Back to Archived Journals » ChronoPhysiology and Therapy » Volume 5
Nerve growth factor, brain-derived neurotrophic factor, and the chronobiology of mood: a new insight into the "neurotrophic hypothesis"
Authors Tirassa P, Quartini A, Iannitelli A
Received 4 June 2015
Accepted for publication 5 August 2015
Published 16 October 2015 Volume 2015:5 Pages 51—64
DOI https://doi.org/10.2147/CPT.S54526
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Marc Hébert
Paola Tirassa,1 Adele Quartini,2 Angela Iannitelli2–4
1National Research Council (CNR), Institute of Cell Biology and Neurobiology (IBCN), 2Department of Medical-Surgical Sciences and Biotechnologies, Faculty of Pharmacy and Medicine – "Sapienza" University of Rome, 3Italian Psychoanalytical Society (SPI), Rome, Italy; 4International Psychoanalytical Association (IPA), London, UK
Abstract: The light information pathways and their relationship with the body rhythms have generated a new insight into the neurobiology and the neurobehavioral sciences, as well as into the clinical approaches to human diseases associated with disruption of circadian cycles. Light-based strategies and/or drugs acting on the circadian rhythms have widely been used in psychiatric patients characterized by mood-related disorders, but the timing and dosage use of the various treatments, although based on international guidelines, are mainly dependent on the psychiatric experiences. Further, many efforts have been made to identify biomarkers able to disclose the circadian-related aspect of diseases, and therefore serve as diagnostic, prognostic, and therapeutic tools in clinic to assess the different mood-related symptoms, including pain, fatigue, sleep disturbance, loss of interest or pleasure, appetite, psychomotor changes, and cognitive impairments. Among the endogenous factors suggested to be involved in mood regulation, the neurotrophins, nerve growth factor, and brain-derived neurotrophic factor show anatomical and functional link with the circadian system and mediate some of light-induced effects in brain. In addition, in humans, both nerve growth factor and brain-derived neurotrophic factor have showed a daily rhythm, which correlate with the morningness–eveningness dimensions, and are influenced by light, suggesting their potential role as biomarkers for chronotypes and/or chronotherapy. The evidences of the relationship between the diverse mood-related disorders, with a specific focus on depression, and neurotrophins are reviewed and discussed herein in terms of their circadian significance, and potential translation into clinical practice.
Keywords: retinal ganglional cells, mesocorticolimbic circuits, chronotherapy, ocular eye drops administration, neurotrophins
Neuroanatomical correlates of time, light, and mood
In humans, circadian (from the Latin word “circa diem”, meaning “about a day”) variations characterize multiple physiological and psychological functions, including core body temperature, endocrine and autonomic functions, sleep, mood, alertness, and cognitive performance (Figure 1).1,2 Sleep and wakefulness are the most obvious manifestations of the mammalian circadian system: during the day, light supports all the activities, while during the night, sleep is crucial for restoring the body and mind (cellular repair and mental recovery).2 The coordinator center of this system is the suprachiasmatic nucleus (SCN) located in the ventral hypothalamus (HYP) which receives direct projections from the retina and represents the master clock. All SCN neurons are coupled by autocrine/paracrine signals and by synaptic signals, and oscillate in coordinate manner to regulate the peripheral oscillators directly through the sympathetic and parasympathetic pathways, and indirectly by hormones, cytokines, and growth factors secretion.3,4 Conversely, signals arising from the periphery reach the brain and drive feedback information from the entire body in order to adapt the SCN activity, and generate a coherent functional network to regulate behaviors and physiology.
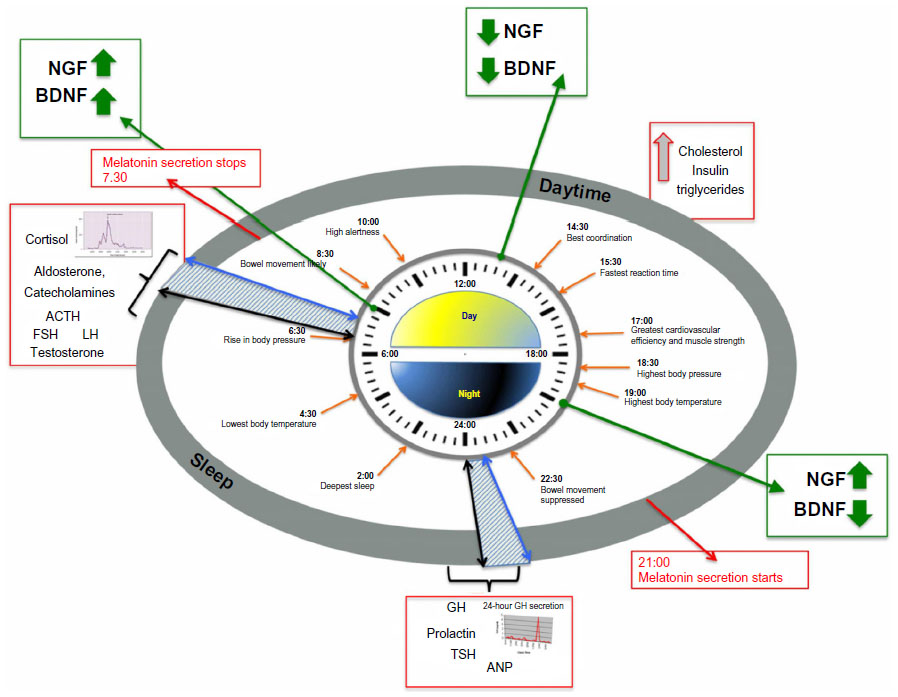 | Figure 1 Circadian time structure in persons adhering to a normal routine of daytime activity (from ~6–7 am to ~10–11 pm) alternating with nighttime sleep and a pre- and post-sleeping time (blue dashed pattern areas). |
Important features of the SCN clock are its resilience to photic cues and the shifting phase during the dark phase, which guarantee the adaptation to geophysical time or environmental changes without generating a constant modification during the light phase. This is possible because although light is the most potent entraining cue, not all the light signals from the retina are capable of phase entrainment. Indeed, only a restricted number of retinal ganglion cells (RGCs), the intrinsically photosensitive retinal ganglion cells (ipRGCs) in the inner retina layer, contribute to regulate the circadian system.5,6 These cells express the light receptor melanopsin also in the absence of any other retinal input, and regenerate their chromophore without involving other cells.7,8 They constitute ~4%–5% of all RGCs, projecting to image-forming brain areas (M2-4-5 ipRGC types), also serving as a relay station for the rods and cones, and nonimage-forming (NIF) brain areas (M1 ipRGC type). Further, ipRGCs are most sensitive to short-wavelength (~480 nm) blue light, remain functional in the absence of rods and cones, and are resistant to injury.8 These properties guarantee that even in blindness or severe ocular pathological conditions, the ipRGCs might convey light signals to the brain, and indicate that the ipRGCs-mediated effects do not require the fine spatial acuity necessary for image formation.9
The NIF effects of light include heart rate and pupil diameter, the entrainment of circadian rhythms, and modulation of locomotor activity, as well as high-level cognitive and emotional processes.10
Retrograde tracing experiments in animals and neuroimaging analysis in humans helped to identify their neuronal correlates, confirming that ipRGCs project directly to the SCN through the retinohypothalamic tract, but also show a widespread brain projection pattern from and to the SCN, which includes other hypothalamic nuclei, thalamic, striatal, brainstem, and limbic structures (Figure 2).7,11,12 Direct projections of ipRGCs to the amygdala (AMY) have been described in rodents, and a retina–AMY functional pathway, passing through the superior colliculus and thalamus, has also been found in humans.13 More, the AMY, the hippocampus (HIP), and the HYP are secondarily influenced by the NIF system by the locus coeruleus, which also receives projections from the SCN.13,14 These brain areas, which represent the neural circuits for the emotional and nonvisual cognitive information processing, and also for the circadian regulation of arousal, are selectively and wavelength dependently activated by acute light exposure, showing an increase in activity following blue light.10,15
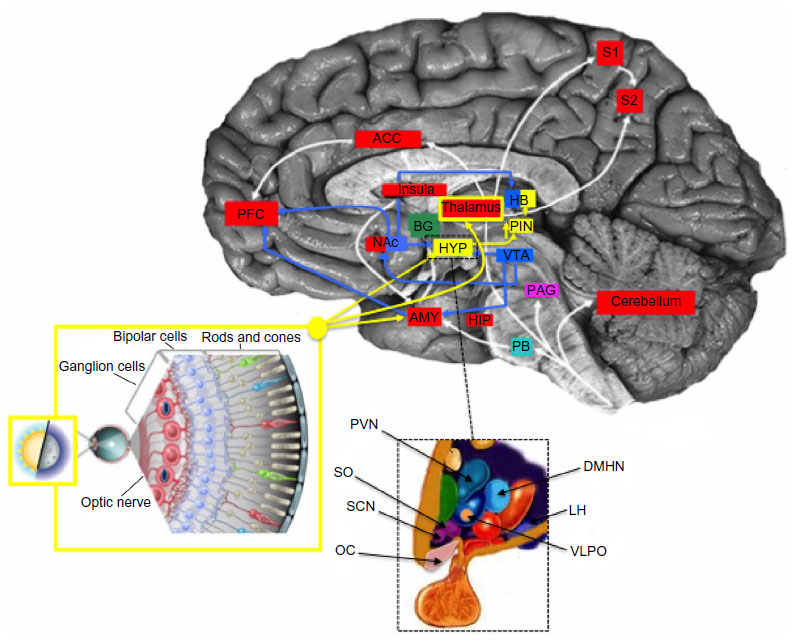 | Figure 2 Circadian, reward, and depression integrated network. |
Speculatively, light inputs to SCN also involve the dopaminergic mesocorticolimbic pathway, also called the reward circuit, which is also indicated as the common neuroanatomical and functional correlate of pain and depression.16 The core of the brain reward/aversion system is the ventral tegmental area and its projections to the nucleus accumbens (NAc), which sends the information to the subcortical limbic areas, like the AMY, the HIP, and the HYP, and then to the prefrontal cortex and the anterior cingulate cortex for processing.17 Brain endogenous opioids and dopamine (DA) pathways mainly regulate the activity of NAc neurons and the release of DA contributing to shape the behavioral response to rewarding or aversive stimuli.18
The diurnal variation of DA transmission in the mesocorticolimbic structures is dependent on the SCN and clock genes expression, thus supporting a functional correlation between the anatomical substrates for mood and mood-related symptoms, reward, and light stimuli.19,20 An integrated view of the anatomical network of NIF, mood, and reward signals is reported in Figure 2.
Brief NGF and BDNF neurobiology
The nerve growth factor (NGF) was discovered in the 1940s by the Nobel Price winners Rita Levi-Montalcini and Stanley Cohen, and it represents the first factor showing survival and differentiative effects on neuronal cells.21 Nowadays, NGF is known to be a member of a group of molecules sharing structural and functional activities, including the brain-derived neurotrophic factor (BDNF), and that are collectively named neurotrophins (NTs).22 Both NGF and BDNF are involved in the regulation of central nervous system development, and extend their survival, protective, and regenerative action on immature and mature neuronal cells during the entire life span, and even in pathological conditions.22
In mammalian brain, these two NTs and their receptors are distributed in all the forebrain areas. Concerning NT mRNAs, they were originally localized in the HIP and cortex, and lately in the striatum, the HYP, the brainstem, and pituitary. This demonstrated the synthesis of NTs in the brain, further suggesting how their local production might serve, beyond other things, to regulate the activities of interneurons, and/or the release of other factors/hormones via autocrine/paracrine mechanism (as reviewed by Sofroniew et al,22 Aloe et al,23 and Cirulli et al24).
Specific tyrosine kinase receptors, the TrkA and TrkB, mediate, respectively, most of the NGF and BDNF actions on their targets, but all the NTs also bind to a membrane glycoprotein p75 receptor (p75NTR), which can activate Trk-convergent or Trk-divergent pathways.25 Indeed, NTs binding to Trks alone or Trk/p75NTR conjointly trigger a complex network of intracellular signaling cascades, including induction of transcription factors (eg, c-fos), different classes of serine/threonine-selective protein kinases (eg, mitogen-activated protein [MAP] kinases), as well as intracellular tyrosine kinases, which result in survival and trophic effects.
Different intracellular signals can be mediated by p75NTR, which can activate survival through the nuclear factor-ºB pathway, and antagonizes the actions of TrkA through the JUN N-terminal kinase and RHOA pathways. Moreover, it has been found that the precursor NGF and BDNF forms also exert biological activity, and chiefly, activate apoptotic signals in neurons by binding the p75NTR/sortilin complex.12,26
In line with this, the increased levels of proNTs associated with an unbalance of Trks/p75NTR are considered as part of a pathological cycle, which induces neuronal degeneration and results in impairment of brain and cognitive functions.27,28
Besides their action as survival factors, NGF and BDNF affect neurotransmitter synthesis and release influencing the activity-dependent synaptic plasticity, but also take part in the reorganization of the neuronal network induced by memory and stress, as well as in depression and following drug administration.29,30
The ability of NTs to stimulate survival of neuronal precursors and neurogenesis and modulate gliogenesis further contributes to support the NGF and BDNF involvement in the regulation of new born cells and connections generated by experiences and adaptation, and in the repair and connectivity rearrangement in pathological conditions.31
It is relevant to note that NGF and BDNF are synthesized in an activity-dependent manner and released upon neuron depolarization, and that they retrogradely and anterogradely act on presynaptic and postsynaptic neurons, respectively, but also exert autocrine and paracrine influence on the surrounding cells.29 Further, it has been observed that NGF is able to stimulate BDNF synthesis and/or release in vivo and to induce BDNF and NT3, another NT, release in vitro as a consequent of TrkA activation.32,33
On the contrary, BDNF-induced release of NT3 in PC12 cells is mediated by p75NTR activation but not by Trks, indicating that the changes in one NT expression might affect the synthesis of the other NTs, and that NTs release is dependent on the relative expression of Trk/p75NTR.34 In addition, the NTs-induced enhancement of their own synthesis occur in autocrine or paracrine manner implying a potential feedback mechanism relevant to synaptic plasticity and activity-dependent functions such as memory formation, learning, and adaptation to environmental change.
To strength this notion, the modification of NTs and NT receptors distribution in the brain, as well as changes in NGF or BDNF concentration in serum, is strictly associated with cognitive and emotional performance in experimental animals and humans, and with antidepressant (AD) and/or physical therapies supporting the concept of the “neurotrophic hypothesis” of mood-related disorders.
Furthermore, in the past years, the emerging contribution of visual system in the regulation of mood and cognition has offered the possibility to prospect a more integrated view of the NTs in affective neuroscience, and in the clinical approach to mood-related disorders, which includes the anatomical and functional interplay between the sensitivity and body response to light and the NTs anatomical and functional pathways.
NGF and BDNF in the retina– brain pathways
A large amount of investigations demonstrated the role played by NTs in the development and functional maintenance of the visual system.35 Both NGF and BDNF and their receptors are expressed in the retina, optic nerve, the visual cortex, and the geniculate nucleus, where they regulate the proliferation, neurite outgrowth, and survival of cells.36 RGCs depend on the retrograde transport of NTs produced by the central targets, although both NGF and BDNF also exert paracrine and autocrine actions in the retina and the retinal recipient areas.37,38 Both exogenous and endogenous NTs can be anterogradely transported and therefore influence the survival of postsynaptic neurons and the development of synapses.38 Peculiarly, in RGCs, the NTs are not rapidly degraded after internalization, but they are differently sorted by a mechanism regulated by the Trks, so that NGF is mainly targeted to lysosomes, while BDNF is recycled to the surface membrane. NT receptors are also rapidly recycled to the cell surface, implying a regulation of receptor densities, and thus having a significant impact on the signals of survival or differentiation.39,40 The anterograde transport and the mechanism of NTs release at the postsynaptic levels also influence NTs produced in the retina, and in turn, the same retina/optic nerve pathways.
In this context, it is relevant that NGF and BDNF are synthesized in the SCN, and changes in circadian rhythmicity are observed when they are injected into the SCN or intracerebroventricularly, suggesting to be implicated in the regulation of the circadian clock.41–45
Historically, the anatomical evidence that demonstrated a dense expression of p75NGFR in the SCN was thought to explain the observed NGF, and subsequently, BDNF effects.46 Actually, the functional relevance of p75NGFR in the SCN is controversial, since the p75NTR in this nucleus is localized on the axon terminals of RGC and basal forebrain neurons and does not identify vasoactive intestinal polypeptide (VIP) neurons as initially suggested by Kiss et al.47 Null mutation of p75 gene in mice does not alter circadian rhythms of behaviors in constant dark but decreases phase shifts induced by brief pulses of light, indicating that the lack of p75NTR might be compensated by other mechanisms.48
Indeed, studies using the lesions of cholinergic projections to the SCN originating in the basal forebrain, and particularly in the nucleus basalis of Meynert and septum – the preferential NTs-responding neurons in the brain – demonstrated a role of p75NGF-cholinergic neurons in the regulation of SCN functions. However, these studies also show that residual p75-immunoreactive terminals from the retina – which might be less accessible to toxin-induced lesion – and/or non-cholinergic retinohypothalamic (RTH) fibers could be necessary to maintain a functional circadian clock.49,50
VIP and Calbidin d28k neurons, for example, are not affected by cholinergic toxin injection in the SCN or intracerebroventricular injection, but since NGF is able to stimulate VIP synthesis and to protect VIP neurons from damage, the possible involvement of Trk-mediated actions is conceivable.49–52
Further, glutamate and gamma-aminobutyric acid transmission have been demonstrated to contribute to the NTs-mediated effects on visual system, and to mediate the light-induced activation of c-fos, extracellular signal-regulated kinase (ERK) 1/2, and clock genes in the SCN.53,54
NTs also affect the response to light by the activation of c-fos, and ERK1/2 in the SCN.55 These data associated with the evidences that both the TrkA and TrkB receptors are expressed in the SCN, and that the K252a – an inhibitor of the Trk family of NT receptors – blocks light-induced phase shifts when injected in the SCN, further support the functional involvement of Trk receptors in the light-induced response in brain.45,55
It is worth to note that the Trk receptor expression is regulated by NTs, and that in turn, Trks can determine the biological outcome of p75NTR signaling, implying that the variation of local NGF and BDNF levels in SCN might correspond to changes in the receptor-mediated light signal transmission.56
To strength this notion, Baeza-Raja et al57 have recently observed that the expression of NTs and their receptors fluctuates in SCN during the light/dark cycle. Chiefly, these authors show that while the expression of NGF, p75NGFR, and TrkA oscillates in phase with clock genes during the 24 hours, the TrkB levels are unchanged. On the contrary, BDNF shows a different pattern with higher expression levels during the subjective night and the lower ones during the subjective day. The circadian NTs signaling pattern is also observed in the liver, indicating a functional link between the SCN activity and the regulation of peripheral NTs.57
Interestingly, the NGF and BDNF levels in the serum and saliva of healthy men and women are also subjected to daily fluctuation, and both the saliva and serum NT levels can be modulated by light.58 These data further indicate a correspondence between the brain and the peripheral release of NTs, and might support their role as physiological markers of the light-induced rhythmicity.
Similar to what occurs in SCN, a circadian pattern with a night peak of BDNF protein is also observable in the retina, geniculate nucleus, and the visual cortex.32,59 These data suggested that the low levels of BDNF during the subjective day might be not sufficient to activate the TrkB cascade, and therefore unable to transmit entraining light signals by retina pathways, or in other words, the BDNF secreted at night may be required for light-induced phase shifts.
On the other hand, the NGF trend shows a pick at the subjective day (CT8), and during the night (CT20), and follows the profile of clock genes, indicating a direct relationship between NGF and the SCN activity. In line with this, NGF induces a phase shift of free-running rhythms similar in both direction and circadian phase dependence to light stimuli, when injected at different time points of the circadian time implying that NGF stimulates neuronal pathways, which are coherent with light stimuli.43,44
The recent evidences that NGF administrated on ocular surface is able to exert effects in the brain might indirectly support this suggestion. Indeed, it has been demonstrated that when ocular administrated in form of eye drops (oNGF), NGF – probably through a trans-conjunctival/trans-scleral route – reaches the retina and the optic nerve and produces effects in the primary visual areas of visual cortex and geniculate nucleus.32
Subsequent studies revealed that oNGF can extent its trophic, differentiative, and regulatory actions on several forebrain structures, and similar to intracerebroventricularly injected NGF, oNGF regulates acetylcholine synthesis, induces recovery of damaged brain cholinergic neurons, and stimulates neurogenesis.28,31,32,60
Although different anatomical connections between the eyes and the brain, including those via nasal and nasolacrimal ducts, could mediate the effects of oNGF, the results of studies using radiolabeled NGF and c-fos expression as markers of neuronal activation support the involvement of retinal pathways. Indeed, NGF-I125 is found in the retina and optic nerve when administrated as eye drops indicating the transport through the RGC axons as also previously reported.32,39 The time-dependent activation of primary visual areas has been confirmed by the analysis of c-fos distribution, which also reveals the activation of several limbic areas, including the SCN, the supraoptic nucleus, and the paraventricular hypothalamic nucleus, demonstrating the involvement of the retinohypothalamic pathways.61
In parallel to increased NGF levels and the effect on the Trk and p75NTR expression, oNGF also stimulates the BDNF at both mRNA and protein levels in the retina and results in changes in BDNF in the HIP, septum, and HYP further supporting the central effects of ocular-applied NGF, and a possible cross talk between BDNF and NGF signaling in the retina and retinal recipient brain areas.28,32,60
Although future studies are necessary to better characterize the effects of oNGF on the SCN, and to disclose its potential role in the regulation of the circadian clock, it is possible to speculate that through its direct or indirect actions on other factors known to regulate light response, including BDNF, treatment with oNGF might also be useful in resetting the alteration of circadian rhythms and behaviors in pathological conditions.
NT-related hypothesis of mood-related disorders
In the past 30 years, a large amount of data from animal neurobehavioral models, human postmortem studies, brain imaging investigations, and genetic researches have demonstrated the involvement of NTs in depression, NGF and BDNF in particular, and generated the “neurotrophic theory” of affective spectrum disorders.
This theory has initially been based on the assumption that the abnormal neuronal and glial densities and architecture observable in patients with psychiatric disturbances, including depression, might depend on reduced neurotrophic support, and thus on the NGF- and BDNF-mediated protective and reparative actions.62–65
Studies on animal models support this hypothesis demonstrating the correlation between NGF and BDNF expression in brain and depressive behaviors, the response to AD treatments, and the sex-related differences in the vulnerability to depression.66
The study of pathological effects of acute and chronic stress, and environmental changes have also highly contributed to disclose the role of NTs as markers and/or risk factor for mood disorders and generate an integrated model in which the NTs-induced brain plasticity and connectivity rearrangement are directly involved in the brain adaptation and resilience and show that stimuli of psychological nature might be implicated in the mechanisms triggering NGF and BDNF release.23,67
The model of early life stress in animals, including maternal separation, has also been used to demonstrate the role of NTs in the development of anxiety-related behaviors, and therefore to identify NGF and BDNF as biomarkers for emotional and mood disorders associated with events occurring during childhood.24,62,68 Studies on humans contribute to consolidate the “neurotrophic model” by linking neurotrophic factors with the mechanisms of action of drugs used for the treatment of these disorders and the epigenetic and genetic susceptibility to develop mood disturbances.69–71 In particular, patients with major depression show significant lower NGF and BDNF levels in serum, and changes in plasma and serum NGF and BDNF occur after AD treatments among major depressive disorder patients.72,73
Genetically, a functional variant of BDNF at codon 66 (val66met) has been identified with the Met allele that results in abnormal intracellular packaging and secretion of BDNF, and it is associated with poorer episodic memory and reduced hippocampal N-acetyl aspartate.74,75 This BDNF val allele is reported to be a possible risk locus for bipolar disorder, but studies in Asian populations have not observed this association.76,77 Jiang et al found that met66 variant is a risk allele for anxiety, while a single-nucleotide polymorphism in the BDNF exon I promoter that decreases promoter activity, -281 C4A, may be protective against anxiety disorders and major depression.78
Like BDNF met66, also, NGF val35 is thought to affect intracellular processing and secretion of the NGF protein, and NGF rs6330 is associated with changes in NGF and NGF receptors in the plasma relatively to the homozygotes CC or CT and TT genotypes, in both psychiatric patients and healthy subjects.79
Sex differences in the daily NGF and BDNF levels in serum of healthy subjects and psychiatric patients further support the role of NTs in the regulation of physiological and psychological dimensions of mood.80–82
Chronopathology and chronopharmacology of mood-related disorders
The first studies on the role of the chronobiological factors in mental diseases were performed in the 1970s.83 However, just in the past 30 years, research was developed, and more relevant results were in the concept of seasonality and the demonstration of a relationship between circadian patterns and psychiatric disorders. Seasonal fluctuation of mood is observed in patients affected by the main psychiatric disorders, even though it is particularly evident in subjects affected by mood-related disorders.84 In Diagnostic and Statistical Manual of Mental Disorders, fifth edition, “seasonal pattern” is a specifier applied to the pattern of major depressive episodes or to the presence of at least one type of episode (mania, hypomania, depression) within the diagnosis of recurrent major depressive disorder or bipolar disorder, respectively.85 Its essential feature is the onset and remission of the various episodes at characteristic times of the year. In most cases, major depressive episodes begin in fall and winter, directly related to day length decreasing, and remit in spring, as day length begins to increase.85,86 These episodes are often characterized by prominent energy, hypersomnia, overeating, weight gain, and a craving for carbohydrates.85 In the study by LeGates et al, a direct association between diminished light exposure and mood functions was demonstrated. What is more, this association was clearly mediated by the ipRGCs.87 With regard to bipolar disorder, a profound switch in mood between periods of mania and depression has also been reported.88 As for major depression, in most cases, shifts to the depressive phase have been observed to begin in autumn as day length decreases and often persist throughout the winter. By March, when day length begins to increase, hypomanic/manic episodes become more prevalent, a phenomenon nicknamed “March madness”.86,89,90 At the basis of these two seasonal forms of depression, two major hypotheses have been postulated: altered pineal gland melatonin daily rhythms and circadian phase shift (for a review, see LeGates et al86). Apart from seasonal forms of depression, monthly and circadian biological clock impairment have also been described in this disorder. Circadian disturbances of the main physiological functions have widely been reported in patients with nonseasonal depression, including increased mean core temperature and decreased period amplitude.91–93 Compared with healthy subjects, patients with depression also show a circadian oscillation in plasma cortisol, norepinephrine, and prolactin, as well as abnormal patterns of melatonin secretion.94,95 With regard to depressive symptomatology, the majority of patients have been shown to present a daily pattern of symptoms, usually more severe in the morning. Up to 90% of these patients report an increase in nocturnal activity, which is accompanied by a decrease in sleeping time and the extended exposure to artificial light at night, while only a minority of them (6%–29%) report hypersomnia.96 Objective sleep measures are also disturbed.97 A phase-advance pattern of rapid-eye-movement activity and changes in dream experience are frequently observed, and as theorized by Freud, “… we must recognize in the dream, the guardian of sleep …”, a biological function and signal of psychophysical balance in sleep maintenance.98,99 Depressed patients also differ in sustained alertness over the 24-hour cycle, specifically with a reduced daytime alertness, compared with normal.100 Moreover, diurnal patterns of motor activity tend to differ between patients with mood disorders and healthy subjects.101 Reaction time and cognitive performance are impaired in morning testing but not evening testing.102
Of note, circadian disturbances have been demonstrated to affect treatment response and clinical outcome, and this is not only in depression.103 Indeed, many drugs show effects/adverse events that vary over the 24 hours of a day, and these variations represent true circadian rhythms in response (eg, they persist in constant environmental conditions). Besides, drug kinetics that govern disposition of drugs (and even target organ sensitivity) show circadian rhythms, and at least some of them are under the control of molecular clocks.104
To date, chronotherapeutics has been shown to be particularly effective in the treatment of allergic rhinitis, arthritis, asthma, cancer, myocardial infarction, peptic ulcer disease, and stroke.105 In the treatment of depression, the word “chronotherapeutics” has taken on a broader significance referring not only to a treatment scheduling corresponding to a specific patient’s biological clock but also to a controlled exposure to various environmental stimuli and medications acting on biological (circadian) rhythms, in order to achieve targeted therapeutic effects.84
A circadian rhythm in the effects/adverse events of a number of mental health medications, including ADs and mood stabilizers, has been demonstrated.106 These rhythms seem to represent endogenous circadian rhythms resulting from the rhythmicity in drug susceptibility of the brain, which is not dependent on drug kinetics but on rhythms of neurotransmitters, receptors, and second messengers.106 Back in 1978, the potent norepinephrine uptake blocker lofepramine was shown to present a greater AD effect during a 3-week course of therapy when administered at 12 am than when administered at 8 am or 4 pm.107 In the study by Nagayama et al, the AD effect of the 5-HT blocker clomipramine during 4 weeks of therapy varied depending on the time of administration.108 In this case, administration at noon was more effective than administration in the morning or in the evening. It has been observed that the norepinephrine and 5-HT systems in the brain present diverse circadian rhythms, with a peak in the release of these two neurotransmitters during the middle dark period for the former and at noon for the latter. This difference could be at the basis of the 24-hour rhythm change in the efficacy of the two tricyclic ADs, although the literature contains only scattered reports that have failed to confirm a circadian rhythm in the effects/adverse events of the various ADs.106,109,110
NGF and BDNF as neuromodulators of chronotherapeutics of depression
As mentioned earlier, in the treatment of depression, chronotherapeutics also refers to interventions known to modulate the circadian clock. In a microarray study including 12,000 transcripts, Li et al observed widespread changes in cyclic gene expression in six regions of postmortem brain tissue of depressed patients matched with controls.111 Specifically, they showed an abnormal phasing of circadian gene expression in patients, with the most robust change seen in the anterior cingulate. In fact, interventions able to induce phase shift (generally a phase advance) in circadian rhythms have been demonstrated to have AD effects.112 These interventions encompass both non-pharmacological and pharmacological strategies. Among the former, sleep deprivation therapy (keeping patients awake for ~36 hours), sleep phase advance (setting sleep time earlier and advancing bedtimes over subsequent nights), and morning bright light therapy (10,000 lux) have been shown to have the most robust AD properties.112–115 Their effect is claimed to be rapid but transient, however, with the possibility to be stabilized by combinations of the different chronotherapeutic interventions among themselves and/or with conventional psychiatric treatments (for a review, see Wirz-Justice113). In this regard, adjunctive triple chronotherapy (combined total sleep deprivation, sleep phase advance, and bright light therapy) has recently been demonstrated to induce a rapid improvement in depressive symptoms in drug-resistant mood disorders and acutely suicidal depressed in-patients without early relapse.116,117 Remarkably, all these interventions have been shown to directly act on clock gene machinery. Studies of clock gene expression in the mice brain suggest that sleep deprivation can produce rapid (within hours) alterations.118,119 Sleep phase advance, morning bright light therapy, and morning “blue” light stimulation have also been reported to affect daily clock gene expression measured in peripheral human blood.120,121 Regarding pharmacological strategies, there is evidence that the selective serotonin reuptake inhibitor drug fluoxetine modulates the activity of the circadian biological clock, via phase advance in the firing of SCN neurons, further increasing the expression of various clock genes in the mice brain.122,123 Agomelatine, a novel dual melatonergic and specific serotonergic AD, can also cause phase-advance shifts in both mice and hamster brain when administered at specific times of day.124–126 Potential actions of agomelatine on clock gene expression have not been reported so far. Using neuronal cell cultures, low doses of the rapid-acting AD ketamine, a noncompetitive N-methyl-d-aspartate receptor antagonist, have been shown to blunt the amplitude of the transcription of different clock genes.127 More recently, one study has reported the effect of escitalopram on circadian genes in subjects with major depressive disorder.128 Of note, phase-advance shift of all these non-pharmacological and pharmacological interventions is contrasted with those of the mood stabilizers lithium and valproate, which can induce a phase delay.129,130 However, in clinical studies, both lithium and valproate have their principal effect on mania (and prevention of manic relapse) rather than acute depressive states.131 Lithium has also been shown to modulate multiple members of clock gene machinery.132 Noteworthy, glycogen synthase kinase-3β (GSK-3β), a key component of the mammalian circadian clock able to affect circadian rhythm generation by modifying the stability of circadian clock molecules, has been indicated as a common molecular target for many of the aforementioned chronotherapeutic interventions.133,134 Sleep deprivation response rates have been reported to be higher in depressed patients who carry a gene promoter polymorphism (rs334558) for decreasing GSK-3β activation.71,113 Lithium, valproate, serotoninergic ADs, as well as agomelatine and low-dose ketamine increase GSK-3β phosphorylation/inactivation.134 Through the inhibition of GSK-3β via multiple signaling cascades such as the phosphatidylinositol-3-kinase (PI3K)/Akt and the MAP kinase kinase (MEK)/ERK pathways, all these treatments are hypothesized to regulate the transcription and expression of different neurotrophic, angiogenic, and neuroprotective proteins (for a review, see Chiu et al135). Valproate has also been shown to act through the inhibition of histone deacetylase.136 Both PI3K/Akt and MEK/ERK pathways have, as a downstream target, the cyclic adenosine monophosphate response element transcription factor (CREB). When activated through phosphorylation, CREB modulated the expression of neurotrophic and cell-protective proteins, such as BDNF, NGF, and Bcl-2.137,138 Interestingly, BDNF and NGF have been reported to function as both downstream molecules resulting from the inhibition of GSK-3β and upstream signals able to inhibit this molecular pathway.139 Based on these evidences, it is possible to hypothesize an integrated AD/NTs cascade which might influence the different aspect of mood disorders, including circadian rhythm alteration, as illustrated in Figure 3. Indeed, a plethora of studies have reported significantly lower BDNF and NGF peripheral levels in patients with major depression.69,140 Some clinical studies have evaluated the changes in plasma or serum BDNF and NGF levels before and after AD treatments among patients with major depressive disorder, and most studies report increases in the BDNF levels following a course of AD treatment.73 With regard to NGF, almost all the researches have revealed no statistically significant difference before and after treatment.72 With regard to pharmacological and non-pharmacological chronotherapeutic interventions, escitalopram, ketamine, lithium, and sleep deprivation have all been demonstrated to increase BDNF peripheral levels in patients with major depression.141–144 The improvement of depressive symptoms with escitalopram coincided with significant improvements in the recall of both the quantity and quality of dreams, and dreaming, therefore, could be considered a biomarker of the efficacy of AD therapy, also for its evidence of chronobiological trends.98,145 Animal models have also confirmed a BDNF change in rat HIP following agomelatine, while treatment with escitalopram affects BDNF expression in HIP and NGF in the cortex but not in other brain areas and serum.146,147 In a rat model of depression, Angelucci et al found that treatment with lithium alters the concentrations of NGF and BDNF in the HIP, frontal cortex, occipital cortex, and striatum, further supporting the role of NTs in the mechanism of action of ADs.148
 | Figure 3 Schematic illustration of the hypothesis of NGF and BDNF involvement in the mechanism of action of ADs. |
With regard to non-pharmacological chronotherapeutic treatments, sleep deprivation in depressed patients has showed to produce a rapid increase in BDNF levels after a single treatment, and affect diurnal serum profile in responding patients.144,149 Light therapy has also showed to affect the diurnal trend of BDNF in the serum and saliva of young healthy women, and serum BDNF concentration in both men and women correlates with the sunshine hours per week throughout the year in both men and women, and with the seasonality of depressive symptoms.58,150 No data on the effects of sleep deprivation on the NGF diurnal and/or nocturnal profile in humans are available at present. However, studies in animals demonstrate that selective sleep deprivation during the rapid-eye-movement sleep phase alters the expression of NGF and BDNF in brain of rodents, and that the effects of sleep deprivation on the NGF expression in somatosensory cortex of rats are influenced by the afferents input.151,152 Recently, Hight et al found that the expression of NGF in somatosensory cortical neurons is high during the dark phase, while it is low during the light phase.153 On the contrary, high levels of NGF are expressed in the visual cortex during the light phase, further supporting the modulation of NGF expression in brain pathways activated by light stimuli, and therefore that changes in circulating or brain NGF and BDNF levels might reflex the light/dark cycle. Remarkably, a daily fluctuation of NGF and BDNF in human serum and plasma, also related to sex, has been found in healthy subjects.82 For example, diurnal BDNF rhythm was recently demonstrated in healthy men, where plasma BDNF and cortisol trends display highest concentrations in the morning, followed by a substantial decrease throughout the day, with lowest values at midnight.154 In women, the BDNF diurnal variations are also associated to the cortisol rhythm, but they are further influenced by ovarian function and contraceptive therapy.155 At variance, Piccinni et al did not find diurnal variation of BDNF in the plasma of women in either the follicular or luteal phase of the menstrual cycle, while variation in plasma BDNF levels was detected in men, with the peak at 8 am and nadir at 10 pm.156 BDNF fluctuation in serum and saliva was also found in healthy women.58 In agreement with Pluchino et al,155 this study shows that both the saliva and the serum BDNF levels in young women in their follicular phase of the ovarian cycle tend to decrease from morning to night and also shows that BDNF trend correlates with morning–evening personality traits and habits, and it is affected by light therapy. These data have recently been reconfirmed by Tirassa and Iannitelli, in a study further exploring the sex difference in the daily trend of NGF and BDNF serum levels (unpublished). Specifically, the study shows that the BDNF levels in men increase from morning to night, while daily NGF presents the “V” shape trend already reported by Bersani et al,82 but serum NGF trend in both man and woman is affected by light exposure. While a number of studies are now available on diurnal variation of NTs levels, at present, only one study has investigated this same issue in patients with depression. In the work by Giese et al, diurnal BDNF oscillations in patients with major depression associated with therapeutic response (in both sexes) after partial sleep deprivation.144 Specifically, subjects identified as responders (after 2 weeks of treatment) were associated with a daily change in serum BDNF at day 1 and even pretreatment, at baseline. This variation of peripheral BDNF concentration revealed characteristics of a diurnal pattern, whereas nonresponders did not exhibit diurnal BDNF variation. Together, all these findings emphasize the importance of a circadian NT rhythm in human health and well-being, while its absence seems to have a negative impact on successful depression treatment outcome. They further support a direct link between depression, biological clock, chronotherapeutics, and brain plasticity.
Conclusion
The data presented support the role of NGF and BDNF in the chronopathology and chronotherapeutics of mood, and therefore suggest these NTs as valuable biomarkers in human studies. Further, the fascinating hypothesis that ocular-applied NTs (by stimulating the retinal pathways associated with NIF functions) might also reset circadian rhythms offers a new interesting field of investigation in neuroscience and psychiatry.
Disclosure
The authors report no conflicts of interest in this work.
References
Albrecht U. Timing to perfection: the biology of central and peripheral circadian clocks. Neuron. 2012;74(2):246–260. | |
Czeisler CA, Buxton OM, Khalsa SB. The human circadian timing system and sleep-wake regulation. In: Kryger MH, Roth T, Dement WC, editors. Principles and Practice of Sleep Medicine. 4th ed. Philadelphia, PA: Elsevier/Saunders; 2005:375–394. | |
Herzog ED. Neurons and networks in daily rhythms. Nat Rev Neurosci. 2007;8(10):790–802. | |
Ueyama T, Krout KE, Nguyen XV, et al. Suprachiasmatic nucleus: a central autonomic clock. Nat Neurosci. 1999;2(12):1051–1053. | |
Hattar S, Liao HW, Takao M, Berson DM, Yao KW. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science. 2002;295(8):1065–1068. | |
Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295(8):1069. | |
Hatori M, Panda S. The emerging roles of melanopsin in behavioral adaptation to light. Trends Mol Med. 2010;16(10):435–446. | |
Cui Q, Ren C, Sollars PJ, Pickard GE, So KF. The injury resistant ability of melanopsin-expressing intrinsically photosensitive retinal ganglion cells. Neuroscience. 2015;284:845–853. | |
Vandewalle G, Collignon O, Hull JT, et al. Blue light stimulates cognitive brain activity in visually blind individuals. J Cogn Neurosci. 2013; 25(12):2072–2085. | |
Vandewalle G, Maquet P, Dijk DJ. Light as a modulator of cognitive brain function. Trends Cogn Sci. 2009;13(10):429–438. | |
Morin LP. Neuroanatomy of the extended circadian rhythm system. Exp Neurol. 2013;243:4–20. | |
Teng HK, Teng KK, Lee R, et al. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and sortilin. J Neurosci. 2005;25(22):5455–5463. | |
Elliott AS, Weiss ML, Nunez AA. Direct retinal communication with the peri-amygdaloid area. Neuroreport. 1995;6(5):806–808. | |
Aston-Jones G, Chen S, Zhu Y, Oshinsky ML. A neural circuit for circadian regulation of arousal. Nat Neurosci. 2001;4(7):732–738. | |
Vandewalle G, Schmidt C, Albouy G, et al. Brain responses to violet, blue, and green monochromatic light exposures in humans: prominent role of blue light and the brainstem. PLoS One. 2007;2(11):e1247. | |
Iannitelli A, Tirassa P. Pain and Depression: the Janus Factor of Human Suffering. In: Battaglia A, editor. Introduction to pain and its relations to nervous system disorders. New York: Wiley-Blackwell: 2015. | |
Borsook D, Becerra L, Carlezon WA Jr, et al. Reward-aversion circuitry in analgesia and pain: implications for psychiatric disorders. Eur J Pain. 2007;11(1):7–20. | |
Navratilova E, Xie JY, Okun A, et al. Pain relief produces negative reinforcement through activation of mesolimbic reward-valuation circuitry. Proc Natl Acad Sci U S A. 2012;109(50):20709–20713. | |
Sleipness EP, Sorg BA, Jansen HT. Diurnal differences in dopamine transporter and tyrosine hydroxylase levels in rat brain: dependence on the suprachiasmatic nucleus. Brain Res. 2007;1129(1):34–42. | |
Hampp G, Ripperger JA, Houben T, et al. Regulation of monoamine oxidase A by circadian-clock components implies clock influence on mood. Curr Biol. 2008;18(9):678–683. | |
Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237(4819):1154–1162. | |
Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24: 1217–1281. | |
Aloe L, Alleva E, Fiore M. Stress and nerve growth factor: findings in animal models and humans. Pharmacol Biochem Behav. 2002;73(1):159–166. | |
Cirulli F, Laviola G, Ricceri L. Risk factors for mental health: translational models from behavioural neuroscience. Neurosci Biobehav Rev. 2009;33(4):493–497. | |
Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4(4):299–309. | |
Chen LW, Yung KK, Chan YS, Shum DK, Bolam JP. The proNGF-p75NTR-sortilin signalling complex as new target for the therapeutic treatment of Parkinson’s disease. CNS Neurol Disord Drug Targets. 2008;7(6):512–523. | |
Cuello AC, Bruno MA, Allard S, Leon W, Iulita MF. Cholinergic involvement in Alzheimer’s disease. A link with NGF maturation and degradation. J Mol Neurosci. 2010;40(1–2):230–235. | |
Tirassa P, Maccarone M, Florenzano F, Cartolano S, De Nicolo S. Vascular and neuronal protection induced by the ocular administration of nerve growth factor in diabetic-induced rat encephalopathy. CNS Neurosci Ther. 2013;19(5):307–318. | |
Lessmann V, Gottmann K, Malcangio M. Neurotrophin secretion: current facts and future prospects. Prog Neurobiol. 2003;69(5):341–374. | |
Castren E. Neurotrophins as mediators of drug effects on mood, addiction, and neuroprotection. Mol Neurobiol. 2004;29(3):289–302. | |
Tirassa P, Maccarone M, Carito V, De Nicolo S, Fiore M. Ocular nerve growth factor administration counteracts the impairment of neural precursor cell viability and differentiation in the brain subventricular area of rats with streptozotocin-induced diabetes. Eur J Neurosci. 2015;41(9):1207–1218. | |
Lambiase A, Tirassa P, Micera A, Aloe L, Bonini S. Pharmacokinetics of conjunctivally applied nerve growth factor in the retina and optic nerve of adult rats. Invest Ophthalmol Vis Sci. 2005;46(10):3800–3806. | |
Canossa M, Griesbeck O, Berninger B, Campana G, Kolbeck R, Thoenen H. Neurotrophin release by neurotrophins: implications for activity-dependent neuronal plasticity. Proc Natl Acad Sci U S A. 1997;94(24):13279–13286. | |
Kruttgen A, Moller JC, Heymach JV Jr, Shooter EM. Neurotrophins induce release of neurotrophins by the regulated secretory pathway. Proc Natl Acad Sci U S A. 1998;95(16):9614–9619. | |
Berardi N, Maffei L. From visual experience to visual function: roles of neurotrophins. J Neurobiol. 1999;41(1):119–126. | |
von Bartheld CS. Neurotrophins in the developing and regenerating visual system. Histol Histopathol. 1998;13(2):437–459. | |
Carmignoto G, Comelli MC, Candeo P, et al. Expression of NGF receptor and NGF receptor mRNA in the developing and adult rat retina. Exp Neurol. 1991;111(3):302–311. | |
Wahle P, Di Cristo G, Schwerdtfeger G, Engelhardt M, Berardi N, Maffei L. Differential effects of cortical neurotrophic factors on development of lateral geniculate nucleus and superior colliculus neurons: anterograde and retrograde actions. Development. 2003;130(3):611–622. | |
Butowt R, von Bartheld CS. Sorting of internalized neurotrophins into an endocytic transcytosis pathway via the Golgi system: ultrastructural analysis in retinal ganglion cells. J Neurosci. 2001;21(22):8915–8930. | |
Zhang Y, Moheban DB, Conway BR, Bhattacharyya A, Segal RA. Cell surface Trk receptors mediate NGF-induced survival while internalized receptors regulate NGF-induced differentiation. J Neurosci. 2000;20(15):5671–5678. | |
Ojeda SR, Hill DF, Katz KH. The genes encoding nerve growth factor and its receptor are expressed in the developing female rat hypothalamus. Brain Res Mol Brain Res. 1991;9(1–2):47–55. | |
Liang FQ, Sohrabji F, Miranda R, Earnest B, Earnest D. Expression of brain-derived neurotrolphic factor and its cognate receptor, TrkB, in the rat suprachiasmatic nucleus. Exp Neurol. 1998;151(2):184–193. | |
Bina KG, Rusak B. Nerve growth factor phase shifts circadian activity rhythms in Syrian hamsters. Neurosci Lett. 1996;206(2–3):97–100. | |
Pizzio GA, Hainich EC, Plano SA, Ralph MR, Golombek DA. Nerve growth factor-induced circadian phase shifts and MAP kinase activation in the hamster suprachiasmatic nuclei. Eur J Neurosci. 2005;22(3):665–671. | |
Liang FQ, Allen G, Earnest D. Role of brain-derived neurotrophic factor in the circadian regulation of the suprachiasmatic pacemaker by light. J Neurosci. 2000;20(8):2978–2987. | |
Sofroniew MV, Isacson O, O’Brien TS. Nerve growth factor receptor immunoreactivity in the rat suprachiasmatic nucleus. Brain Res. 1989;476(2):358–362. | |
Kiss J, Patel AJ, Halasz B. Colocalization of NGF receptor with VIP in rat suprachiasmatic neurones. Neuroreport. 1993;4(12):1315–1318. | |
Golombek DA, Ralph MR. Let there be light: signal transduction in a mammalian circadian system. Braz J Med Biol Res. 1996;29(1):131–140. | |
Beaule C, Amir S. Effect of 192 IgG-saporin on circadian activity rhythms, expression of P75 neurotrophin receptors, calbindin-D28K, and light-induced Fos in the suprachiasmatic nucleus in rats. Exp Neurol. 2002;176(2):377–389. | |
Erhardt C, Galani R, Jeltsch H, et al. Modulation of photic resetting in rats by lesions of projections to the suprachiasmatic nuclei expressing p75 neurotrophin receptor. Eur J Neurosci. 2004;19(7):1773–1788. | |
Paula-Barbosa MM, Silva SM, Andrade JP, Cadete-Leite A, Madeira MD. Nerve growth factor restores mRNA levels and the expression of neuropeptides in the suprachiasmatic nucleus of rats submitted to chronic ethanol treatment and withdrawal. J Neurocytol. 2001;30(3):195–207. | |
Pereira PA, Cardoso A, Paula-Barbosa MM. Nerve growth factor restores the expression of vasopressin and vasoactive intestinal polypeptide in the suprachiasmatic nucleus of aged rats. Brain Res. 2005; 1048(1–2):123–130. | |
Sala R, Viegi A, Rossi FM, et al. Nerve growth factor and brain-derived neurotrophic factor increase neurotransmitter release in the rat visual cortex. Eur J Neurosci. 1998;10(6):2185–2191. | |
Hannibal J. Neurotransmitters of the retino-hypothalamic tract. Cell Tissue Res. 2002;309(1):73–88. | |
Michel S, Clark JP, Ding JM, Colwell CS. Brain-derived neurotrophic factor and neurotrophin receptors modulate glutamate-induced phase shifts of the suprachiasmatic nucleus. Eur J Neurosci. 2006;24(4):1109–1116. | |
Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV. High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature. 1991;350(6320):678–683. | |
Baeza-Raja B, Eckel-Mahan K, Zhang L, et al. p75 neurotrophin receptor is a clock gene that regulates oscillatory components of circadian and metabolic networks. J Neurosci. 2013;33(25):10221–10234. | |
Tirassa P, Iannitelli A, Sornelli F, et al. Daily serum and salivary BDNF levels correlate with morning-evening personality type in women and are affected by light therapy. Riv Psichiatr. 2012;47(6):527–534. | |
Sposato V, Parisi V, Manni L, et al. Glaucoma alters the expression of NGF and NGF receptors in visual cortex and geniculate nucleus of rats: effect of eye NGF application. Vision Res. 2009;49(1):54–63. | |
Tirassa P. The nerve growth factor administrated as eye drops activates mature and precursor cells in subventricular zone of adult rats. Arch Ital Biol. 2011;149(2):205–213. | |
Di Fausto V, Fiore M, Tirassa P, Lambiase A, Aloe L. Eye drop NGF administration promotes the recovery of chemically injured cholinergic neurons of adult mouse forebrain. Eur J Neurosci. 2007;26(9):2473–2480. | |
Fiore M, Angelucci F, Aloe L, Iannitelli A, Korf J. Nerve growth factor and brain-derived neurotrophic factor in schizophrenia and depression: findings in humans and in animal models. Curr Neuropharmacol. 2003;1:1–20. | |
Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59(12):1116–1127. | |
Bersani G, Iannitelli A, Maselli P, et al. Low nerve growth factor plasma levels in schizophrenic patients: a preliminary study. Schizophr Res. 1999;37(2):201–203. | |
Bersani G, Iannitelli A, Fiore M, Angelucci F, Aloe L. Data and hypotheses on the role of nerve growth factor and other neurotrophins in psychiatric disorders. Med Hypotheses. 2000;55(3):199–207. | |
Angelucci F, Mathe AA, Aloe L. Neurotrophic factors and CNS disorders: findings in rodent models of depression and schizophrenia. Prog Brain Res. 2004;146:151–165. | |
Branchi I, D’Andrea I, Fiore M, Di Fausto V, Aloe L, Alleva E. Early social enrichment shapes social behavior and nerve growth factor and brain-derived neurotrophic factor levels in the adult mouse brain. Biol Psychiatry. 2006;60(7):690–696. | |
Zhu SW, Yee BK, Nyffeler M, Winblad B, Feldon J, Mohammed AH. Influence of differential housing on emotional behaviour and neurotrophin levels in mice. Behav Brain Res. 2006;169(1):10–20. | |
Castren E. Neurotrophins and psychiatric disorders. Handb Exp Pharmacol. 2014;220:461–479. | |
Archer T, Oscar-Berman M, Blum K, Gold M. Epigenetic modulation of mood disorders. J Genet Syndr Gene Ther. 2013;4(120):1000120. | |
Benedetti F, Serretti A, Colombo C, Lorenzi C, Tubazio V, Smeraldi E. A glycogen synthase kinase 3-beta promoter gene single nucleotide polymorphism is associated with age at onset and response to total sleep deprivation in bipolar depression. Neurosci Lett. 2004;368(2):123–126. | |
Chen YW, Lin PY, Tu KY, Cheng YS, Wu CK, Tseng PT. Significantly lower nerve growth factor levels in patients with major depressive disorder than in healthy subjects: a meta-analysis and systematic review. Neuropsychiatr Dis Treat. 2015;11:925–933. | |
Brunoni AR, Lopes M, Fregni F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: implications for the role of neuroplasticity in depression. Int J Neuropsychopharmacol. 2008;11(8):1169–1180. | |
Lang UE, Hellweg R, Sander T, Gallinat J. The Met allele of the BDNF Val66Met polymorphism is associated with increased BDNF serum concentrations. Mol Psychiatry. 2009;14(2):120–122. | |
Egan MF, Kojima M, Callicott JH, et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell. 2003;112(2):257–269. | |
Sklar P, Gabriel SB, McInnis MG, et al. Family-based association study of 76 candidate genes in bipolar disorder: BDNF is a potential risk locus. Brain-derived neutrophic factor. Mol Psychiatry. 2002;7(6):579–593. | |
Hong CJ, Huo SJ, Yen FC, Tung CL, Pan GM, Tsai SJ. Association study of a brain-derived neurotrophic-factor genetic polymorphism and mood disorders, age of onset and suicidal behavior. Neuropsychobiology. 2003;48(4):186–189. | |
Jiang X, Xu K, Hoberman J, et al. BDNF variation and mood disorders: a novel functional promoter polymorphism and Val66Met are associated with anxiety but have opposing effects. Neuropsychopharmacology. 2005;30(7):1353–1361. | |
Zakharyan R, Atshemyan S, Gevorgyan A, Boyajyan A. Nerve growth factor and its receptor in schizophrenia. BBA Clin. 2014;1:24–29. | |
Lang UE, Hellweg R, Bajbouj M, Gaus V, Sander T, Gallinat J. Gender-dependent association of a functional NGF polymorphism with anxiety-related personality traits. Pharmacopsychiatry. 2008; 41(5):196–199. | |
Chang CC, Fang WH, Chang HA, Chen TY, Huang SY. Sex-specific association between nerve growth factor polymorphism and cardiac vagal modulation. Psychosom Med. 2014;76(8):638–643. | |
Bersani G, Iannitelli A, Massoni E, et al. Ultradian variation of nerve growth factor plasma levels in healthy and schizophrenic subjects. Int J Immunopathol Pharmacol. 2004;17(3):367–372. | |
Curtis GC. Psychiatry’s need for research in chronobiology. Int J Chronobiol. 1973;1(1):8–10. | |
Benedetti F, Barbini B, Colombo C, Smeraldi E. Chronotherapeutics in a psychiatric ward. Sleep Med Rev. 2007;11(6):509–522. | |
American Psychiatric Associations. Diagnostic and Statistical Manual of Mental of Mental Disorders. 5th ed. Washington, DC/London, England: American Psychiatric Publishing; 2013. | |
LeGates TA, Fernandez DC, Hattar S. Light as a central modulator of circadian rhythms, sleep and affect. Nat Rev Neurosci. 2014; 15(7):443–454. | |
LeGates TA, Altimus CM, Wang H, et al. Aberrant light directly impairs mood and learning through melanopsin-expressing neurons. Nature. 2012;491(7425):594–598. | |
Goodwin FK, Jamison KR. Manic-Depressive Illness: Bipolar Disorders and Recurrent Depression. 2nd ed. New York: Oxford University Press; 2012. | |
Eagles JM. The relationship between mood and daily hours of sunlight in rapid cycling bipolar illness. Biol Psychiatry. 1994;36(6):422–424. | |
Friedman E, Gyulai L, Bhargava M, et al. Seasonal changes in clinical status in bipolar disorder: a prospective study in 1000 STEP-BD patients. Acta Psychiatr Scand. 2006;113(6):510–517. | |
Germain A, Frank E. Chronobiology of the core symptoms of depression. Medicographia. 2008;30(1):30–34. | |
Bersani G, Bersani FS, Prinzivalli E, et al. Premorbid circadian profile of patients with major depression and panic disorder. Riv Psichiatr. 2012;47(5):407–412. | |
Souêtre E, Salvati E, Belugou JL, et al. Circadian rhythms in depression and recovery: evidence for blunted amplitude as the main chronobiological abnormality. Psychiatry Res. 1989;28(3):263–278. | |
Koenigsberg HW, Teicher MH, Mitropoulou V, et al. 24-h Monitoring of plasma norepinephrine, MHPG, cortisol, growth hormone and prolactin in depression. J Psychiatr Res. 2004;38(5):503–511. | |
Claustrat B, Chazot G, Brun J, Jordan D, Sassolas G. A chronobiological study of melatonin and cortisol secretion in depressed subjects: plasma melatonin, a biochemical marker in major depression. Biol Psychiatry. 1984;19(8):1215–1228. | |
Tsuno N, Besset A, Ritchie K. Sleep and depression. J Clin Psychiatry. 2005;66(10):1254–1269. | |
Riemann D, Berger M, Voderholzer U. Sleep and depression – results from psychobiological studies: an overview. Biol Psychol. 2001; 57(1–3):67–103. | |
Quartini A, Anastasia A, Bersani FS, et al. Changes in dream experience in relation with antidepressant escitalopram treatment in depressed female patients: a preliminary study. Riv Psichiatr. 2014; 49(4):187–191. | |
Freud D. Il Sogno. Torino: Bollati Boringhieri; 1990:4.42. | |
Buysse DJ, Nofzinger EA, Germain A, et al. Regional brain glucose metabolism during morning and evening wakefulness in humans: preliminary findings. Sleep. 2004;27(7):1245–1254. | |
Wolff EA 3rd, Putnam FW, Post RM. Motor activity and affective illness. The relationship of amplitude and temporal distribution to changes in affective state. Arch Gen Psychiatry. 1985;42(3):288–294. | |
Moffoot AP, O’Carroll RE, Bennie J, et al. Diurnal variation of mood and neuropsychological function in major depression with melancholia. J Affect Disord. 1994;32(4):257–269. | |
Ohdo S. Chronopharmaceutics: pharmaceutics focused on biological rhythm. Biol Pharm Bull. 2010;33(2):159–167. | |
Levi F, Schibler U. Circadian rhythms: mechanisms and therapeutic implications. Annu Rev Pharmacol Toxicol. 2007;47:593–628. | |
Sukumaran S, Almon RR, DuBois DC, Jusko WJ. Circadian rhythms in gene expression: relationship to physiology, disease, drug disposition and drug action. Adv Drug Deliv Rev. 2010;62(9–10):904–917. | |
Nagayama H. Influences of biological rhythms on the effects of psychotropic drugs. Psychosom Med. 1999;61(5):618–629. | |
Philipp M, Marneros A. Chronobiology and its implications for pharmacotherapy of endogenous depression. Pharmakopsychiatr Neuropsychopharmakol. 1978;11(5):235–240. | |
Nagayama H, Nagano K, Ikezaki A, Tashiro T. Double-blind study of the chronopharmacotherapy of depression. Chronobiol Int. 1991;8(3):203–209. | |
Nakano S, Hollister LE. No circadian effect on nortriptyline kinetics in man. Clin Pharmacol Ther. 1978;23(2):199–203. | |
Usher RW, Beasley CM Jr, Bosomworth JC. Efficacy and safety of morning versus evening fluoxetine administration. J Clin Psychiatry. 1991;52(3):134–136. | |
Li JZ, Bunney BG, Meng F, et al. Circadian patterns of gene expression in the human brain and disruption in major depressive disorder. Proc Natl Acad Sci U S A. 2013;110(24):9950–9955. | |
Czeisler CA, Dijk DK. Use of bright light to treat maladaptation to night shift work and circadian rhythm sleep disorder. J Sleep Res. 1995;4(52):70–73. | |
Wirz-Justice A. From the basic neuroscience of circadian clock function to light therapy for depression: on the emergence of chronotherapeutics. J Affect Disord. 2009;116(3):159–160. | |
Benedetti F. Antidepressant chronotherapeutics for bipolar depression. Dialogues Clin Neurosci. 2012;14(4):401–411. | |
Wirz-Justice A, Terman M. Chronotherapeutics (light and wake therapy) as a class of interventions for affective disorders. Handb Clin Neurol. 2012;106:697–713. | |
Echizenya M, Suda H, Takeshima M, Inomata Y, Shimizu T. Total sleep deprivation followed by sleep phase advance and bright light therapy in drug-resistant mood disorders. J Affect Disord. 2013;144(1–2):28–33. | |
Sahlem GL, Kalivas B, Fox JB, et al. Adjunctive triple chronotherapy (combined total sleep deprivation, sleep phase advance, and bright light therapy) rapidly improves mood and suicidality in suicidal depressed inpatients: an open label pilot study. J Psychiatr Res. 2014;59: 101–107. | |
Wisor JP, O’Hara BF, Terao A, et al. A role for cryptochromes in sleep regulation. BMC Neurosci. 2002;3:20. | |
Wisor JP, Pasumarthi RK, Gerashchenko D, et al. Sleep deprivation effects on circadian clock gene expression in the cerebral cortex parallel electroencephalographic differences among mouse strains. J Neurosci. 2008;28(28):7193–7201. | |
Takimoto M, Hamada A, Tomoda A, et al. Daily expression of clock genes in whole blood cells in healthy subjects and a patient with circadian rhythm sleep disorder. Am J Physiol Regul Integr Comp Physiol. 2005;289(5):R1273–R1279. | |
Zhu Y, Fu A, Hoffman AE, et al. Advanced sleep schedules affect circadian gene expression in young adults with delayed sleep schedules. Sleep Med. 2013;14(5):449–455. | |
Uz T, Ahmed R, Akhisaroglu M, et al. Effect of fluoxetine and cocaine on the expression of clock genes in the mouse hippocampus and striatum. Neuroscience. 2005;134(4):1309–1316. | |
Prouse J, Braselton J, Reynolds L. Fluoxetine modulates the circadian biological clock via phase advances of suprachiasmatic nucleus neuronal firing. Biol Psychiatry. 2006;60(8):896–899. | |
Millan MJ, Gobert A, Lejeune F, et al. The novel melatonin agonist agomelatine (S20098) is an antagonist at 5-hydroxytryptamine2C receptors, blockade of which enhances the activity of frontocortical dopaminergic and adrenergic pathways. J Pharmacol Exp Ther. 2003;306(3):954–964. | |
Bourin M, Mocaer E, Porsolt R. Antidepressant-like activity of S 20098 (agomelatine) in the forced swimming test in rodents: involvement of melatonin and serotonin receptors. J Psychiatry Neurosci. 2004; 29(2):126–133. | |
Van Reeth O, Olivares E, Zhang Y, et al. Comparative effects of a melatonin agonist on the circadian system in mice and Syrian hamsters. Brain Res. 1997;762(1–2):185–194. | |
Bellet MM, Vawter MP, Bunney BG, Bunney WE, Sassone-Corsi P. Ketamine influences CLOCK:BMAL1 function leading to altered circadian gene expression. PLoS One. 2011;6(8):e23982. | |
Li SX, Liu LJ, Xu LZ, et al. Diurnal alterations in circadian genes and peptides in major depressive disorder before and after escitalopram treatment. Psychoneuroendocrinology. 2013;38(11):2789–2799. | |
LeSauter J, Silver R. Lithium lengthens the period of circadian rhythms in lesioned hamsters bearing SCN grafts. Biol Psychiatry. 1993;34(1–2):75–83. | |
Nagayama H. Chronic administration of imipramine and lithium changes the phase-angle relationship between the activity and core body temperature circadian rhythms in rats. Chronobiol Int. 1996;13(4):251–259. | |
Hickie IB, Naismith SL, Robillard R, Scott EM, Hermens DF. Manipulating the sleep-wake cycle and circadian rhythms to improve clinical management of major depression. BMC Med. 2013;11:79. | |
Quiroz JA, Gould TD, Manji HK. Molecular effects of lithium. Mol Interv. 2004;4(5):259–272. | |
Iitaka C, Miyazaki K, Akaike T, Ishida N. A role for glycogen synthase kinase-3beta in the mammalian circadian clock. J Biol Chem. 2005;280(33):29397–29402. | |
Bunney BG, Li JZ, Walsh DM, et al. Circadian dysregulation of clock genes: clues to rapid treatments in major depressive disorder. Mol Psychiatry. 2015;20(1):48–55. | |
Chiu CT, Wang Z, Hunsberger JG, Chuang DM. Therapeutic potential of mood stabilizers lithium and valproic acid: beyond bipolar disorder. Pharmacol Rev. 2013;65(1):105–142. | |
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem. 2001;276(39):36734–36741. | |
Finkbeiner S. CREB couples neurotrophin signals to survival messages. Neuron. 2000;25(1):11–14. | |
Lim S, Moon M, Oh H, Kim HG, Kim SY, Oh MS. Ginger improves cognitive function via NGF-induced ERK/CREB activation in the hippocampus of the mouse. J Nutr Biochem. 2014;25(10):1058–1065. | |
Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65(4):391–426. | |
Hashimoto K, Shimizu E, Iyo M. Critical role of brain-derived neurotrophic factor in mood disorders. Brain Res Brain Res Rev. 2004; 45(2):104–114. | |
Ricci V, Martinotti G, Gelfo F, et al. Chronic ketamine use increases serum levels of brain-derived neurotrophic factor. Psychopharmacology (Berl). 2011;215(1):143–148. | |
Ladea M, Bran M. Brain derived neurotrophic factor (BDNF) levels in depressed women treated with open-label escitalopram. Psychiatr Danub. 2013;25(2):128–132. | |
Ricken R, Adli M, Lange C, et al. Brain-derived neurotrophic factor serum concentrations in acute depressive patients increase during lithium augmentation of antidepressants. J Clin Psychopharmacol. 2013;33(6):806–809. | |
Giese M, Beck J, Brand S, et al. Fast BDNF serum level increase and diurnal BDNF oscillations are associated with therapeutic response after partial sleep deprivation. J Psychiatr Res. 2014;59:1–7. | |
Agnati LF, Barlow PW, BaluŠka F, et al. A new theoretical approach to the functional meaning of sleep and dreaming in humans based on the maintenance of ‘predictive psychic homeostasis’. Commun Integr Biol. 2011;4(6):640–654. | |
Soumier A, Banasr M, Lortet S, et al. Mechanisms contributing to the phase-dependent regulation of neurogenesis by the novel antidepressant, agomelatine, in the adult rat hippocampus. Neuropsychopharmacology. 2009;34(11):2390–2403. | |
Schulte-Herbrüggen O, Fuchs E, Abumaria N, et al. Effects of escitalopram on the regulation of brain-derived neurotrophic factor and nerve growth factor protein levels in a rat model of chronic stress. J Neurosci Res. 2009;87(11):2551–2560. | |
Angelucci F, Aloe L, Jimenez-Vasquez P, Mathe AA. Lithium treatment alters brain concentrations of nerve growth factor, brain-derived neurotrophic factor and glial cell line-derived neurotrophic factor in a rat model of depression. Int J Neuropsychopharmacol. 2003;6(3):225–231. | |
Gorgulu Y, Caliyurt O. Rapid antidepressant effects of sleep deprivation therapy correlates with serum BDNF changes in major depression. Brain Res Bull. 2009;80(3):158–162. | |
Molendijk ML, Haffmans JP, Bus BA, et al. Serum BDNF concentrations show strong seasonal variation and correlations with the amount of ambient sunlight. PLoS One. 2012;7(11):e48046. | |
Sei H, Saitoh D, Yamamoto K, Morita K, Morita Y. Differential effect of short-term REM sleep deprivation on NGF and BDNF protein levels in the rat brain. Brain Res. 2000;877(2):387–390. | |
Brandt JA, Churchill L, Guan Z, Fang J, Chen L, Krueger JM. Sleep deprivation but not a whisker trim increases nerve growth factor within barrel cortical neurons. Brain Res. 2001;898(1):105–112. | |
Hight K, Hallett H, Churchill L, De A, Boucher A, Krueger JM. Time of day differences in the number of cytokine-, neurotrophin- and NeuN-immunoreactive cells in the rat somatosensory or visual cortex. Brain Res. 2010;1337:32–40. | |
Begliuomini S, Lenzi E, Ninni F, et al. Plasma brain-derived neurotrophic factor daily variations in men: correlation with cortisol circadian rhythm. J Endocrinol. 2008;197(2):429–435. | |
Pluchino N, Cubeddu A, Begliuomini S, et al. Daily variation of brain-derived neurotrophic factor and cortisol in women with normal menstrual cycles, undergoing oral contraception and in postmenopause. Hum Reprod. 2009;24(9):2303–2309. | |
Piccinni A, Marazziti D, Del Debbio A, et al. Diurnal variation of plasma brain-derived neurotrophic factor (BDNF) in humans: an analysis of sex differences. Chronobiol Int. 2008;25(5):819–826. |
 © 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2015 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.