;167(12):886–896. doi:10.1016/j.neurol.2011.04.009
7. Hekmatnia A, Ghazavi A, Shooshtari MJM, Hekmatnia F, Basiratnia R. Imaging review of neurofibromatosis: helpful aspects for early detection. Iranian J Radiol. 2011;8(2):63–74.
8. Legius E, Brems H. Genetic basis of neurofibromatosis type 1 and related conditions, including mosaicism. Childs Nerv Syst. 2020;36(10):2285–2295. doi:10.1007/s00381-020-04771-8
9. Ghalayani P, Saberi Z, Sardari F. Neurofibromatosis type 1 (von Recklinghausen's disease): a family case report and literature review. Dental Res J. 2012;9(4):483–488.
10. Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genetics Med. 2010;12(1):1–11. doi:10.1097/GIM.0b013e3181bf15e3
11. Boyd KP, Korf BR, Theos A. NIH public access. J Am Acad Dermatol. 2009;61(1):1–16. doi:10.1016/j.jaad.2008.12.051
12. Available from: https://consensus.nih.gov/1987/1987Neurofibramatosis064html.htm.
13. Ortonne N, Wolkenstein P, Blakeley JO, et al. Cutaneous neurofibromas: current clinical and pathologic issues. Neurology. 2018;91(2):S5–S13. doi:10.1212/WNL.0000000000005792
14. Hassell DS, Bancroft LW, Kransdorf MJ, et al. Imaging appearance of diffuse neurofibroma. AJR Am J Roentgenol. 2008;190(3):582–588. doi:10.2214/AJR.07.2589
15. Liu Y, Jordan JT, Bredella MA, et al. Correlation between NF1 genotype and imaging phenotype on whole-body MRI: NF1 radiogenomics. Neurology. 2020;94(24):e2521–e2531. doi:10.1212/WNL.0000000000009490
16. Ghosh PS, Ghosh D. Teaching NeuroImages: MRI “target sign” and neurofibromatosis type 1. Neurology. 2012;78(9):899–900. doi:10.1212/WNL.0b013e318248df63
17. Ahlawat S, Blakeley JO, Langmead S, Belzberg AJ, Fayad LM. Current status and recommendations for imaging in neurofibromatosis type 1, neurofibromatosis type 2, and schwannomatosis. Skeletal Radiology. 2020;49(2):199–219. doi:10.1007/s00256-019-03290-1
18. Levy AD, Patel N, Dow N, Abbott RM, Miettinen M, Sobin LH. From the archives of the AFIP abdominal neoplasms in patients with neurofibromatosis Type 1: radiologic-pathologic correlation. RadioGraphics. 2005;25:455–480. doi:10.1148/rg.252045176
19. Meyer A, Billings SD. What’s new in nerve sheath tumors. Virchows Archiv. 2020;476(1):65–80. doi:10.1007/s00428-019-02671-0
20. Ahlawat S, Blakeley JO, Rodriguez FJ, Fayad LM. Imaging biomarkers for malignant peripheral nerve sheath tumors in neurofibromatosis type 1. Neurology. 2019;93(11):e1076–e1084. doi:10.1212/WNL.0000000000008092
21. Ahlawat S, Fayad LM, Khan MS, et al. Whole body MRI Committee for the REiNS International Collaboration; REiNS International Collaboration Members 2016. Current whole-body MRI applications in the neurofibromatoses: NF1, NF2, and schwannomatosis. Neurology. 2016;87(7 Suppl 1):S31–9. doi:10.1212/WNL.0000000000002929
22. Fayad LM, Blakeley J, Plotkin S, Widemann B, Jacobs MA. Whole body MRI at 3T with quantitative diffusion weighted imaging and contrast-enhanced sequences for the Characterization of peripheral lesions in patients with neurofibromatosis Type 2 and schwannomatosis. ISRN Radiol. 2013;2013:627932. doi:10.5402/2013/627932
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.