Back to Journals » Cancer Management and Research » Volume 8
Neoadjuvant therapy for early-stage breast cancer: the clinical utility of pertuzumab
Authors Gollamudi J, Parvani J, Schiemann W, Vinayak S
Received 7 August 2015
Accepted for publication 23 November 2015
Published 18 February 2016 Volume 2016:8 Pages 21—31
DOI https://doi.org/10.2147/CMAR.S55279
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 3
Editor who approved publication: Dr Kenan Onel
Jahnavi Gollamudi,1,* Jenny G Parvani,2,* William P Schiemann,3 Shaveta Vinayak3,4
1Department of Internal Medicine, 2Department of Biomedical Engineering, 3Case Comprehensive Cancer Center, Case Western Reserve University, 4Department of Hematology and Oncology, University Hospitals Case Medical Center, Cleveland, OH, USA
*These authors contributed equally to this work
Abstract: Approximately 20% of breast cancer patients harbor tumors that overexpress human epidermal growth factor receptor 2 (HER2; also known as ErbB2), a receptor tyrosine kinase that belongs to the epidermal growth factor receptor family of receptor tyrosine kinases. HER2 amplification and hyperactivation drive the growth and survival of breast cancers through the aberrant activation of proto-oncogenic signaling systems, particularly the Ras/MAP kinase and PI3K/AKT pathways. Although HER2-positive (HER2+) breast cancer was originally considered to be a highly aggressive form of the disease, the clinical landscape of HER2+ breast cancers has literally been transformed by the approval of anti-HER2 agents for adjuvant and neoadjuvant settings. Indeed, pertuzumab is a novel monoclonal antibody that functions as an anti-HER2 agent by targeting the extracellular dimerization domain of the HER2 receptor; it is also the first drug to receive an accelerated approval by the US Food and Drug Administration for use in neoadjuvant settings in early-stage HER2+ breast cancer. Here, we review the molecular and cellular factors that contribute to the pathophysiology of HER2 in breast cancer, as well as summarize the landmark preclinical and clinical findings underlying the approval and use of pertuzumab in the neoadjuvant setting. Finally, the molecular mechanisms operant in mediating resistance to anti-HER2 agents, and perhaps to pertuzumab as well, will be discussed, as will the anticipated clinical impact and future directions of pertuzumab in breast cancer patients.
Keywords: breast cancer, HER2, monoclonal antibody, neoadjuvant, pertuzumab, receptor tyrosine kinase, signal transduction, trastuzumab
Introduction
Breast cancer remains a significant health concern worldwide, accounting for ~1.7 million newly diagnosed cases and 522,000 deaths in 2012.1,2 Despite the implementation of improved screening and early detection protocols, the American Cancer Society still estimates that invasive breast cancer will be diagnosed in ~232,000 women in the USA in 2015, killing more than 40,000 patients in the same time span.3 Breast cancer is a heterogeneous disease that comprises at least five genomically distinct subtypes that coalesce as the second leading cause of cancer death in women.3 Among individual breast cancer subtypes, those classified as human epidermal growth factor receptor 2 (HER2)-positive represent ~20% of all breast cancer cases and are characterized by their dramatic overexpression of HER2, a critical 185 kDa receptor tyrosine kinase (RTK) located at chromosome 17a that drives the aberrant proliferation and survival of breast cancer cells.2,4,5 Historically, HER2-positive (HER2+) breast cancers were considered to be among the most aggressive female cancers, becoming so through the hyperactivation of HER2 and its signaling systems in breast cancer cells. However, with the recent advent of HER2-directed therapies, this breast cancer subtype has become treatable in the neoadjuvant and adjuvant clinical settings.6–8 Indeed, the first clinical trial that combined the anti-HER2 agent, trastuzumab, with chemotherapy showed significantly improved overall survival in patients with metastatic HER2+ breast cancers.9 Likewise, administering trastuzumab to patients with early stage, locally advanced HER2+ breast cancers demonstrated similar survival benefits,7,8,10,11 thereby revolutionizing the management of this breast cancer subtype in adjuvant and neoadjuvant settings.
Despite its overall clinical efficacy, patients treated with trastuzumab are prone to develop resistance to this anti-HER2 agent, an event that paved the way for the formulation of new and mechanistically distinct anti-HER2 agents necessary to circumvent cross-resistance and disease relapse.12,13 Accordingly, the humanized monoclonal antibody, pertuzumab, is a second-generation anti-HER2 agent that binds HER2 and prevents its dimerization. Once bound, trastuzumab prevents HER2 from dimerizing with itself or other epidermal growth factor receptor (EGFR) family members, resulting in the inactivation of oncogenic signaling systems.14 In the succeeding sections, we highlight the pathophysiology associated with HER2+ breast cancers, as well as their ability to be targeted effectively by pertuzumab in both neoadjuvant and adjuvant clinical settings. Finally, we will discuss recent advances in our understanding related to how HER2+ breast cancer cells acquire resistance to anti-HER2 agents, as well as how these untoward events impact clinical practice.
Cell signaling mediated by HER2
The EGFR family of RTKs
HER2 (also known as ErbB2) belongs to the EGFR family of RTKs, which also consists of EGFR (also known as HER1 or ErbB1), HER3 (also known as ErbB3), and HER4 (also known as ErbB4).2,5,15,16 The dramatic overexpression of HER2 in human breast cancers is primarily attributed to gene amplification;17 however, dysregulated expression or activity of numerous transcription factors that govern HER2 mRNA synthesis has also been implicated in eliciting elevated HER2 expression,2 including Foxp3,18 PEA3,19 AP-2α and YY1,20 and a G-quadruplex complex comprises Ku70, Ku80, PURA, nucleolin, and hnRNP K.21 Recent studies also point to a prominent role of post-translational activities in governing aberrant HER2 expression, particularly its ability to be internalized and degraded in breast cancer cells.22–24 For instance, the RING finger E3 ubiquitin ligase neuregulin receptor degradation protein-1 (Nrdp-1) was previously shown to modulate the expression of EGFR family members.22,24 Importantly, the loss of Nrdp-1 expression results in the stabilization and dramatic accumulation of HER2 (and HER3) in human breast cancers, including those lacking amplification of the HER2 locus.25
With the exception of HER2, all additional members of the EGFR family of RTKs have identifiable ligands and growth factors capable of binding and mediating their activation, which transpires following the homo- or heterodimerization of RTK subunits.2,5,15,16 The activation of HER2 likely occurs following its heterodimerization with ligand-bound EGFR, HER3, or HER4; however, amplification of the HER2 locus is sufficient to stimulate the activation of HER2 in a ligand-independent manner.2,5,15,16,26 Once dimerized, members of the EGFR family undergo transphosphorylation on tyrosine residues, resulting in an “open” protein kinase conformation that enables the recruitment and stimulation of downstream signaling effectors (Figure 1). Interestingly, crystallography analyses determined that the dimerization of EGFR family members elicits their activation allosterically through the formation of asymmetric dimers, a mechanism reminiscent of that employed by Src and CDK/cyclin.2,5,16,27 Moreover, the absence of a bona fide ligand-binding domain in HER2 enables this RTK to assume an “open” conformation that is primed to partner with other EGFR family members, particularly HER3.5 Ultimately, HER2 dimers autophosphorylate on C-terminal tyrosine residues, which serve as docking sites for a variety of downstream effector molecules, particularly Shc and its activation of the Ras/MAP kinase pathway, as well as p85 and its activation of the PI3K/AKT pathway (Figure 1).28,29
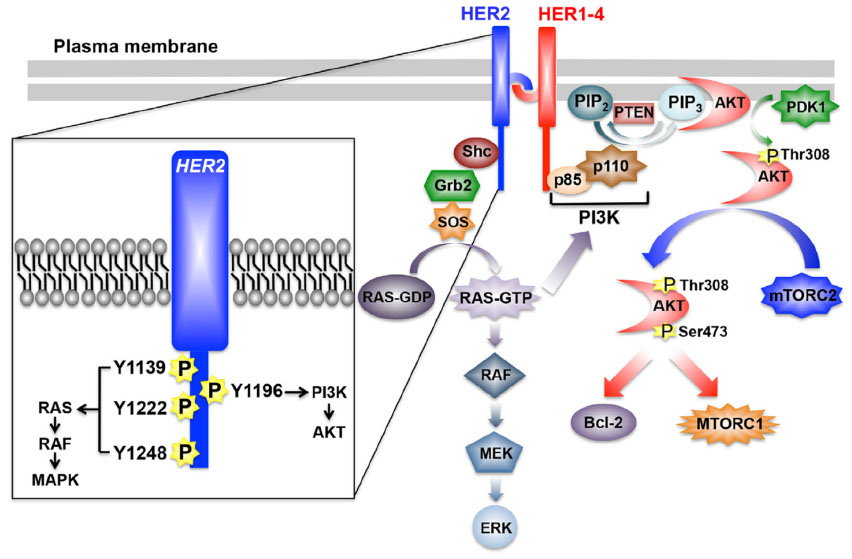 | Figure 1 Schematic of HER2 signaling system. |
The RAS/MAP kinase signaling pathway
Following its activation, HER2 is readily phosphorylated on as many as six distinct tyrosine residues, including Y1005, Y1023, Y1139, Y1196, Y1222, and Y1248, all of which play varying roles in coupling HER2 to the activation of the Ras/MAP kinase pathway.2,5,16 Of these phosphotyrosine docking sites, Y1139 (Grb2), Y1222 (Shc), and Y1248 (Shc) are the most prominently phosphorylated and serve to recruit and activate SOS, which stimulates Ras by catalyzing its exchange of GDP for GTP (Figure 1).2,5,16,28 Once activated, Ras-GTP interacts physically with Raf, leading to its localization and activation at the plasma membrane and, consequently, the sequential activation of MEK1/2 upon its phosphorylation by Raf, and of ERK1/2 upon its phosphorylation by MEK1/2 (Figure 1).30,31 The constitutive activation of the Ras/MAP kinase pathway by HER2 is primarily responsible for driving the dysregulated proliferation of HER2+ breast cancers. The magnitude and extent to which the Ras/MAP kinase pathway is activated is governed by a host of positive and negative feedback loops, and its pathophysiological importance lies beyond the scope of this review. Readers desiring in-depth summaries pertaining to the molecular mechanisms whereby the Ras/MAP kinase pathway promotes tumorigenesis are directed to several comprehensive reviews.32,33
The PI3K/AKT signaling pathway
Breast cancer is a disease that not only reflects defects in the cell cycle but also irregularities in the ability of carcinoma cells to succumb to apoptotic stimuli. HER2+ breast cancers acquire resistance to apoptotic signals primarily through their constitutive activation of the PI3K/AKT pathway. The coupling of HER2 to the activation of PI3K transpires through Ras-dependent and -independent mechanisms.30,31 With respect to the former, activated Ras-GTP can interact physically with the catalytic subunit of PI3K, p110, leading to its localization and activation at the plasma membrane.30,31 Alternatively, the phosphorylation of HER2 at Y1196 facilitates docking of the regulatory subunit of PI3K, p85, which is bound to p110 and inhibits its activation. Importantly, docking of the p85 to Y1196-HER2 relieves the inhibitory constraints placed upon p110, leading to its phosphorylation of PIP2 (phosphatidylinositol-4,5-bisphosphate) to produce the lipid second messenger, PIP3 (phosphatidylinositol-3,4,5-trisphosphate).34–36 Once produced, PIP3 recruits AKT to the plasma membrane to facilitate its phosphorylation at two activating residues: 1) T308, which is phosphorylated by PDK1, and 2) S473, which is primarily phosphorylated by mTORC2, but can also be modified by integrin-linked kinase, PKC, ATM, DNA-PK, and even AKT.36 Activated AKT is then able to regulate various downstream effector molecules, such as mTORC1, Bcl-2, GSK3, and FOXOs, which coalesce in promoting cell survival, proliferation, and metabolism.16 Finally, the activation status of the PI3K/AKT pathway is negatively regulated by two major lipid phosphatases, namely PTEN and INPP4B. Indeed, PTEN is a powerful tumor suppressor whose expression and activity is lost in numerous human cancers, including 40% of human breast cancers; it also dephosphorylates PIP3 at the 3′-phosphate position, thereby inhibiting PDK1-mediated phosphorylation of AKT at T308.37,38 Although INPP4B is also a lipid phosphatase, it preferentially dephosphorylates phosphatidylinositols at the 4′-phosphate position, an event that suppresses AKT phosphorylation at S473. Similar to PTEN, inactivation of INPP4B has recently been associated with the etiology of breast cancer.39 Given the breadth of this subject, readers desiring in-depth summaries pertaining to the molecular mechanisms whereby the PI3K/AKT pathway promotes tumorigenesis are directed to several comprehensive reviews.34–36
Landmark neoadjuvant trials of pertuzumab
Mechanism and rationale for administering pertuzumab
Protein structure-function and crystallography studies established that EGFR family members comprise three major domains: an extracellular ligand binding domain, a transmembrane domain, and an intracellular protein tyrosine kinase (PTK) domain. The notable exceptions to this organizational structure are found in HER2, which lacks a functional ligand binding motif, and in HER3, which possesses severely impaired PTK activity.5,16 The extracellular domains of EGFR family members can be further subdivided into four distinct regions, with Domains I and III functioning to bind bivalent ligands and growth factors. In the absence of ligand, EGFR, HER3, and HER4 adopt a “closed” and inactive conformation through the interaction of Domain II with Domain IV. Ligand binding to Domain I of these RTKs relieves the interaction between Domains II and IV, thereby eliciting an “open” and active conformation that exposes the dimerization motif in Domain II to homo- or heterodimerize with its corresponding partner. The inability of HER2 to bind ligands and growth factors enables this RTK to assume a constitutively active and “open” conformation that is primed to interact with other EGFR family members in either a ligand-dependent manner or a ligand-independent manner.5,16,40 This characteristic coupled with the amplified expression of HER2 in mammary epithelial cells underlies the development and metastatic progression of HER2+ breast cancers, which necessitated the advancement of anti-HER2-targeted therapies. As shown in Figure 2, trastuzumab targets Domain IV and serves to inhibit the formation of ligand-independent HER2 homo- and heterodimers. However, trastuzumab is unable to alleviate the readiness of HER2 to form highly active heterodimers with ligand-bound EGFR family members, thereby contributing to diminished clinical response in a subset of patients. Importantly, pertuzumab targets Domain II and prevents its ability to heterodimerize with ligand-bound EGFR family members (Figure 2). Given the promiscuity of HER2 to form homo- and heterodimeric complexes in the absence and presence of ligand, it stands to reason that the co-administration of pertuzumab with trastuzumab would greatly enhance the clinical effectiveness of either agent administered singly. In the succeeding sections, we highlight the two landmark clinical trials that support the aforementioned hypothesis and led to the rapid approval of pertuzumab in the neoadjuvant clinical settings.
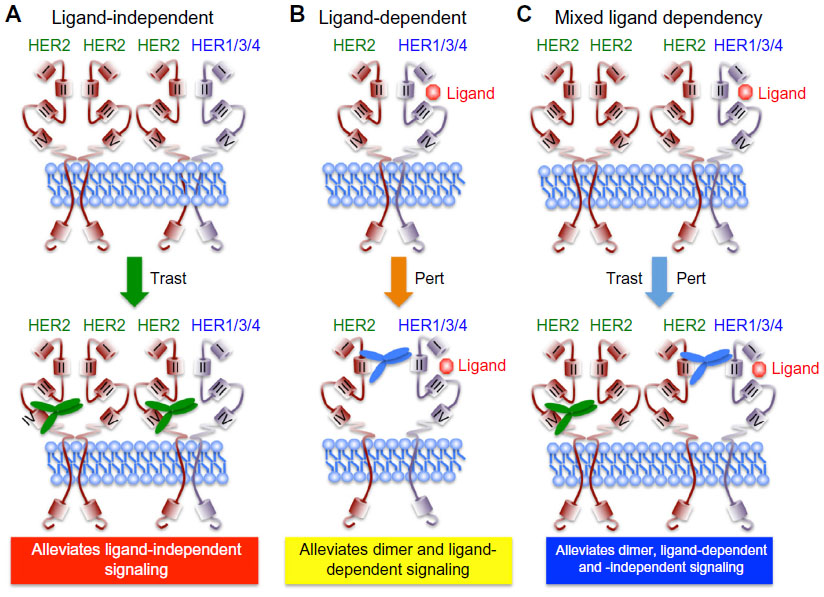 | Figure 2 Schematic of trastuzumab- and pertuzumab-mediated inhibition of HER2 receptor activation. |
NeoSphere trial
The NeoSphere study (Neoadjuvant Study of Pertuzumab and Herceptin in an Early Regimen Evaluation) was a randomized, open-label Phase II clinical trial that enrolled 417 patients. Inclusion in the study required HER2+ breast cancer patients to harbor operable, locally advanced, or inflammatory breast cancer, all of which needed to exceed 2 cm in diameter. Patients were stratified on the basis of their tumor type and its hormone receptor expression status (ie, estrogen and progesterone receptors), and subsequently were randomized in a 1:1:1:1 fashion into one of four 12-week neoadjuvant treatment arms: 1) docetaxel and trastuzumab (DT); 2) docetaxel, trastuzumab, and pertuzumab (DTP); 3) docetaxel and pertuzumab (DP); or 4) trastuzumab and pertuzumab (TP). Upon completion of neoadjuvant treatment, the vast majority of patients underwent surgery, and subsequently received an additional year of adjuvant trastuzumab and anthracycline-based chemotherapy (FEC; 5-fluorouracil, epirubicin, and cyclophosphamide). The primary endpoint was pathologic complete response (pCR), defined as the lack of invasive cancer in the breast at the time of surgery. As shown in Figure 3, the pCR rate achieved was significantly higher for the DTP regimen as compared with the remaining three neoadjuvant arms. Extending the pCR definition to encompass the lack of invasive cancer in all sampled regional lymph nodes, which represents a more conservative definition of pCR resulted in a modest 5%–10% reduction in overall pCR rates across all treatment arms; however, the pCR for the DTP arm (39.3% pCR; 95% confidence interval [CI] 30%–49.2%) remained significantly higher than those observed in the DT (21.5% pCR; 95% CI 14%–30.5%), DP (17.7% pCR; 95% CI 10.7%–26.8%), or TP (11.2% pCR; 95% CI 5.9%–18.8%) arms. Finally, pCR rates were also calculated in relation to hormone receptor expression status (ie, estrogen receptor α and progesterone receptor), which established significantly higher pCR rates for hormone receptor-negative tumors as compared with their hormone receptor-positive counterparts (Figure 4).41
 | Figure 3 Results from the NeoSphere trial comparing pCR rates by experimental treatment arm. |
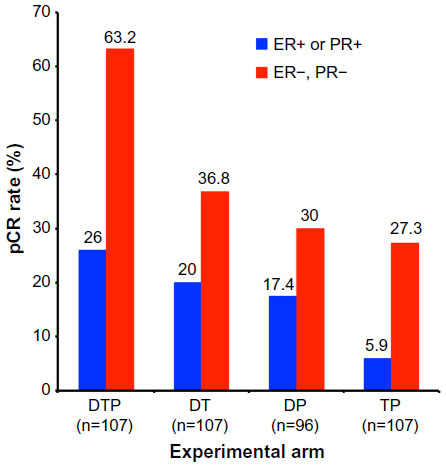 | Figure 4 Results from the NeoSphere trial comparing pCR rates by experimental treatment arm and hormone receptor status. |
Recently, the 5-year follow-up results of the NeoSphere trial were presented at the 2015 Annual Meeting of the American Society of Clinical Oncology (ASCO). Indeed, an increase in pCR positively correlated with improvements in the extent of patient disease-free (DFS) and progression-free (PFS) survival. For instance, the measured 5-year DFS rates were 84% for DTP-treated patients (95% CI 72%–91%), 81% for DT-treated patients (95% CI 72%–88%), 75% for DP-treated patients (95% CI 64%–83%), and 80% for TP-treated patients (95% CI 70%–86%). Similar success was achieved for the 5-year PFS rates, which were 86% for DTP-treated patients (95% CI 77%–91%), 81% for DT-treated patients (95% CI 71%–87%), 73% for DP-treated patients (95% CI 63%–81%), and 73% for TP-treated patients (95% CI 64%–81%).41 It is important to note that while impressive 5-year survival rates were achieved for patients treated with both neutralizing HER2 antibodies (ie, TP arm), the best overall outcomes were always attained by incorporating a chemotherapy regimen with the anti-HER2 agents. Likewise, despite the fact that TP-treated patients were nearly three times as likely to be classified as “nonresponders” as compared with their DTP-treated counterparts (ie, 33% of TP versus 11.9% of DTP), the overall DFS and PFS rates achieved by the TP arm were nevertheless quite remarkable. The contrasting results between these anti-HER2 treatment groups may be explained by the innate heterogeneity of HER2 breast cancers, which span the spectrum of all intrinsic breast cancer subtypes (eg, luminal A, luminal B, HER2+, and basal-like). Because anti-HER2 therapies are most efficacious against HER2-enriched breast cancers, it stands to reason that incorporating PAM50 genetic profiling analyses into future clinical trials may serve as a useful biomarker to predict for response to anti-HER2 therapy, thereby providing a “precision medicine” approach to breast cancer patients.42,43
TRYPHAENA trial
The TRYPHAENA study (Tolerability of Pertuzumab, Herceptin, and Anthracyclines in Neoadjuvant Breast Cancer) was a randomized, open-label Phase II clinical trial that enrolled 225 patients using identical clinical criteria as those employed in the NeoSphere trial. Patients were randomized in a 1:1:1 fashion into one of three 18-week neoadjuvant treatment groups: 1) Arm 1: FEC with concurrent TP, followed by TP (FEC + T + P x3→D + T + P x3); 2) Arm 2: FEC, followed by DPT (FEC x3→D + T + P x3); or 3) Arm 3: DTP with carboplatin (C; DCT plus P x6). Upon completion of neoadjuvant treatment, all patients underwent surgery, and subsequently continued to receive adjuvant trastuzumab treatment to 1 year. Unlike the NeoSphere trial, the primary endpoint of the TRYPHAENA trial aimed to determine the cardiac toxicities associated with co-administering pertuzumab and trastuzumab in the absence or presence of known cardiac toxic agents, anthracyclines. Cardiac tolerability was measured by incidence of left ventricular systolic dysfunction (LVSD), as well as by a decline of >10% in the left ventricular ejection fraction (LVEF). The secondary endpoint was directed at measuring pCR, which again was defined as the absence of histological evidence of invasive cancer in the breast of surgical specimens. Interestingly, the LVSD grades and significant reductions in LVEF measurements were similar across all experimental arms: 1) LVSD grades of 5.6% for arm 1 (95% CI 1.5%–13.6%) versus 4.0% for arm 2 (95% CI 0.8%–11.2%) versus 2.6% for arm 3 (95% CI 0.3%–9.2%) and 2) reduced LVEF measurements of 5.6% for arm 1 (95% CI 1.5%–13.6%) versus 5.3% for arm 2 (95% CI 1.5%–13.1%) versus 3.9% for arm 3 (95% CI 0.8%–11.1%). Surprisingly, pertuzumab was not associated with an increased rate of cardiac dysfunction when used in conjunction with trastuzumab and standard chemotherapy, regardless of the drug or the sequence of administration. Figure 5 shows that the pCR rates were similar across all experimental arms, findings that were mirrored when analyzing pCR rates according to the more conservative definition (ie, no invasive cancer in breast and lymph nodes; data not shown). Likewise, incorporation of anthracyclines also failed to impact the pCR rates achieved by dual administration of TP. Finally, and as expected, pCR rates were significantly higher in hormone receptor-negative tumors as compared with their hormone receptor counterparts (Figure 6).44
 | Figure 5 Results from the TRYPHAENA trial comparing pCR rates by experimental treatment arm. |
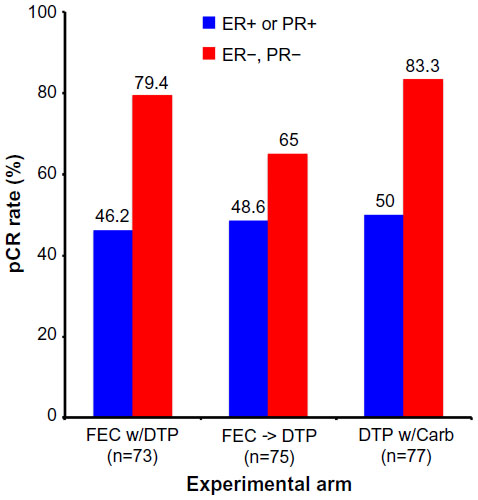 | Figure 6 Results from the TRYPHAENA trial comparing pCR rates by experimental treatment arm and hormone receptor status. |
Lessons learned from NeoSphere and TRYPHAENA
Completion of the NeoSphere and TRYPHAENA neoadjuvant trials was pivotal in demonstrating the enhanced therapeutic response achieved by combining pertuzumab and trastuzumab with chemotherapy as compared with HER2 monotherapy. Moreover, findings from the TRYPHAENA neoadjuvant trial also highlighted the lack of significant cardiac toxicities associated with dual pertuzumab and trastuzumab administration. It is interesting to note that the pCR rates observed in the TRYPHAENA trial were dramatically higher than those reported in the NeoSphere study (after adjusting for definition of pCR), suggesting that longer durations for administering dual HER2 neoadjuvant treatments (18- versus 12-week cycle) may contribute to this improved response. Future studies need to address this hypothesis, as well as to assess the long-term outcomes and effectiveness of incorporating pertuzumab with trastuzumab in adjuvant settings, a question currently being addressed in the ongoing APHINITY clinical trial (NCT01358877).
In light of the favorable outcomes observed in both neoadjuvant trials, a Canadian study was implemented to assess the cost-effectiveness of incorporating dual anti-HER2 therapies for the treatment of breast cancer patients. Cost indices measured included monies necessary to purchase and administer the drugs, as well as those required to manage adverse events, supportive care, and subsequent therapy regimens. In doing so, the authors found that combining pertuzumab with trastuzumab in neoadjuvant settings greatly increased patient life-years, as well as augmented quality-adjusted life-years (QALYs). Indeed, the incremental cost per QALY ranged from $25,388 (CAD; NeoSphere) to $46,196 (CAD; TRYPHAENA), which was deemed to be cost-effective as determined by probabilistic sensitivity analysis.45
Lastly, beyond its obvious therapeutic effectiveness, pertuzumab has also changed the approach to developing and attaining approval for new anticancer agents. Indeed, the overwhelmingly favorable results obtained in the aforementioned trials resulted in the accelerated approval of pertuzumab in 2013 by the US Food and Drug Administration for pCR-related use in the neoadjuvant setting. Importantly, neoadjuvant studies require shorter durations to complete and can provide valuable information regarding the overall efficacy of a drug as evaluated by pCR. Likewise, tissue biopsies obtained for pCR analyses can also be interrogated to identify predictive biomarkers of pertuzumab response and resistance. Thus, neoadjuvant clinical trials represent an attractive setting for studying the effectiveness of novel anticancer agents, and as such, the overwhelming success of pertuzumab has now paved way for several ongoing neoadjuvant clinical trials designed to determine the efficacy of novel therapies in early-stage breast cancers.46
Predictive biomarkers of pertuzumab response and resistance
Science and medicine have accomplished remarkable strides in achieving pCR after neoadjuvant chemotherapy with dual pertuzumab and trastuzumab blockade of HER2 signaling in breast cancers; however, there remains a significant number of patients who fail to exhibit pCRs and eventually manifest disease recurrence. Although the molecular mechanisms responsible for treatment failure remain to be elucidated, significant investigations are currently ongoing to identify novel biomarkers capable of predicting patient response or resistance to targeted HER2 therapies, including those mediated by pertuzumab and trastuzumab. For instance, the TRYPHAENA and NeoSphere neoadjuvant trials, as well as the more recent adjuvant trials have analyzed multiple biomarkers to assess the response of HER2+ breast cancer to anti-HER2 agents, particularly that to pertuzumab. Not surprisingly, the most consistent and robust predictor of pertuzumab response reflects the amplification of the HER2 locus itself within mammary tumors. However, additional biomarkers are also being evaluated, including the presence of tumor infiltrating lymphocytes (TILs). Indeed, histomorphic analyses clearly show an enhanced recruitment of TILs into HER2+ tumors, an event correlated with improved anti-tumor response to trastuzumab. Moreover, TIL levels that exceeded 5% were associated with higher pCR rates, and every additional 1% increase in TIL recruitment was associated with a 3% decrease in the rate of disease recurrence.47 Thus, future studies need to assess the presence of TILs and their value in predicting patient response to pertuzumab-based therapies.
At present, specific biomarkers capable of predicting patient response or resistance to pertuzumab have yet to be identified and validated; however, several candidate markers have recently been linked to poor patient response when treated with pertuzumab. Indeed, the TRYPHAENA trial observed a lower pCR rate in HER2+ breast cancer patients whose tumors housed mutated PIK3CA (49% pCR) as compared with those expressing wild-type PIK3CA (64% pCR); however, this effect did not reach statistical significance (P=0.17).48 As discussed previously, constitutive activation of the PI3K/AKT pathway drives the aberrant growth and survival of HER2+ breast cancers, and as such, future studies need to monitor the mutational status of PIK3CA and other downstream effectors as novel predictors of resistance to pertuzumab-based therapies.48,49
Mechanisms of resistance to anti-HER2 therapy
Compensatory changes in HER2 and co-receptor expression
Resistance to anti-HER2 therapy can transpire at multiple points along the HER2 signaling systems. Indeed, therapeutic resistance to HER2-directed therapies, particularly that to trastuzumab, often reflects dramatic increases50 or decreases51 in HER2 expression, as well as genetic alterations that elicit the 1) synthesis of truncated HER2 isoforms lacking the epitope targeted by trastuzumab52 or 2) generation of constitutively active HER2 molecules resulting either from the formation of highly stable HER2 homodimers (eg, Δ16 splice variants)53,54 or from activating mutations in the PTK domain.55,56 The robust upregulation of the HER2 co-receptors, EGFR and HER3, as well as that of their ligands (eg, TGF-α) has also been linked to the development of resistance in trastuzumab-treated tumors.56,57 Finally, the interaction of HER2 with heterologous receptors and adhesion molecules has also been observed to elicit resistance to anti-HER2 agents. For instance, incorporation of MUC4 into HER2:HER3 complexes occludes trastuzumab from accessing its binding epitope.58,59 Likewise, the production of the MUC1 cleavage product, MUC1* (or MUC1-C) readily interacts with HER2, leading to its constitutive activation and resistance to trastuzumab.60,61 Along these lines, TGF-β-dependent62 and -independent signals can promote the incorporation of α6, β1, and β3 integrins into HER2 complexes, thereby eliciting resistance to anti-HER2 therapies.56 Future studies need to determine the extent to which the aforementioned events directly impact 1) the long-term sensitivity of HER2+ breast cancer cells to pertuzumab and 2) the potential to develop cross-resistance between pertuzumab and trastuzumab in HER2+ breast cancer patients.
Compensatory changes in signaling systems activated by HER2 and other growth factors
The primary outcome of anti-HER2 therapy results in the inactivation of proliferative and survival signals propagated by the coupling of HER2 to the RAS/MAP kinase and PI3K/AKT pathways, respectively. Interestingly, resistance to anti-HER2 therapies can be elicited by the acquisition of activating mutations in key positive regulators of these pathways, including the catalytic domains of PIK3CA, RAS, Src, and NF-κB, and by the acquisition of inactivating mutations in key negative regulators, such as PTEN.56,57,63 Likewise, therapeutic resistance may arise from the stimulation of parallel signaling systems coupled to the activation of the RAS/MAP kinase and PI3K/AKT pathways. Indeed, targeted inactivation of several signaling systems, including the MET, AXL, FGFR, IGF-1R, EphA2, and Notch pathways, have been shown to alleviate resistance to trastuzumab and anti-HER2 therapies.63–67 Interestingly, all of the aforementioned signaling systems can induce epithelial–mesenchymal transition (EMT) programs, which not only enhance disease dissemination and the expansion of cancer stem cells but also promote the acquisition of chemoresistant phenotypes in breast cancer cells.68 Thus, the initiation of EMT phenotypes may also elicit resistance to anti-HER2-targeted therapies, a supposition bolstered by the finding that compensatory Wnt/β-catenin signaling confers resistance to trastuzumab in an EMT-dependent manner.69 Along these lines, reduced expression of the Ser/Thr phosphatase, PPM1H enhances the proteosomal degradation of the cell-cycle inhibitor, p27Kip1 and, consequently, renders HER2+ breast cancer cells insensitive to trastuzumab.70,71 Constitutive expression of the pro-survival factors Bcl-2 and Bcl-XL in response to PRKACA72 or artemin73 also elicit resistance to trastuzumab and contribute to disease progression. Thus, future studies need to assess the relative importance of the aforementioned parallel pathways in eliciting resistance to pertuzumab, as well as to identify novel biomarkers capable of streamlining and prioritizing novel dual inhibitor regimens necessary to restore breast cancer sensitivity to anti-HER2 agents.
Conclusion
The remarkable success of pertuzumab in neoadjuvant trials to significantly impact tumor pCR resulted in its unprecedented accelerated drug approval by the Food and Drug Administration, an event that has forever altered the landscape of drug development and clinical testing in the USA. Indeed, the 2015 National Comprehensive Cancer Network clinical guidelines now recommend the use of pertuzumab in neoadjuvant settings to treat HER2+ tumors that are >2 cm in size, as well as those that have disseminated to lymph nodes. Moreover, including this recommendation into the therapeutic guidelines for pertuzumab has led to its widespread use in neoadjuvant settings. Despite its tremendous clinical success thus far, numerous questions remain regarding the broader therapeutic applications of pertuzumab in treating breast and other carcinoma patients, including how to best identify appropriate patient populations and a repertoire of effective biomarkers capable of predicting patient response and resistance. Ongoing studies are clearly aimed at answering these questions, as well as provide eagerly awaited evidence related to the long-term effectiveness of combining pertuzumab with trastuzumab in adjuvant trial settings.
Acknowledgments
Members of the Schiemann and Vinayak laboratories are thanked for providing helpful comments and suggestions. Research support was provided in part by the Department of Defense to JGP (BC133808), and by the National Institutes of Health to WPS (CA129359, CA177069, and CA194518) and SV (CA076917).
Disclosure
The authors report no conflicts of interest in this work.
References
Tao Z, Shi A, Lu C, Song T, Zhang Z, Zhao J. Breast cancer: epidemiology and etiology. Cell Biochem Biophys. Epub 2014 Dec 28. | |
Dittrich A, Gautrey H, Browell D, Tyson-Capper A. The HER2 signaling network in breast cancer-like a spider in its web. J Mammary Gland Biol Neoplasia. 2014;19(3–4):253–270. | |
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. | |
Slamon DJ, Godolphin W, Jones LA, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science. 1989;244(4905):707–712. | |
Baselga J, Swain SM. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat Rev Cancer. 2009;9(7):463–475. | |
Subbiah IM, Gonzalez-Angulo AM. Advances and future directions in the targeting of HER2-positive breast cancer: implications for the future. Curr Treat Options Oncol. 2014;15(1):41–54. | |
Gianni L, Eiermann W, Semiglazov V, et al. Neoadjuvant chemotherapy with trastuzumab followed by adjuvant trastuzumab versus neoadjuvant chemotherapy alone, in patients with HER2-positive locally advanced breast cancer (the NOAH trial): a randomised controlled superiority trial with a parallel HER2-negative cohort. Lancet. 2010;375(9712):377–384. | |
Piccart-Gebhart MJ, Procter M, Leyland-Jones B, et al. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med. 2005;353(16):1659–1672. | |
Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344(11):783–792. | |
Slamon D, Eiermann W, Robert N, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N Engl J Med. 2011;365(14):1273–1283. | |
Tolaney SM, Barry WT, Dang CT, et al. Adjuvant paclitaxel and trastuzumab for node-negative, HER2-positive breast cancer. N Engl J Med. 2015;372(2):134–141. | |
Cardoso F, Piccart MJ, Durbecq V, Di Leo A. Resistance to trastuzumab: a necessary evil or a temporary challenge? Clin Breast Cancer. 2002; 3(4):247–257. | |
Kumler I, Tuxen MK, Nielsen DL. A systematic review of dual targeting in HER2-positive breast cancer. Cancer Treat Rev. 2014; 40(2):259–270. | |
Badache A, Hynes NE. A new therapeutic antibody masks ErbB2 to its partners. Cancer Cell. 2004;5(4):299–301. | |
Eroglu Z, Tagawa T, Somlo G. Human epidermal growth factor receptor family-targeted therapies in the treatment of HER2-overexpressing breast cancer. Oncologist. 2014;19(2):135–150. | |
Roskoski R, Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. | |
Ross JS, Fletcher JA. The HER-2/neu oncogene in breast cancer: prognostic factor, predictive factor, and target for therapy. Oncologist. 1998;3(4):237–252. | |
Zuo T, Wang L, Morrison C, et al. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell. 2007;129(7):1275–1286. | |
Myers E, Hill AD, Kelly G, McDermott EW, O’Higgins NJ, Young LS. A positive role for PEA3 in HER2-mediated breast tumour progression. Br J Cancer. 2006;95(10):1404–1409. | |
Powe DG, Akhtar G, Habashy HO, et al. Investigating AP-2 and YY1 protein expression as a cause of high HER2 gene transcription in breast cancers with discordant HER2 gene amplification. Breast Cancer Res. 2009;11(6):R90. | |
Zhang T, Zhang H, Wang Y, McGown LB. Capture and identification of proteins that bind to a GGA-rich sequence from the ERBB2 gene promoter region. Anal Bioanal Chem. 2012;404(6–7):1867–1876. | |
Fry WH, Kotelawala L, Sweeney C, Carraway KL 3rd. Mechanisms of ErbB receptor negative regulation and relevance in cancer. Exp Cell Res. 2009;315(4):697–706. | |
Sweeney C, Carraway KL 3rd. Negative regulation of ErbB family receptor tyrosine kinases. Br J Cancer. 2004;90(2):289–293. | |
Sweeney C, Miller JK, Shattuck DL, Carraway KL 3rd. ErbB receptor negative regulatory mechanisms: implications in cancer. J Mammary Gland Biol Neoplasia. 2006;11(1):89–99. | |
Yen L, Cao Z, Wu X, et al. Loss of Nrdp1 enhances ErbB2/ErbB3-dependent breast tumor cell growth. Cancer Res. 2006;66(23):11279–11286. | |
Olayioye MA. Update on HER-2 as a target for cancer therapy: intracellular signaling pathways of ErbB2/HER-2 and family members. Breast Cancer Res. 2001;3(6):385–389. | |
Zhang X, Gureasko J, Shen K, Cole PA, Kuriyan J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell. 2006;125(6):1137–1149. | |
Badache A, Goncalves A. The ErbB2 signaling network as a target for breast cancer therapy. J Mammary Gland Biol Neoplasia. 2006; 11(1):13–25. | |
Schulze WX, Deng L, Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol. 2005;1:2005.0008. | |
Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22. | |
Vigil D, Cherfils J, Rossman KL, Der CJ. Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat Rev Cancer. 2010;10(12):842–857. | |
Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014; 13(12):928–942. | |
Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13(11):828–851. | |
Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer. 2015;15(1):7–24. | |
Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov. 2014;13(2):140–156. | |
Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009; 8(8):627–644. | |
Hollander MC, Blumenthal GM, Dennis PA. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat Rev Cancer. 2011;11(4):289–301. | |
Song MS, Salmena L, Pandolfi PP. The functions and regulation of the PTEN tumour suppressor. Nat Rev Mol Cell Biol. 2012; 13(5):283–296. | |
Agoulnik IU, Hodgson MC, Bowden WA, Ittmann MM. INPP4B: the new kid on the PI3K block. Oncotarget. 2011;2(4):321–328. | |
Metzger-Filho O, Winer EP, Krop I. Pertuzumab: optimizing HER2 blockade. Clin Cancer Res. 2013;19(20):5552–5556. | |
Gianni L, Pienkowski T, Im YH, et al. Efficacy and safety of neoadjuvant pertuzumab and trastuzumab in women with locally advanced, inflammatory, or early HER2-positive breast cancer (NeoSphere): a randomised multicentre, open-label, phase 2 trial. Lancet Oncol. 2012; 13(1):25–32. | |
Parker JS, Mullins M, Cheang MC, et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol. 2009; 27(8):1160–1167. | |
Prat A, Bianchini G, Thomas M, et al. Research-based PAM50 subtype predictor identifies higher responses and improved survival outcomes in HER2-positive breast cancer in the NOAH study. Clin Cancer Res. 2014;20(2):511–521. | |
Schneeweiss A, Chia S, Hickish T, et al. Pertuzumab plus trastuzumab in combination with standard neoadjuvant anthracycline-containing and anthracycline-free chemotherapy regimens in patients with HER2-positive early breast cancer: a randomized phase II cardiac safety study (TRYPHAENA). Ann Oncol. 2013;24(9):2278–2284. | |
Attard CL, Pepper AN, Brown ST, et al. Cost-effectiveness analysis of neoadjuvant pertuzumab and trastuzumab therapy for locally advanced, inflammatory, or early HER2-positive breast cancer in Canada. J Med Econ. 2015;18(3):173–188. | |
Bardia A, Baselga J. Neoadjuvant therapy as a platform for drug development and approval in breast cancer. Clin Cancer Res. 2013; 19(23):6360–6370. | |
Salgado R, Denkert C, Campbell C, et al. Tumor-Infiltrating lymphocytes and sssociations with pathological complete response and event-free survival in HER2-positive early-stage breast cancer treated with lapatinib and trastuzumab: a secondary analysis of the NeoALTTO Trial. JAMA Oncol. 2015;1(4):448–454. | |
Schneeweiss A, Chia S, Hegg R, et al. Evaluating the predictive value of biomarkers for efficacy outcomes in response to pertuzumab- and trastuzumab-based therapy: an exploratory analysis of the TRYPHAENA study. Breast Cancer Res. 2014;16(4):R73. | |
Majewski IJ, Nuciforo P, Mittempergher L, et al. PIK3CA mutations are associated with decreased benefit to neoadjuvant human epidermal growth factor receptor 2-targeted therapies in breast cancer. J Clin Oncol. 2015;33(12):1334–1339. | |
Vazquez-Martin A, Colomer R, Brunet J, Menendez JA. Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int J Oncol. 2007;31(4):769–776. | |
Koninki K, Barok M, Tanner M, et al. Multiple molecular mechanisms underlying trastuzumab and lapatinib resistance in JIMT-1 breast cancer cells. Cancer Lett. 2010;294(2):211–219. | |
Arribas J, Baselga J, Pedersen K, Parra-Palau JL. p95HER2 and breast cancer. Cancer Res. 2011;71(5):1515–1519. | |
Castiglioni F, Tagliabue E, Campiglio M, Pupa SM, Balsari A, Menard S. Role of exon-16-deleted HER2 in breast carcinomas. Endocr Relat Cancer. 2006;13(1):221–232. | |
Mitra D, Brumlik MJ, Okamgba SU, et al. An oncogenic isoform of HER2 associated with locally disseminated breast cancer and trastuzumab resistance. Mol Cancer Ther. 2009;8(8):2152–2162. | |
Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3(2):224–237. | |
Rimawi MF, Schiff R, Osborne CK. Targeting HER2 for the treatment of breast cancer. Annu Rev Med. 2015;66:111–128. | |
Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2012;9(1):16–32. | |
Funes M, Miller JK, Lai C, Carraway KL 3rd, Sweeney C. The mucin Muc4 potentiates neuregulin signaling by increasing the cell-surface populations of ErbB2 and ErbB3. J Biol Chem. 2006; 281(28):19310–19319. | |
Price-Schiavi SA, Jepson S, Li P, et al. Rat Muc4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int J Cancer. 2002; 99(6):783–791. | |
Raina D, Uchida Y, Kharbanda A, et al. Targeting the MUC1-C oncoprotein downregulates HER2 activation and abrogates trastuzumab resistance in breast cancer cells. Oncogene. 2014;33(26):3422–3431. | |
Fessler SP, Wotkowicz MT, Mahanta SK, Bamdad C. MUC1* is a determinant of trastuzumab (Herceptin) resistance in breast cancer cells. Breast Cancer Res Treat. 2009;118(1):113–124. | |
Wang SE, Xiang B, Zent R, Quaranta V, Pozzi A, Arteaga CL. Transforming growth factor beta induces clustering of HER2 and integrins by activating Src-focal adhesion kinase and receptor association to the cytoskeleton. Cancer Res. 2009;69(2):475–482. | |
Yamaguchi H, Chang SS, Hsu JL, Hung MC. Signaling cross-talk in the resistance to HER family receptor targeted therapy. Oncogene. 2014;33(9):1073–1081. | |
Baker AT, Zlobin A, Osipo C. Notch-EGFR/HER2 bidirectional crosstalk in breast cancer. Front Oncol. 2014;4:360. | |
Haluska P, Carboni JM, TenEyck C, et al. HER receptor signaling confers resistance to the insulin-like growth factor-I receptor inhibitor, BMS-536924. Mol Cancer Ther. 2008;7(9):2589–2598. | |
Nahta R, Yu D, Hung MC, Hortobagyi GN, Esteva FJ. Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat Clin Pract Oncol. 2006;3(5):269–280. | |
Nahta R, Yuan LX, Zhang B, Kobayashi R, Esteva FJ. Insulin-like growth factor-I receptor/human epidermal growth factor receptor 2 heterodimerization contributes to trastuzumab resistance of breast cancer cells. Cancer Res. 2005;65(23):11118–11128. | |
Taylor MA, Parvani JG, Schiemann WP. The pathophysiology of epithelial-mesenchymal transition induced by transforming growth factor-beta in normal and malignant mammary epithelial cells. J Mammary Gland Biol Neoplasia. 2010;15(2):169–190. | |
Wu Y, Ginther C, Kim J, et al. Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes EMT-like phenotype in trastuzumab-resistant HER2-overexpressing breast cancer cells. Mol Cancer Res. 2012;10(12):1597–1606. | |
Elster N, Collins DM, Toomey S, Crown J, Eustace AJ, Hennessy BT. HER2-family signalling mechanisms, clinical implications and targeting in breast cancer. Breast Cancer Res Treat. 2015;149(1):5–15. | |
Lee-Hoeflich ST, Pham TQ, Dowbenko D, et al. PPM1H is a p27 phosphatase implicated in trastuzumab resistance. Cancer Discov. 2011;1(4):326–337. | |
Moody SE, Schinzel AC, Singh S, et al. PRKACA mediates resistance to HER2-targeted therapy in breast cancer cells and restores anti-apoptotic signaling. Oncogene. 2015;34(16):2061–2071. | |
Ding K, Banerjee A, Tan S, et al. Artemin, a member of the glial cell line-derived neurotrophic factor family of ligands, is HER2-regulated and mediates acquired trastuzumab resistance by promoting cancer stem cell-like behavior in mammary carcinoma cells. J Biol Chem. 2014;289(23):16057–16071. |
 © 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2016 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.