Back to Archived Journals » Research and Reports in Neonatology » Volume 4
Neonatal diabetes mellitus: current perspective
Authors Kataria A, Palliyil Gopi R, Mally P, Shah B
Received 30 September 2013
Accepted for publication 8 November 2013
Published 25 March 2014 Volume 2014:4 Pages 55—64
DOI https://doi.org/10.2147/RRN.S38206
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Video abstract presented by Anglina Kataria
Views: 2428
Anglina Kataria,1 Resmy Palliyil Gopi,2 Pradeep Mally,3 Bina Shah2
1Department of Pediatrics, 2Pediatric Endocrinology, Department of Pediatrics, 3Neonatology, Department of Pediatrics, NYU School of Medicine, New York, NY, USA
Abstract: Neonatal diabetes mellitus (NDM) is a rare form of diabetes characterized by hyperglycemia occurring in the first few months of life. It can present as either transient NDM (TNDM), which resolves by a few months, or permanent NDM (PNDM), which continues throughout life. The etiology of this disease remained unclear until recently, when advances in molecular genetic techniques illuminated the mechanisms involved in the pathogenesis of the disease. Having delineated the genes involved in insulin production and secretion and their association with NDM, we currently understand the molecular basis of this disease. While most TNDM cases are caused by the overexpression of chromosome 6q24, the majority of PNDM cases are due to mutations in the adenosine triphosphate-sensitive potassium (KATP) channel. The improved understanding of the etiology of the disease had revolutionized the diagnosis and its management with oral sulfonylureas. The primary objective of this study was to review the current understanding of neonatal diabetes, including its genetic etiologies, clinical presentation, diagnosis, acute treatment, and long-term management.
Keywords: hyperglycemia, intrauterine growth retardation, KATP channel mutations, sulfonylurea
Introduction
Neonatal diabetes mellitus (NDM) is defined as insulin-requiring persistent hyperglycemia occurring in first 6 months of life. Although NDM is considered a rare disease and is usually estimated to affect one in 300,000 to 400,000 newborns, its incidence has been reported to be as high as 1:89,000 in some European countries.1,2 Based on the disease course, NDM is stratified as transient NDM (TNDM) and permanent NDM (PNDM). TNDM, accounting for 50% to 60% of all NDM cases, requires initial insulin treatment but appears to resolve spontaneously by a median of 12 weeks of age, only to relapse years later. Permanent NDM is less common and is also characterized by early hyperglycemia; however, unlike TNDM, PDM has no period of remission and must be treated lifelong. Unlike autoimmune diabetes, which is extremely rare before 6 months of age, NDM is a monogenic form of diabetes, with insulinopenia resulting from abnormal pancreatic islet development, decreased B-cell mass, or B-cell dysfunction.3
Types of neonatal diabetes mellitus
Transient neonatal diabetes mellitus
Recent studies have implied that the deficit in insulin output in TNDM can arise from a delayed maturation of the pancreatic islets and B-cells, as a consequence of altered expression of genes on chromosome 6. It was first associated with paternal uniparental disomy of chromosome 6 (UPD6) in 1995, suggesting it was disorder of imprinting.4 Over the years, studies confirmed that TNDM was the result of the inappropriate overexpression of the chromosome region 6q24, which is now known to contain two major TNDM gene candidates, ZAC 1 (zinc finger, apoptosis, and cell cycle) and HYMA1 ([Fe] hydrogenase subunit HymA).5,6 Normally, this locus at chromosome 6q24 is imprinted such that only the paternal allele is actively expressed, while the maternal allele remains silent. It is estimated that 70% of TNDM is the result of a loss of imprinting in the chromosome 6q24 region and the subsequent overexpression of both gene products.7 The overexpression of the genes in the 6q24 locus has three known genetic etiologies: 1) paternal UPD6 (partial or complete), which accounts for the majority of sporadic cases; 2) duplication of the TNDM1 region of the paternal chromosome 6, which is common in familial cases; and 3) maternal hypomethylation of the TNDM1 region and thus activation of the maternal allele, which is also found in sporadic cases (Table 1).6 Recent studies have identified, in a small number of cases, TNDM is a result of mutations in the KCNJ11 (potassium inwardly-rectifying channel, subfamily j, member 11) and ABCC8 (ATP-binding cassette transporter subfamily C, member 8) genes, which encode the KATP channel (Table 1).8 Another novel but rare genetic cause of TNDM involves mutations in the HNF-1B (hepatocyte nuclear factor-1B gene),9,10 which was previously known to be responsible for maturity-onset diabetes of the young 5 (MODY5). Although hyperglycemia remits in TNDM patients by the age of 18 months, a relapse of diabetes occurs in 50% of the patients, usually during adolescence or early adulthood, at median age of 14 years. Relapse has been reported as early as 4 years of age, but usually coincides with a period of increased insulin demand, such as puberty or pregnancy. Thus, this transient form of disease is probably a permanent B-cell defect with variable expression during growth and development.3,6,11
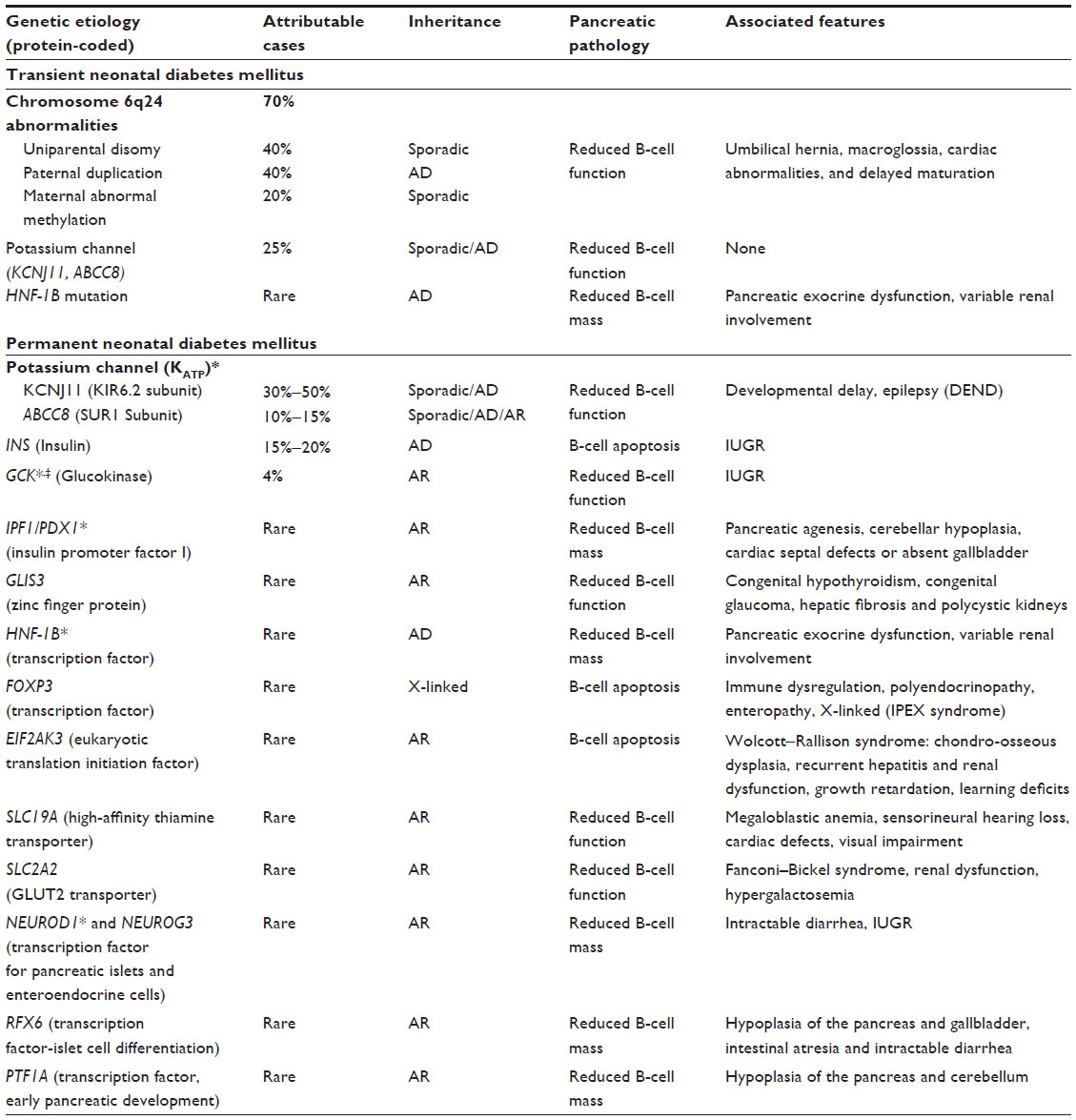 | Table 1 Known genetic mutations causing transient and permanent neonatal diabetes mellitus |
Permanent neonatal diabetes mellitus
To date, permanent NDM is known to be caused by approximately a dozen genes involved in pancreatic development, B-cell apoptosis, or dysfunction. Mutations in the KCNJ11 or ABCC8 genes, encoding the KATP channel subunits KIR6.2 (ATP-sensitive inward rectifier potassium channel) and SUR (sulfonylurea receptor)1, respectively, account for the majority of PNDM cases.11 Other mutations leading to PNDM include the genes that encode GCK, insulin promoter factor 1, pancreas transcription factor 1α, FOXP3 (forkhead box P3 protein), or EIF2AK3 (eukaryotic translation initiation factor 2-alpha kinase).12 The KATP channel is a heterooctameric complex composed of four SUR1 and pore-forming KIR6.2 subunits; maintenance of the 1:1 ratio of SUR1: KIR6.2 is essential to both the assembly and function of KATP.12,13 In normal pancreatic B-cells, glucose metabolism results in an increased production of intracellular ATP, which binds to the KIR6.2 subunit, causing the potassium channel to close and inhibiting potassium efflux; this subsequently results in cell membrane depolarization, triggering an influx of calcium and initiating insulin secretion (Figure 1). Mutations of the KCNJ11 gene compromises the sensitivity of inhibitory ATP by the KIR6.2 subunit, causing permanent opening of the potassium channel and preventing cell depolarization and insulin secretion.13 Similarly, activating mutations of the ABCC8 gene decrease the channel closure and compromise insulin release.12 Although mutations in the KCNJ11 gene and ABCC8 gene result in distinct molecular mechanisms, at the physiological level, they both manifest as insulin deficiency and remain clinically indistinguishable. The identification of these mutations in NDM patients is imperative as it not only helps establish diagnosis but also, directs the long-term medical management and genetic counseling. Oral sulfonylurea agents are highly effective in patients with KCNJ11 gene and ABCC8 gene mutations, providing better glycemic control than insulin.11,14 In a minority of patients, PNDM secondary to KATP channel mutations has also been associated with neurological features, representing the most severe clinical form of PNDM, Developmental delay, Epilepsy, and Neonatal Diabetes (DEND) syndrome. This is believed to be the result of mutated KATP channels in the brain since both the KCNJ11 gene and the ABCC8 gene are expressed in neuronal tissue. Fortunately, more common is a milder picture of this spectrum, with less severe developmental delay and without generalized epilepsy, known as intermediate DEND (iDEND).11,15
 | Figure 1 Pancreatic beta cell depicting the role of genetic defects in neonatal diabetes mellitus. |
Heterozygous mutations in the insulin gene (INS) have been identified and could be the culprits in 15%–20% of PNDM cases; patients with these mutations have permanent diabetes without extrapancreatic features except for a low birth weight, a feature of all NDM subtypes as a result of the insulin deficiency. A complete deficiency of the glycolytic enzyme GCK is another cause of PNDM. GCK acts as the glucose sensor of the pancreatic B-cell since the conversion of glucose to glucose-6-phosphate by GCK is the rate-limiting step of the glycolytic pathway and ultimately determines the threshold for glucose-induced insulin secretion. Inactivating homozygous mutations of the GCK gene have a dramatic effect on the phosphorylation, blocking glucose metabolism and ATP production and hence, diminishing insulin secretion. Homozygous inactivating mutations in the insulin promoter factor-1 (IPFI) gene, a master regulator of exocrine and endocrine pancreatic development, are associated with marked exocrine and endocrine failure secondary to pancreatic agenesis. It is worth noting that mutations in two genes, IPF-1 and GCK cause MODY in the heterozygous state but PNDM in the homozygous state (Table 1).3
A number of other syndromes have been associated with PNDM, reflecting that the roles played by the genes extend beyond pancreatic B-cell dysfunction. For instance, mutations in the NEUROD1 (neuronal differentiation 1) and NEUROG3 (neurogenin-3) genes, which encode transcription factors, result in the loss of enteroendocrine cells and are associated with intractable diarrhea and intrauterine growth retardation (IUGR) in PNDM patients. Other examples include Immune dysregulation, Polyendocrinopathy, Enteropathy, X-linked (IPEX) syndrome and Wolcott−Rallison syndrome, caused by mutations in the genes encoding transcription factors FOXP3 and EIF2AK3, respectively (Table 1). Other rare causes of NDM include mutations in genes encoding transporters such as glucose transporter (GLUT)2 and thiamine transporter (see Table 1). It is worth noting that in cases where the cause of NDM remains unknown, mitochondrial diabetes may also be considered, especially in presence of other organ manifestations. Maternally inherited diabetes associated with early-onset bilateral sensorineural deafness is one such example of mitochondriopathy secondary to a messenger deoxyribonucleic acid (mDNA) A3243G point mutation. A suspicion of mitochondrial diabetes is provided by a strong familial clustering of diabetes.16
Although our focus rests on neonatal diabetes, it is important to acknowledge that many of the genes discussed here are implicated in other disorders, most notably congenital hyperinsulinism and MODY. In fact, inactivating mutations in KCNJ11, ABCC8, and GCK are among the common genetic causes of congenital hyperinsulinism. On the other hand, MODY presents later in life and is associated with mutations in GCK, HNF1B, PDX1 (pancreatic and duodenal homeobox 1), and NEUROD1.
Presenting features
The cardinal symptoms of NDM include hyperglycemia at birth, IUGR, failure to thrive, dehydration, and, rarely, ketoacidosis. The hyperglycemia is markedly severe and is accompanied by low or undetectable insulin and C-peptide levels. Although hyperglycemia has been reported to manifest as the typical diabetic symptoms of polyuria and polydipsia in NDM patients, the clinical picture may mimic sepsis in a newborn as the young age of the patients often makes these parameters difficult to assess.12,17 Hyperglycemia can also, rarely, progress to ketoacidosis but more so in PNDM than in TNDM patients. It can occur as early as 3 days of age and has been reported as the presenting symptom in multiple cohort studies and case reports.17−21
IUGR is seen in >95% of TNDM patients, typically developing in the third trimester, such that the birth weight is typically 1.5−2.5 kg.6 In fact, low birth weight can be regarded as a bioassay for in utero insulin secretion since insulin’s role as growth factor is an important component of fetal growth. The incidence of neonatal macrosomia in the setting of uncontrolled maternal diabetes or gestational diabetes is further evidence for insulin-mediated growth. It is important to note that maternal insulin cannot cross the placental barrier, and therefore, the failure of the developing fetus to produce insulin severely affects fetal growth parameters, including weight, length, and head circumference.21 In contrast, IUGR is not as prevalent or severe in cases of PNDM. A 2002 French cohort study found that IUGR was present in 74% cases of TNDM versus 36% cases of PNDM. Moreover, the TNDM patients were diagnosed at an earlier age (median age 6 days; range 1−81 days) relative to the PNDM patients (median age 27 days; range 1−127 days). The birth weight was also significantly lower in the TNDM patients (1,987±510 g) compared with the PNDM patients (2,497±610 g). Although the reasons are not clear, it is hypothesized that a defect in B-cell function and/or development is present in early postnatal life in TNDM, while satisfactory fetal B-cell function and/or development followed by a rapid B-cell failure after birth occurs in PNDM.21 A recent (2007) study by Flanagan et al illustrated that birth weight was significantly lower in TNDM patients with chromosome 6q abnormalities than in TNDM patients with KATP channel mutations.8
Diagnosis
Hyperglycemia is commonly defined as blood glucose more than 6.9 mmol/L or plasma glucose 8.3 mmol/L. While NDM is important in the differential diagnoses of neonatal hyperglycemia, it is also quite rare and therefore, other reversible causes of hyperglycemia should first be excluded. Neonatal hyperglycemia has been most commonly associated with high rates of glucose infusion (exceeding 10 to 12 mg/kg/min) in preterm or extremely low-birth-weight infants since prematurity and IUGR are associated with immature B-cell and reduced B-cell mass respectively, resulting in relative insulin deficiency.22,23 Other iatrogenic factors include use of inotropes, glucocorticoid therapy, lipid infusions, theophylline, and caffeine.24,25 Sepsis, surgical procedure, anesthesia, and other critical illnesses can lead to elevated blood sugar levels, potentially secondary to a stress response and concomitant rise in epinephrine and cortisol, decreased insulin release, and impaired glucose utilization.24 Inborn errors of amino acid metabolism, including methylmalonic aciduria and alaninuria, can also cause secondary hyperglycemia.
Differentiating between monogenic diabetes and autoimmune diabetes can be a potential diagnostic dilemma given the variable age of presentation for NDM. In a retrospective study with 111 children requiring insulin before 1 year of age, it was found that children diagnosed before 180 days of age had a greater frequency of protective human leukocyte antigen (HLA) alleles against Type 1 diabetes and lower prevalence of autoimmune markers than did those diagnosed after 180 days of age.26 Hence, all patients presenting with hyperglycemia within the first 6 months of life and low birth weight should raise the suspicion for NDM and should be subsequently screened for genetic causes. Sequencing of the KCNJ11 gene and chromosome 6q24 should be considered in all patients, while testing for ABCC8 gene mutations should only be done if the former tests are negative. Other rare causes of NDM includes INS and GCK gene mutations (Table 1) and should be considered if other tests remain negative, and in presence of other features.
A prenatal diagnosis of NDM by a deoxyribonucleic acid (DNA) analysis of fetal cells obtained through amniocentesis or chorionic villus sampling is possible for pregnancies at increased risk of NDM and if the disease-causing mutation(s) in the family are previously known. Preimplantation genetic diagnosis may be an option when the disease-causing mutation has been identified.27 Molecular genetic testing is offered in the USA and most European countries. Many labs also test patients from other countries. Some genetic tests for NDM are available as research tests, with the result reported within a few weeks of receipt of the sample. These tests are expensive but have a huge impact on the management of the proband and other family members.
Genetic counseling
Genetic counseling for NDM patients depends on the genetic etiology as the mode of inheritance widely varies for the genes involved. The majority of TNDM cases result from UPD6, which is sporadic and therefore has a low probability of recurrence in siblings and offspring. In familial cases with paternal duplication of the chromosome 6q24 region, affected males have a 50% chance of passing TNDM to their offspring. In the case of maternal duplication, children will not be affected, but the sons can potentially transmit TNDM to their offspring.11
On the other hand, most cases of KATP channel mutations in PNDM are the result of spontaneous, de novo heterozygous mutations, affecting children with a negative family history. However, activating mutations in the KCNJ11 gene and ABCC8 gene may confer an autosomal dominant mode of inheritance, and the offspring of affected individuals have a 50% chance of inheriting the mutation.28 Moreover, unlike KCNJ11 gene mutations, ABCC8 gene mutations have also been reported to be inherited in an autosomal recessive pattern. Knowing the mode of inheritance is vital and changes the dynamic of genetic counseling since the risk of unaffected parents having a child with recessive ABCC8 gene mutations dramatically rises to 25%, when compared with heterozygous de novo mutations. However, the offspring of the affected proband have a lower probability of being affected, unlike the case of de novo heterozygous mutations, which confer 50% risk of transmission to the next generation.11,13,29 PNDM secondary to INS, GCK, or PDX1 gene mutations is inherited in an autosomal recessive fashion, which implies that both parents of the affected individual are heterozygote for the mutation. In this case, siblings of the affected child have a 25% risk of having PNDM, while the offspring of an affected child will be heterozygote carriers.
Treatment of neonatal diabetes
The mainstay of the management of NDM in the immediate newborn period is insulin, regardless of the etiology.3 Another available treatment option is oral sulfonylurea, which is administered based on the genetic diagnosis of KATP channel mutations.30,31 Treatment with insulin should be started immediately after diabetes is diagnosed by persistent hyperglycemia or elevated glycated hemoglobin (HbA1c).
Management of acute hyperglycemia
Neonates with diabetes mellitus can present with significant hyperglycemia, electrolyte disturbance, dehydration, and ketoacidosis.18 The initial management includes fluid resuscitation with isotonic electrolyte solutions, to treat the dehydration that results from osmotic diuresis. The appropriate fluid therapy is calculated on an individual basis and administered slowly over a period of 24−48 hours to avoid cerebral edema that results from too rapid correction.18,24 Neonates and infants presenting with diabetic ketoacidosis should be managed in an intensive care unit, under the supervision of a pediatric endocrinologist, with frequent monitoring of blood glucose, electrolytes, and neurological status. Neonatal diabetic ketoacidosis is managed according to the same principles guiding the therapy for children and adolescents with diabetes mellitus.32,33 Insulin therapy should be started with careful attention as newborns are very sensitive to insulin and therefore in danger of severe hypoglycemia. A continuous intravenous infusion of regular insulin is started at 0.05−0.1 U/kg/hr and titrated up or down as needed based on the blood glucose levels.34 The goal of therapy is to allow normal energy utilization by tissues as well as to replace fluids and electrolytes.
Insulin therapy
Insulin therapy is crucial to obtain satisfactory weight gain and growth, especially in babies with IUGR, but the treatment of NDM is complex due to the paucity of subcutaneous fat and the need for the low doses of insulin. Insulin is administered as an intravenous infusion, intermittent subcutaneous therapy, or as continuous subcutaneous insulin infusion (CSII).24,35 It is often delivered initially by an intravenous infusion as this enables better titration of doses based on the blood glucose levels. After the initial treatment of the diabetic ketoacidosis, the intravenous infusion of regular insulin should be continued in infants with persistent hyperglycemia, despite reductions in glucose infusion rates, and in those with persistent glucose excursions, while others are transitioned to an appropriate regimen of subcutaneous insulin.36 Finding a suitable regimen is usually not an easy task as few data are available on the most appropriate insulin preparations for young infants. Patients are transitioned to injections of basal insulin or a combination basal/bolus insulin regimen.
Basal insulin
Basal insulin may be intermediate-acting insulin, like NPH (isophane) insulin or long-acting insulin, like insulin glargine or insulin detemir. Intermediate-acting insulin, like NPH, does not provide insulin delivery profiles that complement the feeding patterns and results in significant swings in glucose levels and hypoglycemia.35 Insulin glargine, with an almost flat action profile and 24-hour action, would overcome the problems of intermediate acting insulin, but the safety and effectiveness of glargine insulin have not been established in patients less than 6 years of age.37 However, it is being used in the treatment of toddlers and children with type 1 diabetes mellitus and has shown to be suited for treating newborns and infants, with better glucose control and less hypoglycemia.37 Glargine is started at a dose of 0.5−1 U/day and is often divided into twice-daily dosing to provide an adequate basal effect. Another long-acting insulin analogue that has been used in NDM is insulin detemir. Unlike insulin glargine, insulin detemir has a peak, which results in hypoglycemia. The prolonged action of detemir is due to the binding of fatty acid residues to serum albumin, and the lack of subcutaneous fat and low albumin in many newborns also affects the pharmacokinetics of this insulin. Another long-acting insulin, ultralente, provides good control of blood glucose, but is not available in the USA.38 Some infants can maintain an appropriate blood glucose level on just small doses of basal insulin without requiring any bolus insulin.
Bolus insulin
Bolus is given with either short-acting insulin, like regular insulin, or rapid-acting insulin, like insulin lispro or insulin aspart. The groups of insulins should be used cautiously as their peak action can result in significant hypoglycemia, even in very small doses. As in children with type 1 diabetes, an insulin to carbohydrate ratio and a ratio of insulin to blood glucose level (insulin sensitivity factor) can be used in newborns and infants to give the bolus for carbohydrate and the high-blood glucose correction.
Diluted insulin
Newborns and infants usually require very small doses of insulin, with gradations in fractions of a unit, which can be difficult to measure with a standard insulin syringe, hence there is a need to use diluted insulin. Diluents appropriate for each insulin are provided by the pharmaceutical companies.39 Eli Lilly (Eli Lilly and Co, Indianapolis, IN, USA) provides a common diluent for Humalog®, Humulin N®, and Humulin R® and Novo Nordisk (Novo Nordisk A/S, Bagsvaerd, Denmark) provides the insulin diluting medium for Novolog®. Dilute insulin should be prepared under aseptic conditions by a pharmacist who is familiar with the dilution technique. Usually insulin is diluted to a concentration of 1:10 (equivalent to U-10) or 1:2 (equivalent to U-50). Due to instability issues, diluted insulin loses its potency more quickly than standard insulin and should be discarded 30 days after preparation or earlier, based on the manufacturer’s recommendation.40 Diluted insulin should be used with caution in newborns or infants with impaired liver function due to the presence of some preservatives. The long-acting insulins glargine and detemir usually should not be diluted due to the risk of alteration of the pharmacokinetic profile. However, due to the requirement of very small doses in newborns, these insulin preparations may need to be diluted with normal saline. But studies have not evaluated the efficacy or stability of diluted long-acting insulin products. Extreme caution should be observed when using diluted insulin preparation, in order to avoid dose errors.27
Continuous subcutaneous insulin infusion
The delivery of small doses of insulin and variable insulin requirements, together with the frequent monitoring of blood glucose, remains a major challenge in NDM. CSII allows for low rates of insulin delivery that are suited for newborns with diabetes. In addition, CSII provides greater flexibility, to adjust for the variability in oral intake as well as changes in energy expenditure as the child grows.36 When compared with subcutaneous insulin injections, CSII therapy has also been effective in reducing both HbA1c and hypoglycemic episodes, in patients managing it appropriately.41 In children on continuous enteral or parenteral nutrition, the total insulin dose is administered as a basal rate, while in orally fed neonates, basal rates combined with boluses can better mimic physiological insulin delivery.36 Some centers recommend CSII for young infants as it is safe, more physiological, easier to manage than injections, and offers the possibility of very low rates of insulin delivery.1
Continuous glucose monitoring sensor
A continuous glucose monitoring sensor (CGMS) consists of an electrode sensor that catalyzes glucose oxidation, generating an electric current that is recorded by a monitor. The CGMS is inserted subcutaneously and allows continuous measurement of glucose in the interstitial fluid. The sensor is especially useful in preterm babies and in small for gestational age babies who are at risk for wide excursions in blood glucose levels. The CGMS is programmed to give alerts when glucose levels are either lower or higher than acceptable limits, and this feature enables the caregiver to respond promptly. The prompt care is clinically important as both hypoglycemia and hyperglycemia are associated with acute neurophysiological abnormalities and later neurodevelopmental impairment. However, despite its proposed advantages for continuous blood glucose measurement, there is limited experience with its use, or accuracy in newborns.42 Further studies are required to validate the use of CGMS in newborns.
Oral sulfonylurea
Patients with NDM were previously believed to require lifelong insulin treatment as they have little or no endogenous insulin. The identification of KATP channel mutations in these patients has revolutionized the treatment of NDM. The oral sulfonylureas commonly used in adults with type 2 diabetes mellitus have been used with success in cases of NDM caused by mutations in the KCNJ11 and ABCC8 genes. Sulfonylurea binds to the SUR1 subunits of the KATP channel and closes the channel in an ATP-independent manner, causing membrane depolarization and insulin secretion.43 In the same way, sulfonylurea causes insulin secretion in the mutated KATP channels by binding to the SUR subunits. Of the known genetic subtypes of NDM, only patients with activating KCNJ11 or ABCC8 gene mutations respond to treatment with a sulfonylurea. Sulfonylurea can be started in a patient as soon as the genetic diagnosis is established, and a trial of sulfonylurea in NDM without a genetic diagnosis is not recommended.44 Patients on insulin can be transitioned to oral sulfonylurea over a period of a few weeks to months as an outpatient or over few days as an inpatient, based on specific protocols.30 In both settings, the dose of sulfonylurea is increased with a concomitant reduction in insulin dose, based on the blood glucose levels. About 90% of patients with KCNJ11 gene mutations and 85% of patients with ABCC8 gene mutations can successfully transition from insulin to sulfonylurea.30,31 It has been shown that patients with ABCC8 gene mutation require lower doses of sulfonylurea than those with KCNJ11 gene mutation and hence, require a different treatment protocol.31 Infants as young as 1 month have been successfully transitioned to sulfonylurea therapy.45 Long-term insulin therapy is required for all other causes of PNDM, although a mild beneficial effect of oral sulfonylurea has been reported in patients with GCK gene mutations.46
Treatment with sulfonylurea reduces fluctuations in blood glucose and improves the glycemic control by reducing HbA1c levels without concomitant hypoglycemia.13,14 This improvement in glycemic control reduces the risk of diabetic complications. In addition to the diabetes control, nonspecific sulfonylurea drugs, like glibenclamide (glyburide), improves neurological function by improving both muscle strength and cognitive function.47,48 Improved neurodevelopmental outcome has been reported in children with intermediate and complete DEND syndrome treated with sulfonylurea, but the best outcome is seen if transition occurs early in the first 6 months of life.49
A small group (10%−15%) of patients with KCNJ11 and ABCC8 gene mutations do not respond to sulfonylurea therapy. This is attributed to the characteristic of the specific mutation, to B-cell glucotoxicity from prolonged poor control before transfer, or to noncompliance.30 In fact, patients should be given a trial of sulfonylurea for a period of at least 3 months, with a dose as high as 1.5 mg/kg/day, before considering it as a treatment failure as B-cell function improves with time on sulfonylureas.50 Sometimes, even identical mutations within the same family can produce a variable clinical picture and different responses to treatment.51
Most patients with KATP channel mutations are treated with glibenclamide. Other first and second generation sulfonylurea drugs, like tolbutamide, glipizide, gliclazide, and glimepiride, have been used with success in patients with KATP channel mutation.30,31 The doses required in the treatment of NDM are usually higher than those used in patients with type 2 diabetes mellitus.49 With time, many patients have been able to reduce their doses of sulphonylureas and maintain excellent glycemic control. Despite the requirement of high doses, the side effects of sulfonylurea are mild and transitory. The most common side effects are diarrhea, nausea, vomiting, and abdominal pain.52 Hypoglycemia, transient allergic skin reactions, and tooth discoloration are some of the less common side effects.14,53 Also, the unknown interactions of sulfonylurea with the extrapancreatic KATP channels found in the cardiac, smooth and skeletal muscles, adipocytes, and brain are of concern for long-term use in children.
Follow up of patients on sulfonylurea for more than 68 months has showed that chronic sulfonylurea therapy retains its efficacy in patients with PNDM, contrary to the findings reported in type 2 diabetes, where secondary sulfonylurea failure has been reported.54 Capillary blood glucose monitoring and close follow up is needed in all patients treated with sulfonylurea. Parents and patients should understand that the risk of hypo- and hyperglycemia exists on sulfonylurea treatment, and education in their prevention and treatment should be given. Parents should also be cautioned that insulin may be needed, at times, for eg, to control high blood sugars during illness.55
Diet
All newborns with NDM should receive a high caloric diet along with an adequate amount of insulin to promote satisfactory weight gain and growth.1 Any reduction of glucose and calories to improve hyperglycemia in these babies significantly affects their weight gain.1 A high caloric diet can be achieved with the help of the dietician, who can assist in calculating the calories and in determining the carbohydrate content of breast milk or formula. The carbohydrate content of term human milk and the standard infant formula are similar (approximately 70−75 g/L), and the carbohydrate content of human milk fortifier is negligible (<0.1 g/packet). Dietary management requires a team approach to address the caloric needs and carbohydrate counting.
Follow up
Frequent monitoring of blood glucose and the maintenance of blood glucose in the appropriate range is essential to prevent acute and long-term complications. The HbA1c should be monitored every 3 months and the level maintained between 7.5%−8.5%.56 All patients with TNDM need-long-term follow up due to the potential for recurrence of diabetes in later life. If there is recurrence, the treatment varies between diet, oral hypoglycemic agents, and insulin.57 Patients need annual screening for the chronic complications of diabetes, including urinalysis for microalbuminuria and ophthalmologic examination for retinopathy.27 While the course and severity of the disease varies greatly and is dependent on genetic etiology, patients can have good health and normal intellectual development.58
In conclusion, NDM is a rare disease that presents as either the TNDM or PNDM form. With advances in molecular genetics, various genetic subtypes have been identified, and treatment varies with the type of disease. Molecular genetic diagnosis is recommended in all patients with NDM. The majority of patients with KATP channel mutation respond successfully to oral sulfonylurea therapy, which markedly decreases the complexity of diabetes management and provides a better quality of life.
Disclosure
The authors report no conflicts of interest in this work.
References
Polak M, Cavé H. Neonatal diabetes mellitus: a disease linked to multiple mechanisms. Orphanet. J Rare Dis. 2007;2:12. | |
Grulich-Henn J, Wagner V, Thon A, et al. Entities and frequency of neonatal diabetes: data from the diabetes documentation and quality management system (DPV). Diabet Med. 2010;27(6):709–712. | |
Aguilar-Bryan L, Bryan J. Neonatal diabetes mellitus. Endocr Rev. 2008;29(3):265–291. | |
Temple IK, James RS, Crolla JA, et al. An imprinted gene(s) for diabetes? Nat Genet. 1995;9(2):110–112. | |
Gardner RJ, Mackay DJ, Mungall AJ, et al. An imprinted locus associated with transient neonatal diabetes mellitus. Hum Mol Genet. 2000;9(4):589–596. | |
Mackay DJ, Temple IK. Transient neonatal diabetes mellitus type 1. Am J Med Genet C Semin Med Genet. 2010;154C(3):335–342. | |
Abdollahi A. LOT1 (ZAC1/PLAGL1) and its family members: mechanisms and functions. J Cell Physiol. 2007;210(1):16–25. | |
Flanagan SE, Patch AM, Mackay DJ, et al. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes. 2007;56(7):1930–1937. | |
Yorifuji T, Kurokawa K, Mamada M, et al. Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: Phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor-1beta gene due to germline mosaicism. J Clin Endocrinol Metab. 2004;89(6):2905–2908. | |
Edghill EL, Bingham C, Slingerland AS, et al. Hepatocyte nuclear factor-1 beta mutations cause neonatal diabetes and intrauterine growth retardation: support for a critical role of HNF-1beta in human pancreatic development. Diabet Med. 2006;23(12):1301–1306. | |
Murphy R, Ellard S, Hattersley AT. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat Clin Pract Endocrinol Metab. 2008;4(4):200–213. | |
Babenko AP, Polak M, Cavé H, et al. Activating mutations in the ABCC8 gene in neonatal diabetes mellitus. N Engl J Med. 2006;355(5):456–466. | |
Hattersley AT, Ashcroft FM. Activating mutations in Kir6.2 and neonatal diabetes: new clinical syndromes, new scientific insights, and new therapy. Diabetes. 2005;54(9):2503–2513. | |
Shah B, Breidbart E, Pawelczak M, Lam L, Kessler M, Franklin B. Improved long-term glucose control in neonatal diabetes mellitus after early sulfonylurea allergy. J Pediatr Endocrinol Metab. 2012;25(3–4):353–356. | |
Zwaveling-Soonawala N, Hagebeuk EE, Slingerland AS, Ris-Stalpers C, Vulsma T, van Trotsenburg AS. Successful transfer to sulfonylurea therapy in an infant with developmental delay, epilepsy and neonatal diabetes (DEND) syndrome and a novel ABCC8 gene mutation. Diabetologia. 2011;54(2):469–471. | |
Maassen JA, ‘T Hart LM, Van Essen E, et al. Mitochondrial diabetes: molecular mechanisms and clinical presentation. Diabetes. 2004;53 Suppl 1:S103–S109. | |
Woolley SL, Saranga S. Neonatal diabetes mellitus: A rare but important diagnosis in the critically ill infant. Eur J Emerg Med. 2006;13(6):349–351. | |
Abaci A, Razi CH, Ozdemir O, et al. Neonatal diabetes mellitus accompanied by diabetic ketoacidosis and mimicking neonatal sepsis: a case report. J Clin Res Pediatr Endocrinol. 2010;2(3):131–133. | |
Gloyn AL, Pearson ER, Antcliff JF, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med. 2004;350(18):1838–1849. | |
Lee JH, Tsai WY, Chou HC, Tung YC, Hsieh WS. Permanent neonatal diabetes mellitus manifesting as diabetic ketoacidosis. J Formos Med Assoc. 2003;102(12):883–886. | |
Metz C, Cavé H, Bertrand AM, et al; NDM French Study Group. Neonatal diabetes mellitus: chromosomal analysis in transient and permanent cases. J Pediatr. 2002;141(4):483–489. | |
Meetze W, Bowsher R, Compton J, Moorehead H. Hyperglycemia in extremely- low-birth-weight infants. Biol Neonate. 1998;74(3):214–221. | |
Ogilvy-Stuart AL, Beardsall K. Management of hyperglycaemia in the preterm infant. Arch Dis Child Fetal Neonatal Ed. 2010;95(2):F126–F131. | |
Fosel S. Transient and permanent neonatal diabetes. Eur J Pediatr. 1995;154(12):944–948. | |
Beardsall K, Vanhaesebrouck S, Ogilvy-Stuart AL, et al. Prevalence and determinants of hyperglycemia in very low birth weight infants: cohort analyses of the NIRTURE study. J Pediatr. 2010;157(5):715–719. e1. | |
Iafusco D, Stazi MA, Cotichini R, et al; Early Onset Diabetes Study Group of the Italian Society of Paediatric Endocrinology and Diabetology. Permanent diabetes mellitus in the first year of life. Diabetologia. 2002;45(6):798–804. | |
De León DD, Stanley CA. Permanent neonatal diabetes mellitus. In: Pagon RA, Adam MP, Bird TD, Dolan CR, Fong CT, Stephens K, editors. GeneReviews™. Seattle, WA: University of Washington; 1993. | |
Rubio-Cabezas O, Klupa T, Malecki MT; CEED3 Consortium. Permanent neonatal diabetes mellitus – the importance of diabetes differential diagnosis in neonates and infants. Eur J Clin Invest. 2011;41(3):323–333. | |
Ellard S, Flanagan SE, Girard CA, et al. Permanent neonatal diabetes caused by dominant, recessive, or compound heterozygous SUR1 mutations with opposite functional effects. Am J Hum Genet. 2007;81(2):375–382. | |
Pearson ER, Flechtner I, Njølstad PR, et al; Neonatal Diabetes International Collaborative Group. Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations. N Engl J Med. 2006;355(5):467–477. | |
Rafiq M, Flanagan SE, Patch AM, Shields BM, Ellard S, Hattersley AT; Neonatal Diabetes International Collaborative Group. Effective treatment with oral sulfonylureas in patients with diabetes due to sulfonylurea receptor 1 (SUR1) mutations. Diabetes Care. 2008;31(2):204–209. | |
Wolfsdorf J, Glaser N, Sperling MA; American Diabetes Association. Diabetic ketoacidosis in infants, children, and adolescents: A consensus statement from the American Diabetes Association. Diabetes Care. 2006;29(5):1150–1159. | |
Wolfsdorf J, Craig ME, Daneman D, et al. Diabetic ketoacidosis in children and adolescents with diabetes. Pediatr Diabetes. 2009;10(Suppl 12):S118–S133. | |
Martin RJ, Fanaroff AA, Walsh MC, editors. Neonatal-Perinatal Medicine: Diseases of the Fetus and Infant. 9th ed. St Louis, MO: Mosby; 2011:1497–1620. | |
Sarici SU, Alpay F, Dündaryz MR, Gyknay E. Neonatal diabetes mellitus: patient report and review of the literature. J Pediatr Endocrinol Metab. 2001;14(4):451–454. | |
Bharucha T, Brown J, McDonnell C, et al. Neonatal diabetes mellitus: Insulin pump as an alternative management strategy. J Paediatr Child Health. 2005;41(9–10):522–526. | |
Jeha GS, Venkatesh MP, Edelen RC, Kienstra KA, Karaviti L, Fernandes CJ. Neonatal diabetes mellitus: patient reports and review of current knowledge and clinical practice. J Pediatr Endocrinol Metab. 2005;18(11):1095–1102. | |
Mitamura R, Kimura H, Murakami Y, Nagaya K, Makita Y, Okuno A. Ultralente insulin treatment of transient neonatal diabetes mellitus. J Pediatr. 1996;128(2):268–270. | |
Rosenzweig JL. The use of diluted insulin. Diabetes Technol Ther. 2000;2(1):67–68. | |
Stickelmeyer MP, Graf CJ, Frank BH, Ballard RL, Storms SM. Stability of U-10 and U-50 dilutions of insulin lispro. Diabetes Technol Ther. 2000;2(1):61–66. | |
Wintergerst KA, Hargadon S, Hsiang HY. Continuous subcutaneous insulin infusion in neonatal diabetes mellitus. Pediatr Diabetes. 2004;5(4):202–206. | |
Beardsall K, Ogilvy-Stuart AL, Ahluwalia J, Thompson M, Dunger DB. The continuous glucose monitoring sensor in neonatal intensive care. Arch Dis Child Fetal Neonatal Ed. 2005;90(4):F307–F310. | |
Gribble FM, Reimann F. Sulphonylurea action revisited: the post-cloning era. Diabetologia. 2003;46(7):875–891. | |
Chakera AJ, Flanagan SE, Ellard S, Hattersley AT. Comment on: Khurana et al. The diagnosis of neonatal diabetes in a mother at 25 years of age. Diabetes Care. 2012;35:e59. Diabetes Care. 2013;36(2):e31. | |
Ješić MM, Ješić MD, Maglajlić S, Sajić S, Necić S. Successful sulfonylurea treatment of a neonate with neonatal diabetes mellitus due to a new KCNJ11 mutation. Diabetes Res Clin Pract. 2011;91(1):e1–e3. | |
Turkkahraman D, Bircan I, Tribble ND, Akçurin S, Ellard S, Gloyn AL. Permanent neonatal diabetes mellitus caused by a novel homozygous (T168A) glucokinase (GCK) mutation: initial response to oral sulphonylurea therapy. J Pediatr. 2008;153(1):122–126. | |
Slingerland AS, Nuboer R, Hadders-Algra M, Hattersley AT, Bruining GJ. Improved motor development and good long-term glycaemic control with sulfonylurea treatment in a patient with the syndrome of intermediate developmental delay, early-onset generalised epilepsy and neonatal diabetes associated with the V59M mutation in the KCNJ11 gene. Diabetologia. 2006;49(11):2559–2563. | |
Slingerland AS, Hurkx W, Noordam K, et al. Sulphonylurea therapy improves cognition in a patient with the V59M KCNJ11 mutation. Diabet Med. 2008;25(3):277–281. | |
Greeley SA, Tucker SE, Worrell HI, Skowron KB, Bell GI, Philipson LH. Update in neonatal diabetes. Curr Opin Endocrinol Diabetes Obes. 2010;17(1):13–19. | |
Babiker T, Shepherd MH, Hattersley AT. Continue with long term sulfonylureas in patients with mutations in the KCNJ11 gene when there is evidence of response even if insulin treatment is still required. Diabetes Res Clin Pract. 2013;100(3):e63. | |
Russo C, Salina A, Aloi C, Iafusco D, Lorini R, d’Annunzio G. Mother and daughter carrying the same KCNJ11 mutation but with a different response to switching from insulin to sulfonylurea. Diabetes Res Clin Pract. 2011;94(2):e50–e52. | |
Codner E, Flanagan S, Ellard S, García H, Hattersley AT. High-dose glibenclamide can replace insulin therapy despite transitory diarrhea in early-onset diabetes caused by a novel R201L Kir6.2 mutation. Diabetes Care. 2005;28(3):758–759. | |
Kumaraguru J, Flanagan SE, Greeley SA, et al. Tooth discoloration in patients with neonatal diabetes after transfer onto glibenclamide: a previously unreported side effect. Diabetes Care. 2009;32(8):1428–1430. | |
Iafusco D, Bizzarri C, Cadario F, et al. No beta cell desensitisation after a median of 68 months on glibenclamide therapy in patients with KCNJ11-associated permanent neonatal diabetes. Diabetologia. 2011;54(10):2736–2738. | |
Codner E, Flanagan SE, Ugarte F, et al. Sulfonylurea treatment in young children with neonatal diabetes: dealing with hyperglycemia, hypoglycemia, and sick days. Diabetes Care. 2007;30(5):e28–e29. | |
Silverstein J, Klingensmith G, Copeland K, et al; American Diabetes Association. Care of children and adolescents with type 1 diabetes: a statement of the American Diabetes Association. Diabetes Care. 2005;28(1):186–212. | |
Temple IK, Shield JP. Transient neonatal diabetes, a disorder of imprinting. J Med Genet. 2002;39(12):872–875. | |
von Mühlendahl K, Herkenhoff H. Long-term course of neonatal diabetes. N Engl J Med. 1995;333(11):704–708. |
 © 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2014 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.