Back to Journals » Drug Design, Development and Therapy » Volume 15
Network Pharmacology Prediction and Molecular Docking-Based Strategy to Discover the Potential Pharmacological Mechanism of Huai Hua San Against Ulcerative Colitis
Authors Liu J, Liu J, Tong X, Peng W ![]() , Wei S, Sun T, Wang Y, Zhang B, Li W
, Wei S, Sun T, Wang Y, Zhang B, Li W ![]()
Received 13 May 2021
Accepted for publication 5 July 2021
Published 28 July 2021 Volume 2021:15 Pages 3255—3276
DOI https://doi.org/10.2147/DDDT.S319786
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Professor Jianbo Sun
Jiaqin Liu,1,2 Jian Liu,1,2 Xiaoliang Tong,3 Weijun Peng,4 Shanshan Wei,1,2 Taoli Sun,5 Yikun Wang,1,2 Bikui Zhang,1,2 Wenqun Li1,2
1Department of Pharmacy, The Second Xiangya Hospital, Central South University, Changsha, Hunan, 410011, People’s Republic of China; 2Institute of Clinical Pharmacy, Central South University, Changsha, Hunan, 410011, People’s Republic of China; 3Department of Dermatology, The Third Xiangya Hospital, Central South University, Changsha, Hunan, 410013, People’s Republic of China; 4Department of Integrated Traditional Chinese & Western Medicine, The Second Xiangya Hospital, Central South University, Changsha, Hunan, 410011, People’s Republic of China; 5School of Pharmacy, Hunan University of Chinese Medicine, Changsha, Hunan, 410208, People’s Republic of China
Correspondence: Wenqun Li
Department of Pharmacy, The Second Xiangya Hospital, Central South University, No. 139 Renmin Road, Changsha, 410011, People’s Republic of China
Tel +86-731-85292093
Email [email protected]
Background: Huai Hua San (HHS), a famous Traditional Chinese Medicine (TCM) formula, has been widely applied in treating ulcerative colitis (UC). However, the interaction of bioactives from HHS with the targets involved in UC has not been elucidated yet.
Aim: A network pharmacology-based approach combined with molecular docking and in vitro validation was performed to determine the bioactives, key targets, and potential pharmacological mechanism of HHS against UC.
Materials and Methods: Bioactives and potential targets of HHS, as well as UC-related targets, were retrieved from public databases. Crucial bioactive ingredients, potential targets, and signaling pathways were acquired through bioinformatics analysis, including protein–protein interaction (PPI), as well as the Gene Ontology (GO) and the Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis. Subsequently, molecular docking was carried out to predict the combination of active compounds with core targets. Lastly, in vitro experiments were conducted to further verify the findings.
Results: A total of 28 bioactive ingredients of HHS and 421 HHS-UC-related targets were screened. Bioinformatics analysis revealed that quercetin, luteolin, and nobiletin may be potential candidate agents. JUN, TP53, and ESR1 could become potential therapeutic targets. PI3K-AKT signaling pathway might play an important role in HHS against UC. Moreover, molecular docking suggested that quercetin, luteolin, and nobiletin combined well with JUN, TP53, and ESR1, respectively. Cell experiments showed that the most important ingredient of HHS, quercetin, could inhibit the levels of inflammatory factors and phosphorylated c-Jun, as well as PI3K-Akt signaling pathway in LPS-induced RAW264.7 cells, which further confirmed the prediction by network pharmacology strategy and molecular docking.
Conclusion: Our results comprehensively illustrated the bioactives, potential targets, and molecular mechanism of HHS against UC. It also provided a promising strategy to uncover the scientific basis and therapeutic mechanism of TCM formulae in treating diseases.
Keywords: Huai Hua San, ulcerative colitis, network pharmacology, quercetin, JUN, PI3K/AKT signal pathway
Graphical Abstract:

Introduction
Ulcerative colitis (UC) is a form of inflammatory bowel disease (IBD) characterized by inflammation of the intestinal mucosa, whose pathogenesis consists of related immuno-inflammatory pathways.1 Although some traditional therapeutic drugs, for instance, corticosteroids, can be applied to treat UC, there still exist some adverse effects.2,3 Moreover, the incidence of the disease has been increasing over the past decade in both industrialized and developing countries, while the burden will continue to rise worldwide.4–7 Hence, it is urgent to explore alternative novel therapies of UC with low toxicity, low cost and high quality.
After thousands of years of development, numerous herbal formulae of Traditional Chinese Medicine (TCM) have been widely applied in multiple diseases, including cancer,8 diabetes mellitus,9 and so on. Besides, over the last few decades, TCM has been playing a more and more significant role in Chinese clinical practice especially during the COVID-19 pandemic in 2019.10–14 However, due to the complexity of the interactions between components and components, components and targets, targets and the disease, as well as targets and targets, how to uncover the mysterious veil of the underlying molecular mechanism of TCM remains a considerable challenge, which has largely hindered the universal acceptance of TCM worldwide.
Huai Hua San (HHS) is a classic TCM formula first mentioned in a famous ancient medicine treatise, “Pu-Ji-Ben-Shi-Fang”, of the Song dynasty. The formula comprises four herbs, including Aurantii Fructus (Chinese name: Zhiqiao), Schizonepetae Spica (Chinese name: Jingjiesui), Sophorae Flos (Chinese name: Huaihua), and Platycladi Cacumen (Chinese name: Cebaiye) at a weight ratio of 1:1:2:2. Massive evidence has demonstrated that HHS alone or HHS plus other herbs can exert favorable anti-UC effects. HHS was reported to effectively treat UC in rats mainly by reducing the infiltration of inflammatory cells and decreasing the level of inflammation.15 Besides, HHS has the potential to significantly regulate the composition and structure of colonic microbial and prevent DSS-induced dysbiosis, thereby ameliorating colitis in rats.16 Furthermore, according to further classification of UC by TCM theory, HHS could be used in combination with other TCM formulae including Taohuatang, Danggui Shaoyaosan, and Xianfang-Huoming Decoction, all of which exerted good anti-UC effects in clinical practice, with the efficacy consistent with or even better than the treatment with classic modern medicines of UC.17–19 Despite HHS’s long history of use and abundant experimental and clinical support, its active ingredients, putative target genes, and underlying mechanism have not been fully clarified.
Network pharmacology, as a brand new subject based on system biology, bioinformatics, and high throughput histology, is getting more and more motivated.20–22 In practice, the general routes of network pharmacology include research network construction based on databases, network analysis to search the key compounds and targets, and experimental verification to ensure the reliability of the predicted results.23 Therefore, by integrating TCM with network pharmacology analysis, a novel approach, TCM network pharmacology, was established, which can be applied to prioritize disease-associated genes, reveal drug-gene-disease associations, and illustrate the network regulation effects of TCM formulae.24,25 Additionally, molecular docking is a theoretical method to study the interaction and recognition between protein receptors and small molecule ligands owing to its ability to predict the binding mode and affinity strength.26–28 Collectively, network pharmacology and molecular docking can be complementarily integrated for studying TCM, which can provide novel insights in active compounds screening and mechanism exploration, and may even trigger a revolution in the process of TCM modernization and internationalization.
In the present study, a network pharmacology-based strategy, combined with molecular docking and in vitro validation, was performed to discover the active ingredients, potential targets, and molecular mechanism of HHS against UC. The workflow is shown in Figure 1
 |
Figure 1 Schematic representation of the proposed mechanism in HHS against UC. Network pharmacology was used to analyze the crucial ingredients and key targets of HHS in the treatment of UC; molecular docking revealed that three candidate compounds could bind well with three candidate targets, respectively; cell experiments confirmed that candidate compound quercetin can alleviate inflammation in LPS-induced RAW264.7 cells by inhibiting inflammatory factors, phosphorylated c-Jun, as well as downregulating PI3K-Akt signaling pathway. |
Materials and Methods
Data Preparation
Composition of HHS
The compounds of four herbs in HHS were obtained from four public databases including Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform (TCMSP, https://tcmspw.com/tcmsp.php), A Bioinformation Analysis Tool for Molecular mechanism of Traditional Chinese Medicine (BATMAN-TCM, http://bionet.ncpsb.org.cn/batman-tcm/), The Encyclopedia of Traditional Chinese Medicine (ETCM, http://www.tcmip.cn/ETCM/index.php/Home/Index/index.html), and Traditional Chinese Medicine Database@Taiwan (TCM Database@Taiwan, http://tcm.cmu.edu.tw/zh-tw/review.php?menuid=3), all of which can provide information of ingredients in TCM herbs and have been widely reported to be available for compounds exploration in network pharmacology.29–32
Selection of Active Compounds by Pharmacokinetic ADME Prediction
The majority of compounds in TCM cannot reach the specific protein targets in cells owning to their unsuitable pharmacological properties. An appropriate oral bioavailability (OB) is one of the most significant pharmacokinetic parameters of the ADME (absorption, distribution, metabolism, and excretion) properties of drugs due to the primacy and convenience of oral administration, especially for TCM.33 Observation of drug-likeness (DL) is valuable to pre-evaluate plenty of compounds in a short time, and a widely used filter is Lipinski’s Rule-of-five. Compounds that do not violate any criteria as Lipinski’s Rule-of-five are more likely to become oral drugs.34,35
In this study, the specific OB and DL values of compounds in HHS were collected from the TCMSP database. Only those meeting the ADME criteria (ie, OB≥30% and DL≥0.18) were retained for further research.
Collection of Targets of Active Compounds in HHS and UC-Related Targets
Putative targets of active compounds in HHS were obtained from five online databases containing TCMSP, BATMAN-TCM (score cutoff was set to 20), ChemMapper (http://lilab-ecust.cn/chemmapper/, similarity threshold was set to 1.2 and DrugBank was chosen as the bioactivity database), PharmMapper Server (http://www.lilab-ecust.cn/pharmmapper/index.html, normalized fit score ≥0.9), and TargetNet (http://targetnet.scbdd.com/home/index/, Prob value ≥ 0.5), all of which can provide information of multiple proven or predicted targets for the given molecules.29,30,36–38 The UniProt database (https://www.uniprot.org/), which offers a comprehensive, high-quality, and freely accessible resource of protein sequence and functional information,39 was utilized to convert protein target names to their corresponding official gene symbols. Besides, UC-related targets were retrieved by searching the keyword “ulcerative colitis” from five public databases: GeneCards (https://www.genecards.org/, Relevance score ≥1), Online Mendelian Inheritance in Man (OMIM, https://omim.org/), PharmGKB (https://www.pharmgkb.org/), Therapeutic Target Database (TTD, http://db.idrblab.net/ttd/), and DrugBank (https://go.drugbank.com/), all of which can be applied to collect disease-related targets.40–44
Bioinformatics Analysis
Protein–Protein Interaction (PPI) Data
To further identify the core regulatory targets, PPI analysis was performed by submitting overlapping targets of active compounds in HHS and UC to the STRING database (https://string-db.org/), which currently owns the largest number of organisms and proteins, as well as broad and diverse benchmarked data sources.45 The species was limited to Homo sapiens, the minimum required interaction score was set to 0.97, and the independent target protein nodes were hidden. Subsequently, the PPI results were exported from STRING and imported into Cytoscape 3.8.0, which is widely applied to construct and visualize the network, especially in the field of network pharmacology analysis.
The CytoNCA plugin in Cytoscape software was utilized to calculate six topological parameters of nodes: betweenness centrality (BC), closeness centrality (CC), degree centrality (DC), eigenvector centrality (EC), network centrality (NC), and local average connectivity (LAC), all of which can provide an in‑depth analysis of the attributes of nodes in the interactive network. Higher quantitative values of parameters stand for the greater significance of nodes in the network. Only target nodes with all six parameters higher than the corresponding median values in PPI network were retained to construct a new PPI network for further research. The NetworkAnalyzer plugin for Cytoscape software was used for node degree distribution diagrams, and the network was treated as non-directed graph. Likewise, in the new network, six parameters were calculated and nodes with all six parameters higher than the corresponding median values were retained to construct the core PPI network.
Enrichment Analysis
After transferring official gene symbols of identified HHS-UC target genes to associated Entrez IDs, Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were carried out to further study the functions of the identified potential anti-UC target genes of HHS based on R 4.0.2 and related R packages (colorspace, stringi, DOSE, clusterProfiler, ggplot2, enrichplot, pathview, BiocManager, and org.Hs.eg.db). Only functional terms and pathways with q values <0.05 were considered statistically significant and retained.
Network Construction
Data of HHS-UC targets, bioactive compounds, disease, herbs, or signaling pathways were imported into the Cytoscape 3.8.0 software. After being polished, three networks were obtained as follows: (1) Compounds-disease-targets (C-D-T) network of treatment with HHS for UC; (2) PPI network of treatment with HHS for UC; and (3) Compounds-pathways-targets (C-P-T) network of treatment with HHS for UC.
Validation
Molecular Docking
Molecular docking is a frequently used method in drug discovery for its capacity to accurately predict the conformation of small molecule ligands within the appropriate target binding site and to assess the binding affinity.46,47 In the present study, candidate target proteins were selected for molecular docking analysis based on being from the human species, owning higher degree values in the core PPI network (s 2D and 3), and associated with more significant bioactive compounds in C-D-T network (Figure 4); candidate compounds for docking were selected based on owning higher values of degree in C-D-T network (Figure 4) and associated with more key targets in the core PPI network (Figure 2D). The docking process combined AutoDock tools 1.5.6 with Vina (ie, AutoDock Vina), which is a new and widely used strategy for molecular docking and has been validated to improve the speed and accuracy of molecular docking with a new scoring function, efficient optimization and multithreading.48,49 Parameters of the mating box were obtained from AutoDock tools 1.5.6, and the docking was carried out by Vina. The specific process was as follows:
 |
Figure 2 PPI network of HHS-UC. (A) The interactive PPI network obtained from STRING database with the minimum required interaction score set to 0.97. It comprises 268 nodes and 637 edges. Each node represents relevant targets, and edges stand for protein-protein associations, including known interactions (azure represents curated databases, purple indicates experimentally determined), predicted interactions (green represents gene neighborhood, red stands for gene fusions, and blue indicates gene co-occurrence), and others (light green represents text mining, black stands for co-expression, and light blue indicates protein homology). (B) PPI network imported from STRING database to Cytoscape 3.8.0. (C) PPI network of more significant proteins extracted from (B) by filtering 6 parameters: BC, CC, DC, EC, NC and LAC. This network is made up of 75 nodes and 307 edges. (D) Core PPI network of core proteins extracted from (C); this network contains 18 nodes and 57 edges. Higher degree values indicate larger node sizes. Red indicates a higher degree, and yellow represents a lower degree. |
 |
Figure 3 (A–C) The node degree distribution diagrams of Figure 2 B–D; (D) The degree of 18 core targets of the PPI network in Figure 2D. The y-axis displays the degree values of related targets, and the x-axis shows gene names. |
 |
Figure 4 C-D-T network of HHS against UC. It contains 454 nodes and 2569 edges. Blue ellipse nodes stand for the target genes; the purple, orange, green, and rose-red colors in ellipse nodes indicate bioactive compounds from Schizonepetae Spica, Sophorae Flos, Platycladi Cacumen, and Aurantii Fructus, respectively; and the red diamond nodes represent four herbs in HHS. |
(1) The preparation of macromolecule receptor files: 3D structures of proteins were retrieved from the Protein Data Bank (PDB) (http://www.rcsb.org/) and then inputted into software Pymol to be modified by removal of water molecules, co-crystallized ligand and ions. Subsequently, in AutoDock tools 1.5.6, missing hydrogens and Kollman partial charges were added, non-polar hydrogens were merged to their corresponding carbons. The structures were then saved as PDBQT protein receptor files.
(2) The preparation of small molecule ligand files: After downloaded from PubChem database (https://pubchem.ncbi.nlm.nih.gov/), the 2D structures of compounds were then imported into Chem3D software to minimize energy (MM2 force field) and transferred to 3D structures. Subsequently, in AutoDock tools 1.5.6, the obtained 3D structures were modified by the addition of hydrogens and protonation. Non-polar hydrogens were merged to their corresponding carbons rotations, and torsions were set automatically in the software. Then structures were saved as PDBQT ligand files.
(3) The construction of mating pockets: The PDBQT structures of receptors and ligands were imported to AutoDock tools 1.5.6 to construct mating pockets of docking. When constructing a mating box, spacing (angstrom) was set to 1, center was set on the macromolecule, and numbers of points in x-, y-, and z-dimension were set to keep the protein completely covered by mating box. For each protein, these parameters of the mating box were defined by a configuration (config) file for further docking by Vina, which included the following: the center of the mating box (centre_x, centre_y, centre_z), the size of the mating box (size_x, size_y, size_z), the maximum number of binding modes to output (num modes) (set to 20), energy_range (set to 5) and exhaustiveness (set to 8).
(4) Docking and visualization: Vina software was utilized to conduct molecular docking and calculate docking affinity. During the process, PDBQT files of protein and ligand were also used in addition to the config file, as the PDBQT files determine the information of ligands and proteins for docking. After Vina finished running, the results were output as .pdbqt* and log*.txt files and scored according to docking binding free energy to establish the binding affinity score between the target protein receptors and small molecule ligands. Docking models with the lowest binding affinity were visualized and hydrogen bonds were displayed by Pymol (3D) and LigPlus (2D), both of which have been widely applied to visualize the docking results including docking site and hydrogen-bond interaction patterns between the ligand and the main-chain or side-chain elements of the protein.50,51
Cell Culture
Mouse mononuclear macrophage leukemia cells (RAW264.7), purchased from Beijing Beina Chuanglian Institute of Biotechnology (Beijing, China), were cultured in Dulbecco’s Minimum Essential Medium (DMEM) medium supplemented with 10% fetal bovine serum, 100μg/mL streptomycin, and 100U/mL penicillin, at 37°C with 5% CO2 and 95% air.
Enzyme-Linked Immunosorbent Assay
The RAW264.7 macrophage cells were seeded in 6-well plates at 1.0–2.0×105 cells/well and incubated for 24 h. Subsequently, the experiment was divided into six groups, including control group, Que (H) group (quercetin: 20μM), LPS group (model group, LPS: 1μg/mL), LPS + Que (L) group (LPS: 1μg/mL, quercetin: 5μM), LPS + Que (M) group (LPS: 1μg/mL, quercetin: 10μM), LPS + Que (H) group (LPS: 1μg/mL, quercetin: 20μM). The culture supernatant was collected and then centrifuged at 3000 rpm for 20 minutes. Finally, the levels of TNF-α, IL-6, and IL-1β, and relative enzymatic activities of iNOS and COX2 were detected in the culture media through corresponding ELISA kits according to the manufacturer’s instructions.
Western Blot Analysis
Cell lysates were collected by using RIPA buffer with 0.1% PMSF (BOSTER Biological Technology; Wuhan, China). The protein samples were separated by SDS-PAGE using 8–10% gradient gel. The separated protein on the gel was transferred onto 0.45 μm PVDF membranes. The expression of target protein was normalized to GAPDH protein. Antibodies against iNOS (#13120), COX-2 (#12282), p-c-Jun (#9261), c-Jun (#9165), p-PI3K (#4228), PI3K (#4249), p-Akt (#2965), Akt (#4691), GAPDH (#2118), and horseradish peroxidase-linked anti-mouse IgG (#7076) or anti-rabbit IgG (#7074) were purchased from Cell Signaling Technology (Boston, USA).
Statistical Analysis
The results were presented as means ± S.E.M and data comparison of multiple groups was adopted by one-way ANOVA and Student-Newman-Keuls test. The results were considered statistically significant if the P value was less than 0.05.
Results
Data Statistics
The Acquisition of Active Compounds in HHS Using ADME Screening
HHS consists of Aurantii Fructus, Schizonepetae Spica, Sophorae Flos, and Platycladi Cacumen. After searching four databases and performing ADME screening (OB≥30 and DL≥0.18), a total of 28 bioactive compounds were recognized in HHS (Table 1). The 28 compounds included 10 species from Schizonepetae Spica, 10 species from Aurantii Fructus, 8 species from Sophorae Flos, and 7 species from Platycladi Cacumen. Among them, four compounds (hesperetin, kaempferol, quercetin, and β-sitosterol) were derived from more than one herb in HHS and may be the crucial compounds with the therapeutic potential in HHS for UC. The contents of some compounds in related herbs and HHS are shown in Supplementary Table 1.
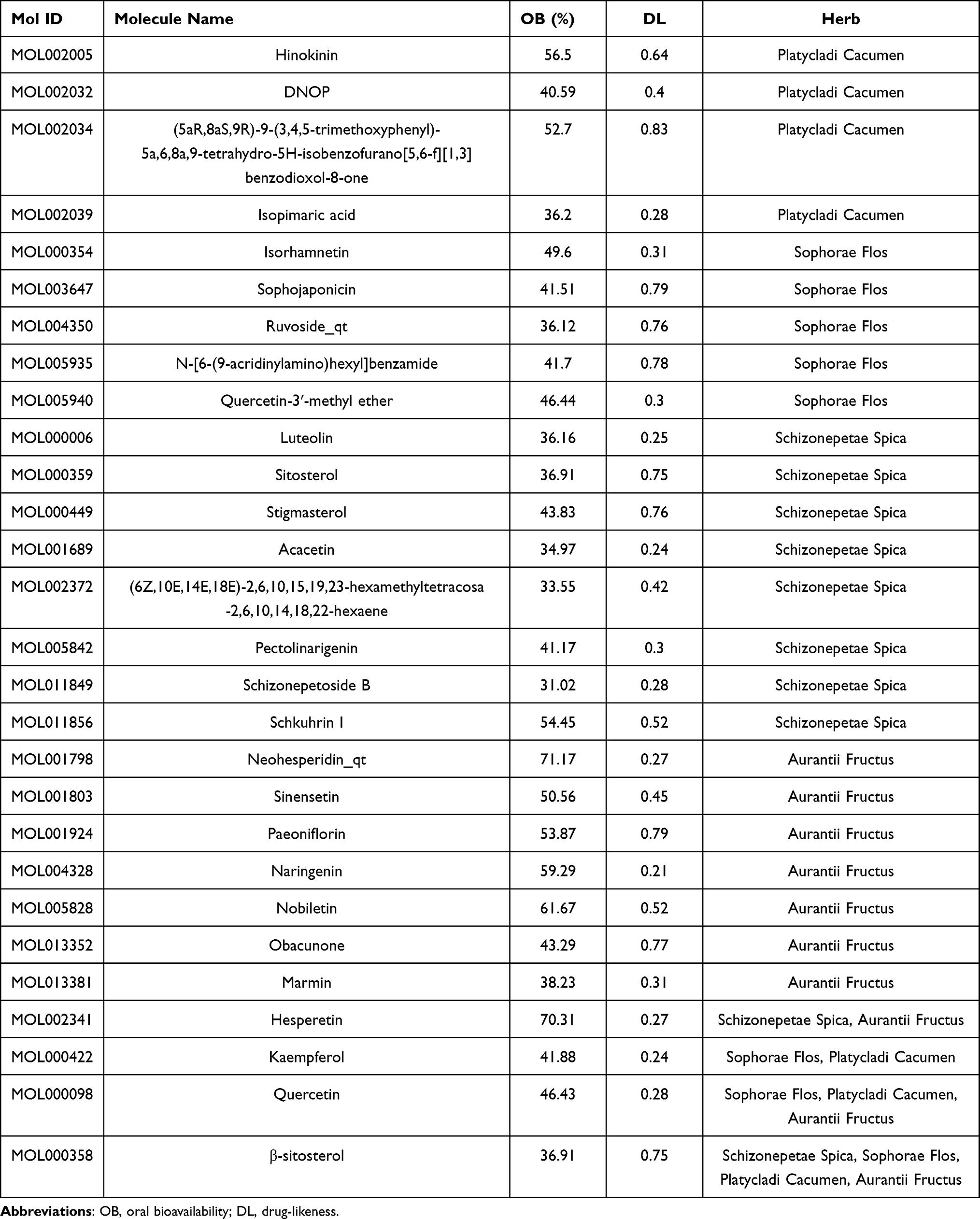 |
Table 1 28 Bioactive Compounds of HHS |
Targets of Bioactive Compounds in HHS and UC-Related Targets
A total of 1052 reviewed or predicted target genes of the bioactives in HHS were retrieved from five databases after we removed duplication values and transferred protein names to gene symbols (Supplementary Table 2). Besides, target genes related to UC were collected from GeneCards (Relevance score≥1, Supplementary Table 3), PharmGkb, Therapeutic Target Database (TTD), DrugBank, and Online Mendelian Inheritance in Man (OMIM) databases, which included 4100 genes in GeneCards, 15 in PharmGkb, 52 in TTD, 182 in DrugBank, and 1 in OMIM by using “ulcerative colitis” as the keyword. A total of 4153 related target genes for UC were collected after being taken for intersection (Figure 5A and Supplementary Table 4). Among 1052 HHS-related targets and 4153 UC-related targets, there were 421 overlapping identified targets, considered as the hub targets for subsequent study (Figure 5B and Supplementary Table 5).
 |
Figure 5 Venn diagrams showing UC targets obtained from 5 databases (A) and the intersection of identified target genes of active compounds and UC (B), respectively. |
The Construction of C-D-T Network and Topological Network Analysis
The association between 28 active compounds in HHS and 421 HHS-UC target genes was visualized by C-D-T network containing 454 nodes and 2569 edges (Figure 4). In C-D-T network, blue ellipse nodes stand for the target genes; the purple, orange, green, and rose-red colors in ellipse nodes indicate bioactive compounds from Schizonepetae Spica, Sophorae Flos, Platycladi Cacumen, and Aurantii Fructus, respectively; the red diamond nodes represent four herbs in HHS. Nodes with a greater number of edges have higher degree values and larger node sizes in the network, indicating greater significance and requiring more attention. The top 8 compound nodes with the largest size of degree were quercetin (MOL000098, n=180), isopimaric acid (MOL002039, n=114), kaempferol (MOL000422, n=104), DNOP (MOL002032, n=102), luteolin (MOL000006, n=102), naringenin (MOL004328, n=92), (6Z,10E,14E,18E)-2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene (MOL002372, n=91), and nobiletin (MOL005828, n=90). The top five gene nodes with the largest size of degree were CA2, RIPK2, TLR9, ALOX15, and PTGS1, with the degree values of 29, 27, 27, 26, and 25, respectively. These top compounds and genes may be the critical nodes in the network and possess an important anti-UC effect.
Construction and Analysis of HHS-UC-Related PPI Network
PPI network which contains 268 nodes and 637 edges was primitively constructed by STRING database, in which nodes represent proteins and edges stand for protein–protein interactions (Figure 2A). In order to further visualize and analyze the protein–protein interactions, the retrieved PPI data were subsequently imported into Cytoscape 3.8.0 to construct a new PPI network, which also includes 268 nodes and 637 edges (Figure 2B). Subsequently, the CytoNCA plugin in Cytoscape software was utilized for mining the core goals. The selection criteria based on corresponding median values were set as follows: BC >17.73, CC >0.01776, DC >3.00, EC >0.01394, NC >1.55, and LAC >1.00. Only gene targets that met the criteria were retained and colored yellow in Figure 2B, and a new PPI network made up of 75 nodes and 307 edges was extracted (Figure 2C). Likewise, in the network in Figure 2C, only targets that met the selection criteria (BC >36.71, CC >0.4111, DC >7.00, EC >0.08690, NC >3.80, and LAC >2.80) were retained and colored yellow in Figure 2C. Finally, a core PPI network comprising 18 nodes and 57 edges was screened out (Figure 2D). Node degree distribution diagrams of PPI networks were retrieved by a Cytoscape plug-in, Network analyzer (Figure 3A–C). In the core PPI network in Figure 2D, a node with larger size and redder color possesses a higher degree value, likewise, a node with smaller size and more yellow color has a smaller degree value. The target nodes with the highest degree values were JUN (n=12), MAPK1 (n=10), MYC (n=9), TP53 (n=8), MAPK3 (n=8), ESR1 (n=6), AKT1 (n=6), FOS (n=6), HSP90AA1 (n=6), IL6 (n=6), CDKN1A (n=6), and TNF (n=6) (Figure 3D), which may play a crucial role in the anti-UC effect of HHS.
Enrichment Analysis
In order to elucidate the functions and the enriched pathways of the potential anti-UC genes of HHS, GO enrichment analysis and KEGG pathway enrichment analysis were carried out.
The GO analysis consists of biological processes (BP), cellular component (CC), and molecular function (MF). In this study, a total of 3963 statistically significant GO terms, including 3528 of BP, 112 of CC, and 323 of MF, were obtained. The top 20 significant enrichment terms of BP, CC and MF with the highest gene counts were visualized in a bubble diagram in Figure 6. The redder color of a dot stands for the lower the q value and greater enrichment of the GO term. The results showed that the targets of HHS in the treatment of UC were mainly enriched in response to molecule of bacterial origin (GO: 0002237), response to lipopolysaccharide (GO: 0032496), response to nutrient levels (GO: 0031667), and other biological processes; in membrane raft (GO: 0045121), membrane microdomain (GO: 0098857), membrane region (GO: 0098589), and other cellular components; in protein serine/threonine kinase activity (GO: 0004674), DNA-binding transcription factor binding (GO: 0140297), cytokine receptor binding (GO: 0005126), and other molecular functions.
 |
Figure 6 The bubble diagram of GO enrichment analysis of HHS-UC genes, including the top 20 significant enrichment terms of BP, CC, and MF. The abscissa represents the proportion of genes of interest in the entry, and the ordinate represents each entry. The larger size of a dot indicates the larger number of genes annotated in the entry, and the redder color of a dot stands for the lower the q value. |
The KEGG pathways were applied to explore the functions and signaling pathways of the identified anti-UC targets of HHS. There were 190 statistically significant HHS-UC-related pathways, among them, the top 30 significant enrichment potential pathways with the highest gene counts were presented in a bar plot diagram (Figure 7), indicating that HHS plays an important role in the treatment of UC through multiple targets and multiple pathways. Furthermore, to elucidate the interrelationships of ingredients, targets and the top 30 pathways, C-P-T network containing 483 nodes and 3404 edges was constructed (Figure 8). The pathways with the top 5 highest gene counts were PI3K-Akt signaling pathway (n=68), Human cytomegalovirus infection (n=58), MAPK signaling pathway (n=57), Kaposi sarcoma-associated herpesvirus infection (n=56), and Hepatitis B (n=55), which could be the key pathways in the effect of HHS against UC. Among them, PI3K-Akt signaling pathway, associated with the highest number of genes, might be the most important HHS-UC pathway (Figure 9). Interestingly, we found that several target genes were involved in multiple pathways, for example, JUN, mentioned above as the most important target in HHS for UC, was involved in 54 HHS-UC-related KEGG pathways, including Inflammatory bowel disease, Kaposi sarcoma-associated herpesvirus infection, Hepatitis B, etc. Collectively, these results demonstrated that HHS acts on UC through multiple pathways, multiple targets, and overall cooperation.
 |
Figure 7 Bar plot diagram of KEGG enrichment pathways (top 30). The x-axis represents the number of target genes in each pathway and the ordinate represents each entry. Redder means lower q value. |
 |
Figure 8 C-P-T network of top 30 pathways. Blue ellipse nodes stand for the target genes. The ellipse nodes in purple, orange, green and rose-red stand for compounds from Schizonepetae Spica, Sophorae Flos, Platycladi Cacumen and Aurantii Fructus. The representatives of red V nodes indicate four herbs in HHS. Each yellow diamond stands for each pathway. The size of the nodes represents the size of the degree. |
 |
Figure 9 PI3K/AKT signaling pathway map. Nodes in red represent HHS-UC-related genes. |
Molecular Docking Analysis
Three candidate target proteins, including JUN (PDB ID: 1jun), TP53 (PDB ID: 1qkt), and ESR1 (PDB ID: 2vuk), were conducted molecular docking with three candidate bioactive compounds (quercetin, luteolin, and nobiletin; the corresponding 2D-chemical structures drawn using ChemDraw software are presented in Figure 10), whose results were visualized by Pymol (2D) and LigPlus (3D) as shown in Figure 11.
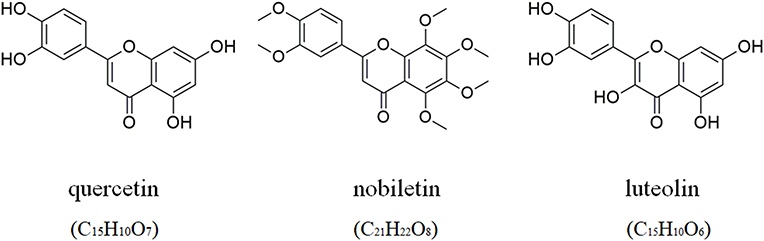 |
Figure 10 The molecular formulae and chemical structures of quercetin, luteolin, and nobiletin. |
 |
Figure 11 The docking model of quercetin, luteolin and nobiletin with JUN, TP53, and ESR1, respectively. The action model of JUN with quercetin, luteolin and nobiletin: (A) JUN with quercetin; (B) JUN with luteolin; (C) JUN with nobiletin. The action model of TP53 with quercetin, luteolin and nobiletin: (D) TP53 with quercetin; (E) TP53 with luteolin; (F) TP53 with nobiletin. The action model of ESR1 with quercetin, luteolin and nobiletin: (G) ESR1 with quercetin; (H) ESR1 with luteolin; (I) ESR1 with nobiletin. The hydrogen bonds were indicated by dashed lines and the length was added around the lines. |
The results revealed that JUN interacted with quercetin, luteolin, and nobiletin, as shown in Figure 11A–C, respectively. As shown in Figure 11A, the structure of quercetin could interact with Gln290 and Asn299 through 2 hydrogen bonds, respectively, and Ser292 through one hydrogen bond in JUN. As shown in Figure 11B, the structure of luteolin could form one hydrogen bond, one hydrogen bond, and 2 hydrogen bonds with Arg302, Gln310 and Gln304, respectively, in JUN. As shown in Figure 11C, nobiletin could interact with Arg302 and Gln304 in JUN via one hydrogen bond and 2 hydrogen bonds, respectively.
The results indicated that TP53 interacted with quercetin, luteolin, and nobiletin, as shown in Figure 11D–F, respectively. As shown in Figure 11D, the structure of quercetin could form one hydrogen bond, one hydrogen bond and 2 hydrogen bonds with Ser166, Ala138 and Asn200 in TP53, respectively. As shown in Figure 11E, the structure of luteolin could interact with Ala138, Ser166, Arg196, Gln100, and Lys101 through one hydrogen bond, respectively, and Ser99 through 2 hydrogen bonds in TP53. As shown in Figure 11F, one hydrogen bond could combine nobiletin with Gly199 in TP53.
The results demonstrated that ESR1 interacted with quercetin, luteolin, and nobiletin, as shown in Figure 11G–I, respectively. As shown in Figure 11G, the structure of quercetin was linked to His524, Thr347, and Leu346 through one hydrogen bond, respectively. As shown in Figure 11H, the structure of luteolin could interact with His524, Thr347, Glu353, and Leu387 through one hydrogen bond, respectively, in ESR1. As shown in Figure 11I, nobiletin could form one hydrogen bond with His377 in ESR1. Therefore, there exists an interplay between these compounds and protein targets with hydrogen bonds.
The binding energy was frequently calculated to evaluate the affinity degree of ingredients with protein targets. It is generally accepted that binding energy which is less than −4.25 kcal/mol, −5.0 kcal/mol or −7.0 kcal/mol indicates a certain, good or strong binding activity between the ligand and the receptor, respectively. The binding energy reflects the possibility of binding between the receptor and the ligand. The lower the binding energy, the higher the affinity of the receptor and the ligand, the more stable the conformation.52 The results of docking binding energy in Table 2 revealed that these ingredients could bind well to the active sites of protein targets. Among them, the docking of ESR1 and luteolin had the lowest binding energy (−9.0 kcal/mol), the docking of JUN and nobiletin had the highest binding energy (−5.4 kcal/mol), and the average binding energy was −6.96 kcal/mol, mirroring a good or even strong binding activity in the molecular docking between all three candidate compounds and each of the candidate protein targets, respectively. Together, three representative compounds (quercetin, luteolin, and nobiletin) of HHS could bind well with three core targets of UC (JUN, TP53, and ESR1), all of which might play key roles in the treatment of UC.
 |
Table 2 HHS Molecular Docking Binding Energy Results (Kcal/Mol) |
Quercetin Suppressed Inflammation in LPS-Induced RAW264.7 Cells
To investigate the anti-inflammatory effects of quercetin against UC, lipopolysaccharide (LPS)-induced RAW264.7 cells were used as the inflammatory model and three concentrations (5μM, 10μM, 20μM) of quercetin were chosen for further cell experiments according to related references. As shown in Figure 12A–C, the levels of pro-inflammatory cytokines TNF-α, IL-6, and IL-1β increased in LPS-induced cells compared with unstimulated cells, while the levels of TNF-α, IL-6, and IL-1β decreased after quercetin treatment in a dose-dependent manner compared with LPS group. As shown in Figure 12D, the intracellular enzyme activities of iNOS and COX-2 increased significantly in LPS-induced RAW264.7 cells compared with control group. However, quercetin with different concentrations significantly reduced the intracellular enzyme activities of iNOS and COX-2 in LPS-induced RAW264.7 cells compared with LPS group, among which the treatment of 20μM quercetin showed the best effect. As shown in Figure 12E, the expression levels of inflammation proteins (iNOS and COX-2) in LPS-induced RAW264.7 cells were significantly higher than those in the control group, whereas the protein expression levels of iNOS and COX-2 in LPS-induced RAW264.7 cells treated with quercetin at different concentrations were significantly inhibited in a dose-dependent manner, of which the treatment effect of 20μM quercetin group was the best. Taken together, quercetin showed anti-UC effects by inhibiting inflammation, which further validated the prediction of network pharmacology that quercetin is the most important component in HHS against UC.
 |
Figure 12 The effect of quercetin on the secretory levels of TNF-α (A), IL-6 (B) and IL-1β (C), the enzyme activity of iNOS and COX-2 (D), protein expression of iNOS and COX-2 (E) in RAW264.7 cells. Date are mean±S.E.M. n=3. **P < 0.01 vs Control; #P < 0.05, ##P < 0.01 vs LPS. |
Quercetin Inhibited the Protein Expression of c-Jun and PI3K/AKT Pathway in LPS-Induced RAW264.7 Cells
Bioinformatics analysis has indicated that c-Jun is the most important target and PI3K/Akt signaling pathway is the most important pathway in the treatment of HHS against UC. Thus, to further investigate the underlying mechanism of how quercetin exhibits its anti-UC activities, the protein levels of p-c-Jun, c-Jun, p-PI3K, PI3K, p-Akt, and Akt in RAW264.7 were examined. As shown in Figure 13, the phosphorylation expression of c-Jun, PI3K, and Akt elevated markedly in RAW 264.7 induced by LPS compared with unstimulated cells, whereas the treatment with different concentrations of quercetin significantly inhibited the high expression of phosphorylation levels of c-Jun, PI3K, and Akt compared with LPS group, among which RAW264.7 cells treated with 20μM quercetin had the best inhibitory effect. In view of the crucial roles of c-Jun and PI3K/Akt signaling pathway in inflammatory responses,53–57 it could be concluded that quercetin exerts anti-inflammatory action through the suppression of the phosphorylated expression of c-Jun and PI3K/Akt signaling pathway in the treatment of UC, which further verified the network pharmacology results of the important functions of c-Jun and PI3K/Akt signaling pathway in HHS for UC.
 |
Figure 13 The effect of quercetin on the protein secretion of c-Jun and PI3K/AKT pathway in LPS-induced RAW264.7. (A) Representative immunoblotting images of p-c-Jun, c-Jun, p-PI3K, PI3K, p-Akt, Akt, and GAPDH; (B–D) Gray value statistics of corresponding proteins. Date are mean±S.E.M. n=3. **P < 0.01 vs Control; #P < 0.05, ##P < 0.01 vs LPS. |
Discussion
HHS has been reported to possess a good pharmacological role and therapeutic effect in the modern clinical treatment of UC. HHS could reduce the levels of tumor necrosis factor (TNF-α) and medullary peroxidase (MPO) inflammatory factors in serum, relieve inflammatory cell infiltration, improve experimental colitis, and reduce the local and systemic inflammatory responses in UC rats.15,58 The therapeutic effect of HHS involved the regulation of levels of inflammatory factors and expression of related proteins through signal transduction pathways.59
UC is a serious medical issue endangering the health of people worldwide, whose optimal disease control is warranted to be achieved.4,60 However, the more detailed mechanism of HHS against UC has not been fully elucidated. Network pharmacology is a promising approach frequently applied to reveal related mechanism and discover new important bioactive compounds from TCM formulae.61 Therefore, we performed network pharmacology and data analysis to explore bioactives, core therapeutic target genes, and potential mechanism of HHS in UC. Moreover, molecular docking analysis and in vitro experiments were conducted to further explore potential mechanism of HHS against UC.
In the present study, a total of 28 bioactive compounds in HHS and 421 HHS-UC target genes were obtained after collecting and screening from multiple databases. Nowadays, a substantial of natural compounds have attracted considerable attention due to their high therapeutic value, low systemic toxicity, and a wide range of pharmacological activities.62–66 Emerging evidence has demonstrated that rutin appears to be the major bioactive compound in HHS with pharmacological effects against UC, whose potential mechanism contains antioxidant effects, suppression of pro-inflammatory mediators’ release and expression of inflammatory proteins.67–69 However, in this study, we found that quercetin, associated with the highest number of genes in C-D-T network, is the most important compound in HHS for UC. Quercetin is a polyhydroxyflavonoid with a variety of biological functions such as antioxidant, anti-inflammatory, anti-viral, and so on.70 Quercetin could offer dose-dependent protection for IBD rats through antioxidant-like effect.71 Moreover, quercetin could exert anti-inflammatory effects in the treatment of UC, which is congruent with the fact that inflammatory responses are important pathological mechanism of UC.1 Both antioxidant and anti-inflammatory effects of quercetin appeared to be mediated via interference in signal transduction pathways including the AP-1 and NF-κB, as well as kinase systems.1,69 Besides, quercetin could enhance epithelial barrier in Caco-2 cells via inducing a strong increase of tight junctions protein claudin-4.72 Interestingly, rutin, mentioned earlier as the major bioactive compound of HHS, exerted a therapeutic potential for UC by liberating its bioactive aglycone quercetin in vivo, which indicates the role of rutin as a quercetin deliverer to the large intestine and further emphasizes the extremely pivotal functions of quercetin against UC.69,73 Taken together, all these studies and our findings highlight the great significance of quercetin in the function of HHS against UC.
In addition to quercetin, other bioactive ingredients screened from HHS were also revealed to possess anti-inflammatory properties and show pharmacological activities for UC. Luteolin (3,4,5,7-tetrahydroxy flavone) with a higher degree value in C-T-D network is a flavonoid with anti-inflammatory effects and has potential clinical use for patients with inflammatory bowel conditions by upregulating upstream proteins (eg, SOCS3 in JAK-STAT signaling), inhibiting ATP binding into the Syk/Src binding pocket as an upstream regulator of NF-κB and AP-1 signaling, and altering transcriptional activation of the STAT3/IRF-1, NF-κB, and AP-1 pathways.74,75 Additionally, luteolin played a good therapeutic role in acute colitis in mice, whose potential mechanism was to activate the Nrf2 signaling pathway, promote Nrf2 to enter the nucleus, up-regulate the levels of mRNA levels of downstream target genes HO-1 and NQ01, inhibit the expression of pro-inflammatory factors TNF-α and IL-6 mRNA, enhance the antioxidant activity of the colon, and regulate oxidation/anti-oxidation balance.76 Likewise, nobiletin with a higher degree value in C-T-D network exerted anti-inflammatory effects via the downregulation of inducible expression of nitric oxide synthase and cyclooxygenase 2 in colitic rats and could restore barrier function through the inhibition of the Akt-NF-κB-MLCK pathway.77 Besides, nobiletin could decrease inflammatory symptoms and markers as well as fibrotic collagen deposition and expression in colitis mice.78 What’s more, the related anti-UC target genes or pathways of quercetin, luteolin, and nobiletin mentioned in these studies are partially consistent with the identified anti-UC target genes and pathways in the present research, indicating the interaction between multi-components and multi-targets of HHS in treating UC. All of these reports provide evidence for the therapeutic potential of luteolin and nobiletin in HHS against UC.
To explore core targets in HHS for UC, the PPI network, which indicates protein–protein interaction, was constructed, revealing that JUN, MAPK1, MYC, TP53, MAPK3 and ESR1, especially JUN, TP53, and ESR1, might be the core targets in the anti-UC process of HHS. Among them, JUN, associated with the largest number of target nodes in the core PPI network, could be the most important target in HHS against UC. Proto-oncogene JUN (c-Jun) is an immediate-early gene and plays an important role in inflammatory responses, in other words, when faced with external stimuli, the expression of c-Jun increases rapidly in cells, enabling cells to adapt to environmental changes.53–56 Given that the inflammatory responses are important pathogenesis of UC,1 it is of great significance to strictly control the expression of c-Jun. Together, these studies elucidated the crucial effect of c-Jun in inflammatory responses. Moreover, the expression of c-Jun proteins increased in UC model groups compared to control groups and decreased in treatment groups compared to UC groups.79–81 These findings are congruent with our results that JUN is extremely important and may participate in the process of HHS against UC.
In addition to JUN, we found that TP53 and ESR1, which showed a higher degree value in the core PPI network and associated with more core ingredients in C-T-D network, might also play a certain anti-UC role in the function of HHS for UC. Several studies have supported our view. TP53 codon 72 Arg/Arg polymorphism was associated with a higher risk for IBD development with the potential roles as biomarkers for cancer and dysplasia screening among patients with IBD, which might provide targeted therapy in patients with IBD-associated colorectal cancer.82,83 Moreover, TP53 mutations represented an initial step in the progression from inflamed colonic epithelium to colorectal cancer, demonstrating the connection between TP53 and the prognosis of UC.84 Besides, the methylation of ESR1 was related to dysplasia/cancer and inflammatory status in UC patients, suggesting the involvement of the methylation of ESR1 in UC carcinogenesis due to chronic active inflammation.85,86 These data further support our findings on the great significance of TP53 and ESR1 in HHS for UC.
To annotate the functions of 421 protein targets and related pathways, the GO enrichment analysis and KEGG pathway enrichment analysis were further conducted. GO results suggested that the target genes were mainly enriched in sites of membrane raft, membrane microdomain, and membrane region, as well as in the biological functions of protein tyrosine kinase activity, heme binding, and monooxygenase activity. KEGG data suggested that 213 of the 421 target proteins significantly enriched in 190 signal pathways, highlighting a therapeutic effect of HHS on UC through multiple pathways. Among them, PI3K-AKT signaling pathway, exhibiting the highest gene count enrichment among the UC-related signal pathways, appeared to be the most critical pathway involved in the treatment of UC. AKT1, as a critical node in PI3K/Akt-mTOR signaling pathway, is involved in the progress and development of UC.87 Besides, PI3K/Akt-mTOR signaling pathway is an important cell signal transduction pathway with a vital role in mediating numerous processes, including cell proliferation, apoptosis, necrosis, inflammation, and so forth. The abnormality of PI3K/Akt-mTOR cell signaling pathway is considered to be involved in the onset of UC with great impacts on UC-induced cell apoptosis and inflammation, as well as colon cancer induced by long-term UC.88–92 Additionally, by up-regulating the PI3K/Akt-mTOR signaling pathway cell apoptosis and inflammation in UC rats could be triggered.57 These findings are congruent with our view that PI3K-AKT signaling pathway is the most important HHS-UC pathway and may be involved in the anti-UC mechanism of HHS.
Based on the findings of network pharmacology, we acquired three crucial bioactive ingredients and three representative targets. Thus, molecular docking was carried out to further validate the prediction by network pharmacology analysis. The results of docking showed that quercetin, luteolin and nobiletin could combine well with JUN, TP53, and ESR1, respectively, and the docking of luteolin and ESR1 showed the lowest binding energy, suggesting that the combination was the most stable. Based on our previous findings that in HHS against UC, quercetin and JUN might be the most important compound and the most important target, respectively, in vitro experiments were performed to further study the action of quercetin on JUN in the treatment of UC. As several reports found that the expression of c-Jun proteins decreased in treatment groups compared to UC groups,79,80 our cell experiments also proved that quercetin inhibited the expression of phosphorylated c-Jun and had an anti-inflammatory effect in LPS-induced RAW264.7 cells. PI3K-AKT signaling pathway was also found to be involved in the anti-UC effects of quercetin, further validating the KEGG results. These findings confirmed the prediction by the network pharmacology approach.
However, the present study has several limitations according to the corresponding guidance.93 Firstly, the information from online databases was based on the reviewed and predicted data; thus, those unproven and undocumented compounds or targets may not be included in our analysis. Secondly, studies on the quantitative determination of the content of 28 compounds are currently incomplete; therefore, future research on content determination should be carried out. Thirdly, absorption pathways, effective parts, markedly effective components, metabolic forms of bioactives in HHS should be studied. Moreover, quercetin, though determined as the most important bioactive ingredient of HHS against UC, could not completely stand for HHS. Thus, additional research is required to further explore the potential molecular mechanism of HHS in the treatment of UC in vitro and in vivo. Lastly, the anti-UC effect of quercetin needs to be further verified in animal models and clinical trials.
In conclusion, we firstly performed network pharmacology to elucidate that in the anti-UC effects of HHS, the most important bioactive compound was quercetin, JUN was the most important target, and PI3K-AKT signaling pathway was the most critical pathway. Additionally, molecular docking analysis validated the prediction by the network pharmacology-based method. Besides, in vitro experiments further demonstrated the molecular mechanism of quercetin against UC. Based on a multidisciplinary strategy, the present study provides evidence for the therapeutic role of HHS in UC, as well as a comprehensive and innovative approach to search for active compounds, core target genes and potential mechanisms in TCM.
Acknowledgments
This study was supported by grants of the National Natural Scientific Foundation of China (No. 81703518, 81973406), Hunan Provincial Natural Scientific Foundation (No. 2019JJ50849, 2020JJ4823), Scientific Research Project of Hunan Provincial Health and Family Planning Commission (No. 202113050843), and Bethune Quest- Pharmaceutical Research Capacity Building Project (No. B-19-H-20200622)
Disclosure
The authors declare no conflicts of interest for this work.
References
1. Tatiya-Aphiradee N, Chatuphonprasert W, Jarukamjorn K. Immune response and inflammatory pathway of ulcerative colitis. J Basic Clin Physiol Pharmacol. 2018;30(1):1–10. doi:10.1515/jbcpp-2018-0036
2. Matsuoka K, Kobayashi T, Ueno F, et al. Evidence-based clinical practice guidelines for inflammatory bowel disease. J Gastroenterol. 2018;53(3):305–353.
3. Gallo G, Kotze PG, Spinelli A. Surgery in ulcerative colitis: When? How? Best Pract Res Clin Gastroenterol. 2018;32–33:71–78. doi:10.1016/j.bpg.2018.05.017
4. Ng SC, Shi HY, Hamidi N, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. 2017;390(10114):2769–2778. doi:10.1016/S0140-6736(17)32448-0
5. Coward S, Clement F, Benchimol EI, et al. Past and future burden of inflammatory bowel diseases based on modeling of population-based data. Gastroenterology. 2019;156(5):1345–1353. doi:10.1053/j.gastro.2019.01.002
6. Kaplan GG, Ng SC. Understanding and preventing the global increase of inflammatory bowel disease. Gastroenterology. 2017;152(2):313–321.e312. doi:10.1053/j.gastro.2016.10.020
7. Cohen RD, Yu AP, Wu EQ, Xie J, Mulani PM, Chao J. Systematic review: the costs of ulcerative colitis in Western countries. Aliment Pharmacol Ther. 2010;31(7):693–707. doi:10.1111/j.1365-2036.2010.04234.x
8. Xiang Y, Guo Z, Zhu P, Chen J, Huang Y. Traditional Chinese medicine as a cancer treatment: modern perspectives of ancient but advanced science. Cancer Med. 2019;8(5):1958–1975. doi:10.1002/cam4.2108
9. Wang J, Ma Q, Li Y, et al. Research progress on Traditional Chinese Medicine syndromes of diabetes mellitus. Biomed Pharmacother. 2020;121:109565. doi:10.1016/j.biopha.2019.109565
10. Corson TW, Crews CM. Molecular understanding and modern application of traditional medicines: triumphs and trials. Cell. 2007;130(5):769–774. doi:10.1016/j.cell.2007.08.021
11. Stone R. Biochemistry. Lifting the veil on traditional Chinese medicine. Science. 2008;319(5864):709–710. doi:10.1126/science.319.5864.709
12. Zhao Z, Li Y, Zhou L, et al. Prevention and treatment of COVID-19 using Traditional Chinese Medicine: a review. Phytomedicine. 2020;153308.
13. Zhang L, Yu J, Zhou Y, Shen M, Sun L. Becoming a faithful defender: traditional Chinese medicine against coronavirus disease 2019 (COVID-19). Am J Chin Med. 2020;48(4):763–777. doi:10.1142/S0192415X2050038X
14. Wu XV, Dong Y, Chi Y, Yu M, Wang W. Traditional Chinese medicine as a complementary therapy in combat with COVID-19-A review of evidence-based research and clinical practice. J Adv Nurs. 2020;77(4):1635–1644. doi:10.1111/jan.14673
15. Guo J, Xia Q, Dun W, Wu K, Huang S. A study on the effect and mechanism of huaihua powder in the treatment of ulcerative colitis by emulating the intestinal subwind blood syndrome. Modern Traditional Chinese Medicine. 2020;40(05):
16. Liu P, Bian Y, Liu T, et al. Huai hua san alleviates dextran sulphate sodium-induced colitis and modulates colonic microbiota. J Ethnopharmacol. 2020;259:112944. doi:10.1016/j.jep.2020.112944
17. Wang X, Zhang Y. Clinical study on the clinical effect of Sophora powder combined with Xianfang-Huoming decoction for ulcerative colitis. Int J Tradit Chin Med. 2017;39(08):701–704.
18. Liu F, Lei N, Tang X. Clinical observation of addition and subtraction therapy of Danggui Shaoyaosan combined with Huaihuasan to ulcerative colitis with syndrome of dampness-heat in large intestine during active stage. Chin J Exp Tradit Med Formulae. 2019;25(20):82–87.
19. Lei N, Kong P, Chen S, Tang X. Regulatory effect of Huaihuasan combined with Taohuatang on immune inflammation during active period of ulcerative colitis with cold-heat syndrome. Chin J Exp Tradit Med Formulae. 2020;26(07):86–91.
20. Zhang Y, Li S. Progress in network pharmacology for modern research of traditional Chinese medicine. Chin J Pharmacol Toxicol. 2015;29(06):883–892.
21. Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabási AL. The human disease network. Proc Natl Acad Sci U S A. 2007;104(21):8685–8690. doi:10.1073/pnas.0701361104
22. Liu Z, Sun X. Network pharmacology: new opportunity for the modernization of traditional Chinese medicine. Acta Pharmaceutica Sinica. 2012;47(6):696–703.
23. Luo TT, Lu Y, Yan SK, Xiao X, Rong XL, Guo J. Network pharmacology in research of Chinese medicine formula: methodology, application and prospective. Chin J Integr Med. 2020;26(1):72–80. doi:10.1007/s11655-019-3064-0
24. Liang X, Li H, Li S. A novel network pharmacology approach to analyse traditional herbal formulae: the Liu-Wei-Di-Huang pill as a case study. Mol Biosyst. 2014;10(5):1014–1022. doi:10.1039/C3MB70507B
25. Li S, Zhang B. Traditional Chinese medicine network pharmacology: theory, methodology and application. Chin J Nat Med. 2013;11(2):110–120. doi:10.3724/SP.J.1009.2013.00110
26. Xue LC, Dobbs D, Bonvin AM, Honavar V. Computational prediction of protein interfaces: a review of data driven methods. FEBS Lett. 2015;589(23):3516–3526. doi:10.1016/j.febslet.2015.10.003
27. Villoutreix BO, Bastard K, Sperandio O, et al. In silico-in vitro screening of protein-protein interactions: towards the next generation of therapeutics. Curr Pharm Biotechnol. 2008;9(2):103–122. doi:10.2174/138920108783955218
28. Vakser IA. Protein-protein docking: from interaction to interactome. Biophys J. 2014;107(8):1785–1793. doi:10.1016/j.bpj.2014.08.033
29. Ru J, Li P, Wang J, et al. TCMSP: a database of systems pharmacology for drug discovery from herbal medicines. J Cheminform. 2014;6(1):13. doi:10.1186/1758-2946-6-13
30. Liu Z, Guo F, Wang Y, et al. BATMAN-TCM: a bioinformatics analysis tool for molecular mechANism of traditional Chinese medicine. Sci Rep. 2016;6:21146. doi:10.1038/srep21146
31. Xu HY, Zhang YQ, Liu ZM, et al. ETCM: an encyclopaedia of traditional Chinese medicine. Nucleic Acids Res. 2019;47(D1):D976–d982. doi:10.1093/nar/gky987
32. Chen CY. TCM database@Taiwan: the world’s largest traditional Chinese medicine database for drug screening in silico. PLoS One. 2011;6(1):e15939. doi:10.1371/journal.pone.0015939
33. Tian S, Wang J, Li Y, Xu X, Hou T. Drug-likeness analysis of traditional Chinese medicines: prediction of drug-likeness using machine learning approaches. Mol Pharm. 2012;9(10):2875–2886. doi:10.1021/mp300198d
34. Giménez BG, Santos MS, Ferrarini M, Fernandes JP. Evaluation of blockbuster drugs under the rule-of-five. Pharmazie. 2010;65(2):148–152.
35. Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev. 2001;46(1–3):3–26. doi:10.1016/S0169-409X(00)00129-0
36. Gong J, Cai C, Liu X, et al. ChemMapper: a versatile web server for exploring pharmacology and chemical structure association based on molecular 3D similarity method. Bioinformatics. 2013;29(14):1827–1829. doi:10.1093/bioinformatics/btt270
37. Wang X, Shen Y, Wang S, et al. PharmMapper 2017 update: a web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017;45(W1):W356–w360. doi:10.1093/nar/gkx374
38. Yao ZJ, Dong J, Che YJ, et al. TargetNet: a web service for predicting potential drug-target interaction profiling via multi-target SAR models. J Comput Aided Mol Des. 2016;30(5):413–424. doi:10.1007/s10822-016-9915-2
39. UniProt Consortium. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019;47(D1):D506–D515. doi:10.1093/nar/gky1049
40. Stelzer G, Rosen N, Plaschkes I, et al. The geneCards suite: from gene data mining to disease genome sequence analyses. Curr Protoc Bioinformatics. 2016;54:
41. Amberger JS, Hamosh A. Searching online mendelian inheritance in man (OMIM): a knowledgebase of human genes and genetic phenotypes. Curr Protoc Bioinformatics. 2017;58:
42. Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther. 2012;92(4):414–417. doi:10.1038/clpt.2012.96
43. Wang Y, Zhang S, Li F, et al. Therapeutic target database 2020: enriched resource for facilitating research and early development of targeted therapeutics. Nucleic Acids Res. 2020;48(D1):D1031–D1041. doi:10.1093/nar/gkz981
44. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46(D1):D1074–d1082. doi:10.1093/nar/gkx1037
45. Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607–D613. doi:10.1093/nar/gky1131
46. López-Vallejo F, Caulfield T, Martínez-Mayorga K, et al. Integrating virtual screening and combinatorial chemistry for accelerated drug discovery. Comb Chem High Throughput Screen. 2011;14(6):475–487. doi:10.2174/138620711795767866
47. Meng XY, Zhang HX, Mezei M, Cui M. Molecular docking: a powerful approach for structure-based drug discovery. Curr Comput Aided Drug Des. 2011;7(2):146–157. doi:10.2174/157340911795677602
48. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455–461. doi:10.1002/jcc.21334
49. Clyne A, Yang L, Yang M, May B, Yang AWH. Molecular docking and network connections of active compounds from the classical herbal formula Ding Chuan Tang. PeerJ. 2020;8:e8685. doi:10.7717/peerj.8685
50. Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51(10):2778–2786. doi:10.1021/ci200227u
51. Martinez X, Krone M, Alharbi N, et al. Molecular graphics: bridging structural biologists and computer scientists. Structure. 2019;27(11):1617–1623. doi:10.1016/j.str.2019.09.001
52. Pei C, Shao L, Liu J, Shi H, Feng J. Study on the mechanism of Carthami Flos in treating retinal vein occlusion based on network pharmacology and molecular docking technology. Nat Prod Res Dev. 2020;32(11):
53. Mechta-Grigoriou F, Gerald D, Yaniv M. The mammalian Jun proteins: redundancy and specificity. Oncogene. 2001;20(19):2378–2389. doi:10.1038/sj.onc.1204381
54. Lamph WW, Wamsley P, Sassone-Corsi P, Verma IM. Induction of proto-oncogene JUN/AP-1 by serum and TPA. Nature. 1988;334(6183):629–631. doi:10.1038/334629a0
55. Wisdom R. AP-1: one switch for many signals. Exp Cell Res. 1999;253(1):180–185. doi:10.1006/excr.1999.4685
56. Jochum W, Passegué E, Wagner EF. AP-1 in mouse development and tumorigenesis. Oncogene. 2001;20(19):2401–2412. doi:10.1038/sj.onc.1204389
57. Jiang W, Han YP, Hu M, Bao XQ, Yan Y, Chen G. A study on regulatory mechanism of miR-223 in ulcerative colitis through PI3K/Akt-mTOR signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23(11):4865–4872. doi:10.26355/eurrev_201906_18074
58. Liu P, Zhong J, Bian Y, Zhong Y, Liu Z. The protective effect of Huaihua San on rat experimental colitis. Chin J Vet Med. 2020;56(12):
59. Chen H. Based on Intestinal sIgA and Inflammatory Factor Change to Explore the Huaihua San on the Treatment Mechanism of UC Model Rats. Hunan University Of Chinese Medicine; 2020.
60. Kobayashi T, Siegmund B, Le Berre C, et al. Ulcerative colitis. Nat Rev Dis Primers. 2020;6(1):74. doi:10.1038/s41572-020-0205-x
61. Zuo H, Zhang Q, Su S, Chen Q, Yang F, Hu Y. A network pharmacology-based approach to analyse potential targets of traditional herbal formulas: an example of Yu Ping Feng decoction. Sci Rep. 2018;8(1):11418. doi:10.1038/s41598-018-29764-1
62. Zhang H, Zhang H, Zai X, et al. Study on the effect of three natural compounds in Spartina alterniflora on uric acid. Chin Wild Plant Res. 2019;38(03):9–12.
63. Yuan X, Ji J, Xu J, et al. Natural herbs, potential autophagy inducers in cancer therapy. J Modern Oncol. 2019;27(05):879–885.
64. Cao Y, Hu Y, Zhang D, Yang J. Research advances in anti-leukemia activities of natural compounds. J Southwest Med Univ. 2020;43(03):300–305.
65. Azab A, Nassar A, Azab AN. Anti-inflammatory activity of natural products. Molecules. 2016;21(10):1321. doi:10.3390/molecules21101321
66. An J, Chen B, Kang X, et al. Neuroprotective effects of natural compounds on LPS-induced inflammatory responses in microglia. Am J Transl Res. 2020;12(6):2353–2378.
67. Zhang X, Yao G, Liu Y, Jin P. Analysis on effect of temperature to rutin content in preparing Huaihua powders by HPLC. Lishizhen Med Materia Medica Res. 2002;03:136–137.
68. Yao G, Zhang X, Zhu M, Zhao Z. Content determination of Rutin in Huaihuasan (Sophora Powder) with HPLC. Herald Med. 2003;02:115–116.
69. Habtemariam S, Belai A. Natural therapies of the inflammatory bowel disease: the case of rutin and its aglycone, quercetin. Mini Rev Med Chem. 2018;18(3):234–243. doi:10.2174/1389557517666170120152417
70. Liu S, Liu J. Advances in the pharmacological effects of quercetin. Chin J Lung Dis. 2020;13(01):104–106.
71. Dodda D, Chhajed R, Mishra J, Padhy M. Targeting oxidative stress attenuates trinitrobenzene sulphonic acid induced inflammatory bowel disease like symptoms in rats: role of quercetin. Indian J Pharmacol. 2014;46(3):286–291. doi:10.4103/0253-7613.132160
72. Amasheh M, Schlichter S, Amasheh S, et al. Quercetin enhances epithelial barrier function and increases claudin-4 expression in Caco-2 cells. J Nutr. 2008;138(6):1067–1073. doi:10.1093/jn/138.6.1067
73. Kim H, Kong H, Choi B, et al. Metabolic and pharmacological properties of rutin, a dietary quercetin glycoside, for treatment of inflammatory bowel disease. Pharm Res. 2005;22(9):1499–1509. doi:10.1007/s11095-005-6250-z
74. Franza L, Carusi V, Nucera E, Pandolfi F. Luteolin, inflammation and cancer: special emphasis on gut microbiota. Biofactors. 2021;47(2):181–189. doi:10.1002/biof.1710
75. Aziz N, Kim MY, Cho JY. Anti-inflammatory effects of luteolin: a review of in vitro, in vivo, and in silico studies. J Ethnopharmacol. 2018;225:342–358. doi:10.1016/j.jep.2018.05.019
76. Zhou Z, Liu B, Wu W. Effect of luteolin on ulcerative colitis mice. Chin J Clin Pharmacol Therap. 2005;10:1152–1155.
77. Xiong Y, Chen D, Yu C, et al. Citrus nobiletin ameliorates experimental colitis by reducing inflammation and restoring impaired intestinal barrier function. Mol Nutr Food Res. 2015;59(5):829–842. doi:10.1002/mnfr.201400614
78. Hagenlocher Y, Gommeringer S, Held A, et al. Nobiletin acts anti-inflammatory on murine IL-10(-/-) colitis and human intestinal fibroblasts. Eur J Nutr. 2019;58(4):1391–1401. doi:10.1007/s00394-018-1661-x
79. Li J. TLR2 and TLR4 Monoclonal Antibodies Blockade Suppress Murine Dextran-Sulfate-Sodium-Induced Acute Colitis. Fudan University; 2010.
80. Long M. To Investigate the Function of c-Jun in Ulcerative Colitis in Rat by Using Uyghur’s Medicine Kui Jiean and RNA Interference Technique. Xinjiang Medical University; 2007.
81. Long M, Abulaiti·Ahemaiti K, Kurexi·yunusi HJ. Expression and significance of interleukin-6 and c-jun in colon tissues of rats with ulcerative colitis. J Xinjiang Med Univ. 2007;5:446–448.
82. Volodko N, Salla M, Eksteen B, Fedorak RN, Huynh HQ, Baksh S. TP53 codon 72 Arg/Arg polymorphism is associated with a higher risk for inflammatory bowel disease development. World J Gastroenterol. 2015;21(36):10358–10366. doi:10.3748/wjg.v21.i36.10358
83. Du L, Kim JJ, Shen J, Chen B, Dai N. KRAS and TP53 mutations in inflammatory bowel disease-associated colorectal cancer: a meta-analysis. Oncotarget. 2017;8(13):22175–22186. doi:10.18632/oncotarget.14549
84. Hirsch D, Gaiser T. [Crohn’s disease-associated colorectal carcinogenesis: TP53 mutations and copy number gains of chromosome arm 5p as (early) markers of tumor progression]. Pathologe. 2018;39(Suppl 2):253–261. [German]. doi:10.1007/s00292-018-0496-9
85. Krela-Kaźmierczak I, Skrzypczak-Zielińska M, Kaczmarek-Ryś M, et al. ESR1 gene variants are predictive of osteoporosis in female patients with Crohn’s disease. J Clin Med. 2019;8(9):1306. doi:10.3390/jcm8091306
86. Saito S, Kato J, Hiraoka S, et al. DNA methylation of colon mucosa in ulcerative colitis patients: correlation with inflammatory status. Inflamm Bowel Dis. 2011;17(9):1955–1965. doi:10.1002/ibd.21573
87. Zhu Y, Shi Y, Ke X, Xuan L, Ma Z. RNF8 induces autophagy and reduces inflammation by promoting AKT degradation via ubiquitination in ulcerative colitis mice. J Biochem. 2020;168(5):445–453. doi:10.1093/jb/mvaa068
88. Baek SH, Ko JH, Lee JH, et al. Ginkgolic acid inhibits invasion and migration and TGF-β-induced EMT of lung cancer cells through PI3K/Akt/mTOR inactivation. J Cell Physiol. 2017;232(2):346–354. doi:10.1002/jcp.25426
89. Yang X, Song X, Wang X, Liu X, Peng Z. Downregulation of TM7SF4 inhibits cell proliferation and metastasis of A549 cells through regulating the PI3K/AKT/mTOR signaling pathway. Mol Med Rep. 2017;16(5):6122–6127. doi:10.3892/mmr.2017.7324
90. Wang H, Zhang C, Xu L, et al. Bufalin suppresses hepatocellular carcinoma invasion and metastasis by targeting HIF-1α via the PI3K/AKT/mTOR pathway. Oncotarget. 2016;7(15):20193–20208. doi:10.18632/oncotarget.7935
91. Ghayad SE, Cohen PA. Inhibitors of the PI3K/Akt/mTOR pathway: new hope for breast cancer patients. Recent Pat Anticancer Drug Discov. 2010;5(1):29–57. doi:10.2174/157489210789702208
92. Guerrero-Zotano A, Mayer IA, Arteaga CL. PI3K/AKT/mTOR: role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev. 2016;35(4):515–524. doi:10.1007/s10555-016-9637-x
93. Li S. Network pharmacology evaluation method guidance-draft. World J Tradit Chin Med. 2021;7(1):165–166. doi:10.4103/wjtcm.wjtcm_11_21
 © 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2021 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.