")
Back to Journals » Clinical Ophthalmology » Volume 10
Neurofibromatosis: an update of ophthalmic characteristics and applications of optical coherence tomography
Authors Abdolrahimzadeh B, Carmen Piraino D, Albanese G, Cruciani F, Rahimi S
Received 19 December 2015
Accepted for publication 6 February 2016
Published 13 May 2016 Volume 2016:10 Pages 851—860
DOI https://doi.org/10.2147/OPTH.S102830
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Dr Scott Fraser
Barmak Abdolrahimzadeh,1 Domenica Carmen Piraino,2 Giorgio Albanese,2 Filippo Cruciani,2 Siavash Rahimi3
1Polimed Beltramelli Medical Center, Rome, Italy; 2Section of Ophthalmology, Department of Sense Organs, University of Rome “Sapienza”, Rome, Italy; 3Pathology Centre, Queen Alexandra Hospital, Portsmouth, UK
Abstract: Neurofibromatosis (NF) is a multisystem disorder and tumor predisposition syndrome caused by genetic mutation on chromosome 17-17q11.2 in NF type 1 (NF1), and on chromosome 22-22q12.2 in NF type 2. The disorder is characterized by considerable heterogeneity of clinical expression. NF1 is the form with the most characteristic ocular manifestations. Lisch nodules of the iris are among the well-known diagnostic criteria for the disease. Glaucoma and associated globe enlargement have been described in a significant proportion of patients with NF1 and orbital–facial involvement. Optic nerve glioma may cause strabismus and proptosis, and palpebral neurofibroma may reach considerable size and occasionally show malignant transformation. Near infrared reflectance has greatly contributed to enhancing our knowledge on choroidal alterations in NF1. Indeed, some authors have proposed to include these among the diagnostic criteria. Optical coherence tomography has given new insight on retinal alterations and is a noninvasive tool in the management of optic nerve gliomas in children. Ocular manifestations in NF type 2 can range from early-onset cataracts in up to 80% of cases to optic nerve hamartomas and combined pigment epithelial and retinal hamartomas.
Keywords: neurofibromatosis, ophthalmic, optical coherence tomography, infrared reflectance, choroideal nodules, Lisch nodules
Introduction
Neurofibromatosis (NF) is considered one of a group of multisystem hereditary diseases that also include tuberous sclerosis, von Hippel–Lindau disease, and Sturge–Weber syndrome. This heterogeneous group of diseases has been alternatively defined as phakomatoses, neurocutaneous syndromes, tumor predisposition syndromes, and ectodermal dysplasia by different authors over the years. They include a vast range of afflictions of the skin and nervous system. The eye and the ocular adnexa may be affected by characteristic lesions that are relatively frequent, and are, therefore, considered as diagnostic hallmarks of the disease, while other ocular manifestations are rare and inconstant. A number of authors have chosen the name “disseminated hamartomatosis” because of findings of hamartoma and tumors in various organs. The associated tumors may lead to morbidity due to their progressive growth and mass effect, and although initially not characterized by aggressive histological characteristics, they can show malignant transformation in a small proportion of cases.
Since NF is characterized by considerable variability of clinical features, Riccardi1 proposed the classification of NF into seven types, but a more recent classification only includes NF type 1 (NF1) and NF type 2 (NF2), while the other types are now considered as variants. The differentiation between the types also has a genetic base. In fact, the genetic alteration in the main two variants is different, being on chromosome 17-17q11.2 in NF12 and chromosome 22-22q12.2 in NF2.2
The prevalence of NF1 is about one in 3,500 live births.1 The transmission modality is autosomal dominant; however, approximately 50% of cases are caused by spontaneous mutations, and there is considerable variability of expression between affected families. Diagnosis is usually performed clinically and is based on specific diagnostic criteria. Mutation analysis using multiple molecular diagnostic techniques for genomic DNA and RNA can also be considered in some cases, for example, in very young children when the diagnosis is suspected but not clinically confirmed. This is because many clinical features of NF1 are age dependent, and diagnosis is occasionally more challenging in the pediatric population.3
The genetic modification in NF1 leads to alteration of expression of the cytoplasmatic protein normally encoded by the NF1 gene known as neurofibromin, a 220 kDa guanosine triphosphate (GTP)ase-activating cytoplasmatic protein that is involved in cell growth regulation mechanisms by regulating the RAS protein, specifically by converting RAS-GTP active form to RAS-GDP inactive form.3,4 The consequence of the lack of neurofibromin results in an excess of the RAS-GTP active form, which promotes excessive cell growth, leading to deregulation and tumorigenesis. For this reason, NF1 is also considered as an RASopathy, which is defined as a disorder caused by a germline mutation that causes deregulation of the RAS mitogen-activated protein kinase pathway.3,4
NF1 is characterized by considerable clinical heterogeneity and age-related nature of clinical expression. The disease may also be seen as mosaicism, also called segmental NF1. In these cases, NF1 maintains the usual characteristics of the disease but is localized only in a body segment.5–7 Segmental NF1 is approximately ten times less frequent than NF1.8 In fact, prevalence of these forms is 1/36,000–40,000.9 Patients with segmental NF1 are also at relatively lower risk for life-threatening complications. As for other features of NF1, ocular findings in segmental NF, such as Lisch nodules, are found only when the ocular district is involved.
Diagnostic criteria
In NF1, the various clinical features affecting the nervous, dermatologic, ocular, and skeletal systems are usually absent at birth but develop shortly after. Approximately 95% of NF1 patients meet diagnostic criteria by age 8, and all do so by age 20. The diagnostic criteria currently used for diagnosis were presented by the National Institute of Health statement in 1987.10 An individual with at least two of the following criteria is considered affected by the disease: at least six café au lait spots with a minimum diameter of 0.5 cm prior to puberty or a minimum diameter of 1.5 cm following puberty, inguinal and/or axillary freckling, at least two Lisch nodules, optic pathway glioma, typical bony lesions such as sphenoid wing dysplasia, pseudoarthritis of the tibia, and a first-degree relative with NF1.10
Cutaneous alterations
Café au lait spots and freckling are frequently seen in NF1 patients. Café au lait spots are not present at birth but become evident at about 1 year of age; they are pigmentary lesions of the skin with well-characterized, uniform pigmentation and defined margins. Freckling is found in approximately 80% of patients under the age of 6 and in 90% of adults over age 30.11 They appear as small freckles less than 5 mm in size and are localized usually in the axillary or inguinal area (Crowes sign).
Neurofibromas, found in almost all NF1 patients over 30 years of age, are prevalently located over the trunk, with 20% seen in the head and neck region.12 They are divided into cutaneous, subcutaneous, and plexiform neurofibromas; the latter are observed in 25%–30% of cases.12,13 Histologically, neurofibromas are formed by Schwann cells, nonneoplastic fibroblast-like cells, mast cells and macrophages, and endothelial and perineural cells.11,14
Clinically, cutaneous neurofibromas are asymptomatic, soft, brown-colored tumors that range from a few millimeters to several centimeters in size. The subcutaneous variety is characterized by firm consistency, tenderness on manipulation, and a size of approximately 3–4 cm. The plexiform neurofibroma shares characteristics similar to the other types, but is more prone to transformation into variants of malignant nerve sheath tumors (7%–13% of cases – also known as MPNST tumors) such as neurofibrosarcoma or malignant schwannoma.15 With advancing age, these tumors can continue to grow to significant sizes and may physically hinder functionality. In fact, enormous neurofibromas, invading the palpebrae, have been described, and these can create functional disturbances.
Systemic and skeletal alterations
Rare systemic alterations of NF1 include cardiovascular abnormalities with hypertension, increased incidence of pheochromocytoma and aortic coarctation, neurocognitive deficits of various severities in 60% of cases,13 leukemia, and lymphoma.16
In NF1, various skeletal anomalies may be found: scoliosis in 10%–15% of patients, thinning of the long bones, pseudoarthrosis of tibia, and absence of the greater wing of the sphenoid bone in 1%–6% of patients.16
Ophthalmic and adenexal manifestations
Palpebral plexiform neurofibroma is usually monolateral, frequently affects the upper eyelid, and appears usually after 2 years of age; it has the tendency to grow and cause asymmetric ptosis associated with deformation of the palpebral margin (Figure 1). In some cases, enormous neurofibroma of the palpebra, defined palpebral elephantiasis, may result.15 The hypertrophy of the nerve fibers of the nerve sheath tumor can be occasionally felt by digital palpation during physical examination of the eyelids. Ptosis associated with facial deformation, megalophthalmous, and NF1 is known as Francois–Katz syndrome.15 An unusually rapid growth, increase in tenderness, or bleeding, warrants further investigation to rule out malignant transformation. These palpebral plexiform neurofibromas have a propensity to relapse, and surgical operations are complicated with profuse bleeding with disappointing results.17–19
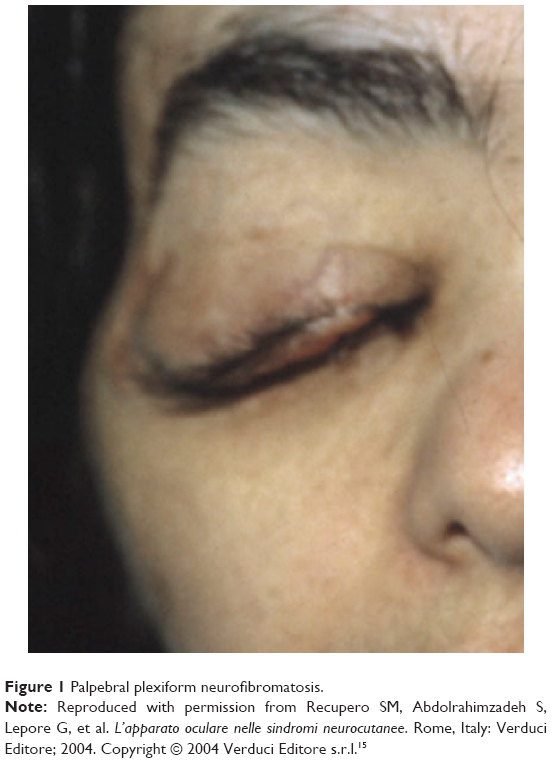 | Figure 1 Palpebral plexiform neurofibromatosis. |
Other rare ocular findings in NF1 include conjunctival neurofibroma (Figure 2) and a characteristic hypertrophy of corneal intrastromal nerves known as “lignes grise”.20
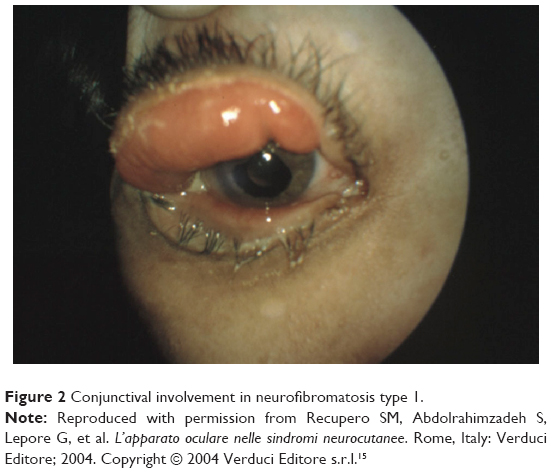 | Figure 2 Conjunctival involvement in neurofibromatosis type 1. |
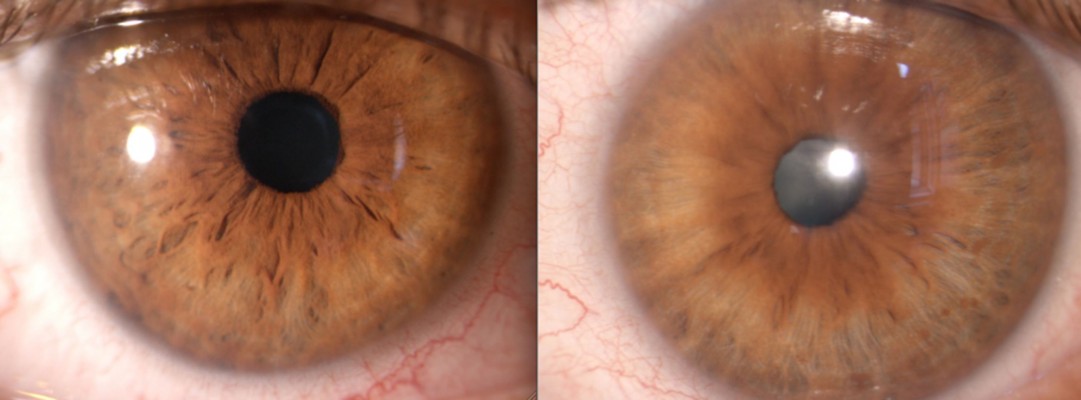
Iris hamartoma in NF1 known as Lisch nodules21 are unusual before 2 years of age, may be occasionally observed before 6 years, but their prevalence, number, and dimensions in individual cases increase considerably with age.22,23 Lewis and Riccardi24,25 described the case of a woman with Lisch nodules as the sole clinical manifestation of NF1 other than having two sons with the disorder. Histologically, Lisch nodules are melanocytic hamartomas composed of melanocytes, elongated fibroblasts, and mast cells. Mast cells have been found to be present both in neurofibromas and Lisch nodules.26 Lisch nodules do not represent a cause of morbidity or disability, but they are diagnostically significant, being one of the principal hallmark manifestations of NF1.8 Slit-lamp observation reveals characteristic nodules with dimensions of approximately 2 mm, absence of vasculature, and a certain chromatic variability ranging from white to yellow or brown15,23 (Figure 3).
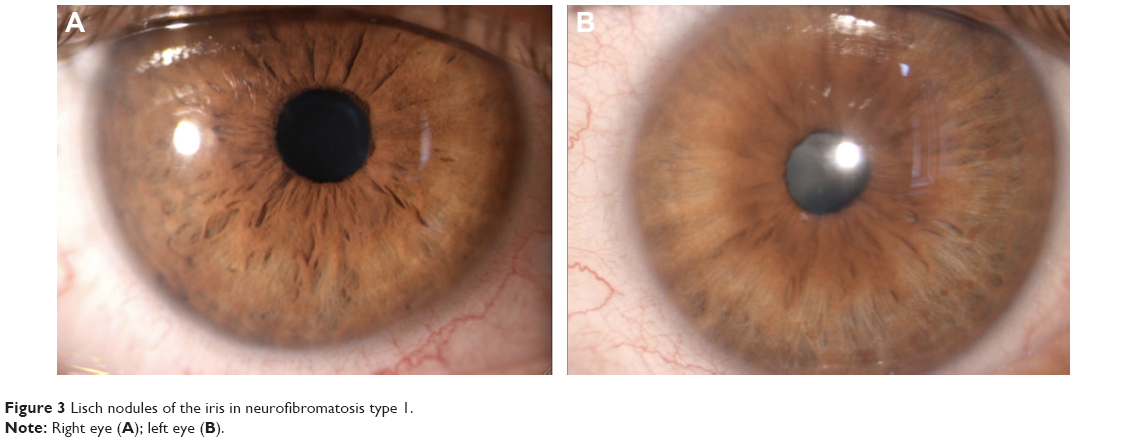 | Figure 3 Lisch nodules of the iris in neurofibromatosis type 1. |
Glaucoma associated with NF112,27,28 must be evaluated by the ophthalmologist during clinical examination. It has been reported in 1/300 NF1 patients.29 Recent studies have made a distinction between NF1 patients with or without orbital–facial involvement (such as plexiform palpebral neurofibroma), and a finding of glaucoma was seen in 23% of the patients with orbital–facial NF1.30 Asymmetric globe enlargement in ipsilateral orbital–facial NF1 was found in patients with glaucoma, with axial lengths ranging from 26 to 36 mm; difference with the contralateral unaffected eye was of 4–6 mm.29,30 Interestingly, this study revealed slight ocular enlargement in oculofacial NF1, even in eyes with normotensive intraocular pressure; indeed, it has been speculated that megalophthalmous may be present in NF1 irrespective of ocular hypertension.30 During clinical evaluation of patients with glaucoma, ultrasound biomicroscopy and anterior chamber optical coherence tomography (OCT) is fundamental to evaluate the ciliary body and anterior chamber31–34 (Figure 4).
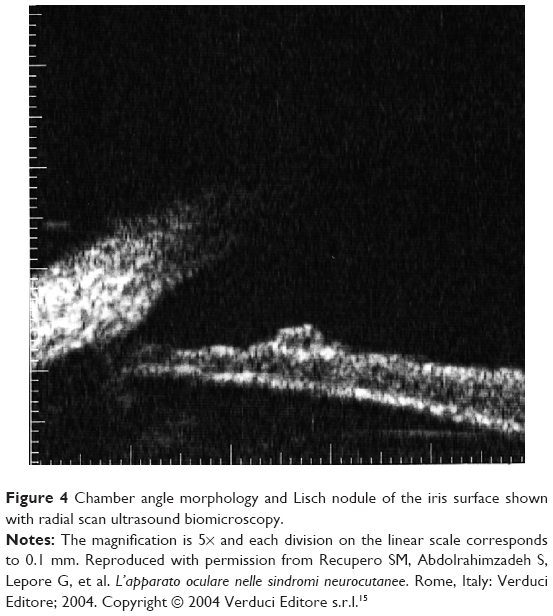 | Figure 4 Chamber angle morphology and Lisch nodule of the iris surface shown with radial scan ultrasound biomicroscopy. |
The ciliary body may show thickening, and there may be signs of angle infiltration by neurofibroma or Lisch nodules.31–33 Glaucoma evident at birth usually suggests congenital abnormality of the angle, while onset at a later date suggests involvement of the anterior angle secondary to NF115,35,36 (Figure 4).
Assessment for glaucoma, including intraocular pressure evaluation, gonioscopy, visual field examination, and optic nerve assessment, is advisable in patients with NF1.37–40
Choroidal abnormalities of NF1 are undetectable by fundus biomicroscopic examination or fluorescein angiography. Rescaldani et al41 used indocyanine green angiography to study the choroid in NF1 patients and showed that there were hypofluorescent areas in the early phases of angiography corresponding to choroidal nodules and speculated that there could be delayed perfusion of the choriocapillaris in these areas. Yasunari et al42 first described the choroidal findings with the use of confocal microscopy using infrared light and found numerous bright patchy areas, especially localized in the posterior pole of the fundus. These authors also performed indocyanine green angiography and found that these bright areas corresponded to hypofluorescence.
More recently, the use of new noninvasive technology such as near infrared reflectance (NIR) and OCT has made it possible to better evaluate choroidal alterations. Viola et al43 evaluated choroidal abnormalities in NF1 patients using NIR in a large group of patients and found nodules in 82% of patients. They found a diagnostic sensitivity of 83% and a specificity of 96%, with a cut-off value of 1.5 nodules (Figure 5). Nakakura et al44 found that most choroidal nodules were located at the posterior pole especially in pediatric patients and also found that there was a higher number of choroidal alterations in the arcade regions and that the extent of choroidal alterations increased with age.
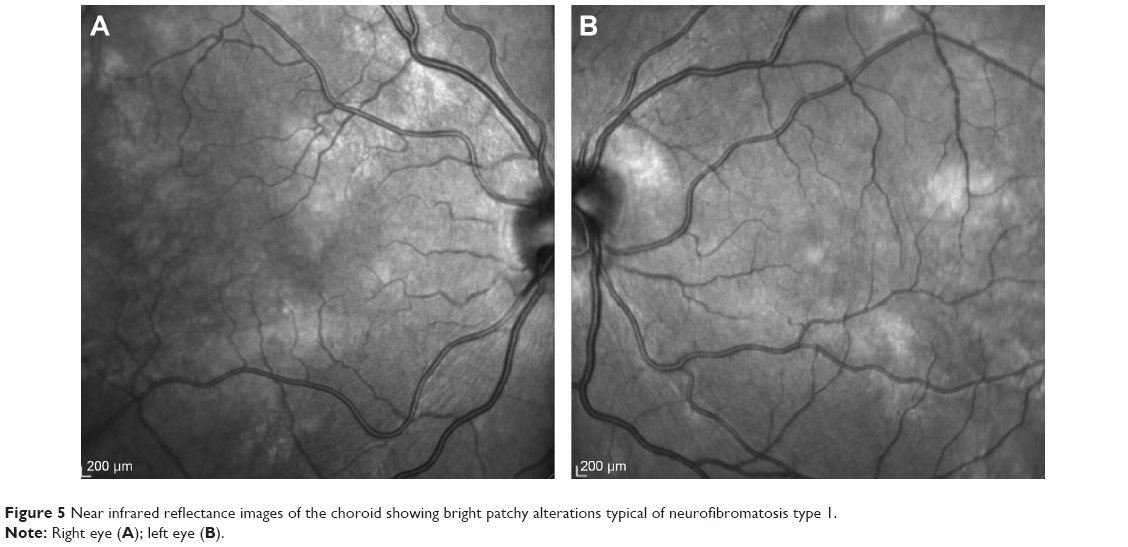 | Figure 5 Near infrared reflectance images of the choroid showing bright patchy alterations typical of neurofibromatosis type 1. |
OCT uses an 830 nm near infrared light source and measures light waves reflected from and scattered by the tissues of the eye, enabling the acquisition of cross-sectional, high-quality in vivo images with accurate differentiation of the retinal layers.45 Furthermore, enhanced depth imaging (EDI) in spectral domain OCT sets the peak sensitivity, or zero-delay line, behind the retinal pigment epithelium and enables the study of the choroid.46
In a recent investigation, using spectral domain technology, choroidal nodules were studied both with NIR imaging and cross-sectional EDI-OCT images, and the authors showed that NIR detected choroidal alterations corresponding to two types of hyperreflective choroidal nodules that were either “dome-shaped” or had “placoid” morphology.47 Furthermore, as the spectral domain software enables segmentation measurements, the authors evaluated choroidal and individual retinal layer thicknesses in patients with NF1 and reported a reduction of the mean choroidal thickness. Interestingly, the neuroepithelium, photoreceptor-retinal pigment epithelium, and outer nuclear layer were also reduced in thickness in NF1 patients compared to healthy control subjects. The authors speculated that altered choroidal circulation, due to the presence of nodules, could be the cause of retinal thinning. A number of other case reports have also shown choroidal and retinal thickness alterations in NF1.48,49
Retinal alterations in NF1 have not been frequently reported. There are a few reports on cases with retinal hamartomas also involving the retinal pigment epithelium.50 Muci-Mendoza et al51 reported retinal microvessel alterations or “corkscrew” vessels in 38% of 32 patients with NF1. These were described as second- or third-order microvenules more frequently found close to the temporal veins (Figure 6). These alterations, confirmed with fluorescein angiography, did not show leakage phenomena. They had a morphological spectrum from very small subtle microvascular alterations to corkscrew-shaped vessels or larger microvessel venous anastomoses. Similar retinal alterations were also recently reported in another study in 35% of 17 patients; the microvascular abnormalities were found overlying choroidal nodules as demonstrated with NIR imaging of the fundus. The authors speculated that neural crest alterations in NF1 can cause functional abnormalities in autonomic vasomotor nerve cells, leading to the formation of retinal microvessel abnormalities.52 Makino et al53 reported a patient with retinal microvascular abnormalities linked with congenital retinal macrovessels across the macula, showing dynamic changes over time.
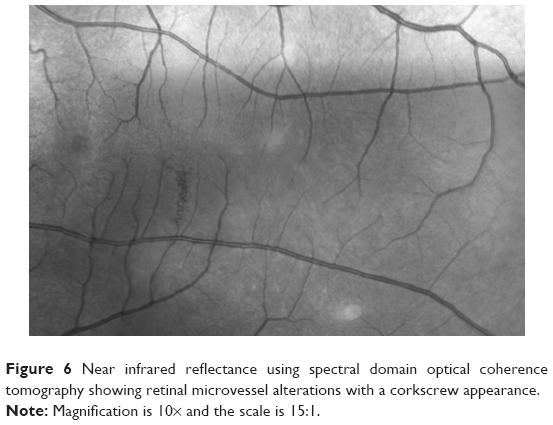 | Figure 6 Near infrared reflectance using spectral domain optical coherence tomography showing retinal microvessel alterations with a corkscrew appearance. |
Optic pathway glioma has been described in 15%–25% of patients with NF1; they usually develop at an early age, typically from age 3 to 5 years. These are actually low-grade tumors and do not possess malignant histological characteristics, nor do they show tendency to malignant transformation. They are classified as WHO Grade 1 pilocytic astrocytoma.54 These frequently arise within the optic pathway, optic nerve, optic chiasma, and postchiasmatic radiations, but may arise in 25% of cases in the brainstem.54–56 Essentially, clinical consequences of these tumors are caused by their volume-occupying characteristics, so depending on location and size, various consequences can follow. Orbital localization at the optic nerve may cause proptosis, strabismus, edema of the papilla, visual field defects, and reduction of visual acuity. Intracranial localization may cause general neurological symptoms of mass-occupying lesions associated with more specific signs based on the localization of tumor, such as visual field alterations, pupillary defects, and puberal precocity. Magnetic resonance imaging is, therefore, indicated in these NF1 cases to rule out progression and to follow-up intracranial/intraorbital tumors.
Visual acuity and visual field analysis cannot be immediately evaluated in young and uncooperative children.57,58 Some authors have used visual-evoked potentials to objectively evaluate the visual pathways and to detect vision variations in NF1.59 However, this testing is not sensitive or specific and is technically stringent; thus, evidence that is reliable in evaluating vision loss in children with NF1 is lacking.55
OCT also allows evaluation of the peripapillary retinal nerve fiber layer (RNFL). Novel research suggests that peripapillary RNFL alterations are detected with OCT earlier than with conventional methods such as visual field testing.60 Furthermore, the peripapillary RNFL can be compared with the macular RNFL thickness and ganglion cell layer thickness to potentially monitor disease progression. Using OCT RNFL thickness analysis, some authors have reported a reduced thickness of the peripapillary RNFL in children affected with NF1 who have optic nerve gliomas.61–63 Gu et al64 also showed a thinning of the ganglion cell layer-inner plexiform layer in children with optic nerve gliomas. Furthermore, Avery et al65 showed that handheld OCT gives highly reproducible thickness measurement of the ganglion cell layer-inner plexiform layer in children with optic pathway gliomas.
Recently, spectral domain OCT was used in adult NF1 patients without optic pathway glioma to evaluate the peripapillary RNFL using the circular diameter scan on the optic disc. The authors also evaluated the thickness of the macular RNFL and the ganglion cell-inner plexiform layer using the spectral domain software, which allows manual segmentation thickness measurements. These authors reported thinning of the peripapillary and macular RNFL and the ganglion cell-inner plexiform layer in adult patients with NF1 compared to healthy controls, which suggested neuronal and axonal loss.66 Figure 7 shows evaluation of the peripapillary RNFL in a patient with NF1.
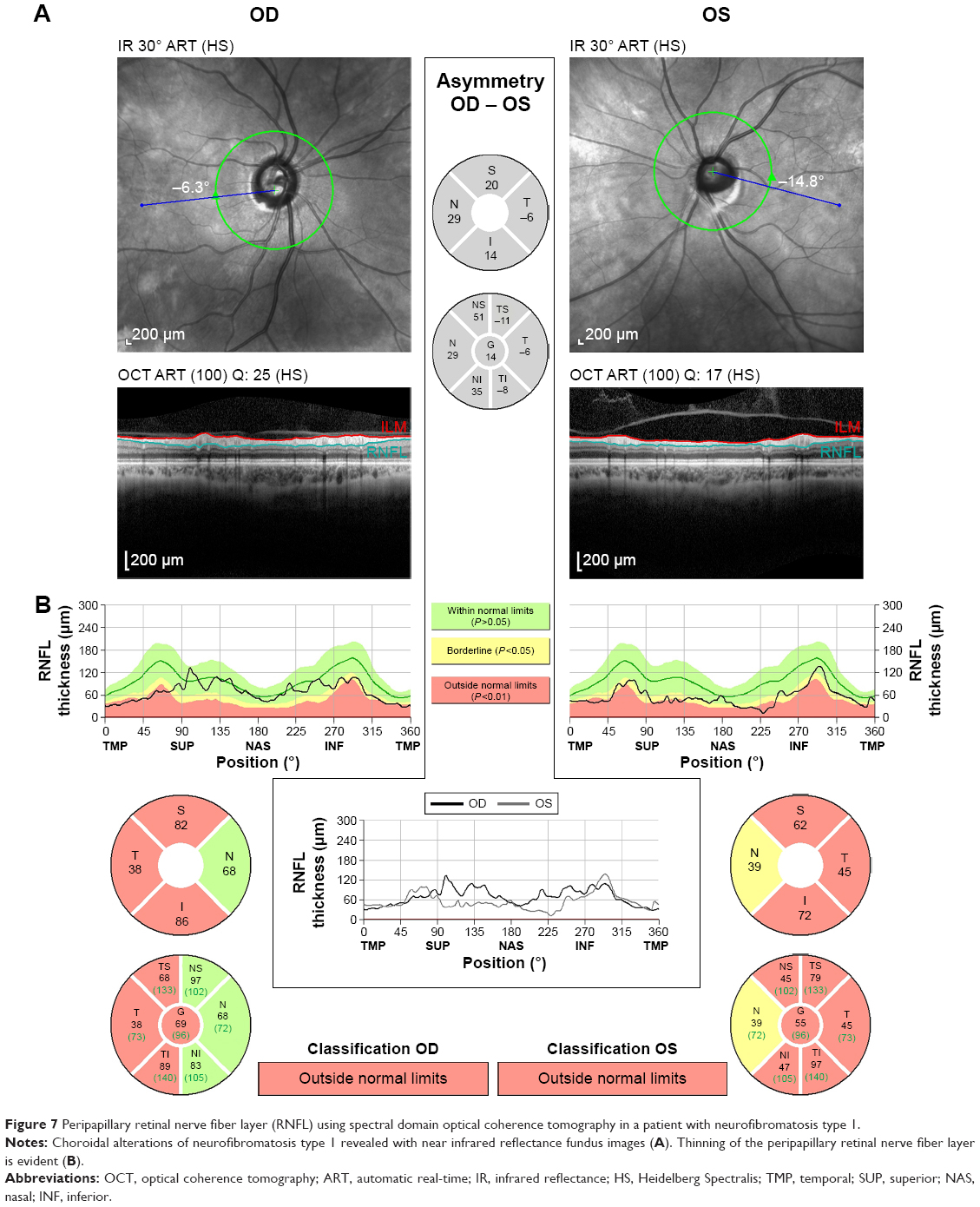 | Figure 7 Peripapillary retinal nerve fiber layer (RNFL) using spectral domain optical coherence tomography in a patient with neurofibromatosis type 1. |
Segmental NF1
NF1 patients may present different phenotypes such as Noonan syndrome and segmental NF1. Noonan syndrome is characterized by ocular hypertelorism, low-set ears, downslanting palpebral fissures and may occur in almost 12% of NF1 patients.67 Segmental NF1 is diagnosed in patients with both parents unaffected and with clinical NF1 features localized to one part of the body in a circumscribed, patchy, or linear arrangement, or in localized regions or organs.68–70 Unilateral Lisch nodules with no other clinical manifestations have also been described in isolated case reports of segmental NF1.71
Neurofibromatosis type 2
NF2 is also an autosomal dominant disease caused due to a mutation in the gene for NF2, which is located on chromosome 22-22q12.2.72 This gene encodes for merlin, which is a protein that assists in linking actin cytoskeleton to the surface of glycoproteins and has an important function in remodeling cells and regulating growth.73 There is a 50% chance of transmission from an affected individual to their offspring, but 50%–60% of patients have no family history, having instead de novo mutations.16 Clinical characteristics of the condition may include bilateral vestibular schwanomas, multiple meningiomas, cranial nerve tumors, spinal tumors, and eye abnormalities.
The most serious ocular findings are optic nerve hamartomas and hamartomas of the retina (combined pigment epithelial and retinal hamartomas)16,74 (Figures 8 and 9).
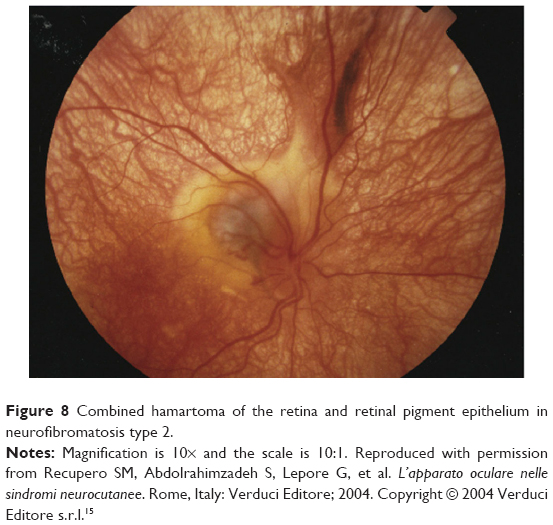 | Figure 8 Combined hamartoma of the retina and retinal pigment epithelium in neurofibromatosis type 2. |
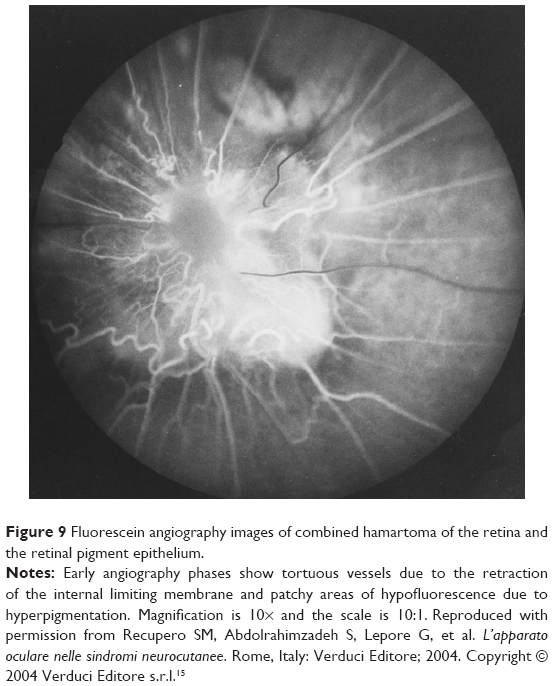 | Figure 9 Fluorescein angiography images of combined hamartoma of the retina and the retinal pigment epithelium. |
A large proportion (60%–80%) of patients have early-onset cataracts, usually with an onset of posterior subcapsular lenticular opacities or cortical wedge opacities.74
Fluorescein angiography images of combined hamartomas of the retina and the retinal pigment epithelium can provide further details. Early angiography phases show tortuous vessels due to the retraction of the internal limiting membrane and patchy areas of hypofluorescence due to hyperpigmentation.16
Other sporadic findings in NF2 are as follows: dystrophic keratitis due to fifth cranial nerve involvement or facial paresis due to seventh cranial nerve involvement.75
Patients with a diagnosis of NF2 should undergo magnetic resonance imaging of the brain to better evaluate any tumor involvement. Over the past decades, radiation therapy was used in the management of NF2-related tumors; today, surgery is considered the standard treatment even if it is not always possible or advisable to remove all lesions.76 Since the introduction of anti-vascular endothelial growth factors (anti-VEGF), this treatment has been used in a broad range of pathologies.77–80 Studies with bevacizumab suggest that anti-VEGF therapies may cause significant reductions in tumor volume and significant improvement in hearing function in NF2 patients.81
Conclusion
Since the description by von Recklinghausen in 1882,82 NF1 has been extensively studied. NF1 together with other phakomatoses such as von Hippel’s disease and sclerosis tuberous complex are now known to be caused by alterations of tumor suppressor genes. However, many questions about these diseases remain unanswered; the role of the neural crest and why different types of neurocutaneous conditions occur together in the same individual, still need clarification. Recent novel molecular genetics studies have excluded Sturge–Weber syndrome from the group of phakomatoses,83–85 but further advances in the field of molecular genetics are necessary and can certainly contribute to the diagnosis and management of NF.
In ophthalmology, we believe that the advent of new diagnostic instruments such as near infrared imaging and OCT have provided new insight to study the ophthalmic manifestations of the disease.
Disclosure
The authors report no conflicts of interest in this work.
References
Riccardi VM. Neurofibromatosis: past, present and future. N Engl J Med. 1991;324:1238–1285. | ||
Jett K, Friedman MJ. Clinical and genetic aspects of neurofibromatosis 1. Genet Med. 2010;12:1–11. | ||
Schnur RE. Type 1 neurofibromatosis: a genio-oculo- dermatologic update. Ocular Genetics. 2012;23:364–372. | ||
Rauen KA, Huson SM, Burkitt-Wright E, et al. Recent developments in neurofibromatoses and RASopathies: management, diagnosis and current and future therapeutic avenues. Am J Med Genet. 2014;12:1–10. | ||
Abdolrahimzadeh S, Piraino DC, Plateroti R, et al. Ocular alterations in a rare case of segmental neurofibromatosis type 1 with a non-classified mutational variant of the NF1 gene. Ophthalmic Genetics. 2016;37(2):214–216. | ||
Tinschert S, Naumann I, Stegmann E, et al. Segmental neurofibromatosis caused by a somatic mutation of the neurofibromatosis type1 (NF1) gene. Eur J Hum Genet. 2000;8:455–459. | ||
Vandenbroucke I, Van Doom R, Callens T, et al. Genetic and clinical mosaicism in a patient with neurofibromatosis type 1. Hum Genet. 2004;114:284–290. | ||
Ingordo V, D’Andria G, Mendicini S, et al. Segmental neurofibromatosis: is it uncommon or undiagnosed? Arch Dermatol. 1995;131:959–960. | ||
Gammal JA. Localized neurofibromatosis. Arch Dermatol. 1931;24:712–713. | ||
NIH Consensus Development Conference. Neurofibromatosis. Conference statement. Arch Neurol. 1988;45:575–578. | ||
Guttmann DH, Aylswort A, Carey CJC, et al. The diagnosis, evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA. 1997;278:51–57. | ||
Huson SM, Harper PS. Compston DA. Von Recklinghausen neurofibromatosis. A clinical and population study in South-East Wales. Brain. 1988;111:1355. | ||
Kandt RS. Tuberous sclerosis complex and neurofibromatosis type 1: the two most common neurocutaneous diseases. Neurol Clin. 2003;21:983. | ||
Swick B. Floret like multinucleated giant cells in a neurofibromatosis type 1 associated neurofibroma. Am J Dermatopathol. 2008;30:632–634. | ||
Recupero SM, Abdolrahimzadeh S, Lepore G, et al. L’apparato oculare nelle sindromi neurocutanee. [The ocular apparatus in neurocutaneous syndromes]. Rome, Italy: Verduci editore; 2004:7–26. Italian. | ||
Hirbe AC, Gutmann DH. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol. 2014;13:834–843. | ||
Coban-Karatas M, Altan-Yaycioglu R, Bal N, Akova YA. Management of facial disfigurement in orbitotemporal neurofibromatosis. Ophthalm Plas Reconstr Surg. 2010;26:124–126. | ||
Balacco Gabrieli C, Recupero SM, Contestabile MT, et al. Fox’s modified technique in the management of blepharoptosis. Ophthalmic Surg Lasers. 1996;27:1–5. | ||
Scuderi N, Chiummariello S, De Gado F, et al. Surgical correction of blepharoptosis using the levator aponeurosis-Muller’s muscle complex readaptation technique: a 15-year experience. Plast Reconstr Surg. 2008;121:71–78. | ||
Lewis RA, Riccardi VM. Von Recklinghausen neurofibromatosis. Incidence of iris hamartoma. Ophthalmology. 1981;88:348–354. | ||
Lisch K. Ueber Beteiligung der Augen, inbesondere das Vorkommen von irisnotchen bei der Neurofibromatose (Recklinghausen). [Ocular involvement and incidence of iris nodules in neurofibromatosis (Recklinghausen)]. Z Augenheilkd. 1937;93:137–143. German. | ||
Recupero SM, Plateroti R, Abdolrahimzadeh S, et al. Lisch nodules in neurofibromatosis type 1: relationship to age and cutaneous neurofibromas. Ann ophthalmol – Glaucoma. 1996;28:178–183. | ||
Ragge NK, Falk RE, Cohen WE, Murphree AL. Images of Lisch nodules across the spectrum. Eye (Lond). 1993;7:75–101. | ||
Lewis RA, Riccardi VM. Penetrance of von Recklinghausen neurofibromatosis: a distinction between predecessors and descendants. Am J Hum Genet. 1988;42:284–289. | ||
Lewis RA, Riccardi VM. Neurofibromatosis classification clarified. Ophthalmology. 1989;92:1123–1124. | ||
Richetta A, Giustini S, Recupero, et al. Lisch nodules of the iris in neurofibromatosis type 1. J Eur Acad Dermatol Venereol. 2004;18:342–344. | ||
Bardelli AM, Federico A, Trasversi C, Fabrizi GM. L’ Enigma genetico della neuropatia ottica di Leber e della neurofibromatosi di Recklinghausen. [The genetic enigma in Leber’s optic neuropathy and Recklinghausen’s neurofibromatosis]. Boll Ocul. 1992;71:99–114. Italian. | ||
Mantelli F, Abdolrahimzadeh S, Mannino G, Lambiase A. Unusual case of angle closure glaucoma in a patient with neurofibromatosis type 1. Case Rep Ophthalmol. 2014;5:386–391. | ||
Grant WM, Walton DS. Distinctive gonioscopic findings in glaucoma due to neurofibromatosis. Arch Ophthalmol. 1968;79:127–134. | ||
Morales J, Imtiaz AC, Bosley TM. Glaucoma and globe enlargement associated with neurofibromatosis type1. Ophthalmology. 2009;116:1725–1730. | ||
Nolan W. Anterior segment imaging: ultrasound biomicroscopy and anterior segment optical coherence tomography. Curr Opin Ophthalmol. 2008;19:115–121. | ||
Mannino G, Malagola R, Abdolrahimzadeh S, et al. Ultrasound biomicroscopy of degenerative retinoschisis and allied alterations of the ciliary body. Br J Ophthalmol. 2001;85:976–982. | ||
Fries U, Makabe R, Ohrloff C. Ultrasound biomicroscopy (UBM) in the ciliary body region. Spektrum der Augenheilkunde. 1995;9(3):131–133. | ||
Cruciani F, Lorenzatti M, Nazzaro V, Abdolrahimzadeh S. Bilateral acute angle closure glaucoma and myopia induced by topiramate. La Clinica Terapeutica. 2009;160:215–216. | ||
Emre S, Palamar M, Ulusoy MO, Gençoğlan G. Ciliary body cysts in neurofibromatosis: a new coexistence? Graefes Arch Clin Exp Ophthalmol. 2012;250:857–861. | ||
Al Freihi SH, Edward DP, Nowilaty SR, Abouammoh MA, Morales J. Iris neovascularization and neovascular glaucoma in neurofibromatosis type 1: report of 3 cases in children. J Glaucoma. 2013;22:336–341. | ||
Feng CS, Jin K, Yi K, Choi DG. Comparison of intraocular pressure measurements obtained by rebound, noncontact, and Goldman applanation tonometry in children. Am J Ophthalmol. 2015;160:937–943. | ||
Patel DE, Cumberland PM, Walters BC, Russell-Eggitt I, Cortina-Borja M, Rahk JS; OPTIC Study Group. Study of optimal perimetric testing in children (OPTIC): normative visual field values in children. Ophthalmology. 2015;122:1711–1717. | ||
Recupero SM, Contestabile MT, Taverniti L, et al. Open-angle glaucoma: variations in the intraocular pressure after visual field examination. J Glaucoma. 2003;12:114–118. | ||
Iester M, Perdicci A, Capris E, et al. Comparison between discriminant analysis models and glaucoma probability score for the detection of glaucomatous optic nerve head changes. J Glaucoma. 2008;17:535–540. | ||
Rescaldani C, Nicolini P, Fatigati G, Bottoni FG. Clinical application of digital indocyanine green angiography in choroidal neurofibromatosis. Ophthalmologica. 1998;212:99–104. | ||
Yasunari T, Shiraki H, Hattori K, Miki T. Frequency of choroidal abnormalities in neurofibromatosis type 1. Lancet. 2000;356:988–992. | ||
Viola F, Villani E, Natacci F, et al. Choroidal abnormalities detected by near-infrared reflectance imaging as a new diagnostic criterion for neurofibromatosis 1. Ophthalmology. 2012;119:369–375. | ||
Nakakura S, Shiraki K, Yasunari T, et al. Quantification and anatomic distribution of choroidal abnormalities in patients with type I neurofibromatosis. Graphes Arch Clin Exp Ophthalmol. 2005;243:980–984. | ||
Geitzenauer W, Hitzenberger CK, Schmidt-Erfurth UM. Retinal optical coherence tomography: past, present and future perspectives. Br J Ophthalmol. 2011;95:171–177. | ||
Margolis R, Spaide RF. A pilot study of enhanced depth imaging optical coherence tomography of the choroid in normal eyes. Am J Ophthalmol. 2009;147:811–815. | ||
Abdolrahimzadeh S, Felli L, Plateroti R, et al. Morphologic and vasculature features of the choroid and associated choroid-retinal thickness alterations in neurofibromatosis type 1. Br J Ophthalmol. 2015;99:789–793. | ||
Ayata A, Unal M, Ersauli D, Tatlipinar S. Near infrared fluorescence and OCT features of choroidal abnormalities in type 1 neurofibromatosis. Child Exp Ophthalmol. 2008;36:390–392. | ||
Rao RC, Choudry N. Enhanced depth imaging spectral domain optical coherence tomography finding in choroidal neurofibromatosis. Ophthalmic Surg Lasers Imaging. 2014;45:466–468. | ||
Destro M, D’ Amico DJ, Gragoudas ES, et al. Retinal manifestations of Neurofibromatosis diagnosis and management. Arch Ophthalmol. 1991;109:662–666. | ||
Muci-Mendoza R, Ranella M, Fuenmayor-rivera D. Cork screw retinal vessels in neurofibromatosis type 1: report of 12 cases. Br J Ophthalmol. 2002;86:282–284. | ||
Abdolrahimadeh S, Felli L, Piraino DC, et al. Retinal microvascular abnormalities overlying choroidal nodules in neurofibromatosis type 1. BMC Ophthalmol. 2014;14:146. | ||
Makino S, Hendo K, Tampo H. Retinal microvascular abnormalities in neurofibromatosis type 1 associated with congenital retinal macrovessels. Case Rep Ophthalmol Med. 2013;2013:604191. | ||
Chen YH, Gutmann DH. Review – the molecular and cell biology of pediatric low grade glioma. Oncogene. 2014;33:2019–2026. | ||
Listernick R, Ferner RE, Liu GT, Gutmamm DH. Optic pathway glioma in neurofibromatosis 1. Controversies and recommendations. Am Neurol. 2007;61:189–198. | ||
Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 classification of tumors of the central nervous system. Acta Neuropathol. 2007;114:97–109. | ||
Koenraads Y, Braun KP, van der Linden DG, et al. Perimetry in young and neurologically impaired children: the Behavioral Visual Field (BEFIE) Screening Test revisited. JAMA Ophthalmol. 2015;133:319–325. | ||
Contestabile MT, Perdicchi A, Amodeo S, et al. The influence of learning effect on frequency doubling technology perimetry (Matrix). J Glaucoma. 2007;16:297–301. | ||
Wolsey DH, Larson SA, Creel D, Hoffman R. Can screening for optic nerve gliomas in patients with neurofibromatosis type 1 be performed with visual-evoked potential testing? J AAPOS. 2006;10:307–311. | ||
Enomoto N, Anaraku A, Ishida K, et al. Size of the optic nerve head and its relationship with the thickness of the macular ganglion cell complex and peripapillary retinal nerve fiber layer in patients with primary open angle glaucoma. J Ophthalmol. 2015;2015:186249. | ||
Chang L, El-Dairi MA, Frempong TA, et al. Optical coherence tomography in the evaluation of neurofibromatosis type-1 subjects with optic pathway gliomas. J AAPOS. 2010;14:511–517. | ||
Topcu-Yilmaz P, Kasim B, Kiratli H. Investigation of retinal nerve fiber layer thickness in patients with neurofibromatosis-1. Jpn J Ophthalmol. 2014;88:172–176. | ||
Parrozzani R, Clementi M, Kotsafti O, et al. Optical coherence tomography in the diagnosis of optic pathway gliomas. Invest Ophthalmol Vis Sci. 2013;54:8112–8118. | ||
Gu S, Glaug N, Cnaan A, et al. Ganglion cell layer-inner plexiform layer thickness and vision loss in young children with optic pathway gliomas. IOVS. 2014;55:1402–1408. | ||
Avery RA, Cnaan A, Schuman JS, et al. Intra- and inter-visit reproducibility of ganglion cell-inner plexiform layer measurements using handheld optical coherence tomography in children with optic pathway gliomas. Am J Ophthalmol. 2014;158:916–923. | ||
Abdolrahimzadeh S, Felli L, Plateroti AM, et al. Spectral domain optical coherence tomography evidence of retinal nerve fiber layer and ganglion cell loss in adult patients with neurofibromatosis type 1. Retina. 2015;36:75–81. | ||
De Luca A, Bottillo I, Sarkozy A, et al. NF1 gene mutations represent the major molecular event underlying neurofibromatosis-Noonan syndrome. Am J Hum Genet. 2005;77:1092–1101. | ||
Ruggieri M, Pavone P, Polizzi A, et al. Ophthalmological manifestation in segmental neurofibromatosis type 1. Br J Ophthalmol. 2004;88:1429–1433. | ||
Tanito K, Arihito Ota, Ryoichi K, et al. Clinical features of 58 Japanese patients with mosaic neurofibromatosis 1. J Dermatol. 2014;41:724–728. | ||
Pisani V, Bonifazi E, Caniglia A. Segmental neurofibromatosis type 1. Differences between the childhood and adult form. Eur J Ped Ophthalmol. 2012;22:257–262. | ||
Nicita F, Iannetti L, Spalice A, et al. Unilateral Lisch nodules in a 47-year-old woman without other stigmata of neurofibromatosis type I: an example of segmental neurofibromatosis? Ophthalmic Genet. 2013;34:178–179. | ||
Wertelecki W, Rouleau GA, Superneau DW, et al. Neurofibromatosis 2: clinical and DNA linkage studies of a large kindred. N Eng J Med. 1988;319:278–283. | ||
Chan JW. Neuro-ophthalmic features of the neurocutaneous syndromes. Int Ophthalmol Clin. 2012;52:73–85. | ||
Evans GR, Lloyd SK, Ramsden RT. Neurofibromatosis type 2. Adv Otorhinolaryngol. 2001;70:91–98. | ||
Yu CBO, Lyons CJ, Dorrell ED, Taylor D. Congenital cavernous sinus syndrome in a child with neurofibromatosis type 2. Can J Ophthalmol. 1999;34:101–103. | ||
Ulmann EJ, Plotkin SR. Neurofibromatoses. Adv Exp Med Biol. 2012;724:266–277. | ||
Schmidt-Erfurth U, Chong V, Loewenstein A, et al. Guidelines for the management of age related macular degeneration by the European Society of Retina Specialists (EURETINA). Br J Ophthalmol. 2014;78:1144–1167. | ||
Gharbiya M, Parisi F, Cruciani F, et al. Intravitreal antivascular endothelial growth factor for retinal angiomatous proliferation in treatment-naive eyes: long term functional and anatomical results using a modified PrONTO-style regimen. Retina. 2014;34:298–305. | ||
Lambiase A, Abdolrahimzadeh S, Recupero SM. An update on intravitreal implants in use for eye disorders. Drugs Today. 2014;50:239–249. | ||
Fenicia V, Abdolrahimzadeh S, Mannino G, et al. Intravitreal bevacizumab in the successful management of choroidal metastases secondary to lung and breast cancer unresponsive to systemic therapy: a case series. Eye (Lond). 2014;28:888–891. | ||
Plotkin SR, Stemmer-Rachamimov AO, Barker FG II, et al. Hearing improvement after bevacizumab in patients affected by neurofibromatosis type 2. N Engl J Med. 2009;361:358–367. | ||
von Recklinghausen FD. Ueber die multiplen Fibrome der Haut und ihre Beziehung zu den multiplken Neuromen. Festschrift zur Feier des funfundzwanzigjahrigen Bestchens des pathologischen Institutes zu Berlin; Herrn Rudolf Virchow dargebracht. [On multiple fibromas of the skin and their relationship to multiple neuromas. 25th anniversary of the founding of the pathological institute of Berlin, presented to professor Rudolf Virchow]. Berlin: Hirschwald; 1882. German. | ||
Recupero SM, Abdolrahimzadeh S, DeDominicis M, Mollo R. Sturge-Weber syndrome associated with nevus of Ota. Eye. 1998;12:212–213. | ||
Riley FC, Campbell RJ. Double phakomatosis. Arch Ophthalmol. 1979;97(3):518–520. | ||
Rodriguez-Bujaldon AL, Vazquez-Bayo C, Jimenez-Puya RJ, Moreno-Gimenez JC. Sturge-Weber syndrome and type1 neurofibromatosis. Acta Dermosifiliogr. 2008;99:313–314. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The full terms of this license are available at https://www.dovepress.com/terms.php and incorporate the Creative Commons Attribution - Non Commercial (unported, v3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted without any further permission from Dove Medical Press Limited, provided the work is properly attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.