Back to Journals » Eye and Brain » Volume 13
Neuro-Ophthalmological Manifestations of Craniosynostosis: Current Perspectives
Authors Duan M, Skoch J ![]() , Pan BS
, Pan BS ![]() , Shah V
, Shah V
Received 16 September 2020
Accepted for publication 12 November 2020
Published 29 January 2021 Volume 2021:13 Pages 29—40
DOI https://doi.org/10.2147/EB.S234075
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Margaret Wong-Riley
Michael Duan,1 Jesse Skoch,2 Brian S Pan,3 Veeral Shah4,5
1Baylor College of Medicine, School of Medicine, Houton, TX, USA; 2Cincinnati Children’s Hospital Medical Center, Division of Pediatric Neurosurgery, Cinicinnati, OH, USA; 3Cincinnati Children’s Hospital Medical Center, Division of Plastic Surgery, Cinicinnati, OH, USA; 4Cincinnati Children’s Hospital Medical Center, Division of Pediatric Ophthalmology, Cinicinnati, OH, USA; 5University of Cincinnati, Department of Ophthalmology, Cincinnati, OH, USA
Correspondence: Veeral Shah
Cincinnati Children’s Hospital Medical Center, Abrahamson Pediatric Eye Institute/Division of Pediatric Ophthalmology, Cinicinnati, OH, USA
Tel +1 513-803-2230
Fax +1 513-558-351
Email [email protected]
Abstract: Craniosynostosis, a premature fusion of cranial sutures that can be isolated or syndromic, is a congenital defect with a broad, multisystem clinical spectrum. The visual pathway is prone to derangements in patients with craniosynostosis, particularly in syndromic cases, and there is a risk for permanent vision loss when ocular disease complications are not identified and properly treated early in life. Extensive advancements have been made in our understanding of the etiologies underlying vision loss in craniosynostosis over the last 20 years. Children with craniosynostosis are susceptible to interruptions in visual input arising from strabismus, refractive errors, and corneal damage; any of these aberrations can result in understimulation of the visual cortex during childhood neurodevelopment and permanent amblyopia. Elevated intracranial pressure resulting from abnormal cranial shape or volume can lead to papilledema and, ultimately, optic atrophy and vision loss. A pediatric ophthalmologist is a crucial component of the multidisciplinary care team that should be involved in the care of craniosynostosis patients and consistent ophthalmologic follow-up can help minimize the risk to vision posed by such entities as papilledema and amblyopia. This article aims to review the current understanding of neuro-ophthalmological manifestations in craniosynostosis and explore diagnostic and management considerations for the ophthalmologist taking care of these patients.
Keywords: syndromic craniosynostosis, non-syndromic craniosynostosis, optic nerve atrophy, pediatric ophthalmology, papilledema
Introduction
The cranial bones of the newborn skull are typically divided by patent sutures characterized by undifferentiated mesenchymal cells. New bone can be deposited along the leading edge of these sutures as cells differentiate, accommodating the developing brain as it grows rapidly in the first years of life. The major sutures include the metopic suture, which separates the paired frontal bones; the sagittal suture, which separates the paired parietal bones; the coronal suture, which separate the frontal bones from the parietal bones; and the lambdoid suture, which separates the parietal bones from the occipital bone. Sutures progressively begin to close in early childhood and, as the brain completes its growth in adulthood, the sutures fuse to complete cranial development.
Craniosynostosis occurs when one or multiple sutures fuse prematurely, inhibiting skull growth perpendicular to the fused suture and encouraging compensatory growth parallel to the suture; this process produces abnormalities in skull morphology and may have deleterious effects on the development of the brain and other neurological structures. It is a relatively common congenital defect thought to affect 1 in 2000 to 2500 births worldwide1 and can occur either as an isolated defect or, in around 15% of cases, exist as part of a genetic syndrome featuring additional anomalies of the limbs, heart, and central nervous system.2 Isolated craniosynostosis typically affects a single suture (most frequently the sagittal suture, followed by the coronal, metopic, and lambdoid sutures),3 while syndromic craniosynostosis is more likely to involve either the coronal suture alone or multiple sutures.4 Over 180 distinct syndromic entities associated with craniosynostosis have been identified,5 with some of the most common forms including the Crouzon, Pfeiffer, Apert, Saethre-Chotzen, and Muenke syndromes. Most craniosynostosis syndromes exhibit autosomal dominant inheritance and frequently involve gain-of-function mutations in the fibroblast growth factor receptor (FGFR) genes or related genes such as TWIST1, allowing FGFR to remain constitutively active and resulting in unopposed osteoblastic differentiation leading to premature suture fusion; in general, the coronal suture is the primary suture involved.6 Furthermore, secondary craniosynostosis can occur in association with such conditions as inborn errors of metabolism, hematological disorders, cephalic malformations, and teratogenic malformations.7 Characteristic cranial appearances produced by isolated synostoses of each suture are reviewed in Table 1 and Figure 1, while Table 2 summarizes the common craniosynostosis syndromes along with their clinical features and known genetic bases.
 |
Table 1 Isolated Cranial Synostoses and Their Associated Alterations in Head Shape |
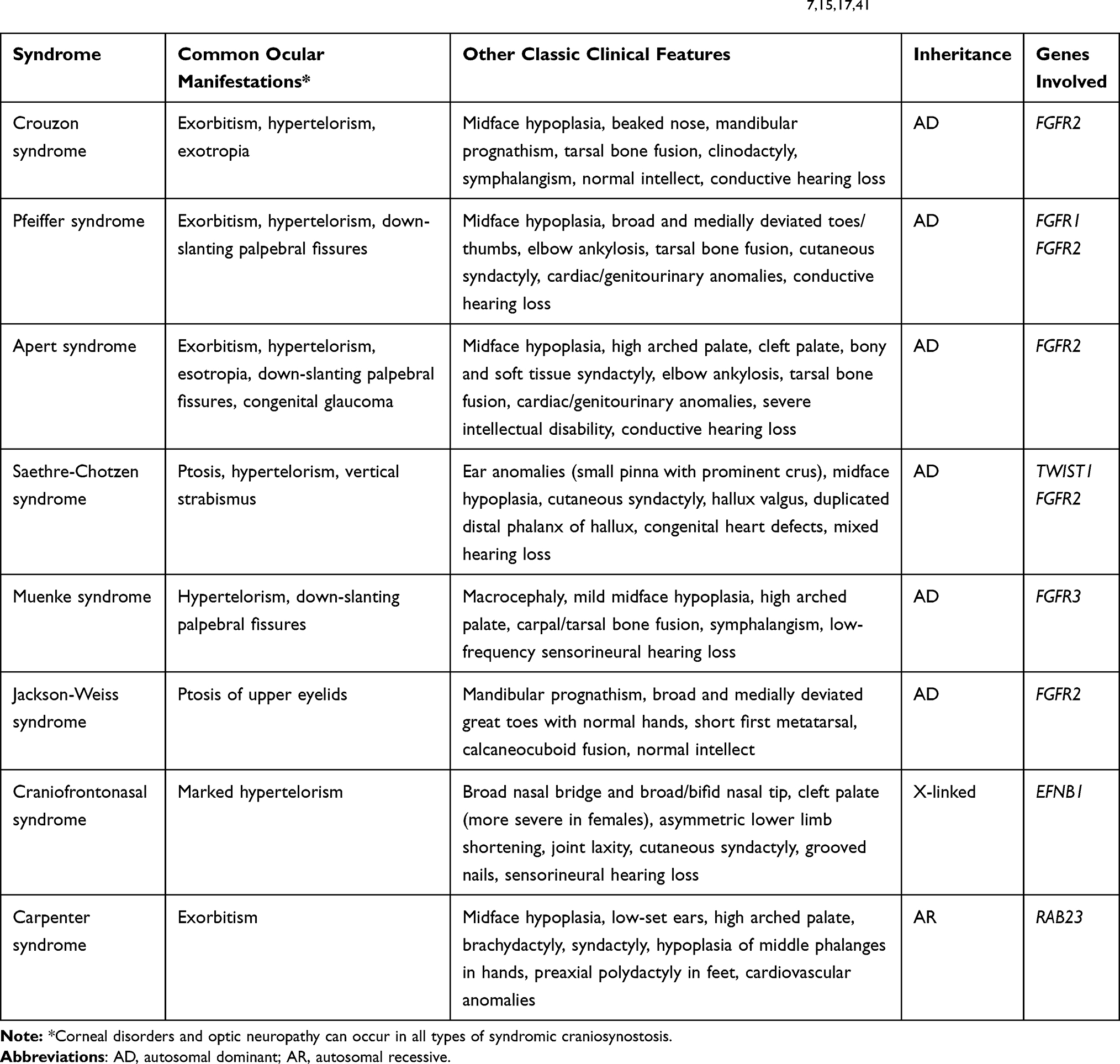 |
Table 2 Common Craniosynostosis Syndromes with Clinical Manifestations and Genetic Bases7,15,17,41 |
 |
Figure 1 (A–C) A child with sagittal craniosynostosis demonstrating scaphocephaly with a long, narrow skull and increased AP (anterior-posterior) skull length. (D and E) A child with unicoronal craniosynostosis with notable anterior plagiocephaly. (F and G) A child with bicoronal craniosynostosis demonstrating brachycephaly with broad forehead and reduced AP skull length. (H–J) A child with metopic craniosynostosis demonstrating trigonocephaly or triangular skull shape and a prominent, vertical forehead ridge. |
The manifestations of craniosynostosis span multiple craniofacial systems, and a multidisciplinary team of specialists is accordingly required to ensure adequate care. Visual complications arising from craniosynostosis are common and include such entities as optic neuropathy secondary to elevated intracranial pressure (ICP), strabismus, refractive errors, exposure keratopathy from exorbitism, and amblyopia resulting from any of the above. Ophthalmologic manifestations are more common and typically more severe in syndromic craniosynostosis due to the cumulative burden of numerous anomalies. In a review of 141 patients with syndromic craniosynostosis, Khan et al documented that 65% of cases were found to involve visual impairment of at least one eye (acuity equal to or less than 20/40) and 40% in both eyes.8 It has also been recognized that visual field deficits are extremely common in syndromic craniosynostosis, implying that visual dysfunction cannot be monitored with tests of central acuity alone.9 Less common etiologies of visual impairment, such as optic nerve hypoplasia and congenital glaucoma, have also been documented in association with craniosynostosis.10 The role of the ophthalmologist in the care of patients with craniosynostosis is, therefore, critical and involves prompt detection of elevated ICP, expeditious diagnosis and treatment of amblyopia in early childhood, and specific management for other ocular manifestations that may be present. This review aims to explore currently recognized neuro-ophthalmological symptoms of craniosynostosis and discuss evaluation and management considerations for the pediatric ophthalmologist seeking to maximize the preservation of vision.
Manifestations
Amblyopia
During the first 8 years of life, proper development of the visual cortex requires equal stimulation from both eyes. Amblyopia occurs when deprivation of vision in one or both eyes results in understimulation of the corresponding cortical neurons; this is thought to the most common cause of visual impairment in patients with craniosynostosis. It is, however, difficult to precisely pinpoint the prevalence of amblyopia in craniosynostosis, as statistics from individual studies vary widely. In a study of patients with various etiologies of craniosynostosis, Hertle et al found that amblyopia was the primary cause of visual loss in 37/43 (86%) of eyes.11 Conversely, Tay et al found that only 4/63 (6.3%) of patients with syndromic craniosynostosis had visual loss attributable to amblyopia.12 Gray et al identified that amblyopia led to visual impairment in 12/56 (21%) of patients with Crouzon syndrome.13 For comparison, longitudinal population data for children ages 4–10 yields a 2.9% prevalence rate for amblyopia.14
Any process that impairs vision, including strabismus, refractive errors, and corneal scarring, represents a risk factor for developing amblyopia. When the underlying cause of amblyopia is not detected and treated early, the window for cortical development may close and visual compromise can be permanent. Management depends on the etiology of amblyopia: strabismus can be treated with patching or surgery depending on severity, corrective lenses should be prescribed to patients with refractive errors, and corneal pathology is managed primarily via prevention. An interesting consideration is that children with midface hypoplasia arising from syndromic craniosynostosis may have increased perspiration due to narrow airways and chronically increased respiratory effort; in such patients, adhesive occlusive patches may be ineffective and atropine penalization may be preferred.15
Strabismus
Strabismus is one of the most common underlying causes of amblyopia, with reported prevalence ranging from 39–76% in patients with craniosynostosis versus the 2.7% incidence of manifest strabismus in the general population.8,12,14 It is most common in unicoronal synostosis, where the eye ipsilateral to the fused suture is affected, and in syndromic craniosynostosis, where there is typically bilateral eye involvement.16 The classic strabismus of craniosynostosis is defined by a characteristic “excyclotorsional syndrome” that involves excyclotorsion, elevation on adduction, and depression on abduction of the affected eye alongside a V-pattern exotropia that is more pronounced in upgaze (see Figure 2).17 The pathogenesis of strabismus in these patients is related to abnormal orbital shape and derangements in the function or number of extraocular muscles that are common in craniosynostosis.18
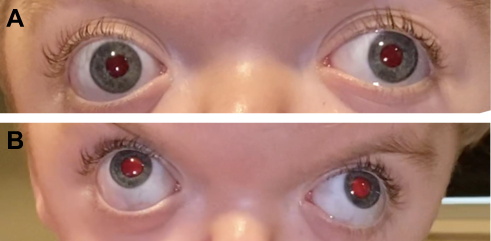 |
Figure 2 A 2-year-old child with syndromic craniosynostosis demonstrating “excylotorsional syndrome” in the (A) primary gaze and with (B) V-pattern exotropia that is prominent in upgaze. |
Corrective surgery can play an important role in the management of strabismus, improving binocular vision and preventing amblyopia. There is debate, however, regarding both the optimal timing and overall efficacy of strabismus surgery in craniosynostosis. Patients with craniosynostosis are classically advised to delay strabismus surgery beyond the first 2 years of life because their ocular deviations may not remain constant after cranial vault reconstruction.5 Conversely, Diamond et al found in a retrospective study of 140 craniosynostosis cases that only 9 patients demonstrated changes in ocular position after undergoing reconstructive operations; this led to the proposal that strabismus surgery should be pursued earlier in life, as the benefit of achieving binocularity may outweigh the comparatively small risk of future shifts in ocular position.19 Further studies are needed to definitively compare outcomes between early and late surgery in the management of craniosynostotic strabismus. Even when strabismus surgery is performed, however, the complex anatomy of these patients makes a curative outcome difficult to achieve. In a retrospective review of 14 craniosynostosis patients that underwent strabismus surgery, Coats et al found that all patients had significant postoperative residual oculomotor dysfunction.20 Orbital imaging, preferably MRI, can be helpful in preparation for surgery and is particularly useful in cases when there are positional or numerical abnormalities in the extraocular muscles.21
Refractive Error
Ametropia, particularly astigmatism, is an extremely common finding in patients with craniosynostosis and serves as another major risk factor for amblyopia. Khan et al found a 40% prevalence of astigmatism greater than 1 diopter (D) and 18% prevalence of anisometropia in their review of 141 syndromic craniosynostosis cases, compared to the 3.5% prevalence in the general population;8 these findings were echoed in a review of 63 patients by Tay et al (40% prevalence of astigmatism and 16% prevalence of anisometropia).12 Interestingly, the incidence of significant astigmatism (more than 1.25 D) was high in Apert, Pfeiffer, Crouzon, craniofrontonasal dysplasia, and non-syndromic unicoronal synostosis while it was dramatically lower with isolated sagittal and metopic synostosis.8 Hypermetropia is also a common refractive error associated with craniosynostosis, although prevalence numbers vary widely: Gray et al found a hypermetropia prevalence of 57% in a review of 51 patients, while Tay et al documented a prevalence of 18%.12,13 This discrepancy may be accounted for by the fact that the Gray et al study included only patients with Crouzon syndrome, a condition in which shallow orbital depth results in a shortened axial length and predisposition to hypermetropia.
Children with craniosynostosis should be regularly screened for refractive errors, as undetected anisometropia results in a high likelihood of developing amblyopia. Patients with unicoronal synostosis are at particularly high risk for amblyopia, as the unilateral pattern of orbital deformation results in a high prevalence of asymmetrical astigmatism.22
Exposure Keratopathy
Exposure keratopathy is the major etiology of corneal pathology encountered in craniosynostosis, particularly in cases of syndromic craniosynostosis where exorbitism due to shallow orbits and resultant lagophthalmos (incomplete palpebral closure) are common manifestations.17 Chronic corneal exposure and lack of adequate tear film protection can lead to a range of manifestations including superficial punctate keratitis, corneal ulceration, corneal scarring, and (in extreme cases) endophthalmitis and eyeball perforation. In Crouzon syndrome, subluxation of the eyeball during infant exertion is possible; such a presentation is a surgical emergency due to risk of damage to the cornea and optic nerve.23
Prevention and treatment of corneal damage are yet another important consideration in the prevention of amblyopia in patients with craniosynostosis. When lagophthalmos is identified, lubricating eye drops and vitamin A ointment confer a protective effect against exposure keratopathy. Lateral tarsorrhaphy, a procedure where the lateral palpebrae are sutured to decrease palpebral fissure width and reduce exorbitism, may be indicated in some cases. Furthermore, postoperative orbital edema after craniofacial surgery can result in exophthalmos and heightened risk for exposure keratopathy; patients should be monitored closely for corneal risk factors in the postoperative period.17
Optic Neuropathy from Elevated ICP
Optic neuropathy is a common ophthalmological complication of craniosynostosis and an important cause of vision loss. Chronically elevated ICP in these patients results in papilledema and ultimately may lead to neuronal death, optic nerve atrophy, and permanent loss of vision; the etiology arises from axoplasmic flow stasis at the optic nerve head leading to intraneuronal ischemia.24 Clinical manifestations include loss of visual acuity and visual field defects.8,9 The highest risk of elevated ICP is seen in patients with multi-sutural involvement and in those with FGFR2 mutations (Apert, Crouzon, and Pfeiffer syndromes), with the overall risk of elevated ICP estimated to be around 30–40% in syndromic craniosynostosis and 15–20% in non-syndromic craniosynostosis (reflecting increased risk with multi-sutural involvement).25–27 Papilledema is also more common in patients with a known causative genetic mutation, although it may still occur in cases where no mutation is identified; it is thought to be present in 10–15% of children with craniosynostosis.12,13,28 Chronic compression or elongation of the optic nerve from synostosis-induced anomalies of the orbit or optic canal may also contribute to optic atrophy.17
A variety of mechanisms have been proposed to explain the prominence of elevated ICP in craniosynostosis. Classically, elevated ICP was thought to arise from a mismatch between cerebral growth and synostotic cranial volume (termed craniocerebral disproportion or CCD). It has been demonstrated, however, that there is little to no correlation between intracranial volume and intracranial pressure29 and that CCD may account for elevated ICP only in patients under 1 year of age or in those with decelerating skull growth following decompressive surgery.30–32 Numerous studies have explored the role of a number of entities including hydrocephalus, respiratory pathologies, and venous hypertension in the development of elevated ICP and optic neuropathy.33–36 Craniosynostotic hydrocephalus, which occurs due to CSF obstruction or malabsorption related to the osseous or associated derangements of craniosynostosis, has been documented in up to 40% of syndromic craniosynostosis cases; it is rarely implicated in non-syndromic craniosynostosis.34 The presence of hydrocephalus is related to the extent and timeline of suture fusion and may result in elevated ICP. However, craniosynostosis patients with evidence of hydrocephalus may exhibit normal pressures if sufficient compensation occurs and, conversely, many patients with severe elevated ICP have normal-appearing ventricles on imaging.36
Recently, it has been increasingly posited that respiratory obstruction and venous hypertension may account for the majority of elevated ICP in children with craniosynostosis. In a study of children with complex craniosynostosis, Hayward et al described interactions between common findings in these children of elevated ICP, reduced cerebral perfusion pressure (CPP, defined as the mean systemic arterial blood pressure minus the ICP and used as a proxy for cerebral blood flow), and respiratory obstruction (common in syndromic craniosynostosis due to maxillary hypoplasia). Among the 11 patients included in the study, all experienced obstructive breathing problems that were temporally related to plateau elevations in ICP and marked decreases in CPP during active sleep. The high incidence of upper airway obstruction, obstructive sleep apnea (OSA), and resultant nighttime hypoxia/hypercapnia in these patients is thought to produce the frank ICP elevations and CPP reductions, which in turn may directly account for optic nerve morbidity. When CPP remains persistently low, compensatory cerebral vasodilation attempting to increase intracranial blood volume can subsequently exacerbate ICP elevations.37 Abnormal patterns of intracranial venous drainage arising from anomalous or stenotic venous sinuses may also contribute to elevated ICP, particularly in syndromic craniosynostosis.35 A study by Taylor et al analyzing 24 angiography studies from 23 patients with both syndromic and non-syndromic craniosynostosis found that >50% stenosis in the sigmoid-jugular sinus complex was found in 18 studies; the authors suggest that venous hypertension produced by this anomalous venous drainage may be the primary factor responsible for the majority of elevated ICP associated with craniosynostosis.36 Respiratory pathology and venous sinus anomalies may even be linked or have compounding effects in the pathogenesis of papilledema: venous collaterals produced to bypass stenotic cerebral sinuses are thought to lack typical vascular autoregulation and may, in conditions of respiratory obstruction-induced hypercapnia and venous sinus vasodilation, prevent a compensatory increase in pulse rate and thereby permit cerebral hypoperfusion.15
The relationship between clinical finding of papilledema and diagnosis of elevated ICP has also been the subject of much scrutiny. By definition, papilledema is the manifestation of elevated ICP transmitting across the lamina cribrosa and optic nerve sheath to induce axoplasmic flow stasis and swelling in the optic nerve head.38 When it is present and other causes of optic disc swelling (ie pseudo-papilledema) have been ruled out, elevated ICP is extremely likely; Tuite et al found in a review of 122 craniosynostosis cases that the presence of papilledema was 98% specific for elevated ICP.28 The sensitivity, however, was highly age-dependent and was only 22% in children less than 8 years of age. Another study of 37 craniosynostosis patients undergoing procedures found that preoperative finding of papilledema was only 17% sensitive for elevated ICP.27 This is concerning given that papilledema has historically been considered a ubiquitous indicator for elevated ICP. The negative predictive value of absent papilledema is further diminished in cases where extensive optic atrophy has already occurred, as destroyed axons no longer swell and may fail to produce disc edema even with high pressures.17 A diagnosis of elevated ICP therefore can not be ruled out on the basis of papilledema not being apparent.
While fundoscopy should not be considered a reliable screening tool for elevated ICP, it remains a critically important element of the ophthalmologic exam in patients with craniosynostosis. Papilledema is the primary driver of optic neuropathy in these patients and the early detection of nerve swelling can facilitate important treatment decisions. As with all etiologies of papilledema, true papilledema arising from elevated ICP must be differentiated from pseudo-papilledema (most commonly arising from optic disc drusen). Drusen consist of cytoplasmic material accumulations within the optic disc that are commonly buried in children, as opposed to their superficial location in adults which make them easier to diagnose; this may appear on fundoscopy as an elevated optic disc that does not obscure underlying retinal vessels, in contrast to the vessel obscuration that commonly occurs with true papilledema. A variety of imaging modalities including fundus autofluorescence, B-scan, and fluorescein angiography can further help distinguish between papilledema and pseudo-papilledema.17 When papilledema has been identified, it can be staged according to the Frisén scale (Table 3).39 Optic atrophy risk depends on both staging and duration: low-stage (Frisén stage 1 or 2) papilledema can subsist for months or years without visual morbidity, while Frisén stage 5 papilledema can result in permanent optic atrophy in a matter of days and thus necessitates emergent ICP reduction.17
 |
Table 3 Papilledema Grading Scale (Frisén Scale)39 |
A definitive diagnosis of elevated ICP can be made by direct intracranial monitoring, although this is an invasive modality that is associated with hemorrhagic complications.40 Other clinical signs that may indicate elevated ICP include headache, vomiting, acute behavioral changes, lethargy, and a “copper beaten skull appearance” on radiograph of the cranium,41 although papilledema may be the only symptom of elevated ICP in some children.42 Due to the low sensitivity of papilledema in detecting elevated ICP, there has been significant interest in a number of non-invasive ICP measurement modalities including optical coherence tomography, visual-evoked potential testing, transcranial Doppler ultrasound, and optic nerve sheath diameter measurement. Fundus imaging with spectral domain optical coherence tomography (SD-OCT) with retinal nerve fiber layer (RNFL) thickness is a sensitive, noninvasive study to detect early papilledema and should be combined with clinical fundoscopic examination to screen for optic nerve pathology.43 Serial documentation with fundus photographs alongside the quantitative information provided by OCT is instrumental in charting the clinical progression of disease. Visual-evoked potentials (VEP) have also shown promise as a tool for monitoring ICP; in a study comparing the ability of visual acuity measurement, optic disc appearance, and VEPs to gauge visual dysfunction, abnormal pattern VEPs were found to be the most sensitive indicator of elevated ICP.44 A study of 114 cases by Thompson et al found that 60% of patients had abnormal pattern VEPs, implying that there is electrophysiological evidence of visual dysfunction in the majority of craniosynostosis patients even when clinical signs may not be apparent.45 When electrophysiological monitoring is pursued, a baseline VEP with serial subsequent measurements may have more interpretive value than an isolated VEP reading. Transcranial Doppler ultrasonography is another modality that has seen recent application in evaluating intracranial pressure; Wang et al found that certain hemodynamic parameters of the middle cerebral artery assessed via transcranial color Doppler were predictively correlated with intracranial pressure, although the majority of patients in this study had traumatic or hypertensive etiologies of elevated ICP.46 Finally, a recent study has demonstrated the potential of MRI- or CT-derived measurement of the optic nerve sheath diameter (ONSD) as an indirect assessment of ICP in craniosynostosis patients. In a study of 56 patients with craniosynostosis compared to 49 controls, Haredy et al found a significant elevation of mean OSND in patients with intracranial hypertension compared to those with normal ICP or controls. There was no significant discrepancy between MRI versus CT measurements, suggesting that either could have utility as a screening tool to monitor ICP in patients with symptoms of elevated ICP and normal fundus examination.47 Craniofacial imaging with CT or MRI may also be helpful in cases where the etiology of optic neuropathy is unclear.17
Identifying the appropriate management approach to elevated ICP can be complicated. There is a lack of consensus regarding normal ICP ranges in children, and intracranial pressure monitoring in young patients is less reliable due to variable intracranial compliance.48 Procedures for treatment of elevated ICP include cranial vault expansion surgery (Figure 3), CSF shunting, and endoscopic third ventriculostomy. Decompressive surgery is the first-line, definitive treatment for papilledema (even in patients with a prior history of vault expansion surgery) and should be given urgent consideration when papilledema is identified, as outcomes can be quite favorable; several studies have demonstrated complete resolution of papilledema in patients undergoing craniofacial decompression.12,49,50 While CSF diversion has classically served as a standard treatment for elevated ICP with papilledema, concerns specific to children with complex craniosynostosis (including persistent venous hypertension and CSF over-drainage removing a growth stimulus for the skull) limit its application.50 Continued long-term monitoring for papilledema is still recommended for patients after surgery, as the detection of recurrent elevated ICP may inform the necessity of additional decompressive procedures. Adenoid tonsillectomy may be effective in treating visual dysfunction in children with ischemic papilledema secondary to respiratory obstruction.51
 |
Figure 3 A 12-month-old with Crouzon syndrome who presented (A and B) with bilateral optic nerve edema (papilledema) and subsequent resolution of papilledema (C and D) with posterior cranial vault distraction followed by staged fronto-orbital advancement. Note: Reproduced from LoPresti M, Buchanan E, Shah V, Hadley C, Monson L, Lam S. Complete resolution of papilledema in syndromic craniosynostosis with posterior cranial vault distraction. Journal of Pediatric Neurosciences. 2017;12(2):199. |
Diagnostic and Management Considerations
The approach to any patient with suspected craniosynostosis begins with thorough clinical evaluation. Items of particular importance on initial evaluation include family history of craniosynostosis or other congenital/genetic conditions, birth and developmental history, past medical history with attention to any completed imaging studies, and surgical history. Physical examination should focus, in addition to sutural involvement and craniofacial anomalies, on identifying classic findings of craniosynostosis syndromes.
Genetic testing should be considered in patients with craniosynostosis, particularly when multiple suture involvement, additional anomalies, or family history point to a syndromic etiology. When the diagnosis is suspected but it is difficult to distinguish between phenotypically overlapping syndromes, testing can establish a specific diagnosis. Syndrome-specific molecular gene studies are useful in confirming clinically apparent presentations of known craniosynostosis syndromes, while chromosomal microarray or karyotyping can be considered when profound developmental delay or extensive additional malformations raise concern for more complex chromosomal derangements.52 With currently available testing, a genetic cause for up to 45% of patients with multisuture craniosynostosis can be identified.41 Non-syndromic craniosynostosis (which simply refers to the absence of concomitant anomalies) can also have a genetic basis and is familial with autosomal dominant inheritance in around 8% of cases.53 One retrospective genetic study demonstrated that causative mutations could be identified in 37.5% of patients with isolated bicoronal synostosis, 17.5% with isolated unicoronal synostosis, and 11% with multisuture synostosis, but no patients with isolated metopic, sagittal, or lambdoid synostosis.54 It is therefore recommended that patients with isolated, non-syndromic coronal craniosynostosis undergo genetic testing, while testing is not currently recommended for patients with isolated, non-syndromic metopic, sagittal, or lambdoid craniosynostosis. Advantages to identifying the genetic basis in cases of craniosynostosis beyond efficient diagnosis include prognostic value, optimizations in care such as guided surgical timing, and the facilitation of genetic counseling.
Consistent ophthalmic care is a crucial element of craniosynostosis management, and as such these patients should be regularly seen by a pediatric ophthalmologist. An ophthalmologist should examine any patient with syndromic or non-syndromic craniosynostosis at the time of diagnosis and be consulted both before and after any craniofacial surgery. It is recommended that comprehensive ophthalmologic examination be performed biannually until 7–9 years of age and yearly thereafter, for children with syndromic craniosynostosis, and annually until 7–9 years and as needed thereafter for non-syndromic craniosynostosis.55 Exam should include assessment of visual acuity, ocular alignment, refractive error, and examination of the cornea and optic nerve; routine fundoscopic exam is particularly important to evaluate for papilledema in these patients.
Surgical intervention in craniosynostosis is typically pursued in early life, aiming to release growth restriction and maximize the potential for normal cerebral development. Operative techniques differentiate broadly into two distinct themes: strip craniectomy, which involves excision of the fused suture to correct growth restriction, and whole-vault cranioplasty (WVC), which incorporates remodeling of all the cranial bones and addresses compensatory deformities of the entire cranial vault.56 WVC, which has historically been considered the hallmark of surgical intervention in craniosynostosis, is typically performed in the first 6–8 months of life to maximize the potential for re-ossification.57 Endoscopic strip craniectomy (ESC), a minimally invasive procedure in which the fused suture is released and a helmet is subsequently used to guide brain growth, has recently gained prominence and can also be performed by 6 months of life. Some evidence exists that ESC, in addition to producing less bleeding and shortened operative/recovery times, may lead to improved ophthalmological outcomes: a retrospective non-randomized study of 37 children with unilateral coronal synostosis treated with either ESC or fronto-orbital advancement (FOA) found greater severity of strabismus, excyclotorsion, and aniso-astigmatism in the FOA group after 14 months of age.58 More controversy exists regarding the optical choice of surgery for isolated sagittal craniosynostosis: in a retrospective study of 70 patients who had undergone either ESC or WVC, Hashim et al found that patients undergoing early WVC had improved long-term neuropsychological outcomes as measured by performance on neurodevelopmental testing compared to those undergoing strip craniectomy.59 Thomas et al also found that strip craniectomy was significantly associated with increased risk of eventual elevated ICP compared to vault remodeling, although there is a variety of definitions and techniques underlying the label of “strip craniectomy” that may be at play.60 Another retrospective study of 300 patients by Garber et al, however, found that ESC patients had significant reductions in transfusion requirements, ICU/hospital stay duration, and need for surgical revision compared to WVC.61 These authors concluded that ESC for sagittal synostosis leads to improved morbidity and lower costs compared to WVC. Regardless of procedure choice, patients may continue to have recurrent deformity or elevated ICP after surgery and (especially in syndromic cases) should be regularly followed until at least 6-8 years of age when brain bulk growth is completed. Post-operative ophthalmologic surveillance should occur every 3 months in multi-sutural synostosis and every 6 months in single-suture synostosis in first follow-up year .57
Conclusion
Craniosynostosis is a common congenital anomaly that frequently features ophthalmologic manifestations. Amblyopia is responsible for the majority of patients with craniosynostosis suffering from vision loss, and in many cases can be alleviated with early detection of causative factors and treatment via occlusion or corrective lenses. Optic atrophy represents a less prevalent but nevertheless significant cause of visual impairment in these patients; regular ocular examination is critical to detect early signs of papilledema and inform the need for decompressive surgery. Genetic testing can assist with the definitive diagnosis and management of craniosynostosis syndromes.
Disclosure
The authors report no conflicts of interest in this work.
References
1. Slater BJ, Lenton KA, Kwan MD, Gupta DM, Wan DC, Longaker MT. Cranial sutures: a brief review. Plast Reconstr Surg. 2008;121(4):170e–8e. doi:10.1097/01.prs.0000304441.99483.97
2. Wilkie AOM, Byren JC, Hurst JA, et al. Prevalence and complications of single-gene and chromosomal disorders in craniosynostosis. Pediatrics. 2010;126(2):e391–e400. doi:10.1542/peds.2009-3491
3. Shillito J, Matson DD. Craniosynostosis: a review of 519 surgical patients. Pediatrics. 1968;41(4):829–853.
4. Hehr U, Muenke M. Craniosynostosis syndromes: from genes to premature fusion of skull bones. Mol Genet Metab. 1999;68(2):139–151. doi:10.1006/mgme.1999.2915
5. Cohen MM
6. Ethylin Wang J, Xiang L, Alan FS, et al. Jackson-Weiss and Crouzon syndromes are allelic with mutations in fibroblast growth factor receptor 2. Nat Genet. 1994;8(3):275. doi:10.1038/ng1194-275
7. Jabs EW, Lewanda AF. Craniosynostosis. Elsevier Inc; 2014.
8. Khan SH, Nischal KK, Dean F, Hayward RD, Walker J. Visual outcomes and amblyogenic risk factors in craniosynostotic syndromes: a review of 141 cases. Br J Ophthalmol. 2003;87(8):999. doi:10.1136/bjo.87.8.999
9. Liasis A, Walters B, Thompson D, Smith K, Hayward R, Nischal KK. Visual field loss in children with craniosynostosis. Childs Nerv Syst. 2011;27(8):1289–1296. doi:10.1007/s00381-010-1378-5
10. Schultz KP, Wiggins CJ, Streff H, Shah VS, Buchanan EP. Syndromic multisuture craniosynostosis with associated anterior segment dysgenesis, optic nerve hypoplasia, and congenital glaucoma. Cleft Palate Craniofac J. 2019;56(6):823–826. doi:10.1177/1055665618820481
11. Hertle RW, Quinn GE, Minguini N, Katowitz JA. Visual loss in patients with craniofacial synostosis. J Pediatr Ophthalmol Strabismus. 1991;28(6):344–349.
12. Tay T, Martin F, Rowe N, et al. Prevalence and causes of visual impairment in craniosynostotic syndromes. Clin Experiment Ophthalmol. 2006;34(5):434–440. doi:10.1111/j.1442-9071.2006.01242.x
13. Gray TL, Casey T, Selva D, Anderson PJ, David DJ. Ophthalmic sequelae of Crouzon syndrome. Ophthalmology. 2005;112(6):1129–1134. doi:10.1016/j.ophtha.2004.12.037
14. Kvarnström G, Jakobsson P, Lennerstrand G. Visual screening of Swedish children: an ophthalmological evaluation. Acta Ophthalmol Scand. 2001;79(3):240–244. doi:10.1034/j.1600-0420.2001.790306.x
15. Ganesh A, Edmond J, Forbes B, et al. An update of ophthalmic management in craniosynostosis. J AAPOS. 2019;23(2):66–76. doi:10.1016/j.jaapos.2018.10.016
16. Dagi LR, MacKinnon S, Zurakowski D, Prabhu SP. Rectus muscle excyclorotation and V-pattern strabismus: a quantitative appraisal of clinical relevance in syndromic craniosynostosis. Br J Ophthalmol. 2017;101(11):1560. doi:10.1136/bjophthalmol-2016-309996
17. Touzé R, Bremond-Gignac D, Robert MP. Ophthalmological management in craniosynostosis. Neurochirurgie. 2019;65(5):310–317. doi:10.1016/j.neuchi.2019.09.016
18. Diamond GR, Katowitz JA, Whitaker LA, Quinn GE, Schaffer DB. Variations in extraocular muscle number and structure in craniofacial dysostosis. Am J Ophthalmol. 1980;90(3):416–418. doi:10.1016/S0002-9394(14)74927-X
19. Diamond RG, Whitaker RL, Diamond GR. Ocular motility in craniofacial reconstruction. Plast Reconstr Surg. 1984;73(1):31–35. doi:10.1097/00006534-198401000-00007
20. Coats DK, Paysse EA, Stager DR. Surgical management of V-pattern strabismus and oblique dysfunction in craniofacial dysostosis. J AAPOS. 2000;4(6):338–342. doi:10.1067/mpa.2000.110337
21. Clark RA. Orbital imaging in strabismus. J Binocul Vis Ocul Motil. 2018;68(3):87–98. doi:10.1080/2576117X.2018.1486678
22. Tarczy-Hornoch K, Smith B, Urata M. Amblyogenic anisometropia in the contralateral eye in unicoronal craniosynostosis. J AAPOS. 2008;12(5):471–476. doi:10.1016/j.jaapos.2008.03.008
23. Kreiborg S, Cohen MM. Ocular manifestations of apert and Crouzon syndromes: qualitative and quantitative findings. J Craniofac Surg. 2010;21(5):1354–1357. doi:10.1097/SCS.0b013e3181ef2b53
24. Rigi M, Almarzouqi SJ, Morgan ML, Lee AG. Papilledema: epidemiology, etiology, and clinical management. (report). Eye Brain. 2015;7:47. doi:10.2147/EB.S69174
25. Greene AK, Mulliken JB, Proctor MR, Meara JG, Rogers GF. Phenotypically unusual combined craniosynostoses: presentation and management. Plast Reconstr Surg. 2008;122(3):853–862. doi:10.1097/PRS.0b013e31817f45f0
26. de Jong T, Bannink N, Bredero-Boelhouwer HH, et al. Long-term functional outcome in 167 patients with syndromic craniosynostosis; defining a syndrome-specific risk profile. J Plast Reconstr Aesthet Surg. 2010;63(10):1635–1641. doi:10.1016/j.bjps.2009.10.029
27. Judy B, Swanson J, Yang W, et al. Intraoperative intracranial pressure monitoring in the pediatric craniosynostosis population. J Neurosurg Pediatr. 2018;22:1–6. doi:10.3171/2018.5.PEDS1876
28. Tuite FG, Chong KW, Evanson FJ, et al. The effectiveness of papilledema as an indicator of raised intracranial pressure in children with craniosynostosis. Neurosurgery. 1996;38(2):272–278. doi:10.1097/00006123-199602000-00009
29. Fok H, Jones BM, Gault DG, Andar U, Hayward R. Relationship between intracranial pressure and intracranial volume in craniosynostosis. Br J Plast Surg. 1992;45(5):394–397. doi:10.1016/0007-1226(92)90013-N
30. Sgouros S. Skull vault growth in craniosynostosis. Childs Nerv Syst. 2005;21(10):861–870. doi:10.1007/s00381-004-1112-2
31. Sgouros S, Hockley AD, Goldin JH, Wake MJ, Natarajan K. Intracranial volume change in craniosynostosis. J Neurosurg. 1999;91(4):617–625. doi:10.3171/jns.1999.91.4.0617
32. Spruijt B, Joosten KFM, Driessen C, Rizopoulos D, Naus NC, Schroeff M. Algorithm for the management of intracranial hypertension in children with syndromic craniosynostosis. Plast Reconstr Surg. 2015;136(2):331–340. doi:10.1097/PRS.0000000000001434
33. Marucci D, Dunaway D, Jones B, Hayward R. Raised intracranial pressure in apert syndrome. Plast Reconstr Surg. 2008;122(4):1162–1168. doi:10.1097/PRS.0b013e31818458f0
34. Collmann H, Sörensen N, Krauss J. Hydrocephalus in craniosynostosis: a review. Childs Nerv Syst. 2005;21(10):902–912. doi:10.1007/s00381-004-1116-y
35. Hayward R. Venous hypertension and craniosynostosis. Childs Nerv Syst. 2005;21(10):880–888. doi:10.1007/s00381-004-1114-0
36. Taylor WJ, Hayward RD, Lasjaunias P, Britto JA, Thompson DN, Jones BM. Enigma of raised intracranial pressure in patients with complex craniosynostosis: the role of abnormal intracranial venous drainage. J Neurosurg. 2001;94(3):377–385. doi:10.3171/jns.2001.94.3.0377
37. Hayward R, Gonsalez S. How low can you go? Intracranial pressure, cerebral perfusion pressure, and respiratory obstruction in children with complex craniosynostosis. J Neurosurg. 2005;102(1 Suppl):16–22. doi:10.3171/ped.2005.102.1.0016
38. Hayreh SS. Optic disc edema in raised intracranial pressure: V. pathogenesis. Arch Ophthalmol. 1977;95(9):1553–1565. doi:10.1001/archopht.1977.04450090075006
39. Frisén L. Swelling of the optic nerve head: a staging scheme. J Neurol Neurosurg Psychiatry. 1982;45(1):13. doi:10.1136/jnnp.45.1.13
40. Blaha M, Lazar D, Winn RH, Ghatan S. Hemorrhagic complications of intracranial pressure monitors in children. Pediatr Neurosurg. 2003;39(1):27–31. doi:10.1159/000070877
41. Agochukwu NB, Solomon BD, Muenke M. Impact of genetics on the diagnosis and clinical management of syndromic craniosynostoses. Childs Nerv Syst. 2012;28(9):1447–1463. doi:10.1007/s00381-012-1756-2
42. Bartels M, Vaandrager J, Jong T, Simonsz H. Visual loss in syndromic craniosynostosis with papilledema but without other symptoms of intracranial hypertension. J Craniofac Surg. 2004;15(6):1019–1022. doi:10.1097/00001665-200411000-00026
43. Swanson JW, Aleman TS, Xu W, et al. Evaluation of optical coherence tomography to detect elevated intracranial pressure in children. JAMA Ophthalmol. 2017;135(4):320–328. doi:10.1001/jamaophthalmol.2017.0025
44. Liasis A, Nischal KK, Walters B, et al. Monitoring visual function in children with syndromic craniosynostosis: a comparison of 3 methods. Arch Ophthalmol. 2006;124(8):1119–1126. doi:10.1001/archopht.124.8.1119
45. Thompson DA, Liasis A, Hardy S, et al. Prevalence of abnormal pattern reversal visual evoked potentials in craniosynostosis. Plast Reconstr Surg. 2006;118(1):184–192. doi:10.1097/01.prs.0000220873.72953.3e
46. Wang Y, Duan YY, Zhou HY, et al. Middle cerebral arterial flow changes on transcranial color and spectral Doppler sonography in patients with increased intracranial pressure. J Ultrasound Med. 2014;33(12):2131–2136. doi:10.7863/ultra.33.12.2131
47. Haredy M, Zuccoli G, Tamber M, Davis A, Nischal K, Goldstein JA. Use of neuroimaging measurements of optic nerve sheath diameter to assess intracranial pressure in craniosynostosis. Childs Nerv Syst. 2018;34(5):939–946. doi:10.1007/s00381-018-3728-7
48. Thompson D, Harkness W, Jones B, Gonsalez S, Andar U, Hayward R. Subdural intracranial pressure monitoring in craniosynostosis: its role in surgical management. Childs Nerv Syst. 1995;11(5):269–275. doi:10.1007/BF00301758
49. Pollack IF, Losken W, Biglan AW. Incidence of increased intracranial pressure after early surgical treatment of syndromic craniosynostosis. Pediatr Neurosurg. 1996;24(4):202–209. doi:10.1159/000121038
50. LoPresti M, Buchanan E, Shah V, Hadley C, Monson L, Lam S. Complete resolution of papilledema in syndromic craniosynostosis with posterior cranial vault distraction. (case report)(case study). J Pediatr Neurosci. 2017;12(2):199. doi:10.4103/jpn.JPN_15_17
51. Liasis A, Nischal KK, Leighton S, Yap S, Hayward R, Dunaway D. Adenoid-tonsillectomy to treat visual dysfunction in a child with craniosynostosis. Pediatr Neurosurg. 2005;41(4):197–200. doi:10.1159/000086561
52. Miller DT, Adam MP, Aradhya S, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(5):749–764. doi:10.1016/j.ajhg.2010.04.006
53. Boyadjiev SA, Consortium IC. Genetic analysis of non-syndromic craniosynostosis. Orthod Craniofac Res. 2007;10(3):129–137. doi:10.1111/j.1601-6343.2007.00393.x
54. Wilkie AOM, Bochukova EG, Hansen RMS, et al. Clinical Dividends from the Molecular Genetic Diagnosis of Craniosynostosis. Cohen MM, editor. Hoboken:Wiley Subscription Services, Inc., A Wiley Company; 2006:2631–2639.
55. McCarthy JG, Warren SM, Bernstein J, et al. Parameters of care for craniosynostosis. Cleft Palate Craniofac J. 2012;49(Suppl):1s–24s. doi:10.1597/11-138
56. Panchal J, Uttchin V. Management of craniosynostosis. Plast Reconstr Surg. 2003;111(6):2032–2048. doi:10.1097/01.PRS.0000056839.94034.47
57. Muenke M, Kress W, Collmann H, Solomon B. Craniosynostoses: Molecular Genetics, Principles of Diagnosis and Treatment. Vol. 19. Karger Medical and Scientific Publishers; 2011.
58. Mackinnon S, Rogers GF, Gregas M, Proctor MR, Mulliken JB, Dagi LR. Treatment of unilateral coronal synostosis by endoscopic strip craniectomy or fronto-orbital advancement: ophthalmologic findings. J AAPOS. 2009;13(2):155–160. doi:10.1016/j.jaapos.2008.10.011
59. Hashim PW, Patel A, Yang JF, et al. The effects of whole-vault cranioplasty versus strip craniectomy on long-term neuropsychological outcomes in sagittal craniosynostosis. Plast Reconstr Surg. 2014;134(3):491–501. doi:10.1097/PRS.0000000000000420
60. Thomas GPL, Johnson D, Byren JC, et al. Periodical shifts in the surgical correction of sagittal craniosynostosis. J Neurosurg Pediatr. 2015;15(4):348–349.
61. Garber ST, Karsy M, Kestle JRW, Siddiqi F, Spanos SP. Comparing outcomes and cost of 3 surgical treatments for sagittal synostosis: a retrospective study including procedure-related cost analysis. Neurosurgery. 2017;81(4):680–687. doi:10.1093/neuros/nyx209
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.