Back to Journals » Journal of Inflammation Research » Volume 15
Necroptosis-Mediated eCIRP Release in Sepsis
Authors Reilly B, Tan C, Murao A, Nofi C, Jha A, Aziz M, Wang P
Received 13 April 2022
Accepted for publication 7 July 2022
Published 17 July 2022 Volume 2022:15 Pages 4047—4059
DOI https://doi.org/10.2147/JIR.S370615
Checked for plagiarism Yes
Review by Single anonymous peer review
Peer reviewer comments 2
Editor who approved publication: Professor Ning Quan
Bridgette Reilly,1 Chuyi Tan,1 Atsushi Murao,1 Colleen Nofi,1,2 Alok Jha,1 Monowar Aziz,1– 3 Ping Wang1– 3
1Center for Immunology and Inflammation, The Feinstein Institutes for Medical Research, Manhasset, NY, USA; 2Department of Surgery, Zucker School of Medicine at Hofstra/Northwell, Manhasset, NY, USA; 3Department of Molecular Medicine, Zucker School of Medicine at Hofstra/Northwell, Manhasset, NY, USA
Correspondence: Ping Wang; Monowar Aziz, The Feinstein Institutes for Medical Research, 350 Community Dr., Manhasset, NY, 11030, USA, Tel +1 516 562-3411; +1 516 562-2436, Fax +1 516 562-2396, Email [email protected]; [email protected]
Introduction: Extracellular cold-inducible RNA-binding protein (eCIRP) is an endogenous pro-inflammatory mediator that exacerbates injury in inflammation and sepsis. The mechanisms in which eCIRP is released have yet to be fully explored. Necroptosis is a programmed cell death that is dependent on the activation of mixed lineage kinase domain-like pseudo kinase (MLKL) which causes the release of damage-associated molecular patterns. We hypothesize that eCIRP is released through necroptosis and intensifies inflammation in sepsis.
Methods: RAW264.7 cells were treated with pan-caspase inhibitor z-VAD (15 μM) 1 h before stimulation with LPS (1 μg/mL). Necroptosis inhibitor, Necrostatin-1 (Nec-1) (10 μM) was added to the cells with LPS simultaneously. After 24 h of LPS stimulation, cytotoxicity was determined by LDH assay. eCIRP levels in the culture supernatants and phospho-MLKL (p-MLKL) from cell lysates were assessed by Western blot. p-MLKL interaction with the cell membrane was visualized by immunofluorescence. Sepsis was induced in C57BL/6 mice by cecal ligation and puncture (CLP). Mice were treated with Nec-1 (1 mg/kg) or DMSO. 20 h post-surgery, serum and peritoneal fluid levels of eCIRP, TNF-α and IL-6 were determined by ELISA. H&E staining of lung tissue sections was performed.
Results: We found that in RAW264.7 cells, LPS+z-VAD induces necroptosis as evidenced by an increase in p-MLKL levels and causes eCIRP release. Nec-1 reduces both p-MLKL activation and eCIRP release in LPS+z-VAD-treated RAW264.7 cells. Nec-1 also inhibits the release of eCIRP, TNF-α and IL-6 in the serum and peritoneal fluid in CLP-induced septic mice. We predicted a transient interaction between eCIRP and MLKL using a computational model, suggesting that eCIRP may exit the cell via the pores formed by p-MLKL.
Conclusion: Necroptosis is a novel mechanism of eCIRP release in sepsis. Targeting necroptosis may ameliorate inflammation and injury in sepsis by inhibiting eCIRP release.
Keywords: eCIRP, necroptosis, Necrostatin-1, macrophage, sepsis
Corrigendum for this paper has been published.
Introduction
During sepsis, systemic inflammation can be initiated by pathogen-associated molecular patterns (PAMPs) and damaged-associated molecular patterns (DAMPs).1 These molecules are recognized by pattern-recognition receptors (PPRs) expressed in both immune and non-immune cells.1,2 The interaction of PAMPs and DAMPs with PRRs activates several downstream pathways which promote the release of pro-inflammatory mediators such as cytokines and chemokines. Several DAMPs, including extracellular cold-inducible RNA-binding protein (eCIRP), have been classified as alarmin danger signals which trigger inflammatory responses in sepsis.3
Cold-inducible RNA-binding protein (CIRP) is a glycine-rich RNA chaperone which, under normal conditions, is located in the nucleus and functions to facilitate RNA translation.4 Under stressed conditions such as hypothermia and hypoxia, CIRP can migrate from the nucleus to cytoplasm and is then released to the extracellular space.5 Once released extracellularly, eCIRP serves as an endogenous pro-inflammatory mediator and is implicated in exacerbated injury in inflammatory conditions, such as sepsis.3 Pharmacological inhibition and genetic knockdown of CIRP have been reported to ameliorate organ injury in sepsis3 as well as other inflammatory conditions.6–8 eCIRP has been shown to induce inflammatory responses in macrophages,9 neutrophils,10 lymphocytes,11 and endothelial cells.5 However, the mechanisms by which eCIRP is released from cells have yet to be fully elucidated.
Several studies implicate cell death as an active event to trigger or amplify the inflammatory response.12 Previously, it was assumed that necrosis was distinct from apoptosis, in part owing to the lack of molecular programming in the process of necrosis. However, in recent years it has become clear that necrotic cell death can be driven by distinct molecular pathways.13 Necroptosis is a form of programmed necrosis and refers to cell death dependent on receptor-interacting protein kinases 1 and 3 (RIPK1 and RIPK3).14 Necroptosis is induced by the activation of toll-like receptors (TLRs) or tumor necrosis factor receptor 1 (TNFR1). During this process, RIPK1 and RIPK3 are activated and interact to form the necrosome.15 The necrosome phosphorylates mixed lineage kinase domain like pseudokinase (MLKL).16–18 p-MLKL is then oligomerized and inserted into membrane to form a pore. The MLKL pore recruits ion channels and contributes to the rupture of the cell membrane. Necroptosis directly causes inflammation through massive release of DAMPs from the deteriorating cell.19 Some specific DAMPs, such as high-mobility group box 1 (HMGB1), have been identified as essential mediators of necroptosis-induced inflammation in vivo.20 It is also well established that RIPK and MLKL deficiencies prevent cell death and inflammation in vivo.21,22
Because of the association of necroptosis with the release of HMGB1, we hypothesized that necroptosis plays a critical role in the release of eCIRP in inflammation and sepsis. Herein, we have demonstrated that eCIRP is released from macrophages after stimulation by necroptosis inducers lipopolysaccharide (LPS) and z-VAD, and that this release can be inhibited by the necroptosis inhibitor, Necrostatin-1 (Nec-1). Additionally, we have shown that Nec-1 prevents the release of eCIRP and pro-inflammatory cytokines in septic mice. Collectively, our data suggest that necroptosis serves as a route for the release of eCIRP from macrophages in sepsis.
Materials and Methods
Mice
C57BL/6 male mice were obtained from Charles River Laboratories. Healthy mice aged 8–12 weeks were used in all experiments. Mice were given 1 week to acclimate after shipping. Animals were housed at 22oC with access to food and water throughout experimentation. The mice were kept on a 12-h light/dark cycle. All animal experiments were performed by the guidelines for the use of experimental animals by the National Institutes of Health (Bethesda, MD). All the experiments involving mice were approved by the Institutional Animal Care and Use Committees (IACUC) of the Feinstein Institutes for Medical Research.
Cecal Ligation and Puncture (CLP)-Induced Sepsis
Sepsis was induced in C57BL/6 mice by CLP as previously described.23 Mice were anesthetized with 2% isoflurane. A midline incision was made, and the abdomen was opened. The cecum was located and ligated with a doubled 4–0 silk suture. The ligated portion of the cecum was punctured twice with a 22-gauge needle and a small amount of cecal matter was expelled. The cecum was returned to the abdomen and the abdomen was closed in layers. Resuscitative fluid (normal saline, 50 mL/kg) and an analgesic (buprenorphine, 0.05–0.1 mg/kg) was administrated by subcutaneous injection. The sham groups of mice were subjected to the same operation but without ligation and puncture of the cecum.
Treatment of Mice with Necroptosis Inhibitor, Necrostatin-1
Mice were treated with Nec-1 (1 mg/kg) dissolved in DMSO and diluted in sterile PBS immediately after surgery or with sterile 0.5% DMSO in PBS. Treatment was administered intraperitoneally (i.p.). 20 hours post-operatively, the mice were sacrificed, and organs and serum were collected for analyses.
Histology
Animals were perfused with saline prior to the collection of lungs. Samples were stored in 10% formalin prior to use. The tissue was sliced into 5 μm sections and then stained with hematoxylin and eosin. Samples were examined using light microscopy and scored using a scoring system for acute lung injury in experimental animals as outlined by the American Thoracic Society.24 Each field received a score from zero to one with one being the worst. Scoring takes into account neutrophil infiltration in the interstitial and alveolar spaces, the presence or lack thereof hyaline membranes, the amount of proteinaceous debris in the airspaces, and the severity of alveolar septal thickening. Each data point represents the average score of the fields within a section.
Cell Culture
RAW264.7 (RAW) cells were obtained from American Type Culture Collection (ATCC). The cells were cultured in DMEM medium supplemented with 10% heat-inactivated FBS, 1% penicillin-streptomycin, and 2 mM glutamine. Cells were incubated at 37oC and supplemented with 5% CO2. For experiments, cells were seeded in 12 well plates, and the appropriate wells were pretreated for 1 hour with 15 μM of the pan-caspase inhibitor, z-VAD. Cells were then given 1 μg/mL of LPS and/or 10 μM of the necroptosis inhibitor, Nec-1 for 24 hours. After stimulation, the cells were centrifuged at 400 x g for 10 minutes. The cell supernatants were decanted and lysis buffer containing protease and phosphatase inhibitors was added to the wells. Collected supernatants and cell lysates were stored at −20oC.
Western Blotting
To prepare the cell supernatants for Western blotting, 250 μL of a 1.43 g/mL solution of trichloroacetic acid (TCA, Cat. No. BP555-250, Fisher Scientific) was added to each 1 mL cell supernatant sample. The samples were then incubated overnight at 4oC and then centrifuged at 13,000 x g for 30 minutes at 4oC. The supernatant was discarded, and the pellet washed with acetone twice. The protein precipitate was then dissolved in 1× tris buffer saline and 4× SDS sample buffer. Proteins were denatured at 95oC and for 5 minutes prior to loading. Cell lysates were collected as previously described. The protein concentrations were determined using the ABC protein assay kit (Cat. No. 5000002, Bio-Rad). Proteins were separated using NuPAGE 4–12% Bis-Tris gels (Invitrogen) and transferred to nitrocellulose membranes (Invitrogen). The membranes were incubated in the antibodies. Antibodies used in this paper are as follows: β-actin (Cat. No. A5441, 1:10,000, Sigma), CIRP (Cat. No. 10209-2-AP; 1:1000, Proteintech), p-MLKL (Cat. No. 37333S, 1:1000, Cell Signaling), and MLKL (Cat. No. 37705S, 1:1000, Cell Signaling). For supernatants, the amount of bovine serum albumin (BSA) present in each sample was assessed using a Ponceau S (PS) solution. Membranes soaked in PS for 5–10 minutes or until the band for BSA became visible. The membranes were then washed twice with deionized water and allowed to dry. The amount of BSA served as a control for the cell supernatant Western blots to confirm similar loading per sample.
LDH Assay
RAW cells were seeded in 96 well plates and treated as previously described. Spontaneous LDH activity groups and maximum LDH activity groups were added alongside the previously described groups. These two groups either had nothing added or lysis buffer added 45 minutes prior to collection, respectively. CyQUANT LDH Cytotoxicity Assay Kit (Cat. No. C20300) was used to determine the LDH activity of the supernatants. The assay was performed and the % Cytotoxicity was calculated according to the instructions provided in the product information sheet.
Immunofluorescence
RAW cells were cultured on glass bottom petri dishes and treated with LPS and z-VAD-fmk. The nuclei were stained by using NucBlueTM Live cell stain ReadyProbes reagent (Hoechst 33342, Cat. No. 2325910, Invitrogen,) and then rinsed twice with cold PBS. The cell surface membrane was then stained with MemBrite fix Cell Surface staining kit (Biotium, Cat. No. 30093) according to the company’s instructions. Cells were washed with PBS and fixed using 4% methanol for 15 minutes at room temperature. The solution was removed, and the cells were washed three times with PBS and blocked with 5% BSA in PBS for 1 hour at room temperature. After blocking, cells were incubated in rabbit anti-p-MLKL primary antibody (dilution 1:200; Cat. No. 101375, Cell Signaling Technology) overnight at 4oC. Cells were washed three times and incubated in Alexa Fluor 594 Donkey anti-rabbit IgG (Cat. No. A21207, Thermo Fisher Scientific) for 2 hours. After washing three times, the samples were sealed using prolong gold anti-fade (Cat. No. P36934, Thermo Fisher Scientific). Cells were visualized by using Axio Observer. Z1/7 equipped with Zeiss LSM900 confocal microscopy system. Z-stack images were acquired with Plan-Apochromat 63x/1.40 Oil DIC M27 objective lens. SR-4Y fast acquisition mode of Airyscan and 4× averaging was used. The images obtained by confocal microscope were merged and combined by FIJI Image J.
ELISA
TNF-α, IL-6 and eCIRP levels were quantitatively measured from mouse serum by ELISA kits according to the manufacturer’s protocols. TNF-α (Cat. No. 558534) and IL-6 (Cat. No. 555240) ELISA kits were purchased from BD Biosciences. eCIRP ELISA kits (Cat. No. E12931m) were bought from WUHAN EIAAB science co., LTD.
Homology Modeling of CIRP and MLKL
The structure models of CIRP (P60824) and MLKL (Q9D2Y4) were generated by retrieving sequences from UniProt and using Iterative Threading ASSEmbly Refinement (I-TASSER) server25 based on templates identified by threading approach to maximize the sequence coverage, percentage identity, and confidence. The CIRP structure has three different domains including RNA binding domain (aa 6–84), disordered region (aa 70–172) and polar residues (aa 143–172). The MLKL structure has four different domains including a protein kinase domain (aa 192–456), N-terminal bundle and brace, NBB (aa 1–143), coiled coil 1 (aa 61–81) and coiled coil 2 (aa 138–229). The N-terminal 4-helix bundle (HB) domain of MLKL is known as the necroptotic effector domain and is responsible for oligomerization and translocation to the membrane.26,27 The models were refined by repeated relaxations by short molecular dynamics (MD) simulations for 0.6 ns (mild) and 0.8 ns (aggressive) relaxations with 4 fs time step after structure perturbations. This model refinement step enhanced certain parameters including Rama favored residues and decrease in poor rotamers. The structures of MLKL and CIRP were docked using ATTRACT tool,28 which uses an approach of conformational flexibility of binding partners. In the docking process, potential energies on a grid are pre-calculated and then interpolations from the nearest grid points are used to calculate interactions. Moreover, several energy minimizations steps or Monte Carlo simulations are involved in the docking process. The interactions between the CIRP-MLKL complex were analyzed using PDBePISA tool.29 The surface area of interaction interface and other thermodynamic parameters were calculated. The complex structure was visualized using PyMOL and Chimera.30
Statistical Analysis
All statistical analyses were performed with GraphPad Prism version 7.0 software (GraphPad Software, La Jolla, CA). The Shapiro–Wilk test assessed the data for normality. Comparisons between two groups were performed with a two-tailed Student’s t-test. Comparisons between multiple groups were analyzed using a one-way or two-way analysis of variance and Tukey’s multiple comparisons test. The statistical significance was set at p value <0.05.
Results
Necroptosis Induces eCIRP Release in Macrophages
To investigate the role of necroptosis in eCIRP release, we induced necroptosis by stimulating RAW264.7 cells with LPS and z-VAD, a pan-caspase inhibitor. Our results show that LPS+z-VAD significantly increased the levels of eCIRP in the culture supernatants of RAW cells compared to the control group (Figure 1A and B, Supplementary Figure 1). LPS is already known to cause eCIRP release3 and LPS alone increased eCIRP release by 7.73-fold. To confirm that the dosage of LPS and z-VAD was sufficient to induce necroptosis, we checked for the intracellular expression of p-MLKL. The intracellular p-MLKL was markedly increased in the LPS+z-VAD group (Figure 1C and D, Supplementary Figure 1). In addition, cell death maker LDH assays demonstrated a significant increase in the percentage of cell death in the LPS+z-VAD groups compared to the control groups (Figure 1E), suggesting eCIRP release by necroptosis was, at least in part, due to the membrane rupture. Taken together, these data indicate that LPS+z-VAD induces cell necroptosis as well as eCIRP release in RAW264.7 cells.
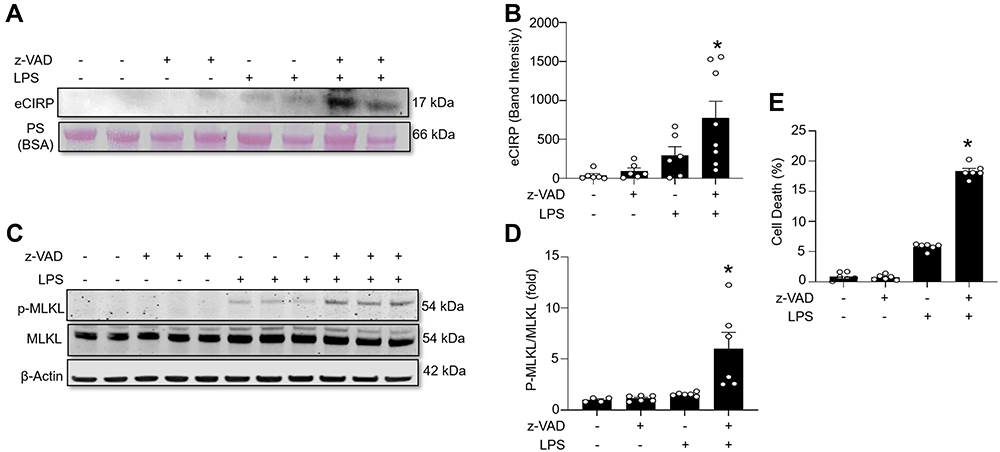 |
Figure 1 Necroptosis induces eCIRP release from macrophages. RAW264.7 cells were pre-treated with or without z-VAD (15 μM) for 30 min and then stimulated with LPS (1 µg/mL) for 24 h. Cell culture supernatants and cell lysates were collected. (A and B) eCIRP present in the culture supernatants was quantified by Western blotting. (C and D) p-MLKL, MLKL, and β-actin were quantified by Western blotting. Representative Western blot images and quantitative bar diagrams are shown. (E) The release of LDH in the cell supernatants was measured by CyQUANTTM LDH cytotoxicity assay kit. Data are expressed as means ± SEM. Groups were compared by one-way ANOVA and Tukey’s multiple comparisons test. n=4–8 samples/group as shown in circles. *p<0.05 vs (-) LPS, (-) z-VAD. PS, Ponceau S red staining. Full blot images are shown in Supplementary Figure 1. |
Necrostatin-1 Inhibits Necroptosis and eCIRP Release in Macrophages
To further confirm the effect of necroptosis on eCIRP release from macrophages, we treated cells with the necroptosis inhibitorNec-1, and measured the amount of eCIRP in the supernatants. We found that LPS+z-VAD significantly increased the levels of eCIRP in the culture supernatants of RAW264.7 cells compared to the control group, while Nec-1 treatment decreased the eCIRP release in LPS+z-VAD treated cells (Figure 2A and B, Supplementary Figure 2). We also observed that Nec-1 treatment inhibited the expression of p-MLKL in LPS+z-VAD treated cells (Figure 2C and D, Supplementary Figure 2). Additionally, Nec-1 treatment suppressed cell death in LPS+z-VAD treated cells (Figure 2E). These data demonstrate the ability of Nec-1 to inhibit eCIRP release from macrophages by suppressing necroptosis.
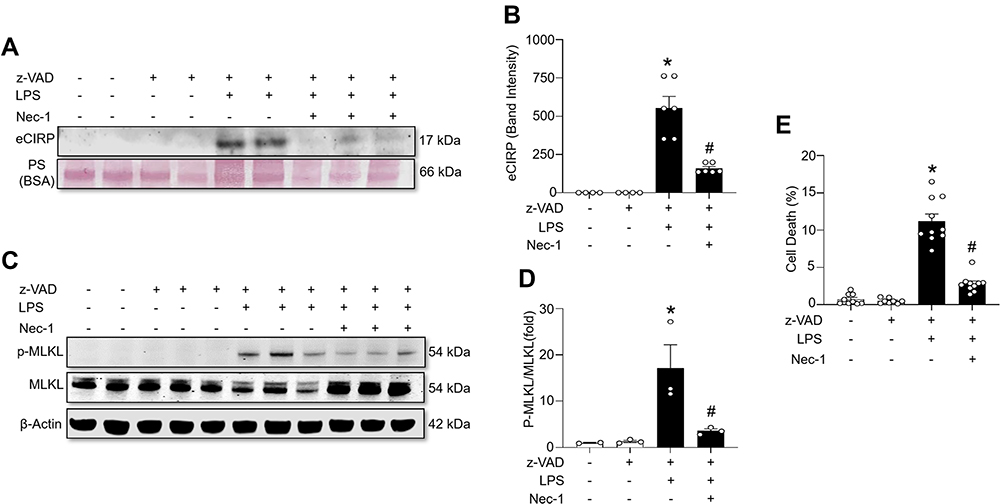 |
Figure 2 Necroptosis inhibitor necrostatin-1 inhibits eCIRP release from macrophages. RAW264.7 cells were pre-treated with z-VAD (15 μM) for 30 min and then stimulated with LPS (1 µg/mL) and Necrostatin-1 (Nec-1, 10 μM) for 24 hours, and cell culture supernatants and cell lysates were collected. (A and B) eCIRP present in the culture supernatants was quantified by Western blotting. (C and D) p-MLKL, and MLKL were quantified by Western blotting with β-actin as a loading control. Representative Western blot images and quantitative bar diagrams are shown. (E) The release of LDH in the cell supernatants was measured by CyQUANTTM LDH cytotoxicity assay kit. Data are expressed as means ± SEM. Groups were compared by one-way ANOVA and Tukey’s multiple comparisons test. n=3–10 samples/group. *p<0.05 vs (-) LPS, (-) z-VAD, (-) Nec-1; #p<0.05 vs (+) LPS, (+) z-VAD, (-) Nec-1. PS, Ponceau S red staining. Full blot images are shown in Supplementary Figure 2. |
Necroptosis Inhibitor Necrostatin-1 Inhibits the Release of eCIRP and Pro-Inflammatory Cytokines in a Mouse Model of Sepsis
To investigate the effect of necroptosis inhibitor Nec-1 on eCIRP release and inflammation in sepsis, sepsis was induced by CLP in C57BL/6 mice, and levels of eCIRP were measured in serum and peritoneal fluid. We found that treatment with Nec-1 significantly decreases the serum level of eCIRP compared with vehicle treated septic mice (Figure 3A). In a previous study, we found a correlation between serum levels of eCIRP and pro-inflammatory cytokines following CLP-induced sepsis.3 To confirm this correlation, serum and peritoneal fluid levels of IL-6 and TNF-α levels were assessed. We found that the serum TNF-α and IL-6 levels were increased in CLP induced septic mice, while Nec-1 decreased those levels compared to those in the vehicle treated septic mice (Figure 3B and C). We found that Nec-1 treatment reduced the level of eCIRP in peritoneal fluid compared to vehicle treated septic mice (Figure 3D). We demonstrate a similar trend of TNF-α and IL-6 in the peritoneal fluid (Figure 3E and F). We also found that Nec-1 significantly decreased the lung injury scores of septic mice (Figure 4A and B). These data suggest that pharmacologically blocking necroptosis significantly decreases eCIRP in CLP models of sepsis and attenuates known downstream consequences of CIRP.
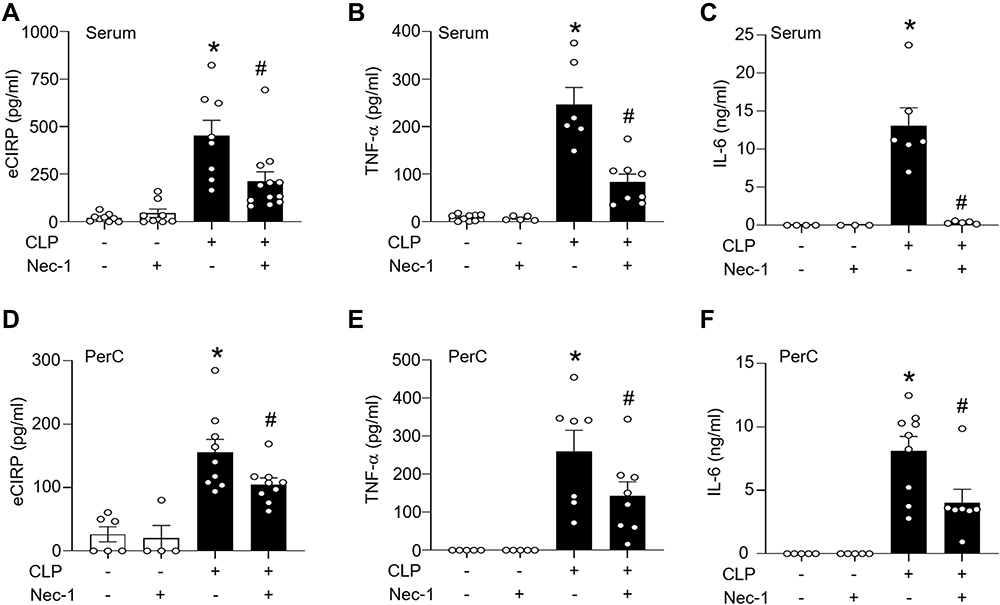 |
Figure 3 Necroptosis inhibitor necrostatin-1 inhibits the release of eCIRP and pro-inflammatory cytokines in sepsis. Sepsis was induced in C57BL/6 mice by CLP. Sham-operated animals served as control mice. C57BL/6 mice were injected i.p. with either necrostatin-1 (1 mg/kg) or vehicle in equivalent volumes immediately following CLP. At 20 h of CLP or sham operation, blood and peritoneal fluid were harvested from mice. (A) Serum levels of eCIRP were assessed by ELISA. (B and C) Serum levels of TNF-α, and Il-6 were assessed by ELISA. (D-F) Peritoneal fluid levels of eCIRP, TNF-α, and IL-6 were assessed by ELISA. n=4–9 mice/group (each circle represents one sample). *p<0.05 vs (-) CLP, (-) Nec-1; #p<0.05 vs (+) CLP, (-) Nec-1. Data are expressed as means ± SEM. Groups were compared by one-way ANOVA and Tukey’s multiple comparisons test. |
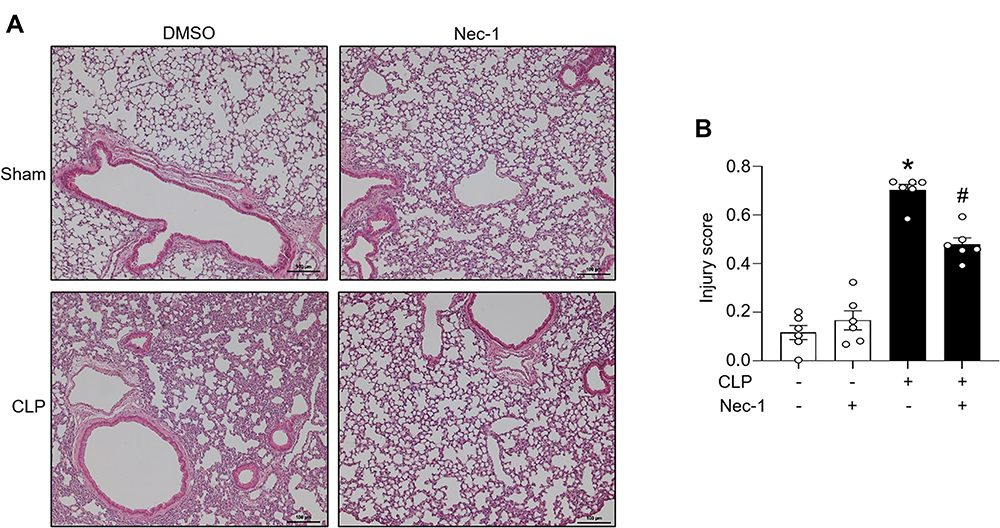 |
Figure 4 Necroptosis attenuates lung injury in CLP-induced sepsis. Sepsis was induced via CLP in mice. Sham operated mice functioned as a control. C57BL/6 mice were injected i.p. with either necrostatin-1 (1 mg/kg) or vehicle in equivalent volumes immediately following CLP. 20 h post sham or CLP operation, lungs were collected. (A) The lungs were sliced into 5 μm sections and then stained with hematoxylin and eosin. Samples were examined using light microscopy. (B) Samples were given an injury score scored using a scoring system for acute lung injury in experimental animals as outlined by the American Thoracic Society. n=6 mice/group (each circle represents the tissue sample obtained from one mouse). *p<0.05 vs (-) CLP, (-) Nec-1; #p<0.05 vs (+) CLP, (-) Nec-1. Data are expressed as means ± SEM. Groups were compared by one-way ANOVA and Tukey’s multiple comparisons test. |
CIRP Interacts Transiently with MLKL
To determine the localization of p-MLKL following LPS+z-VAD stimulation, we performed immunostaining in RAW264.7 cells. We found that p-MLKL was predominantly located in the cytoplasm in the DMSO-treated cells, while in LPS+z-VAD-treated cells, p-MLKL was colocalized in the plasma membrane (Figure 5A). The CIRP-MLKL structure complex modelling showed that the residues from disordered region of CIRP interact with the 4-helix bundle (4HB) domain of MLKL (Figure 5B). The protein-protein interaction interface of CIRP-MLKL structure complex has an interaction interface surface area of 902.5 Ȧ2. The other thermodynamic parameters of the complex include a free energy of binding upon complex formation (∆iG) of −4.3 kcal/mol and a free energy of dissociation (∆Gdisso) of −7.0 kcal/mol. This suggests that the interaction between CIRP-MLKL is transient. The entropy change after dissociation is 12.6 kcal/mol. Moreover, Gln147 of CIRP forms a hydrogen bond (2.43 Ȧ) with the Tyr118 of the 4HB domain of MLKL. Other hydrogen bonds include His119 of the 4HB domain of MLKL with the Arg116 (1.95 Ȧ) and Asn68 (2.67 Ȧ) of CIRP respectively. According to one model, the RIPK3-mediated phosphorylation of the MLKL pseudo kinase domain activation loop (Ser345) can induce a conformational change and lead to unleashing of the executioner 4HB domain or the complete N-terminal domain (aa 1–125) of MLKL, its membrane translocation, and ultimately lead to cell death and release of CIRP.31 Other models suggest that the 4HB domain of MLKL might induce cell death by activation of downstream effectors such as ion channels, direct permeabilization of membranes and formation of transmembrane pores, all of which remain the subject of further analysis.
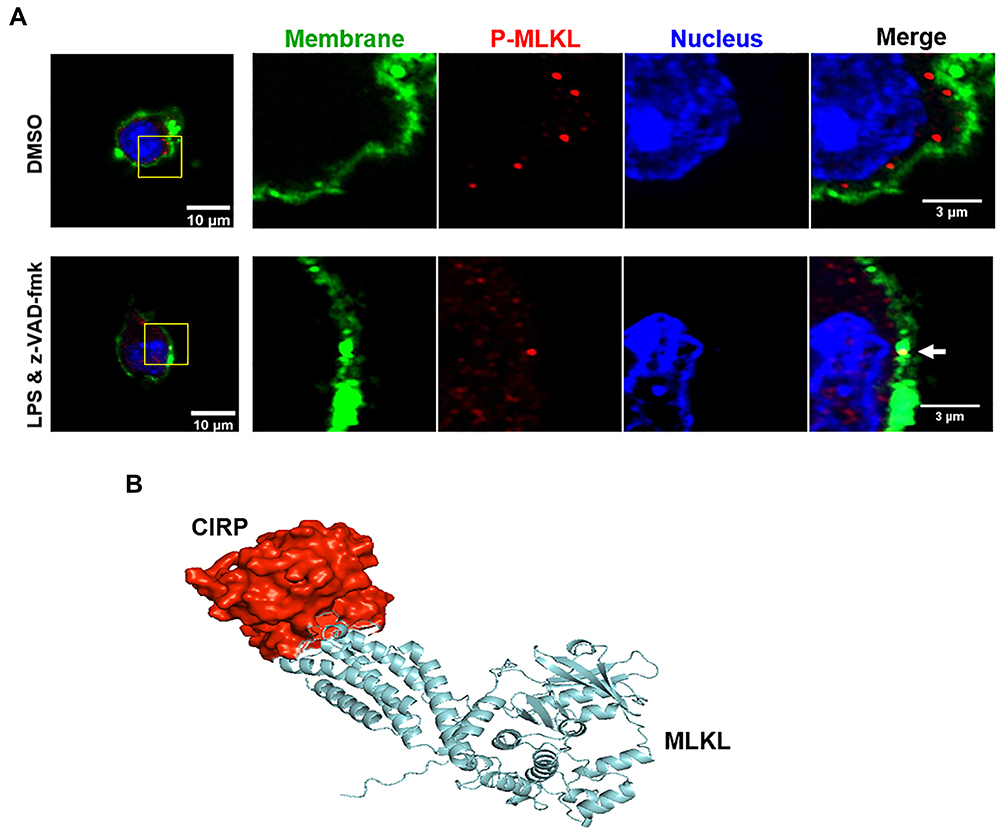 |
Figure 5 p-MLKL co-localizes in the membrane, and MLKL and CIRP interact transiently. (A) RAW264.7 cells were pre-treated with z-VAD (15µM) for 30 min and then stimulated with LPS (1 µg/mL) for 24 h. After LPS stimulation, the cells were fixed and then immunostained with cell surface membrane marker (green), p-MLKL (red), and DNA (Hoechst 3334, blue), and the images were captured by confocal microscopy. Two confocal microscopy images in green and red fluorescence channels were tested for colocalization in ImageJ software with the colocalization finder plug-in. The plug-in calculates the Pearson correlation coefficient of the two original images and displays a correlation diagram, which is highlighted in white on a composite picture (bottom right of control and LPS-treated groups) of the two original images. The duplicated merge images indicated as colocalization panels highlight the areas in white, where green and red signals colocalize, as shown in the white arrow. (B) A computational model of MLKL’s (blue) interaction with CIRP (red) by using Iterative Threading ASSEmbly Refinement (I-TASSER) server. |
Discussion
It is well established that eCIRP functions as an endogenous pro-inflammatory mediator which results in exaggerated injury in inflammatory conditions including sepsis, ischemia/reperfusion injury, and other inflammatory in vivo models.3,32 These effects are mitigated in CIRP genetic knockout animal models of diseases.3,33 eCIRP induces a pro-inflammatory response in several cell types including macrophages, neutrophils, endothelial cells, and epithelial cells.9,10,32,34 Recent studies have demonstrated that eCIRP induces inflammation by binding to TLR4 and TREM-1.3,33 Antagonists of eCIRP targeting its interaction with TLR4 and TREM-1 abrogate inflammation and tissue injury in various inflammatory diseases.3,33 While the pro-inflammatory effects of eCIRP are well established, less is known about the mechanisms of eCIRP release. CIRP lacks the signal peptide sequence necessary to be released through the endoplasmic reticulum (ER)-Golgi pathway.32 Currently, eCIRP has been confirmed to be released both passively by necrosis and actively by secretory lysosomes, exosomes, and gasdermin D (GSDMD) pores.3,32,35,36 However, other possible mechanisms for eCIRP release have yet to be fully explored. Therefore, efforts focused on resolving the mechanisms for eCIRP release are critical for finding novel potential therapies for targeting and preventing eCIRP induced inflammation.
Although programmed cell death is essential for the maintenance of tissue homeostasis and physiological processes like epithelial cell renewal and lymphocyte selection,12 excessive or uncontrolled programmed cell death results in inflammation, tissue damage and disease pathogenesis.19 Sepsis is characterized by increased DAMPs and increased apoptosis, necroptosis, secondary necrosis, pyroptosis, and ferroptosis.37 Therefore, there is a strong connection between DAMP release and these cell death events. Necroptosis is a regulated form of necrosis induced by TLRs or TNFR1 that is mediated by the proteins RIPK3 and MLKL.17,18 Necroptosis induces inflammation by the release of DAMPs as the membrane ruptures and the cell disintegrates.19 HMGB1 and IL-1 family cytokines are known as DAMPs that are released by necroptotic cells.38 Our current study indicates that necroptosis could be another key contributor to eCIRP release during inflammation.
Our data shows that eCIRP is released from macrophages through necroptosis induced by LPS+z-VAD. The levels of p-MLKL in the cell lysates confirm that the LPS and z-VAD concentrations are sufficient to stimulate these pathways as intended. Furthermore, treatment with a necroptosis inhibitor is effective at preventing LPS and z-VAD-induced eCIRP release in macrophages. CIRP could be released passively as the cell membrane becomes ruptured, as indicated by the LDH release. It could also be released via the pore on the cell membrane created by p-MLKL based on the findings of our computational model. This model indicates that CIRP binding, and dissociation are both thermodynamically spontaneous which is suggestive of a transient interaction between CIRP and MLKL. Once MLKL is inserted into the membrane, this transient interaction could serve to navigate CIRP through the channel and outside the cell. Our study reveals the necroptosis pathway as a novel route of CIRP release and provides probable and non-exclusive mechanisms for this to occur (Figure 6). Under normal conditions, limited necroptosis has an important function in maintaining tissue homeostasis.19 However, in excess, necroptosis is known to disrupt homeostasis and is suspected in the mechanism of other inflammatory diseases such as inflammatory bowel disease. One study demonstrated that in mice, intestinal epithelial cell (IEC)-specific knock out of FADD, an apoptotic regulator, induces increased numbers of necrotic in IECs which results in spontaneous colitis. RIPK3 knockout was proven to effectively mediate the pathological effects of this FADD deficiency.21 It has been shown that important regulators of necroptotic cell death, RIPK1, RIPK3 and MLKL, are associated with severe disease progression. Plasma RIPK3 levels are associated with the severity of acute respiratory distress syndrome in sepsis and trauma.39 Elevated serum levels of MLKL are an independent negative predictor of the survival of ICU patients.40 Moreover, compared to healthy individuals, COVID-19 patients consistently exhibit elevated plasma levels of RIPK1, RIPK3, and MLKL throughout their ICU stay.41 Nec-1 is a specific, small-molecule inhibitor of necroptosis and functions by allosterically inhibiting RIPK1. It has demonstrated protective capabilities in an ischemic brain injury model and from TNF-induced systemic inflammatory response syndrome.42–44 Our previous study also demonstrated that Nec-1 increases survival and decreases both systemic inflammation as well as lung injury in neonatal mice with sepsis.45 Studies have also established that Nec-1 has an anti-inflammatory role in different acute conditions such as liver failure,46 ischemia/reperfusion injury47 and endocrine diseases.48
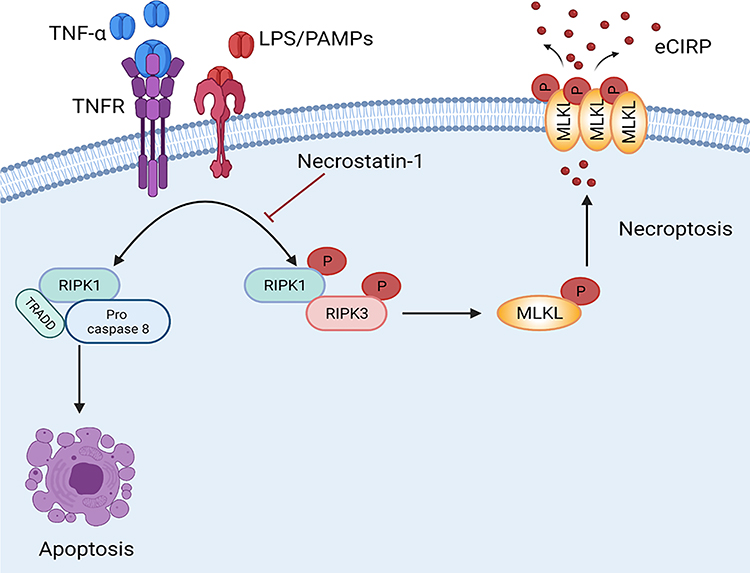 |
Figure 6 Necroptosis induces eCIRP release from macrophage in Sepsis. Bacterial sepsis or endotoxemia induces phosphorylated RIPK1 and RIPK3, respectively, facilitates the activation of MLKL via phosphorylation. Then the p-MLKL oligomerizes and is inserted into the membrane to form a pore. During inflammation, CIRP is translocated from nuclear to cytoplasm, binds to p-MLKL in the cell membrane, and released through the MLKL pores or cell rupture. Necrostatin-1, an inhibitor of cell necroptosis, attenuates eCIRP release in sepsis. Abbreviations: RIPK1/3, receptor-interacting protein kinases 1 and 3; MLKL, mixed lineage kinase domain like pseudo kinase. |
In this study, administration of Nec-1 mitigates the eCIRP release caused by CLP-induced bacterial sepsis. Nec-1 also reduces serum and peritoneal fluid levels of pro-inflammatory cytokines (ie, TNF-α and IL-6) which are known to be associated with eCIRP release. This data implicates necroptosis as a substantial source of eCIRP release in bacterial sepsis. The improvement of cytokine and lung injury suggests that the eCIRP released from necroptosis induces known downstream consequences of eCIRP. It is likely that in sepsis, the release of eCIRP is non-exclusive to macrophages as necroptosis in other cell types, such as IECs, have been shown to cause inflammation in a number of diseases.49 There is also evidence to support that necroptosis also plays a pro-inflammatory role in neutrophil-associated disorders.50 Our study highlights the potential of targeting necroptosis as a novel therapeutic avenue for treating inflammatory diseases.
Besides LPS-mediated cellular necroptosis, the TNF-TNFR1-dependent pathway plays a critical role in necroptosis. A limitation of our study is that we did not directly study the TNF-mediated eCIRP release through necroptosis. However, LPS can indirectly induce TNF release, which may subsequently aid in eCIRP release via necroptosis. Thus, our results of LPS-mediated eCIRP release through necroptosis direct future studies to using other stimulants of necroptosis to replicate our findings. Another limitation of our study is that we did not evaluate the possible interactions between anti-apoptotic proteins, apoptosis and/or necroptosis mediators in sepsis. In order to further pursue the therapeutic potential of necroptosis inhibition, future studies will be necessary to evaluate the interplay between these three pathways during sepsis. Activation of TNFR1 results in the induction of two diverging pathways: caspase-dependent apoptosis and RIPK-mediated necroptosis.51 Growing evidence suggests that the balance between apoptotic and anti-apoptotic pathways is important in determining outcomes for septic patients. A previous study has correlated increased pro-apoptotic-caspase-3/9 and anti-apoptotic survivin expression with higher instances of mortality in septic patients.52 Another study demonstrated an increase in oxidative stress along with the upregulation of these proteins in septic patients compared to both non-septic trauma patients and healthy individuals. It was also found that these factors are related to anti-apoptotic, inflammatory, and innate immunity alterations in sepsis.53 Therefore, the relationship between these three signaling pathways and the subsequent release of eCIRP and eCIRP-induced inflammation, as well as the extent that modulating necroptosis will have on these pathways/inflammatory phenomena remain areas for future investigation.
The two main pathways of apoptosis are the extrinsic pathway, which is initiated by death ligands such as FASL, TNF, TRAIL, and the intrinsic pathway, which involves the release of Bax/Bcl2 proteins from mitochondria upon oxidative stress.54,55 Each pathway activates its initiator caspases, such as caspase-8, −9, and −10, which in turn activate the executioner caspase-3. The execution pathway results in characteristic cytomorphological features including cell shrinkage, chromatin condensation, formation of cytoplasmic blebs and apoptotic bodies. Z-VAD is a pan-caspase inhibitor that irreversibly binds to the catalytic site of several caspase proteases and is a widely used inhibitor of apoptosis.56 Apoptosis from both intrinsic and extrinsic pathways is blocked by z-VAD as the activation of caspase-3 is required in both mechanisms.57 However, despite z-VAD being widely used as an apoptosis inhibitor, a possible drawback to z-VAD is that it requires much higher doses to be effective relative to other apoptotic inhibitors. Additionally, z-VAD-fmk has no pharmacological potential as, in vivo, it is metabolized into of fluoroacetate, which is toxic to the cells.58 In future studies, Q-VD-OPh (quinolyl-valyl-O-methylaspartyl-[−2,6-difluorophenoxy]-methyl ketone), a different pan-caspase inhibitor could be used to further validate our findings of necroptosis-mediated eCIRP release during sepsis as it is effective at significantly lower doses and is nontoxic in vivo.58
In conclusion, our data implicates necroptosis as a method of eCIRP release both in vitro and in vivo. We also provide evidence suggesting eCIRP can be released not only though cell membrane rupture but also through MLKL pores prior to the disintegration of the membrane. This study reveals necroptosis as a potential target for pharmacologically controlling the negative consequences of eCIRP in sepsis.
Acknowledgments
This study was supported by the National Institutes of Health (NIH) grants R35GM118337 (P.W.), U01AI133655 (P.W.) and R01GM129633 (M.A.). We thank Yongchan Lee of the Center for Immunology and Inflammation of the Feinstein Institutes for Medical research for technical assistance.
Author Contributions
All authors made a significant contribution to the work reported, whether that is in the conception, study design, execution, acquisition of data, analysis and interpretation, or in all these areas; took part in drafting, revising or critically reviewing the article; gave final approval of the version to be published; have agreed on the journal to which the article has been submitted; and agree to be accountable for all aspects of the work. Equally contributed first authors: Bridgette Reilly and Chuyi Tan. These senior authors contributed to this work equally: Monowar Aziz and Ping Wang.
Disclosure
The authors declared that they have no conflicts of interest in relation to this work.
References
1. Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81(1):1–5. doi:10.1189/jlb.0306164
2. Denning NL, Aziz M, Gurien SD, Wang P. DAMPs and NETs in sepsis. Front Immunol. 2019;10:2536. doi:10.3389/fimmu.2019.02536
3. Qiang X, Yang WL, Wu R, et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat Med. 2013;19(11):1489–1495. doi:10.1038/nm.3368
4. Nishiyama H, Itoh K, Kaneko Y, et al. A glycine-rich RNA-binding protein mediating cold-inducible suppression of mammalian cell growth. J Cell Biol. 1997;137(4):899–908. doi:10.1083/jcb.137.4.899
5. Yang WL, Sharma A, Wang Z, et al. Cold-inducible RNA-binding protein causes endothelial dysfunction via activation of Nlrp3 inflammasome. Sci Rep. 2016;6:26571. doi:10.1038/srep26571
6. Denning NL, Aziz M, Ochani M, Prince JM, Wang P. Inhibition of a triggering receptor expressed on myeloid cells-1 (TREM-1) with an extracellular cold-inducible RNA-binding protein (eCIRP)-derived peptide protects mice from intestinal ischemia-reperfusion injury. Surgery. 2020;168(3):478–485. doi:10.1016/j.surg.2020.04.010
7. Jacob A, Ma Y, Nasiri E, et al. Extracellular cold inducible RNA-binding protein mediates binge alcohol-induced brain hypoactivity and impaired cognition in mice. Mol Med. 2019;25(1):24. doi:10.1186/s10020-019-0092-3
8. McGinn J, Zhang F, Aziz M, et al. The protective effect of a short peptide derived from cold-inducible RNA-binding protein in renal ischemia-reperfusion injury. Shock. 2018;49(3):269–276. doi:10.1097/shk.0000000000000988
9. Zhou M, Aziz M, Denning NL, et al. Extracellular CIRP induces macrophage endotoxin tolerance through IL-6R-mediated STAT3 activation. JCI Insight. 2020;5(5). doi:10.1172/jci.insight.133715
10. Ode Y, Aziz M, Wang P. CIRP increases ICAM-1(+) phenotype of neutrophils exhibiting elevated iNOS and NETs in sepsis. J Leukoc Biol. 2018;103(4):693–707. doi:10.1002/jlb.3a0817-327rr
11. Royster W, Jin H, Wang P, Aziz M. Extracellular CIRP decreases Siglec-G expression on B-1a cells skewing them towards a pro-inflammatory phenotype in sepsis. Mol Med. 2021;27(1):55. doi:10.1186/s10020-021-00318-y
12. Wallach D, Kang TB, Kovalenko A. Concepts of tissue injury and cell death in inflammation: a historical perspective. Nat Rev Immunol. 2014;14(1):51–59. doi:10.1038/nri3561
13. Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370(5):455–465. doi:10.1056/NEJMra1310050
14. Galluzzi L, Vitale I, Abrams JM, et al. Molecular definitions of cell death subroutines: recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012;19(1):107–120. doi:10.1038/cdd.2011.96
15. Cho YS, Challa S, Moquin D, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137(6):1112–1123. doi:10.1016/j.cell.2009.05.037
16. Weil JV, Byrne-Quinn E, Battock DJ, Grover RF, Chidsey CA. Forearm circulation in man at high altitude. Clin Sci. 1971;40(3):235–246. doi:10.1042/cs0400235
17. Zhang DW, Shao J, Lin J, et al. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325(5938):332–336. doi:10.1126/science.1172308
18. Zhao J, Jitkaew S, Cai Z, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA. 2012;109(14):5322–5327. doi:10.1073/pnas.1200012109
19. Pasparakis M, Vandenabeele P. Necroptosis and its role in inflammation. Nature. 2015;517(7534):311–320. doi:10.1038/nature14191
20. Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity. 2013;38(2):209–223. doi:10.1016/j.immuni.2013.02.003
21. Welz PS, Wullaert A, Vlantis K, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477(7364):330–334. doi:10.1038/nature10273
22. Rickard JA, O’Donnell JA, Evans JM, et al. RIPK1 regulates RIPK3-MLKL-driven systemic inflammation and emergency hematopoiesis. Cell. 2014;157(5):1175–1188. doi:10.1016/j.cell.2014.04.019
23. Hubbard WJ, Choudhry M, Schwacha MG, et al. Cecal ligation and puncture. Shock. 2005;24(Suppl 1):52–57. doi:10.1097/01.shk.0000191414.94461.7e
24. Matute-Bello G, Downey G, Moore BB, et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44(5):725–738. doi:10.1165/rcmb.2009-0210ST
25. Yang J, Yan R, Roy A, et al. The I-TASSER suite: protein structure and function prediction. Nat Methods. 2015;12(1):7–8. doi:10.1038/nmeth.3213
26. Huang D, Zheng X, Wang ZA, et al. The MLKL channel in necroptosis is an octamer formed by tetramers in a dyadic process. Mol Cell Biol. 2017;37(5). doi:10.1128/mcb.00497-16
27. Bansal N, Sciabola S, Bhisetti G. Understanding allosteric interactions in hMLKL protein that modulate necroptosis and its inhibition. Sci Rep. 2019;9(1):16853. doi:10.1038/s41598-019-53078-5
28. Schindler CE, de Vries SJ, Zacharias M. iATTRACT: simultaneous global and local interface optimization for protein-protein docking refinement. Proteins. 2015;83(2):248–258. doi:10.1002/prot.24728
29. Krissinel E, Henrick K. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007;372(3):774–797. doi:10.1016/j.jmb.2007.05.022
30. Sanner MF, Olson AJ, Spehner JC. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers. 1996;38(3):305–320.
31. Tanzer MC, Matti I, Hildebrand JM, et al. Evolutionary divergence of the necroptosis effector MLKL. Cell Death Differ. 2016;23(7):1185–1197. doi:10.1038/cdd.2015.169
32. Aziz M, Brenner M, Wang P. Extracellular CIRP (eCIRP) and inflammation. J Leukoc Biol. 2019;106(1):133–146. doi:10.1002/jlb.3mir1118-443r
33. Denning NL, Aziz M, Murao A, et al. Extracellular CIRP as an endogenous TREM-1 ligand to fuel inflammation in sepsis. JCI Insight. 2020;5(5). doi:10.1172/jci.insight.134172
34. Tan C, Gurien SD, Royster W, Aziz M, Wang P. Extracellular CIRP induces inflammation in alveolar type II cells via TREM-1. Front Cell Dev Biol. 2020;8:579157. doi:10.3389/fcell.2020.579157
35. Murao A, Tan C, Jha A, Wang P, Aziz M. Exosome-mediated eCIRP release from macrophages to induce inflammation in sepsis. Front Pharmacol. 2021;12:791648. doi:10.3389/fphar.2021.791648
36. Tan C, Reilly B, Jha A, et al. Active release of eCIRP via gasdermin D channels to induce inflammation in sepsis. J Immunol. 2022;208(9):2184–2195. doi:10.4049/jimmunol.2101004
37. Murao A, Aziz M, Wang H, Brenner M, Wang P. Release mechanisms of major DAMPs. Apoptosis. 2021;26(3–4):152–162. doi:10.1007/s10495-021-01663-3
38. Simpson J, Loh Z, Ullah MA, et al. Respiratory syncytial virus infection promotes necroptosis and hmgb1 release by airway epithelial cells. Am J Respir Crit Care Med. 2020;201(11):1358–1371. doi:10.1164/rccm.201906-1149OC
39. Shashaty MGS, Reilly JP, Faust HE, et al. Plasma receptor interacting protein kinase-3 levels are associated with acute respiratory distress syndrome in sepsis and trauma: a cohort study. Crit Care. 2019;23(1):235. doi:10.1186/s13054-019-2482-x
40. Vucur M, Roderburg C, Kaiser L, et al. Elevated serum levels of mixed lineage kinase domain-like protein predict survival of patients during intensive care unit treatment. Dis Markers. 2018;2018:1983421. doi:10.1155/2018/1983421
41. Ruskowski K, Neb H, Talbot SR, et al. Persistently elevated plasma levels of RIPK3, MLKL, HMGB1, and RIPK1 in COVID-19 ICU patients. Am J Respir Cell Mol Biol. 2022. doi:10.1165/rcmb.2022-0039LE
42. Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1(2):112–119. doi:10.1038/nchembio711
43. Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–321. doi:10.1038/nchembio.83
44. Duprez L, Takahashi N, Van Hauwermeiren F, et al. RIP kinase-dependent necrosis drives lethal systemic inflammatory response syndrome. Immunity. 2011;35(6):908–918. doi:10.1016/j.immuni.2011.09.020
45. Bolognese AC, Yang WL, Hansen LW, et al. Inhibition of necroptosis attenuates lung injury and improves survival in neonatal sepsis. Surgery. 2018;164(1):110–116. doi:10.1016/j.surg.2018.02.017
46. Kondo T, Macdonald S, Engelmann C, et al. The role of RIPK1 mediated cell death in acute on chronic liver failure. Cell Death Dis. 2021;13(1):5. doi:10.1038/s41419-021-04442-9
47. Liao S, Apaijai N, Luo Y, et al. Cell death inhibitors protect against brain damage caused by cardiac ischemia/reperfusion injury. Cell Death Discov. 2021;7(1):312. doi:10.1038/s41420-021-00698-4
48. Tonnus W, Belavgeni A, Beuschlein F, et al. The role of regulated necrosis in endocrine diseases. Nat Rev Endocrinol. 2021;17(8):497–510. doi:10.1038/s41574-021-00499-w
49. Wang X, Yousefi S, Simon HU. Necroptosis and neutrophil-associated disorders. Cell Death Dis. 2018;9(2):111. doi:10.1038/s41419-017-0058-8
50. Wang X, He Z, Liu H, Yousefi S, Simon HU. Neutrophil necroptosis is triggered by ligation of adhesion molecules following GM-CSF priming. J Immunol. 2016;197(10):4090–4100. doi:10.4049/jimmunol.1600051
51. Ting AT, Bertrand MJM. More to life than NF-κB in TNFR1 signaling. Trends Immunol. 2016;37(8):535–545. doi:10.1016/j.it.2016.06.002
52. Miliaraki M, Briassoulis P, Ilia S, et al. Survivin and caspases serum protein levels and survivin variants mRNA expression in sepsis. Sci Rep. 2021;11(1):1049. doi:10.1038/s41598-020-78208-2
53. Miliaraki M, Briassoulis P, Ilia S, et al. Oxidant/antioxidant status is impaired in sepsis and is related to anti-apoptotic, inflammatory, and innate immunity alterations. Antioxidants. 2022;11(2):231. doi:10.3390/antiox11020231
54. Aziz M, Jacob A, Wang P. Revisiting caspases in sepsis. Cell Death Dis. 2014;5(11):e1526. doi:10.1038/cddis.2014.488
55. Peter ME. Programmed cell death: Apoptosis meets necrosis. Nature. 2011;471(7338):310–312. doi:10.1038/471310a
56. Inoue S, Browne G, Melino G, Cohen GM. Ordering of caspases in cells undergoing apoptosis by the intrinsic pathway. Cell Death Differ. 2009;16(7):1053–1061. doi:10.1038/cdd.2009.29
57. Scoltock AB, Cidlowski JA. Activation of intrinsic and extrinsic pathways in apoptotic signaling during UV-C-induced death of Jurkat cells: the role of caspase inhibition. Exp Cell Res. 2004;297(1):212–223. doi:10.1016/j.yexcr.2004.03.025
58. Keoni CL, Brown TL. Inhibition of apoptosis and efficacy of pan caspase inhibitor, Q-VD-OPh, in models of human disease. J Cell Death. 2015;8:1–7. doi:10.4137/jcd.s23844
 © 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
© 2022 The Author(s). This work is published and licensed by Dove Medical Press Limited. The
full terms of this license are available at https://www.dovepress.com/terms
and incorporate the Creative Commons Attribution
- Non Commercial (unported, 3.0) License.
By accessing the work you hereby accept the Terms. Non-commercial uses of the work are permitted
without any further permission from Dove Medical Press Limited, provided the work is properly
attributed. For permission for commercial use of this work, please see paragraphs 4.2 and 5 of our Terms.
Recommended articles
C-Terminal Fibronectin Exerts Beneficial Effects in Reducing Tissue Damage and Modulating Macrophage Function in a Murine Septic Model
Geng H, Wu Y, Chen Y
Journal of Inflammation Research 2023, 16:1509-1521
Published Date: 10 April 2023
Erbin Regulates Tissue Factors Through Ras/Raf Pathway in Coagulation Disorders in Sepsis
Yang C, Lei C, Jing G, Xia Y, Zhou H, Wu D, Zuo J, Gong H, Wang X, Dong Y, Aidebaike D, Wu X, Song X
Journal of Inflammation Research 2025, 18:1739-1754
Published Date: 5 February 2025
Cell Death Index Predicts Sepsis Outcomes and Highlights Necroptosis as a Therapeutic Target
Gan J, Long Q, Fang S, Song S, Wu J, Li X, Ye H, Gao Z, Zheng Y
Journal of Inflammation Research 2026, 19:577930
Published Date: 19 April 2026